 Elie Naddaf
Elie Naddaf
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 27 September 2022
Sec. Diagnostic and Forensic Neuropathology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.1020113
This article is part of the Research Topic Diagnosis of Neuromuscular Disorders in the Era of Personalized Genomic Medicine View all 6 articles
Inclusion body myositis (IBM) is a progressive muscle disease affecting patients over the age of 40, with distinctive clinical and histopathological features. The typical clinical phenotype is characterized by prominent involvement of deep finger flexors and quadriceps muscles. Less common presentations include isolated dysphagia, asymptomatic hyper-CKemia, and axial or limb weakness beyond the typical pattern. IBM is associated with marked morbidity as majority of patients eventually become wheelchair dependent with limited use of their hands and marked dysphagia. Furthermore, IBM mildly affects longevity with aspiration pneumonia and respiratory complications being the most common cause of death. On muscle biopsy, IBM is characterized by a peculiar combination of endomysial inflammation, rimmed vacuoles, and protein aggregation. These histopathological features are reflective of the complexity of underlying disease mechanisms. No pharmacological treatment is yet available for IBM. Monitoring for swallowing and respiratory complications, exercise, and addressing mobility issues are the mainstay of management. Further research is needed to better understand disease pathogenesis and identify novel therapeutic targets.
Inclusion body myositis (IBM) is a sporadic muscle disease of aging, almost exclusively affecting individuals over the age of 40 (1). Traditionally, IBM is classified as an idiopathic inflammatory myopathy. However, the unique clinical phenotype, the peculiar combination of various histopathological findings, and the slowly progressive, treatment-refractory course, made IBM a hot debate topic regarding its pathogenesis and the best way to treat it. As the clinical and histopathological features are not universally present in all IBM patients, patients are often misdiagnosed, especially earlier in the disease course (2, 3). Depending on the prominent clinical and/or histopathological features in a particular patient, common misdiagnoses include polymyositis or other myositides, hereditary myopathy with rimmed vacuoles or other inherited myopathies, compressive mononeuropathies (especially ulnar neuropathy at the elbow or median neuropathy at the wrist) or radiculopathies (especially L3/4 radiculopathy), or a motor neuron disorder. Hereditary myopathies with rimmed vacuoles, associated with a systemic proteinopathy, are sometimes called hereditary IBM (hIBM). However, the term hIBM (vs. sporadic IBM or sIBM) may be misleading, as it implies it is the hereditary form of the same disease, while indeed these are two different diseases with different clinical phenotype, disease course, and patient demographics. As treatment, prognosis, and implications for other family members is widely variable between these various entities, misdiagnosis can have a major implication on patient care. Furthermore, the lack of a curative treatment often results in loss to follow up and consequently, lack of monitoring for disease complications and providing adequate supportive care. In this review article, we focus on addressing these diagnostic and therapeutic challenges in patients with IBM.
Most epidemiologic studies in IBM focused on estimating the incidence and prevalence of the disease. IBM affects males about twice as common as females, with a prevalence varying from 1 to 182 per million among those aged 50 and older (4–8). This variability in the reported prevalence is at least in part due to variability in case ascertainment methods and the used diagnostic criteria. Regarding associated conditions, patients with IBM are 2.7 times more likely to have a peripheral neuropathy, 6.2 times more likely to have Sjogren syndrome, and 3.9 times more likely to have hematologic malignancies, especially T-cell large granular lymphocytic leukemia, when compared to population controls (9). In contrast, there is no evidence for increased prevalence of neurodegenerative diseases or solid cancers in IBM population (9, 10). Given the predilection to indispensable muscles, IBM is associated with marked morbidity. The muscle weakness steadily progresses over time with a variable decline rate, although progression may be more pronounced earlier in the disease course (11, 12). Despite having predilection to certain muscles at earlier stages, any skeletal muscle can be affected at advanced stages. Almost all patients become wheelchair dependent within 20 years from onset, with a median time from symptom onset to wheelchair dependence about 10.5 years (7, 12, 13). Furthermore, dysphagia is highly prevalent in IBM, with aspiration pneumonia, in addition to respiratory complications of the disease, being the most common cause of death (9, 14–16). As a result, IBM is associated with a modest decrease in longevity, with a 10-year survival of 36–42% compared to 59% in population controls, and a mean age at death of 79.3 years compared to 83.6 in controls (8–10).
IBM is a slowly progressive disease, mimicking the clinical course of neurodegenerative diseases such as Parkinson's or Alzheimer's. Hence, presentation with rapidly progressive weakness, such as going from normal gait to needing a walking aid within a year from onset, should cast doubts about the diagnosis and prompt searching for alternative etiologies. IBM has predilection to finger flexors and knee extensors (Figure 1). As a result, patients most commonly present with either hand grip or lower limb weakness (e.g., difficulty with stairs, or difficult rising from a low seat), often asymmetric. This distinctive pattern of weakness, when present, strongly raises suspicion for the diagnosis. However, manual evaluation of the quadriceps strength can be challenging. Therefore, mild to moderate weakness of this muscle may be overlooked, and patients erroneously labeled as having intact strength despite reporting difficulty with their daily life activities. Manual testing of knee extension should be performed with the knee bent (e.g., 90 degrees), rather than fully extended or locked, so the knee mechanics are at the advantage of the examiner (17). Functional examination of the knee strength, such as kneeling on one knee then getting up without using the hands, should follow especially when weakness is not detected on manual testing. Examiner should keep in mind that patients may have difficulty performing this task without necessarily having quadriceps weakness, for instance in patients with hip or knee osteoarthritis or obesity.
