
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 13 January 2023
Sec. Neurocritical and Neurohospitalist Care
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.1066724
 Felix Neumaier1,2,3
Felix Neumaier1,2,3 Christian Stoppe4,5,6
Christian Stoppe4,5,6 Anzhela Stoykova1
Anzhela Stoykova1 Miriam Weiss1,7
Miriam Weiss1,7 Michael Veldeman1
Michael Veldeman1 Anke Höllig1
Anke Höllig1 Hussam Aldin Hamou1Yasin Temel8
Hussam Aldin Hamou1Yasin Temel8 Catharina Conzen1
Catharina Conzen1 Tobias Philip Schmidt1Rabia Dogan1
Tobias Philip Schmidt1Rabia Dogan1 Martin Wiesmann9
Martin Wiesmann9 Hans Clusmann1
Hans Clusmann1 Gerrit Alexander Schubert1,7
Gerrit Alexander Schubert1,7 Roel Hubert Louis Haeren8
Roel Hubert Louis Haeren8 Walid Albanna1*
Walid Albanna1*Objective: Inflammation is increasingly recognized to be involved in the pathophysiology of aneurysmal subarachnoid hemorrhage (aSAH) and may increase the susceptibility to delayed cerebral ischemia (DCI). Macrophage migration inhibitory factor (MIF) has been shown to be elevated in serum and cerebrospinal fluid (CSF) after aSAH. Here, we determined MIF levels in serum, CSF and cerebral microdialysate (MD) at different time-points after aSAH and evaluated their clinical implications.
Methods: MIF levels were measured in serum, CSF and MD obtained from 30 aSAH patients during early (EPd1−4), critical (CPd5−15) and late (LPd16−21) phase after hemorrhage. For subgroup analyses, patients were stratified based on demographic and clinical data.
Results: MIF levels in serum increased during CPd5−15 and decreased again during LPd16−21, while CSF levels showed little changes over time. MD levels peaked during EPd1−4, decreased during CPd5−15 and increased again during LPd16−21. Subgroup analyses revealed significantly higher serum levels in patients with aneurysms located in the anterior vs. posterior circulation during CPd5−15 (17.3 [15.1–21.1] vs. 10.0 [8.4–11.5] ng/ml, p = 0.009) and in patients with DCI vs. no DCI during CPd5−15 (17.9 [15.1–22.7] vs. 11.9 [8.9–15.9] ng/ml, p = 0.026) and LPd16−21 (17.4 [11.7–27.9] vs. 11.3 [9.2–12.2] ng/ml, p = 0.021). In addition, MIF levels in MD during CPd5−15 were significantly higher in patients with DCI vs. no DCI (3.6 [1.8–10.7] vs. 0.2 [0.1–0.7] ng/ml, p = 0.026), while CSF levels during the whole observation period were similar in all subgroups.
Conclusion: Our findings in a small cohort of aSAH patients provide preliminary data on systemic, global cerebral and local cerebral MIF levels after aSAH and their clinical implications.
Clinical trial registration: ClinicalTrials.gov, identifier: NCT02142166.
Despite aggressive multidisciplinary treatment, aneurysmal subarachnoid hemorrhage (aSAH) is still associated with a high overall morbidity and mortality (1). In addition to early brain injury (EBI), a significant number of aSAH patients develop delayed cerebral ischemia (DCI); a complication that can result in secondary brain injury (2). Although long attributed to angiographic vasospasm, DCI is now recognized to reflect a cascade of events that also comprises microvascular dysfunction due to inflammation, oxidative stress and other factors (3–5). Timely treatment of DCI is essential to prevent long-term impairments but its detection remains a diagnostic challenge, especially in unconscious patients. For example, because DCI also occurs in the absence of angiographic vasospasm (6), measurements utilizing near-infrared spectroscopy (7, 8) or transcranial Doppler (9, 10) often lack sufficient specificity for its faithful detection.
Given the emerging role of inflammation in the pathophysiology of aSAH, inflammatory cytokines are increasingly recognized as potential predictive and/or diagnostic biomarkers for DCI in poor grade aSAH patients (11). Macrophage migration inhibitory factor (MIF) is a cytokine expressed by a broad range of cell types (including immune cells, endocrine cells, epithelial and endothelial cells, as well as neurons and glial cells) and secreted in response to pathophysiological stimuli such as hypoxia, surgical stress or infections. MIF is an upstream regulator of the immune response and key mediator of local and systemic inflammatory responses (12, 13) that has been implicated in numerous immune-mediated pathologies (14–16). Previous studies indicate that elevated levels of MIF in serum and cerebrospinal fluid (CSF) may be associated with DCI and poor functional outcomes after aSAH (17–19). However, no previous studies have examined the relationship between systemic (i.e., serum) and cerebral (i.e., CSF) MIF concentrations in the same group of patients, which could provide insight into the exact origin of MIF released in response to aSAH. In addition, there is still a lack of data on local MIF concentrations in the brain and their potential association with DCI and functional impairments after aSAH. Therefore, we evaluated MIF levels in serum, CSF and cerebral microdialysate (MD) in patients after aSAH and evaluated their clinical significance.
The study protocol was approved by the local ethics committee (EK062/14), and written consent for study inclusion was obtained from all patients or their legal representatives. This cohort observational study has been registered at ClinicalTrials.gov (NCT02142166) as part of a larger scale prospective data collection.
The present study is based on 30 aSAH patients treated at our facility between 12/2014 and 06/2017 who met the following inclusion criteria: (1) age>18 years, (2) availability of an external ventricular drainage for CSF collection and/or cerebral catheters for MD collection (see below), and (3) no foreseeable early mortality due to brain stem injury. Demographic data obtained on admission or during in-hospital treatment included sex, age, body mass index (BMI), blood pressure, smoking status, aneurysm location, treatment modality, clinical condition on admission according to the Hunt & Hess (HH) grading scale, radiological severity on admission according to the modified Fisher Scale (mFS) and clinical outcome after 12 months according to the extended Glasgow Outcome Scale (GOSE). Clinical outcome was assessed by an independent physician based on clinical evaluation in the outpatient clinic or the clinical status compiled from medical reports.