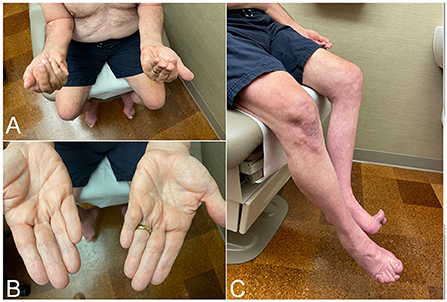
Figure 1. Clinical findings in patients with inclusion body myositis. (A) Patient attempting to make a fist, demonstrating bilateral finger flexion weakness most severely affecting flexion at the distal interphalangeal joint, worse on the left side. (B) Same patient as in (A). Effacement of finger wrinkling over the palmar aspect of the interphalangeal joints, more pronounced on the left side, most noticeable over the distal interphalangeal joints. (C) Patient attempting knee extension, demonstrating bilateral quadriceps weakness, more severe on the left side where there is more noticeable thigh and leg muscle atrophy.
About 14% of IBM patients have an atypical presentation beyond hand grip and quadriceps muscle weakness (3). Such patients may present with dysphagia, foot drop, proximal upper limb weakness, facial diplegia, axial weakness or head drop, or asymptomatic elevated creatine kinase level (hyper-CKemia) (3, 18–22). Respiratory insufficiency usually occurs at advanced stages of the disease. Patients with IBM do not typically have any significant cardiac muscle involvement, or extraskeletal manifestations of the disease.
Despite the distinctive clinical phenotype, a muscle biopsy remains the gold standard for diagnosis. It would be challenging to base the diagnosis solely on the quadriceps weakness, due to technical difficulty with manual muscle testing as mentioned above, and as the quadriceps may be involved to the same extent as hip flexors in some patients. While finger flexion weakness, more than shoulder abduction, can be more characteristic and easier to demonstrate, prominent finger flexion weakness can also be seen in other acquired (amyloidosis, sarcoidosis) and hereditary (especially myotonic dystrophy type 1) myopathies (23). Patients with atypical presentations pose additional diagnostic challenges with further delay in diagnosis (3). Therefore, clinical-pathological correlation remains crucial, and all widely-used diagnostic criteria require fulfillment of certain muscle biopsy features (1, 24).
Nerve conduction studies and electromyography (EMG) help determining the nature of the process underlying the patient's reported weakness: myopathic vs. neuropathic, and rule out a motor neuron disorder or multiple mononeuropathies that can have similar presentation. Furthermore, EMG findings are taken in consideration when selecting a target for a muscle biopsy. Nerve conduction studies are usually within normal limits, or may detect a superimposed length-dependent peripheral neuropathy. Needle EMG typically demonstrates early recruitment of short duration, low amplitude, complex, motor unit potentials (MUP), with fibrillation potentials in almost all patients (25). Up to a third of patients may demonstrate myotonic or myotonic-like discharges (3, 25, 26). The findings are usually more prominent in weaker muscles, such as flexor digitorum profundus and quadriceps. Furthermore, mixed short and long duration MUP, within the same muscle, are often encountered in IBM (3, 25). In such patients, the short MUPs may be overlooked, and the patient may get erroneously diagnosed with a neuropathic process such as an anterior interosseous neuropathy or L3/4 radiculopathy. The long duration MUPs in IBM are often complex in morphology, mimicking a subacute neuropathic process (25). Similar to EMG findings, neuropathic changes (denervation atrophy and/or reinnervation) are seen in vast majority of muscle biopsies from patients with IBM (27).
Our understanding of IBM pathogenesis stemmed from the description of its peculiar histopathological findings (Figure 2). The three canonical features of IBM include: endomysial inflammation, where inflammatory cells surround and invade non-necrotic muscle fibers, also known as autoaggressive inflammation; the presence of rimmed vacuoles; and protein aggregation as witnessed by the accumulation of congophilic deposits and 15/18 nm filaments (tubulofilaments) on electron microscopy (EM) (28, 29). In order to establish the diagnosis of IBM on histological grounds (clinico-pathologically defined IBM), the three canonical features have to be present, in addition to fulfilling clinical and laboratory criteria (1). However, up to 25% of patients with clinical features of IBM do not have rimmed vacuoles or congophilic deposits on biopsy, which results in the erroneous diagnosis of “polymyositis” or “myositis not otherwise specified,” and unnecessary treatment with corticosteroids or other immunosuppressants (2, 30). Furthermore, the lack of congophilic deposits or tubulofilaments was the most common reason why patients with IBM failed to fulfill various diagnostic criteria in one study (31). However, Congo red stain and EM were only performed on a small proportion of patients. Moreover, EM is not widely available for clinical use. In our Muscle laboratory at Mayo clinic, Congo red staining is routinely performed on all muscle biopsies. Slides are reviewed under rhodamine optics, rather than polarized light, which is more sensitive for the detection of amyloid deposits (32). Detecting protein aggregates by alternative methods, such as TDP43 and p62 by immunohistochemistry, can help further increasing the diagnostic yield of a muscle biopsy (33, 34).
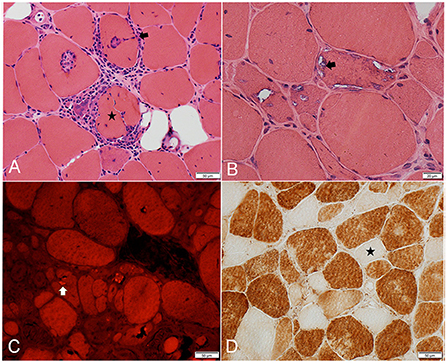
Figure 2. Frozen muscle sections from patients with inclusion body myositis. (A) H&E stain: inflammatory cells surrounding, and focally invading (arrow) muscle fibers. One muscle fiber (star) is completely split apart by inflammatory cells. (B) H&E stain: 3 adjacent muscle fibers harboring multiple vacuoles rimmed by a membranous material (example: arrow). (C) Congo Red stain viewed under Rhodamine optics: several fibers display one or more congophilic (bright red) inclusions (example: arrow). (D) Cytochrome C oxidase reaction: several fibers are devoid of enzyme reactivity (example: star).