After aneurysms were secured by surgical clipping or endovascular treatment, prophylactic enteral nimodipine (6 × 60 mg) was applied as tolerated (20), and the patients were monitored for the following signs of DCI: In awake patients, DCI was diagnosed based on the criteria for clinical deterioration defined by Vergouwen (2), which comprise appearance of new focal neurological deficits or a decrease in Glasgow Coma Scale by ≥2 points for ≥1 h. In unconscious patients, DCI was defined as cerebral metabolic (lactate/pyruvate ratio ≥ 40) or oxygenation (ptiO2< 10 mmHg) crisis or appearance of new CT perfusion deficits if other causes could be ruled out. To this end, invasive neuromonitoring was performed according to recent consensus recommendations (7) by measurement of brain tissue oxygenation with Neurovent PTO catheters (Raumedic AG, Helmbrechts, Germany) and cerebral metabolism with 71 high cut-off microdialysis catheters (μdialysis, Stockholm, Sweden), which were perfused at 0.3 μl/min with standard perfusion fluid. Brain oxygenation probes and microdialysis catheters were advanced about 3–4 cm subcortically via a small craniostomy and after dural perforation, so that they terminated within the vascular territory of the aneurysm, and were held in place by a bolt affixed within the craniostomy (21). Samples were collected at least every 3 h and analyzed as described previously (21).
The first line of treatment for DCI involved induction of euvolemic hypertension by intravenous noradrenaline infusion to raise systolic arterial blood pressure to ≥180 mmHg. When there was no improvement, relevant hypoperfusion and vasoconstriction were verified by cerebral angiography and/or perfusion CT and endovascular rescue treatment was performed as described previously (21, 22). Cerebral infarctions during ongoing DCI or diagnosed as first sign of DCI and confirmed by at least two imaging modalities were considered DCI-related infarctions.
Serum (7 ml) and CSF (1 ml, obtained from the proximal access point of a ventricular drain) were collected from every patient between days 1–21 (day 1–3: once; day 4–21: every third day). Whenever possible, MD (30–120 μl) was collected every 6 hours following a stabilization period of 24 h after catheter insertion. Vials containing MD samples were retained in designated storage racks. Blood samples were immediately centrifuged and serum and CSF were pipetted into suitable polypropylene cryotubes (VWR International, Darmstadt, Germany). All samples were permanently deep-frozen at −80°C, protected from light, and quantitatively analyzed for MIF levels at a “Good Laboratory Practice” (GLP) certified laboratory using a customized U-PLEX® Biomarker Group 1 Multiplex Assay from MSD (Meso Scale Discovery®, Cat-No: K15067L-2).
Based on the typical time-course for occurrence of EBI and DCI, we defined three time windows in reference to the initial insult on day 0 for data danalysis, which comprised an early phase (days 1–4: EPd1−4) for reflection of initial injury and aSAH severity, a critical phase (days 5–15: CPd5−15) characterized by high susceptibility to DCI, and a late phase (days 16–21: LPd16−21) corresponding to the end of ICU and/or DCI treatment. If more than one measurement was available for a patient in a given period, all measurements in this period were used to calculate a mean value. In some cases, the critical phase was further subdivided into an early critical (CPd5−8), intermediate critical (CPd9−12) and late critical (CPd13−15) phase to examine the time-course of changes during days 5–15.
For subgroup analysis, patients were stratified into groups according to sex, age (<59 years vs. ≥59 years based on the median), smoking status (smoker vs. non-smoker), BMI (BMI<26 vs. BMI≥26 based on the median), arterial hypertension (hypertension vs. no hypertension), systemic infection diagnosed based on clinical signs of sepsis (infection vs. no infection), clinical condition (good grade = HH1−3 vs. poor grade = HH4−5) and radiological classification on admission (good grade = mFS1−2 vs. poor grade = mFS3−4), treatment modality (endovascular treatment vs. surgical clipping), aneurysm location (anterior vs. posterior circulation), occurrence of DCI (DCI vs. no DCI) or DCI-related infarction (infarction vs. no infarction), and clinical outcome after 12 months (favorable = GOSE5−8 vs. unfavorable = GOSE1−4).
After testing for normality via the Shapiro-Wilk test, continuous parametric data were compared using T-tests, while the Mann-Whitney U-test or Wilcoxon signed-rank test were used for non-parametric data, as applicable. Nominal data were tested between groups using the Fisher's exact test or Chi2 test. For categorical variables, data are given as numbers and percentages. Parametric values are expressed as mean ± standard deviation (SD), whereas non-parametric values are expressed in terms of median values [25th−75th percentile]. Boxplots show median, 25th and 75th percentile (box), minimum and maximum (whiskers) and individual data points (dots). Because MD levels of MIF often ranged from very small (<0.1 ng/ml) to very large (>37 ng/ml) values, a log scale was used in some of the corresponding figures to better visualize all of the data points shown. Correlation analysis was performed using the Spearman's rank correlation coefficient (rs) as a non-parametric measure of rank correlation. Statistical significance was set at p<0.05 and statistical results with p ≤ 0.1 were considered as trend. All analyses were performed using IBM® SPSS® Statistics V22.0 (IBM, Armonk, NY, USA), Microsoft Excel 2010 (Microsoft, Redmond, USA) or Minitab 17.1.0 (Minitab Inc., Pennsylvania, USA).
A total of 30 aSAH patients with a median age of 59 [50–64] years and a female:male ratio of 22 (73%) to 8 (27%) were included in the present study. A summary of their demographic data is provided in Table 1. Seventeen (57%) of the patients developed DCI, which occurred between day 3 and 10 (median: day 7 [5-9]) and progressed to infarction in six (35%) of these patients. Clinical outcome after 12 months was favorable (GOSE5−8) in 16 (53.3%) and unfavorable (GOSE1−4) in 10 (33.3%) patients, while no follow-up data were available for the remaining four patients (13.3%). Clinical outcome was independent of sex (p = 1.000), age (p = 0.756), BMI (p = 0.248), arterial hypertension (p = 0.701), smoking status (p = 0.340), systemic infection (p = 0.248), treatment modality (p = 0.234), clinical (p = 0.339) or radiological (p = 0.190) status on admission according to HH or mFS, respectively, development of DCI (p = 0.248) or DCI-related infarctions (p = 0.163) and ventilation time (p = 0.516). Anterior circulation aneurysms were more frequent (70%) and tended to be associated with unfavorable clinical outcomes (p = 0.087).
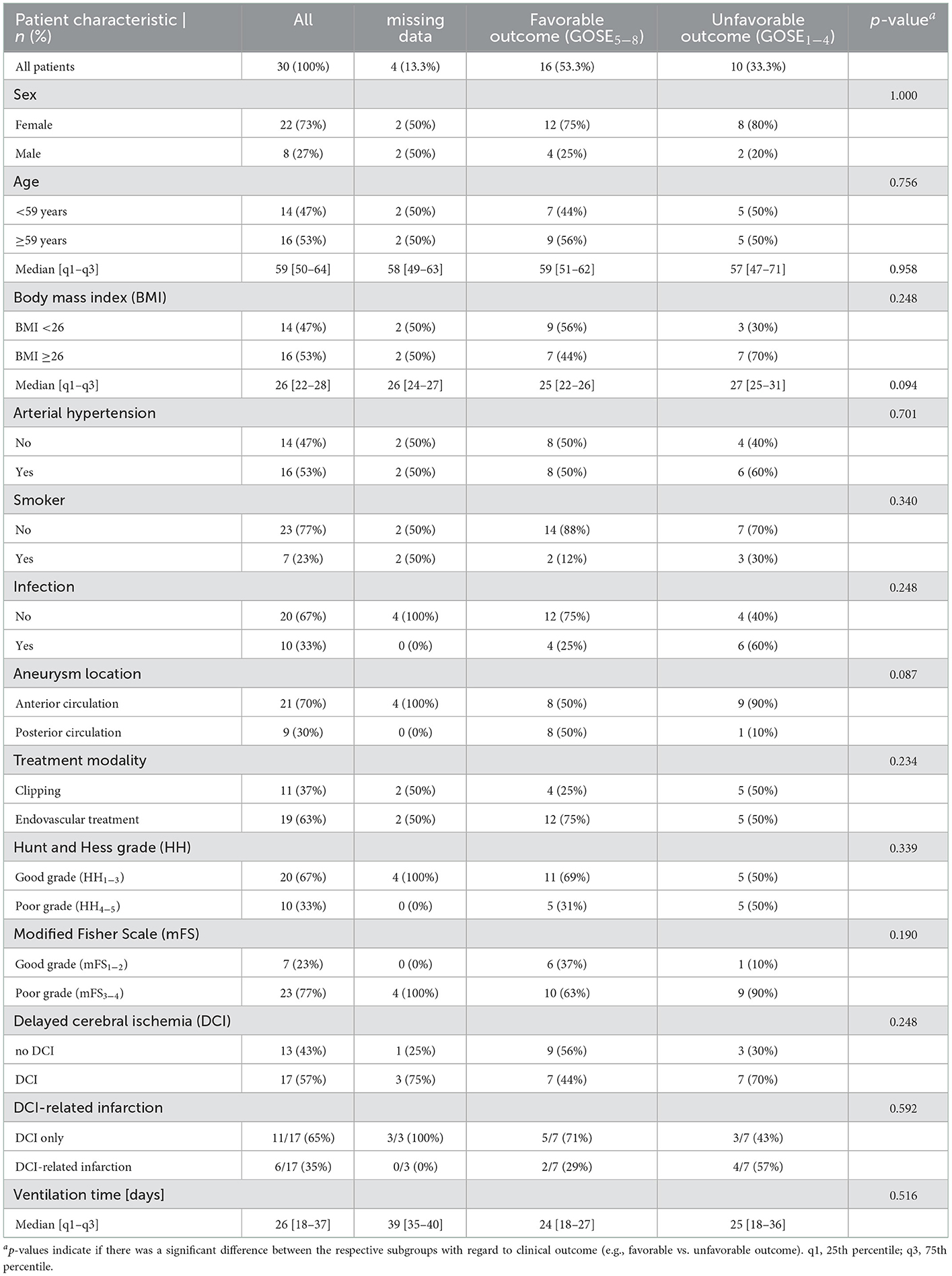
Table 1. Patient characteristics and their relation to clinical outcome after 12 months.
A comparison of MIF levels determined in serum, CSF and MD during the different phases after hemorrhage is provided in Figure 1A. Median serum levels increased from 12.2 [8.7–16.4] ng/ml during EPd1−4 to 15.5 [11.9–19.8] ng/ml during CPd5−15 (p = 0.281), and decreased again to 12.8 [10.1–21.4] ng/ml during LPd16−21 (p = 0.078). The levels in CSF were roughly two-fold higher and showed little changes over time, with median levels of 31.6 [27.1–33.7] ng/ml during EPd1−4, 30.6 [22.1–33.6] ng/ml during CPd5−15 (p = 0.917) and 30.0 [29.0–31.8] ng/ml during LPd16−21 (p = 0.575). Finally, MD levels of the cytokine during EPd1−4 (7.2 [0.2–15.1] ng/ml) were almost five times lower than CSF levels, and further decreased to 2.5 [0.3–5.7] ng/ml during CPd5−15 (p = 0.091), but increased again to 4.2 [1.3–8.1] ng/ml during LPd16−21 (p = 0.180). None of the changes over time in any of the compartments reached statistical significance. However, correlation analysis revealed a significant positive correlation between MIF levels in serum and CSF during EPd1−4 (Spearman rs=0.514, p = 0.012, Figure 1B), while no significant correlations were detected between early serum and MD (rs = 0.429, p = 0.397) or CSF and MD levels (rs = 0.400, p = 0.600). In addition, no significant correlations between MIF levels in different compartments were detected during CPd5−15 or LPd16−21. Further subdivision of CPd5−15 into three time-intervals (Figure 1C) showed little change in CSF MIF levels over time, while the values in serum and MD showed a progressive increase and decrease, respectively, during the first 15 days.