The most common muscle biopsy findings in IBM are the increased sarcolemmal expression of MHC class I, detected by immunohistochemistry, and the presence of cytochrome c oxidase negative fibers, both of which are present in almost all IBM muscle biopsies (Figure 2D) (30, 35, 36). Upregulation of MHC-I expression lacks specificity as it can be seen in other immune-mediated and sometimes inherited myopathies, limiting its diagnostic value. Combining MHC-I with MHC-II is reported to increase specificity to for the diagnosis of an inflammatory myopathy, however, MHC-II staining on muscle is not widely available yet (37). The presence of significant mitochondrial abnormalities in muscle specimens demonstrating endomysial inflammation should strongly raise suspicion for IBM, with one study reporting a 100% sensitivity and 74% specificity in this context (35). Patients with endomysial inflammation and prominent mitochondrial dysfunction, but without rimmed vacuoles or protein aggregates, are sometimes referred to as having “polymyositis with mitochondrial pathology” (PM-Mito) (38). It remains debatable whether this should be considered a separate entity, given the high prevalence of mitochondrial abnormalities in IBM and as a significant proportion of these patients are eventually diagnosed with IBM (39–41). A less common muscle biopsy finding in IBM is the presence of granulomas. IBM and sarcoid myopathy are the most common diagnoses in patients with granulomatous myositis on muscle biopsy (42, 43). Lastly, a component of denervation atrophy is seen in majority of biopsies, manifesting with groups of atrophic angulated fibers overreacting to non-specific esterase (25, 27).
Selecting the target for a muscle biopsy is of utmost importance. It is preferable to choose a clinically-affected muscle, where the weakness is of moderate severity (21). This would limit the chances of a false negative (normal or minimally affected muscle) or non-diagnostic endstage muscle (severe weakness). Using EMG (muscle with spontaneous activity) and muscle imaging data when available, can help optimize the yield of a muscle biopsy. Many institutions only perform muscle biopsies from the quadriceps, especially when performing a punch biopsy. Luckily, the quadriceps is commonly involved in IBM but not all patients have quadriceps involvement at presentation. One caveat with the quadriceps muscle is that the four heads are not usually equally affected. Hence, correlation with clinical examination, electrodiagnostic testing, and imaging would ensure proper selection of biopsy site. Lastly, muscle involvement may be asymmetric in IBM, which should be taken in consideration, especially when using EMG for muscle selection, as it is typically performed on one side saving the contralateral side for biopsy. Occasionally, patients may require a repeat muscle biopsy to establish the diagnosis (3, 21).
Antibodies against cytosolic 5'-nucleotidase 1A (cN-1A) are the only available serum diagnostic test for inclusion body myositis (44, 45). Overall, the sensitivity is limited, around 30–50%, when using ELISA, which is the most commonly used platform for commercial testing (46). Specificity is high, usually more than 90% (44, 45). However, it is much lower in patients with other connective tissue diseases, such as systemic lupus erythematous, Sjögren syndrome, or dermatomyositis, as up to a third of these patients may have positive cN-1A antibodies and not have IBM (47, 48). Given the challenges mentioned above and the variability in the used detection methods, cN-1A results should be cautiously interpreted in light of the patient's clinical and histopathological findings.
Muscle MRI findings in IBM commonly follow the same clinical pattern with preferential involvement, sometimes asymmetrically, of finger flexors, mainly flexor digitorum profundus, and quadriceps. In the quadriceps, the rectus femoris is usually spared and there is a proximal-to-distal gradient, with more pronounced fatty infiltration near the knee (Figure 3) (49, 50). In the legs, the medial gastrocnemius is the most involved with sparing of the tibialis posterior and soleus muscles (51, 52). In contrast to other inflammatory myopathies, fatty infiltration is typically more prominent than increased T2 signal (edema) in patients with IBM. Nevertheless, the use of muscle MRI as a diagnostic tool in inflammatory myopathies remains limited (53). However, this classic MRI pattern should raise suspicion for IBM even if the diagnosis was not considered on clinical grounds, such as in patients with isolated hyper-CKemia or with atypical disease presentation (3).
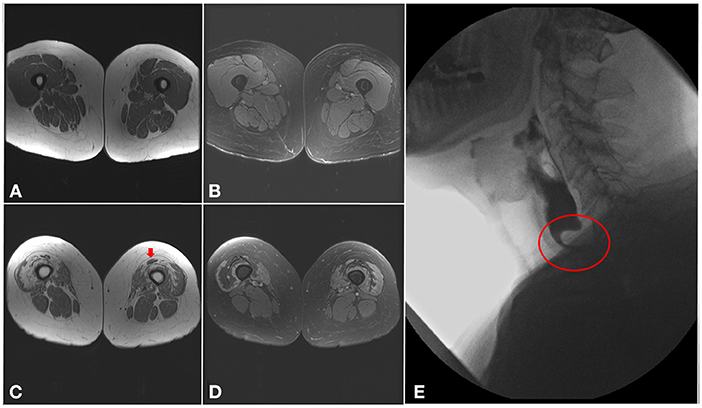
Figure 3. Imaging findings in inclusion body myositis. (A–D) MRI of the thighs from a patient with IBM. Axial T1 images (A,C) showing a proximal-to-distal gradient, with preservation of proximal segments (A) and fatty infiltration of the distal vastus medialis and lateralis bilaterally (C), more pronounced in the right lower limb, with relative sparing of the rectus femoris (arrow). Axial T2 images (B,D) demonstrating sparing of the proximal segments of the quadriceps muscles (B), and only mild T2 hyperintensity surrounding areas of fatty infiltration distally (D). (E) Barium swallow demonstrating a cricopharyngeal bar with severe (more than 75%) luminal narrowing (circle).