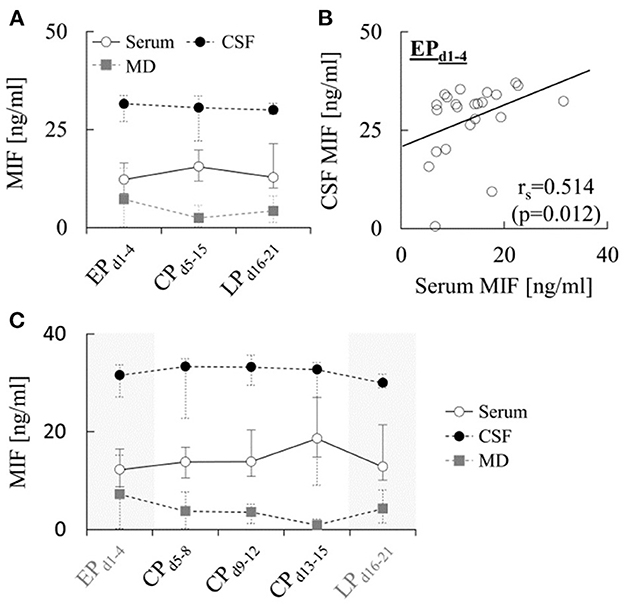
Figure 1. Concentration of MIF in different compartments after aSAH. (A) Concentration of macrophage migration inhibitory factor (MIF) measured in the indicated compartments during the early (EPd1−4), critical (CPd5−15) and late (LPd16−21) phase after aneurysmal subarachnoid hemorrhage (aSAH). In total, at least one sample for quantification of MIF levels during EPd1−4, CPd5−15 and LPd16−21 was available from 26, 19 and 26 (serum), 23, 15 and 11 (CSF) and 9, 20 and 8 (MD) patients, respectively (see Supplementary Tables 1–3). Data are shown as median [25th−75th percentile]. Statistical significance of changes over time was assessed using a repeated-measures ANOVA. (B) Correlation between MIF concentrations in CSF and serum during the early phase. Correlation analysis was performed using the Spearman's rank correlation coefficient. (C) Same data as in A, but with measurements obtained between days 5 and 15 subdivided into three sub-intervals to illustrate temporal changes during the critical phase. Note that the number of patients for which samples in these sub-intervals were available was accordingly lower (n = 10–12, 7–10, and 10–16 per sub-interval for serum, CSF and MD, respectively) than the numbers for CPd5−15 given in A and Supplementary Tables 1–3.
When comparing the different subgroups, a prominent difference in serum MIF levels was observed for patients stratified according to aneurysm location. In particular, during CPd5−15, patients with anterior circulation (AC) aneurysms exhibited significantly (p = 0.009) higher serum levels (17.3 [15.1–21.1] ng/ml) than patients with posterior circulation (PC) aneurysms (10.0 [8.4–11.5] ng/ml) (Figure 2), while CSF and MD levels were similar in both subgroups (Supplementary Tables 2, 3).
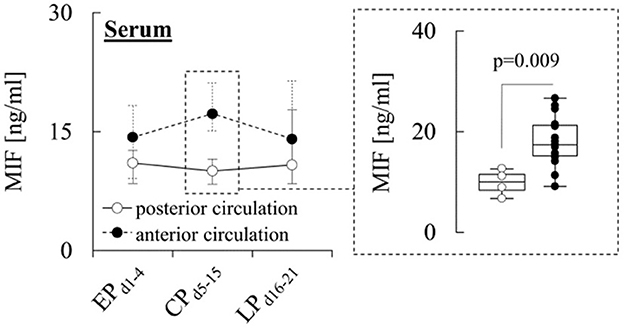
Figure 2. Serum MIF levels and aneurysm location. Concentration of macrophage migration inhibitory factor (MIF) in serum from patients with aneurysms located in the anterior or posterior circulation during the early (EPd1−4), critical (CPd5−15) and late (LPd16−21) phase after aneurysmal subarachnoid hemorrhage (aSAH). Data are shown as median [25th−75th percentile] and (for significant differences) as boxplots and individual values (see inset). Statistical significance was assessed using the Mann-Whitney U-test. For details on the exact number of patients per time-point see Supplementary Table 1.
In Figure 3, MIF levels in patients with and without DCI are illustrated. Serum levels of the cytokine were similar in both subgroups during EPd1−4, but significantly higher in patients with DCI during CPd5−15 (17.9 [15.1–22.7] ng/ml vs. 11.9 [8.9–15.9] ng/ml in patients without DCI, p = 0.026) and LPd16−21 (17.4 [11.7–27.9] ng/ml vs. 11.3 [9.2–12.2] ng/ml in patients without DCI, p = 0.021) (Figure 3A). In addition, MIF levels in MD samples from patients with DCI were significantly higher during CPd5−15 (3.6 [1.8–10.7] ng/ml vs. 0.2 [0.1–0.7] ng/ml in patients without DCI, p = 0.021) (Figure 3C), while there were no differences in CSF levels between the two subgroups (Figure 3B). There were also no differences between MIF levels in any of the compartments when patients with DCI were further stratified into patients with DCI only and patients with DCI-related infarction (Supplementary Tables 1–3).
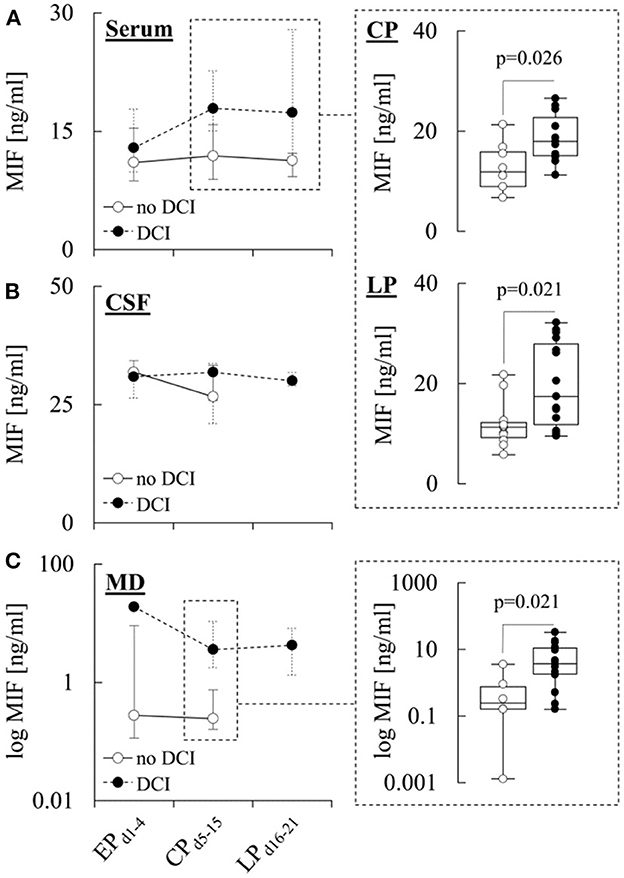
Figure 3. MIF levels in different compartments and the development of DCI. Concentration of macrophage migration inhibitory factor (MIF) in (A) serum, (B) cerebrospinal fluid (CSF), and (C) microdialysate (MD) from patients with or without delayed cerebral ischemia (DCI) during the early (EPd1−4), critical (CPd5−15) and late (LPd16−21) phase after aneurysmal subarachnoid hemorrhage (aSAH). Data are shown as median [25th−75th percentile] and (for significant differences) as boxplots and individual values (see insets). Statistical significance was assessed using the Mann-Whitney U-test. For details on the exact number of patients per time-point see Supplementary Tables 1–3.
A tendency for higher serum levels during CPd5−15 was observed in patients with unfavorable clinical outcomes, although these differences did not reach statistical significance (Supplementary Figure 1A). Interestingly however, after stratification according to outcome, the correlation between serum and CSF MIF levels during EPd1−4 (i.e., Figure 1B) was no longer observed in patients with favorable outcomes but became even stronger in patients with unfavorable outcomes (Supplementary Figure 1A inset). In addition, there were again no differences between CSF MIF levels (Supplementary Figure 1B), but MD levels of the cytokine during CPd5−15 tended to be higher in patients with unfavorable outcomes (Supplementary Figure 1C) and to be negatively correlated with the GOSE score (Supplementary Figure 1C inset).
When the measurements during CPd5−15 were further subdivided into three time-periods, the progressive increase in serum MIF shown in Figure 1C appeared to be almost exclusively attributable to patients with AC aneurysms, while serum levels in patients with PC aneurysms actually decreased over time and were significantly lower on days 9–12 (9.0 [6.8–11.5] ng/ml vs. 18.9 [14.1–21.6] ng/ml in patients with AC aneurysms, p = 0.042, Supplementary Figure 2A). In addition, patients with DCI exhibited consistently higher MD MIF levels during the first 12 days after hemorrhage, although a significant difference was only observed at the start of the critical phase (days 5–8), when MD MIF levels amounted to 5.7 [3.9–16.1] ng/ml in patients with DCI compared to 0.2 [0.1–0.3] ng/ml in patients without DCI (p = 0.007, Supplementary Figure 2D).
Finally, MIF levels in serum, CSF or MD were neither associated with sex, hypertension, smoking status or treatment modality, nor with the clinical or radiological status on admission, although there was a tendency for higher serum levels in patients with poor radiological status throughout the observation period (Supplementary Tables 1–3). Likewise, even though clinical signs of sepsis were associated with a tendency for higher serum levels during CPd5−15 and LPd16−21 (Supplementary Table 1), the levels in CSF and MD were similar in patients with or without infection (Supplementary Tables 2, 3).
In the present study, we examined temporal changes of MIF levels in serum, CSF, and MD during the first 3 weeks after hemorrhage and their relation to disease progression and outcome. Our findings indicate that anterior circulation (AC) aneurysms may be associated with higher MIF levels in serum and that higher serum and especially MD MIF levels during the critical phase may be associated with DCI. In addition, they provide preliminary data on differences between systemic (i.e., serum), global cerebral (i.e., CSF) and local cerebral (i.e., MD) MIF levels, with potential implications for its origin and pathophysiological relevance.
Unlike most other cytokines, MIF is constitutively expressed in various tissues, most notably the pituitary and adrenal glands, from which it can be released into circulation in response to inflammatory, hypoxic and other stress stimuli (13, 23, 24). Previous studies have demonstrated that ischemic stroke (25, 26), intracerebral hemorrhage (27) and aSAH (17, 18) are associated with acute increases of circulatory MIF levels, which is in line with our observation that serum MIF levels during EPd1−4 (12.2 [8.7–16.4] ng/ml) exceeded the upper limit of 10 ng/ml typically observed in healthy subjects (17, 28). Interestingly, previous studies reported median serum MIF levels of 30.1 [19.1–37.9] ng/ml (17) and 22.3 [17.4–27.0] ng/ml (18) in aSAH patients immediately at hospital admission, which is roughly two-fold higher than the values we observed during EPd1−4. Considering that serum MIF levels after ischemic stroke have been shown to peak within the first 24 h after ictus (25, 26, 29), this difference could be attributable to our sampling window for EPd1−4, which comprised the first 4 days after hemorrhage but not the day of aSAH onset.
Furthermore, we found a significant positive correlation between serum and CSF MIF levels during EPd1−4 (Figure 1B), possibly indicating that intrathecal MIF release could be involved in the acute elevation of circulating MIF after aSAH. Thus, assuming a physiological serum concentration of 6–10 ng/ml (17, 28), a pathological CSF concentration of 30 ng/ml, and a 1:10 dilution after CSF flow into venous blood (30), the early serum levels of 12.2 [8.7–16.4] ng/ml we observed could well be accounted for by CSF-related changes of MIF concentrations in blood, especially given that early impairments of blood-brain- and blood-CSF-barrier are well-known features of aSAH (31, 32), which could be further aggravated by MIF itself (33). The fact that after stratification based on outcome, a significant correlation was only observed in patients with poor clinical outcome (as shown in the Supplementary material) is in line with this assumption, since early blood-brain-barrier dysfunction has been shown to predict neurological outcomes after aSAH (34).
In addition, we found that aSAH is associated with a further delayed increase of serum MIF levels during CPd5−15 (Figure 1C). During this phase, there was no longer a correlation between CSF and serum levels. We also noted that differences in serum MIF levels between patients with and without signs of clinical sepsis were limited and non-significant, as shown in the Supplementary material. While this might suggest that systemic infection is not the main stimulus for increased circulating MIF concentrations in aSAH patients, it could also simply reflect the small sample size of our study.
Interestingly, the increase during CPd5−15 appeared to be restricted to patients with AC aneurysms, whereas serum MIF levels in patients with PC aneurysms actually decreased during the first 2 weeks after ictus, as shown in the Supplementary material. Blood supply to the pituitary gland depends on vessels from the AC (35) and the MIF gene has been shown to contain several hypoxia-responsive elements in its promoter (29, 36, 37). This makes it tempting to speculate that hypoxia-induced up-regulation of MIF expression in the pituitary gland could be involved in the increase of serum MIF levels during CPd5−15. However, given that the AC is responsible for more than 80% of the brain's blood supply (38), the increase of serum levels in these patients could also reflect other factors, such as a more extensive hypoperfusion of brain tissue during the initial insult. Moreover, based on the small number of patients, of which the majority (70%) had AC aneurysms, further investigations are needed to firmly establish if there are associations between circulating MIF and aneurysm location.