Creatine kinase (CK) level is elevated in 75−80% of patients, typically < 15 times upper limit of normal (22, 25). Higher CK levels should cast doubt about the diagnosis and prompt searching for alternative acquired or inherited etiologies.
When screened, some IBM patients may have clonal expansion of a large granular T cells (T-LGL) population, and rarely T-LGL leukemia (9, 54). Differentiating T-LGL clonal expansion from T-LGL leukemia and the best therapeutic approach for the latter remain a topic of debate (55, 56). Therefore, from hematological perspective, routine screening for T-LGL leukemia is not recommended. One practical approach would be to obtain a CBC and a peripheral smear. The presence of lymphocytosis, cytopenias, and/or large granular T cells should prompt further investigation via T-cell receptor gene rearrangement and/or flow cytometry, and referral to Hematology. Nevertheless, from neurological perspective, the utility of detecting a T-LGL clone as a diagnostic tool for IBM has not been fully investigated.
Regarding muscle biopsy findings, overall, there is no clear or significant correlation between clinical findings and muscle histopathological features (27). In a recent study, we evaluated the correlation of various muscle biopsy findings with clinical variables including quadriceps strength, summated strength score, modified Rankin scale, severity of dysphagia, and baseline characteristics such as age at biopsy and disease duration (27). The main clinical variable that had a strong [Kendal tau correlation coefficient (k) of 0.62] and statistically-significant correlation with endomysial inflammation was the severity of dysphagia, evaluated by a formal swallow evaluation. This is intriguing as dysphagia is the most anecdotally-reported manifestation of IBM with some response to immunotherapy (57, 58). The most significant correlation with quadriceps strength was the increased endomysial connective tissue, reflective of the chronic loss of muscle fibers (27). Interestingly, modified Rankin scale was inversely (k = 0.39) correlated with inflammation (27). Rimmed vacuoles and congophilic deposits had no notable (k < 2) or statistically significant correlation with any clinical variable (27).
Regarding cN-1A antibodies, there are conflicting results about the association of cN-1A seropositivity with more prominent (degree of severity) or more frequent (present or not) dysphagia, which can be in part due to the variability in the methods used (27, 59–64). Similarly, there are varying results regarding the association of seropositivity with the severity of limb muscle weakness or pattern of weakness (27, 59, 62, 63). Taken altogether, cN-1A seropositive patients may be more likely to have slightly more pronounced dysphagia and/or muscle weakness. However, if such association is indeed present, it is modest at best, and not sufficient to use the antibody to predict severity or disease phenotype in clinical practice.
Lastly, Ck levels do not correlate with clinical findings in IBM (22, 27). Hence, decreased CK levels should not be used as evidence for response to immunotherapy and a reason to keep patients on such treatments.
Traditionally, IBM has been considered an inflammatory muscle disease. The main support for the inflammatory hypothesis is the robust inflammatory infiltrate on muscle biopsy, where inflammatory cells, namely cytotoxic CD8+T cells, surround and invade non-necrotic muscle fibers (28). These T cells are clonally restricted, antigen driven, and express markers of high differentiation such as CD57 and KLRG1 (65–70). Furthermore, there is a strong association between IBM and HLA-DRB1 genes (71–73). However, the refractoriness to immunosuppressive therapy, the lingering disease course, the age of the at-risk population, and the male predominance would all be atypical for an inflammatory disease. In addition to the lingering course and the characteristics of the population at risk, IBM shares common pathological features with other neurodegenerative diseases, namely the accumulation of rimmed vacuoles and protein aggregates such as amyloid-β precursor protein and amyloid- β, as seen in Alzheimer's disease, and p62 and Tar-DNA binding protein 43 (TDP-43) as seen in amyotrophic lateral sclerosis and frontotemporal dementia (29, 74–76). While the role of amyloid deposits in IBM pathogenesis remains uncertain, the accumulation of TDP-43+ inclusions in the sarcoplasm of IBM patients is accompanied by TDP-43 nuclear depletion, resulting in loss of TDP-43 splicing repression of non-conserved cryptic exons, a feature seen in amyotrophic lateral sclerosis and frontotemporal dementia (77). Several non-inflammatory pathways have been briefly described in IBM, although the exact level of dysfunction in each pathway remains poorly defined. Those include disrupted protein homeostasis, excessive and/or impaired autophagy, mitochondrial dysfunction, oxidative stress, disrupted calcium homeostasis, ER stress and extracellular matrix involvement (78–81). A more detailed review of these pathways is out of the scope of this review article, some of them are nicely summarized in (82, 83).
Despite the lack of pharmacological treatment for IBM, there are several items that need to be addressed on a regular basis: swallowing, respiratory function, muscle weakness and mobility.
Dysphagia is often overlooked despite aspiration pneumonia being the most common cause of death. Screening should probably be performed on an annual basis, at least by obtaining a detailed history and keeping a low threshold for a referral to a speech pathologist for a formal evaluation. Questionnaires, such as the “Eating Assessment Tool” (EAT) can be used (84). When present, the dysphagia may be due to a combination of obstruction and weakness. The cricopharyngeus muscle can scar, which results in luminal obstruction, known as a cricopharyngeal band or bar (Figure 3E) (3, 85). If the obstruction is significant, endoscopic dilation or a cricopharyngeal myotomy could be considered. For the oropharyngeal weakness, treatment consists mostly of adaptive strategies such as eating specific consistencies or volumes to avoid aspiration. There is anecdotal evidence for improved dysphagia with immunotherapy, namely IVIG, which may provide temporary relief in selected cases, however there is no evidence that IVIG changes the overall disease course (57, 86, 87).