In contrast to the time-course of changes in serum, MD MIF levels peaked during EPd1−4 and progressively decreased during the first 2 weeks after hemorrhage (Figure 1C), which is reminiscent of the stereotyped response of various brain cytokines to injury observed after aSAH and traumatic brain injury (39). In addition, MD MIF levels were independent of the levels in CSF or serum, suggesting that they could reflect local cytokine release from glial, neuronal and/or immune cells, which have all been demonstrated to constitutively and/or inducibly express MIF (13, 40, 41). Clearance of the cytokine from brain interstitial fluid into CSF could possibly account for the decrease of MD MIF levels during the first 2 weeks. However, CSF levels were at least four to five times higher than the values in MD (Figure 1A), suggesting that cleared MIF either accumulates in the CSF or that another source of MIF release into the CSF exists. For example, previous animal studies found that cells of the choroid plexus exhibit the strongest baseline immunoreactivity for MIF in the rat brain and that stress stimuli result in its immediate release into the CSF (40). Resident or infiltrating monocytes/macrophages could form another source for release of MIF, thereby contributing to the sustained elevation of CSF MIF levels after aSAH.
It should be noted that, in contrast to our findings, a previous smaller observational study observed peak CSF MIF levels on day 9 after aSAH and found them to be significantly higher in patients with systemic infection and/or DCI (19). We can only speculate that this might be related to differences in the study populations, treatment protocols, sample handling and analysis procedures or other factors.
Serum MIF levels during CPd5−15 and LPd16−21 were also significantly higher in patients with DCI (Figure 3A). Interestingly, and in contrast to the changes in MD (see below), serum MIF levels in patients with and without DCI started at similar levels but grew differently during the study period and peaked at the end of CPd5−15, as shown in the Supplementary material. This may suggest that DCI preceded the observed increase in serum levels, and that the higher levels in DCI patients simply reflect their more severe status and/or the requirement for more intense treatments (like induced hypertension or invasive ventilation). In line with this assumption, we found no evidence that the delayed increase of serum MIF levels was due to local or global cerebral release of the cytokine, suggesting that it mainly reflected peripheral release of MIF, which could well be aggravated by invasive treatments and infective or other complications. More importantly, our findings also indicate that local (MD) rather than global (CSF) cerebral levels of the cytokine could be associated with the development of DCI, as reflected in significantly higher MD MIF levels in patients with DCI during CPd5−15 (Figure 3C). When the samples obtained during CPd5−15 were subdivided into three time-periods, significantly higher MD MIF levels in patients with DCI were observed on days 5–8, as shown in the Supplementary material. This coincided almost exactly with the occurrence of DCI in our study (median: day 7 [5-9]).
To the best of our knowledge, data on the pathophysiological role of MIF in the brain is still sparse. For example, previous findings on the effects of MIF on neuronal function in different stroke models are conflicting, with some studies reporting that the cytokine is up-regulated and aggravates neurologic deficits by promoting cell death (42, 43) and/or increasing blood-brain-barrier permeability (33), while others found that it is down-regulated and may have neuroprotective functions (44, 45). With regards to aSAH, our findings argue for a local increase of MIF levels in the brain of patients with DCI, but they provide no clear-cut evidence regarding the pathophysiological relevance. Thus, higher MD MIF levels in these patients could be interpreted in terms of a detrimental role of the cytokine which increases the susceptibility to DCI, or in terms of a compensatory up-regulation that serves neuroprotective functions and is more pronounced in affected patients.
Given the limits of a small exploratory study, several limitations have to be carefully considered for the interpretation of our findings. The sample size in our study was notably small and not based on any formal power calculations, and we did not correct for multiple statistical comparisons. The availability of CSF and/or MD samples was a requirement for inclusion in this study, which may have resulted in an over-representation of more severely affected patients, as reflected in the relatively high incidence of DCI (Table 1). Even more importantly, samples from the different compartments were not always available for all patients, which may have affected the observed time-course of changes, especially with regard to EPd1−4 and LPd16−21. Thus, while during CPd5−15, at least one MD sample was available from the majority (20/30 = 67%) of patients, the number of patients with MD samples during EPd1−4 and LPd16−21 were only 9 (30%) and 8 (27%), respectively. In addition, analysis of MD samples may have been influenced by factors such as variable catheter placement or MIF recovery. As such, our data should be regarded as preliminary and interpreted with due caution until they can be validated by human studies with adequate power. Moreover, our findings provide no information on causal relationships between MIF levels and aneurysm location, DCI or clinical outcome, which needs to be addressed in appropriate animal models.
Our findings in a small cohort of aSAH patients provide first data on differences between systemic, global cerebral and local cerebral MIF levels after aSAH, and their potential relation to aneurysm location and DCI. Larger clinical trials and studies with animal models will be required to validate our findings and explore potential causal relationships between MIF levels and disease progression.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the local ethics committee of the RWTH Aachen (EK062/14). The patients/participants provided their written informed consent to participate in this study.
Conceived, designed, and performed the experiments: FN, CS, AS, and WA. Data acquisition: AS, MWe, MV, RD, CC, and WA. Analysis and interpretation of data: FN, CS, AS, YT, RH, and WA. First drafting of the manuscript and illustrations: FN and WA. Critical review of the manuscript: CS, HH, YT, MWi, HC, GS, RH, AH, TS, CC, FN, and WA. The final manuscript was approved by all authors.
This work was supported by the START-Program of the Faculty of Medicine, RWTH Aachen, Germany. Grant number 691540 and by the German Research Foundation to CS (DFG, STO 1099/8-1).