Similar to dysphagia, respiratory failure is a major source of morbidity and mortality in IBM. It is important to screen via a detailed questionnaire and to have a low threshold to perform additional testing, such as overnight oximetry, pulmonary function tests, or a sleep study, and to refer the patient to a sleep medicine specialist when appropriate. While dysphagia can occur at any disease stage, respiratory failure usually occurs at advanced stages.
Routine evaluation by physical medicine and rehabilitation team is often needed. This includes offering adaptive strategies or assistive devices (walker, bracing, wheelchair etc.) to help with hand dexterity and mobility, evaluating for safety and fall prevention, and providing an exercise program. Exercise, especially resistance training, may help preserving or even improving muscle strength in patients with IBM (88, 89).
There is no evidence-based pharmacological therapy for IBM. Yet, prednisone remains one of the most commonly prescribed drugs in IBM. To note, treatment with corticosteroids could potentially be harmful, although this has not been established with certainty either. In one study, Benveniste et al. reported that treated IBM patients (with corticosteroids or other immunosuppressants) needed a walking aid sooner than untreated patients (12). Comparison of baseline characteristics between treated and untreated groups was not available. In another study, there was a separation in survival curves for IBM patients treated with corticosteroids (lower survival) compared with untreated IBM patients, with no difference in age, sex, the presence of dysphagia, gait difficulty or follow-up duration between the two groups (9). Although confounding factors could not be excluded in both study, it'd be best to avoid empiric treatment with corticosteroids or other immunosuppressants given the lack of evidence to support their efficacy in IBM.
In addition to prednisone and IVIG, other agents targeting the immune system that have been tried in IBM include: azathioprine, methotrexate, antithymotcyte globulin, etanercept, anakinra, alemtuzumab, natalizumab and IFNβ1A (90). Furthermore, agents targeting non-inflammatory pathways included: lithium, oxandrolone, follistatin gene therapy, bimagrumab, arimoclomol, and rapamycin. Lithium was considered because it inhibits glycogen synthase kinase-3 (GSK). GSK is involved in various cell processes, such as autophagy, cell survival/differentiation, and cell cycle regulation, and has been associated with hyperphosphorylation of tau proteins [reviewed in (91)]. Oxandrolone (synthetic androgen), follistatin (myostatin inhibitor) gene therapy, and bimagrumab (myostatin inhibitor) have been considered as enhancers of skeletal muscle mass development (92–94). Arimoclomol is a heat shock protein inducer and was considered for improving protein homeostasis in stressed cells (95, 96). Rapamycin (sirolimus) was considered due to its immunosuppressive effect and ability to enhance autophagy by inhibiting the mammalian target of rapamycin (mTOR) that is an autophagy inhibitor (97). Phase 2 trial was recently completed and the primary outcome was reportedly not met. However, the full results for this study and many of the mentioned trials have not been published yet in peer-reviewed journals.
There are two main ongoing trials in IBM: Sirolimus and ABC008. Sirolimus is currently being tested in a multicenter phase 3 trial (https://clinicaltrials.gov/ct2/show/NCT04789070?recrs=abdf&cond=inclusion$+$body$+$myositis&draw=6&rank=5). ABC008 is a monoclonal antibody targeting KLRG1 receptor, which selectively depletes highly differentiated cytotoxic T cells (https://clinicaltrials.gov/ct2/show/NCT04659031). In an IBM xenograft model, human T cells were depleted from xenografts in 4 mice by treatment with a CD3 monoclonal antibody (OKT3), and compared to 4 untreated xenografts (77). MHC1 expression was subsequently reduced in the treated samples, however, rimmed vacuoles and loss of TDP-43 function persisted when evaluated at 2- and 4-months post treatment (77). The small sample size, the low percentage of muscle fibers displaying rimmed vacuoles, and the limited follow up time were acknowledged as study limitations. It remained unclear if the persistently detected myodegenerative changes reflected ongoing disease activity vs. residua from previous damage. Whether KLRG1+-T- cell depletion will halt disease progression is yet to be determined.
The most critical unmet need in IBM remains the lack of an effective treatment. This is due to several factors, especially poor understanding of the underlying pathogenesis. The complexity of IBM histopathology and disease mechanisms sparked an ongoing debate on whether the disease is primarily inflammatory or neurodegenerative in nature (98). However, determining which is the cause vs. the consequence may not be the most crucial factor to find effective treatments. Similar to other chronic disorders, the various involved immune and non-immune pathways likely form intertwined, irreversible vicious circles, that are sustained over time (99). Further research is needed to better understand the relationship between the innate immune system and neurodegeneration first, define the exact level of dysfunction in the invoked pathways in IBM, and then identify novel therapeutic targets that would help break that destructive loop. Whether systems biology approaches will help define involved pathways on an individual patient level is yet to be determined. Biological and technical variabilities remain a challenge.
From clinical trial design perspective, the main challenges are inherent to IBM's clinical heterogeneity, relative rarity, and slowly progressive course. The clinical phenotype is variable at early stages of the disease, the period where it would be ideal to intervene, whereas patients typically converge into the classic phenotype at more advanced stages (3). Hence, earlier in the disease course, the disability profile can be mostly driven by the difficulty walking, the limited use of the upper limb, the difficulty swallowing or any combination thereof. Furthermore, the weakness is commonly asymmetric differentially affecting the right and left side. This clinical heterogeneity made it challenging to have an outcome measure with sound reliability and content validity. For instance, commonly used lower limb-focused outcome measures (6-min walk distance, quadriceps strength and thigh muscle volume) may have limited validity in patients whose disability is mostly driven by upper limb or swallowing dysfunction (94). In addition to the limited sampling frame in general due to disease rarity, powering clinical trials to detect differences among patient subgroups, such as early vs. late in the disease course, disability profile, race, or sex, would affect feasibility, especially that all currently considered or in-trial drugs are expected to have, at best, a stabilizing or modest effect. Furthermore, the slowly progressive disease course makes it challenging to detect a treatment effect within a 6-to-12-month trial period. It is important to note that the traditional classification of IBM as an idiopathic inflammatory disease has indirectly affected expectations from clinical trials, as “myositis” would be expected to markedly improve or resolve for a drug to be deemed effective.