FN was employed by Forschungszentrum Jülich GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1066724/full#supplementary-material
AC, anterior circulation; aSAH, aneurysmal subarachnoid hemorrhage; CP, critical phase (days 5-15 after hemorrhage); CPd5−8, critical phase (days 5-8 after hemorrhage); CPd9−12, critical phase (days 9-12 after hemorrhage); CPd13−15, critical phase (days 13-15 after hemorrhage); CPd5−15, critical phase (days 5-15 after hemorrhage); CSF, cerebrospinal fluid; DCI, delayed cerebral ischemia; EBI, early brain injury; EPd1−4, early phase (days 1-4 after hemorrhage); GOSE, Glasgow Outcome Scale Extended; HH, Hunt and Hess grading scale; LPd16−21, late phase (days 16-21 after hemorrhage); MD, cerebral microdialysate; MIF, macrophage migration inhibitory factor; mFS, modified Fisher scale; PC, posterior circulation; ptiO2, brain tissue oxygen level.
1. Alleyne CH. Aneurysmal subarachnoid hemorrhage: Have outcomes really improved? Neurology. (2010) 74:1486–7. doi: 10.1212/WNL.0b013e3181e0ef1a
2. Vergouwen MDI, Vermeulen M, van Gijn J, Rinkel GJE, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: Proposal of a multidisciplinary research group. Stroke. (2010) 41:2391–5. doi: 10.1161/STROKEAHA.110.589275
3. Geraghty JR, Testai FD. Delayed cerebral ischemia after subarachnoid hemorrhage: beyond vasospasm and towards a multifactorial pathophysiology. Curr Atheroscler Rep. (2017) 19:50. doi: 10.1007/s11883-017-0690-x
4. Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KTS. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. (2012) 109:315–29. doi: 10.1093/bja/aes264
5. Schenck H, Netti E, Teernstra O, De Ridder I, Dings J, Niemelä M, et al. The role of the glycocalyx in the pathophysiology of subarachnoid hemorrhage-induced delayed cerebral ischemia. Front Cell Dev Biol. (2021) 9:2358. doi: 10.3389/fcell.2021.731641
6. Woitzik J, Dreier JP, Hecht N, Fiss I, Sandow N, Major S, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. (2012) 32:203–12. doi: 10.1038/jcbfm.2011.169
7. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the international multidisciplinary consensus conference on multimodality monitoring in neurocritical care. Intensive Care Med. (2014) 40:1189–209. doi: 10.1007/s00134-014-3369-6
8. Brady KM, Lee JK, Kibler KK, Smielewski P, Czosnyka M, Easley RB, et al. Continuous time-domain analysis of cerebrovascular autoregulation using near-infrared spectroscopy. Stroke. (2007) 38:2818–25. doi: 10.1161/STROKEAHA.107.485706
9. Lavinio A, Schmidt EA, Haubrich C, Smielewski P, Pickard JD, Czosnyka M. Non-invasive evaluation of dynamic cerebrovascular autoregulation using finapres plethysmograph and transcranial doppler. Stroke. (2007) 38:402–4. doi: 10.1161/01.STR.0000254551.92209.5c
10. Toi H, Matsumoto N, Yokosuka K, Matsubara S, Hirano K, Uno M. Prediction of cerebral vasospasm using early stage transcranial doppler. Neurol Med Chir. (2013) 53:396–402. doi: 10.2176/nmc.53.396
11. Watson E, Ding D, Khattar NK, Everhart DE, James RF. Neurocognitive outcomes after aneurysmal subarachnoid hemorrhage: Identifying inflammatory biomarkers. J Neurol Sci. (2018) 394:84–93. doi: 10.1016/j.jns.2018.06.021
12. Larson DF, Horak K. Macrophage migration inhibitory factor: controller of systemic inflammation. Crit Care. (2006) 10:138. doi: 10.1186/cc4899
13. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. (2003) 3:791–800. doi: 10.1038/nri1200
14. Zhang B, Luo Y, Liu M-L, Wang J, Xu D-Q, Dong M-Q, et al. Macrophage migration inhibitory factor contributes to hypoxic pulmonary vasoconstriction in rats. Microvasc Res. (2012) 83:205–12. doi: 10.1016/j.mvr.2011.09.014
15. Bruchfeld A, Carrero JJ, Qureshi AR, Lindholm B, Barany P, Heimburger O, et al. Elevated serum macrophage migration inhibitory factor (MIF) concentrations in chronic kidney disease (CKD) are associated with markers of oxidative stress and endothelial activation. Mol Med. (2009) 15:70–5. doi: 10.2119/molmed.2008.00109
16. Stefaniak J, Schiefer J, Miller EJ, Krenn CG, Baron DM, Faybik P. Macrophage migration inhibitory factor as a potential predictor for requirement of renal replacement therapy after orthotopic liver transplantation. Liver Transplant. (2015) 21:662–9. doi: 10.1002/lt.24103
17. Chen Y-H, Cheng Z-Y, Shao L-H, Shentu H-S, Fu B. Macrophage migration inhibitory factor as a serum prognostic marker in patients with aneurysmal subarachnoid hemorrhage. Clin Chim Acta. (2017) 473:60–4. doi: 10.1016/j.cca.2017.08.018
18. Yang X, Peng J, Pang J, Wan W, Zhong C, Peng T, et al. The association between serum macrophage migration inhibitory factor and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neurotox Res. (2020) 37:397–405. doi: 10.1007/s12640-019-00072-4
19. Kwan K, Arapi O, Wagner KE, Schneider J, Sy HL, Ward MF, et al. Cerebrospinal fluid macrophage migration inhibitory factor: a potential predictor of cerebral vasospasm and clinical outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg. (2019) 1:1–6. doi: 10.3171/2019.6.JNS19613
20. Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G. European stroke organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. (2013) 35:93–112. doi: 10.1159/000346087
21. Albanna W, Weiss M, Müller M, Brockmann MA, Rieg A, Conzen C, et al. Endovascular rescue therapies for refractory vasospasm after subarachnoid hemorrhage: a prospective evaluation study using multimodal, continuous event neuromonitoring. Neurosurgery. (2017) 80:942–9. doi: 10.1093/neuros/nyw132
22. Weiss M, Conzen C, Mueller M, Wiesmann M, Clusmann H, Albanna W, et al. Endovascular rescue treatment for delayed cerebral ischemia after subarachnoid hemorrhage is safe and effective. Front Neurol. (2019) 10:136. doi: 10.3389/fneur.2019.00136
23. Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, et al. is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. (1993) 365:756–9. doi: 10.1038/365756a0
24. Nishino T, Bernhagen J, Shiiki H, Calandra T, Dohi K, Bucala R. Localization of macrophage migration inhibitory factor (MIF) to secretory granules within the corticotrophic and thyrotrophic cells of the pituitary gland. Mol Med. (1995) 1:781–8. doi: 10.1007/BF03401892
25. Li Y-S, Chen W, Liu S, Zhang Y-Y, Li X-H. Serum macrophage migration inhibitory factor levels are associated with infarct volumes and long-term outcomes in patients with acute ischemic stroke. Int J Neurosci. (2017) 127:539–46. doi: 10.1080/00207454.2016.1211648
26. Kadhim MM, Shakir AS. Serum macrophage migration inhibitory factor levels in patients with ischemic stroke in iraqi populations. Medico-Legal Updat. (2020) 20:41772. doi: 10.37506/mlu.v20i4.2116
27. Lin Q, Cai J-Y, Lu C, Sun J, Ba H-J, Chen M-H, et al. Macrophage migration inhibitory factor levels in serum from patients with acute intracerebral hemorrhage: Potential contribution to prognosis. Clin Chim Acta. (2017) 472:58–63. doi: 10.1016/j.cca.2017.07.016
28. Roger T, Schlapbach LJ, Schneider A, Weier M, Wellmann S, Marquis P, et al. Plasma levels of macrophage migration inhibitory factor and d-dopachrome tautomerase show a highly specific profile in early life. Front Immunol. (2017) 8:26. doi: 10.3389/fimmu.2017.00026
29. Wang L, Zis O, Ma G, Shan Z, Zhang X, Wang S, et al. Upregulation of macrophage migration inhibitory factor gene expression in stroke. Stroke. (2009) 40:973–6. doi: 10.1161/STROKEAHA.108.530535