In the era of personalized medicine, individualized outcome measures and clinical trial designs offer an innovative approach to address such difficulties. The n-of-1 trial design concept is intriguing and not yet explored in IBM. N-of-1 trials allow to evaluate treatment response on an individual level using a double-blind, randomized, multiple crossover design, following the same quality standards as traditional trials (100). Results from multiple individuals could be aggregated and analyzed at a group level. The n-of-1 design is best suited for the investigation of chronic or progressive conditions, and one third of such trials have been conducted for neurological disorders (101). Despite challenges, especially logistical and related to data analysis, the n-of-1 trial design is akin to the concept of individualized medicine, and may allow to determine the best treatment regimen for a particular patient, and limit the time spent on suboptimal and/or expensive drugs approved based on results from larger traditional clinical trials (100).
Lastly, the main challenge of translating basic science findings into drug development and clinical trials is the limited availability for disease models. Hereditary inclusion body myopathy (hIBM) with frontotemporal degeneration (IBMPFD) models due to mutations in VCP have been used (102–104). However, these models had major limitations, especially that systemic proteinopathies, such as VCP-myopathy, are clinically distinct from IBM as discussed in the introduction. The IBM xenografts remain thus far the closest to recapitulate disease pathology but preclude functional and behavioral evaluation (77).
IBM is a chronic progressive disease of aging with variable disease onset, decline rate, and disability profile, at least in earlier stages of the disease. A more precise definition of involved, especially non-inflammatory, pathways, more reliable and valid outcome measures, and more variety in identified therapeutic targets are essential in order to make significant advancement in treating the disease.
EN: design and conceptualization of the work, and writing of the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rose MR. 188th ENMC International Workshop: inclusion body myositis, 2-4 december 2011, naarden, The Netherlands. Neuromuscul Disord. (2013) 23:1044–55. doi: 10.1016/j.nmd.2013.08.007
2. Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology. (2008) 70:418–24. doi: 10.1212/01.wnl.0000277527.69388.fe
3. Alamr M, Pinto MV, Naddaf E. Atypical presentations of inclusion body myositis: clinical characteristics and long-term outcomes. Muscle Nerve. (2022). doi: 10.1002/mus.27716. [Epub ahead of print].
4. Oflazer PS, Deymeer F, Parman Y. Sporadic-inclusion body myositis (s-IBM) is not so prevalent in Istanbul/Turkey: a muscle biopsy based survey. Acta Myol. (2011) 30:34–6.
5. Tan JA, Roberts-Thomson PJ, Blumbergs P, Hakendorf P, Cox SR, Limaye V. Incidence and prevalence of idiopathic inflammatory myopathies in South Australia: a 30-year epidemiologic study of histology-proven cases. Int J Rheum Dis. (2013) 16:331–8. doi: 10.1111/j.1756-185X.2011.01669.x
6. Callan A, Capkun G, Vasanthaprasad V, Freitas R, Needham M. A systematic review and meta-analysis of prevalence studies of sporadic inclusion body myositis. J Neuromuscl Dis. (2017) 4:127–37. doi: 10.3233/JND-160198
7. Shelly S, Mielke MM, Mandrekar J, Milone M, Ernste FC, Naddaf E, et al. Epidemiology and natural history of inclusion body myositis: a 40-year population-based study. Neurology. (2021) 96:e2653–e2661. doi: 10.1212/WNL.0000000000012004
8. Lindgren U, Pullerits R, Lindberg C, Oldfors A. Epidemiology, survival, and clinical characteristics of inclusion body myositis. Ann Neurol. (2022) 92:201–12. doi: 10.1002/ana.26412
9. Naddaf E, Shelly S, Mandrekar J, Chamberlain AM, Hoffman EM, Ernste FC, et al. Survival and associated comorbidities in inclusion body myositis. Rheumatology (Oxford, England). (2022) 61:2016–24. doi: 10.1093/rheumatology/keac379
10. Dobloug GC, Garen T, Brunborg C, Gran JT, Molberg Ø. Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum. (2015) 45:301–8. doi: 10.1016/j.semarthrit.2015.06.005
11. Sangha G, Yao B, Lunn D, Skorupinska I, Germain L, Kozyra D, et al. Longitudinal observational study investigating outcome measures for clinical trials in inclusion body myositis. J Neurol Neurosurg Psychiatry. (2021). doi: 10.1136/jnnp-2020-325141
12. Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long-term observational study of sporadic inclusion body myositis. Brain. (2011) 134(Pt 11):3176–84. doi: 10.1093/brain/awr213
13. Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJ, Badrising UA. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. (2011) 134(Pt 11):3167–75. doi: 10.1093/brain/awr217
14. Peng A, Koffman BM, Malley JD, Dalakas MC. Disease progression in sporadic inclusion body myositis: observations in 78 patients. Neurology. (2000) 55:296–8. doi: 10.1212/WNL.55.2.296
15. Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR. Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome. Am J Phys Med Rehabil. (2008) 87:883–9. doi: 10.1097/PHM.0b013e31818a50e2
16. Voermans NC, Vaneker M, Hengstman GJ. ter Laak HJ, Zimmerman C, Schelhaas HJ, et al. Primary respiratory failure in inclusion body myositis. Neurology. (2004) 63:2191–2. doi: 10.1212/01.WNL.0000145834.17020.86
17. Allenbach Y, Benveniste O, Decostre V, Canal A, Eymard B, Herson S, et al. Quadriceps strength is a sensitive marker of disease progression in sporadic inclusion body myositis. Neuromuscul Disord. (2012) 22:980–6. doi: 10.1016/j.nmd.2012.05.004
18. Finsterer J, Stöllberger C, Kovacs GG. Asymptomatic hyper-creatine-kinase-emia as sole manifestation of inclusion body myositis. Neurol Int. (2013) 5:34–6. doi: 10.4081/ni.2013.e11
19. Cox FM, Verschuuren JJ, Verbist BM, Niks EH, Wintzen AR, Badrising UA. Detecting dysphagia in inclusion body myositis. J Neurol. (2009) 256:2009–13. doi: 10.1007/s00415-009-5229-9
20. Ghosh PS, Laughlin RS, Engel AG. Inclusion-body myositis presenting with facial diplegia. Muscle Nerve. (2014) 49:287–9. doi: 10.1002/mus.24060
21. Alhammad RM, Naddaf E. Myopathies presenting with head drop: Clinical spectrum and treatment outcomes. Neuromuscul Disord. (2020) 30:128–36. doi: 10.1016/j.nmd.2019.12.001
22. Badrising UA, Maat-Schieman ML, van Houwelingen JC, van Doorn PA, van Duinen SG, van Engelen BG, et al. Inclusion body myositis. Clinical features and clinical course of the disease in 64 patients. J Neurol. (2005) 252:1448–54. doi: 10.1007/s00415-005-0884-y
23. Nicolau S, Liewluck T, Milone M. Myopathies with finger flexor weakness: not only inclusion-body myositis. Muscle Nerve. (2020) 62:445–54. doi: 10.1002/mus.26914
24. Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Ann Neurol. (1995) 38:705–13. doi: 10.1002/ana.410380504
25. Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. observations in 40 patients. Brain. (1989) 112 (Pt 3):727–47. doi: 10.1093/brain/112.3.727
26. Kazamel M, Sorenson EJ, Milone M. Clinical and electrophysiological findings in hereditary inclusion body myopathy compared with sporadic inclusion body myositis. J Clin Neuromuscul Dis. (2016) 17:190–6. doi: 10.1097/CND.0000000000000113
27. Pinto MV, Laughlin RS, Klein CJ, Mandrekar J, Naddaf E. Inclusion body myositis: correlation of clinical outcomes with histopathology, electromyography and laboratory findings. Rheumatology (Oxford, England). (2022) 61:2504–11. doi: 10.1093/rheumatology/keab754
28. Engel AG, Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol. (1984) 16:209–15. doi: 10.1002/ana.410160207
29. Mendell JR, Sahenk Z, Gales T, Paul L. Amyloid filaments in inclusion body myositis. novel findings provide insight into nature of filaments. Archives Neurol. (1991) 48:1229–34. doi: 10.1001/archneur.1991.00530240033013
30. Dahlbom K, Lindberg C, Oldfors A. Inclusion body myositis: morphological clues to correct diagnosis. Neuromuscul Disord. (2002) 12:853–7. doi: 10.1016/S0960-8966(02)00098-6
31. Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. (2014) 83:426–33. doi: 10.1212/WNL.0000000000000642
32. Spuler S, Emslie-Smith A, Engel AG. Amyloid myopathy: an underdiagnosed entity. Ann Neurol. (1998) 43:719–28. doi: 10.1002/ana.410430606
33. Dubourg O, Wanschitz J, Maisonobe T, Béhin A, Allenbach Y, Herson S, et al. Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol. (2011) 30:103–8.
34. Hiniker A, Daniels BH, Lee HS, Margeta M. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Communications. (2013) 1:29. doi: 10.1186/2051-5960-1-29
35. Brady S, Squier W, Sewry C, Hanna M, Hilton-Jones D, Holton JL, et al. retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open. (2014) 4:e004552. doi: 10.1136/bmjopen-2013-004552
36. Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol. (2015) 41:288–303. doi: 10.1111/nan.12149
37. Rodríguez Cruz PM, Luo YB, Miller J, Junckerstorff RC, Mastaglia FL, Fabian V. An analysis of the sensitivity and specificity of MHC-I and MHC-II immunohistochemical staining in muscle biopsies for the diagnosis of inflammatory myopathies. Neuromuscul Disord. (2014) 24:1025–35. doi: 10.1016/j.nmd.2014.06.436
38. Blume G, Pestronk A, Frank B, Johns DR. Polymyositis with cytochrome oxidase negative muscle fibres. Early quadriceps weakness and poor response to immunosuppressive therapy. Brain. (1997) 120 (Pt 1):39–45. doi: 10.1093/brain/120.1.39
39. Temiz P, Weihl CC, Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci. (2009) 278:25–9. doi: 10.1016/j.jns.2008.11.010
40. Papadimas GK, Kokkinis C, Xirou S, Chrysanthou M, Kararizou E, Papadopoulos C. Polymyositis with mitochondrial pathology or atypical form of sporadic inclusion body myositis: case series and review of the literature. Rheumatol Int. (2019) 39:1459–66. doi: 10.1007/s00296-019-04314-8
41. Winkler M, von Landenberg C, Kappes-Horn K, Neudecker S, Kornblum C, Reimann J. Diagnosis and clinical development of sporadic inclusion body myositis and polymyositis with mitochondrial pathology: a single-center retrospective analysis. J Neuropathol Exp Neurol. (2021) 80:1060–7. doi: 10.1093/jnen/nlab101
42. Dieudonné Y, Allenbach Y, Benveniste O, Leonard-Louis S, Hervier B, Mariampillai K, et al. Granulomatosis-associated myositis: high prevalence of sporadic inclusion body myositis. Neurology. (2020) 94:e910–e20. doi: 10.1212/WNL.0000000000008863