30. Reiber H. Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci. (2003) 21:79–96.
31. Chen S, Xu P, Fang Y, Lenahan C. The updated role of the blood brain barrier in subarachnoid hemorrhage: from basic and clinical studies. Curr Neuropharmacol. (2020) 18:1266–78. doi: 10.2174/1570159X18666200914161231
32. Solár P, Zamani A, Kubíčková L, Dubový P, Joukal M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS. (2020) 17:35. doi: 10.1186/s12987-020-00196-2
33. Liu Y-C, Tsai Y-H, Tang S-C, Liou H-C, Kang K-H, Liou H-H, et al. Cytokine MIF enhances blood-brain barrier permeability: impact for therapy in ischemic stroke. Sci Rep. (2018) 8:743. doi: 10.1038/s41598-017-16927-9
34. Lublinsky S, Major S, Kola V, Horst V, Santos E, Platz J, et al. Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine. (2019) 43:460–72. doi: 10.1016/j.ebiom.2019.04.054
35. Doglietto F, Prevedello DM-S, Belotti F, Ferrari M, Lancini D, Schreiber A, et al. The superior hypophyseal arteries: anatomical study with an endoscopic endonasal perspective. Oper Neurosurg. (2019) 17:321–31. doi: 10.1093/ons/opy393
36. Baugh JA, Gantier M, Li L, Byrne A, Buckley A, Donnelly SC. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochem Biophys Res Commun. (2006) 347:895–903. doi: 10.1016/j.bbrc.2006.06.148
37. Welford SM, Bedogni B, Gradin K, Poellinger L, Broome Powell M, Giaccia AJ. HIF1 delays premature senescence through the activation of MIF. Genes Dev. (2006) 20:3366–71. doi: 10.1101/gad.1471106
38. Boyajian RA, Schwend RB, Wolfe MM, Bickerton RE, Otis SM. Measurement of anterior and posterior circulation flow contributions to cerebral blood flow. J Neuroimaging. (1995) 5:1–3. doi: 10.1111/jon1995511
39. Thelin EP, Tajsic T, Zeiler FA, Menon DK, Hutchinson PJA, Carpenter KLH, et al. Monitoring the neuroinflammatory response following acute brain injury. Front Neurol. (2017) 8:351. doi: 10.3389/fneur.2017.00351
40. Bacher M, Meinhardt A, Lan HY, Dhabhar FS, Mu W, Metz CN, et al. MIF expression in the rat brain: implications for neuronal function. Mol Med. (1998) 4:217–30. doi: 10.1007/BF03401919
41. Ogata A, Nishihira J, Suzuki T, Nagashima K, Tashiro K. Identification of macrophage migration inhibitory factor mRNA expression in neural cells of the rat brain by in situ hybridization. Neurosci Lett. (1998) 246:173–7. doi: 10.1016/S0304-3940(98)00203-1
42. Inácio AR, Ruscher K, Leng L, Bucala R, Deierborg T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow Metab. (2011) 31:1093–106. doi: 10.1038/jcbfm.2010.194
43. Lambertsen KL, Deierborg T. “A detrimental role of mif in ischemic brain damage,” in The MIF Handbook (Singapore: World Scientific). p. 361–75. doi: 10.1142/9789814335362_0019
44. Zhang S, Zis O, Ly PTT, Wu Y, Zhang S, Zhang M, et al. Down-regulation of MIF by NFκB under hypoxia accelerated neuronal loss during stroke. FASEB J. (2014) 28:4394–407. doi: 10.1096/fj.14-253625
Keywords: aneurysmal subarachnoid hemorrhage, delayed cerebral ischemia, macrophage migration inhibitory factor (MIF), cerebral microdialysis, brain
Citation: Neumaier F, Stoppe C, Stoykova A, Weiss M, Veldeman M, Höllig A, Hamou HA, Temel Y, Conzen C, Schmidt TP, Dogan R, Wiesmann M, Clusmann H, Schubert GA, Haeren RHL and Albanna W (2023) Elevated concentrations of macrophage migration inhibitory factor in serum and cerebral microdialysate are associated with delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Front. Neurol. 13:1066724. doi: 10.3389/fneur.2022.1066724
Received: 11 October 2022; Accepted: 29 December 2022;
Published: 13 January 2023.
Edited by:
Sandra Magnoni, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
Adriano Bernini, Centre hospitalier universitaire vaudois (CHUV), SwitzerlandCopyright © 2023 Neumaier, Stoppe, Stoykova, Weiss, Veldeman, Höllig, Hamou, Temel, Conzen, Schmidt, Dogan, Wiesmann, Clusmann, Schubert, Haeren and Albanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walid Albanna,  d2FsaWRhbGJhbm5hQHlhaG9vLmRl
d2FsaWRhbGJhbm5hQHlhaG9vLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.