43. Alhammad RM, Liewluck T. Myopathies featuring non-caseating granulomas: Sarcoidosis, inclusion body myositis and an unfolding overlap. Neuromuscul Disord. (2019) 29:39–47. doi: 10.1016/j.nmd.2018.10.007
44. Pluk H, van Hoeve BJ, van Dooren SH, Stammen-Vogelzangs J, van der Heijden A, Schelhaas HJ, et al. Autoantibodies to cytosolic 5'-nucleotidase 1A in inclusion body myositis. Ann Neurol. (2013) 73:397–407. doi: 10.1002/ana.23822
45. Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, et al. Cytosolic 5'-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. (2013) 73:408–18. doi: 10.1002/ana.23840
46. Salam S, Dimachkie MM, Hanna MG, Machado PM. Diagnostic and prognostic value of anti-cN1A antibodies in inclusion body myositis. Clin Exp Rheumatol. (2022) 40:384–93. doi: 10.55563/clinexprheumatol/r625rm
47. Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, Tiniakou E, Petri M, Baer A, et al. Cytosolic 5'-nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases. Arthritis Care Res. (2016) 68:66–71. doi: 10.1002/acr.22600
48. Herbert MK, Stammen-Vogelzangs J, Verbeek MM, Rietveld A, Lundberg IE, Chinoy H, et al. Disease specificity of autoantibodies to cytosolic 5'-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis. (2016) 75:696–701. doi: 10.1136/annrheumdis-2014-206691
49. Tasca G, Monforte M, De Fino C, Kley RA, Ricci E, Mirabella M. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve. (2015) 52:956–62. doi: 10.1002/mus.24661
50. Ansari B, Salort-Campana E, Ogier A, Le Troter Ph DA, De Sainte Marie B, Guye M, et al. Quantitative muscle MRI study of patients with sporadic inclusion body myositis. Muscle Nerve. (2020) 61:496–503. doi: 10.1002/mus.26813
51. Cox FM, Reijnierse M, van Rijswijk CS, Wintzen AR, Verschuuren JJ, Badrising UA. Magnetic resonance imaging of skeletal muscles in sporadic inclusion body myositis. Rheumatology (Oxford, England). (2011) 50:1153–61. doi: 10.1093/rheumatology/ker001
52. Phillips BA, Cala LA, Thickbroom GW, Melsom A, Zilko PJ, Mastaglia FL. Patterns of muscle involvement in inclusion body myositis: clinical and magnetic resonance imaging study. Muscle Nerve. (2001) 24:1526–34. doi: 10.1002/mus.1178
54. Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain. (2016) 139(Pt 5):1348–60. doi: 10.1093/brain/aww024
55. Ohgami RS, Ohgami JK, Pereira IT, Gitana G, Zehnder JL, Arber DA. Refining the diagnosis of T-cell large granular lymphocytic leukemia by combining distinct patterns of antigen expression with T-cell clonality studies. Leukemia. (2011) 25:1439–43. doi: 10.1038/leu.2011.107
56. Leblanc F, Zhang D, Liu X, Loughran TP. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncology (London, England). (2012) 8:787–801. doi: 10.2217/fon.12.75
57. Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology. (1997) 48:712–6. doi: 10.1212/WNL.48.3.712
58. Cherin P, Pelletier S, Teixeira A, Laforet P, Simon A, Herson S, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology. (2002) 58:326. doi: 10.1212/WNL.58.2.326
59. Goyal NA, Cash TM, Alam U, Enam S, Tierney P, Araujo N, et al. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry. (2016) 87:373–8. doi: 10.1136/jnnp-2014-310008
60. Lucchini M, Maggi L, Pegoraro E, Filosto M, Rodolico C, Antonini G, et al. Anti-cN1A antibodies are associated with more severe dysphagia in sporadic inclusion body Myositis. Cells. (2021) 10:1146. doi: 10.3390/cells10051146
61. Felice KJ, Whitaker CH, Wu Q, Larose DT, Shen G, Metzger AL, et al. Sensitivity and clinical utility of the anti-cytosolic 5'-nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: report of 40 patients from a single neuromuscular center. Neuromuscul Disord. (2018) 28:660–4. doi: 10.1016/j.nmd.2018.06.005
62. Lilleker JB, Rietveld A, Pye SR, Mariampillai K, Benveniste O, Peeters MT, et al. Cytosolic 5'-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis. (2017) 76:862–8. doi: 10.1136/annrheumdis-2016-210282
63. Amlani A, Choi MY, Tarnopolsky M, Brady L, Clarke AE, Garcia-De La Torre I, et al. Anti-NT5c1A autoantibodies as biomarkers in inclusion body myositis. Front Immunol. (2019) 10:745. doi: 10.3389/fimmu.2019.00745
64. Paul P, Liewluck T, Ernste FC, Mandrekar J, Milone M. Anti-cN1A antibodies do not correlate with specific clinical, electromyographic, or pathological findings in sporadic inclusion body myositis. Muscle Nerve. (2020). doi: 10.1002/mus.27157
65. Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain. (2000) 123 (Pt 10):2030–9. doi: 10.1093/brain/123.10.2030
66. Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC, et al. cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology. (2007) 69:1672–9. doi: 10.1212/01.wnl.0000265398.77681.09
67. Tateyama M, Fujihara K, Misu T, Itoyama Y. CCR7+ myeloid dendritic cells together with CCR7+ T cells and CCR7+ macrophages invade CCL19+ nonnecrotic muscle fibers in inclusion body myositis. J Neurol Sci. (2009) 279:47–52. doi: 10.1016/j.jns.2008.12.024
68. Greenberg SA, Pinkus JL, Kong SW, Baecher-Allan C, Amato AA, Dorfman DM. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain. (2019) 142:2590–604. doi: 10.1093/brain/awz207
69. Goyal NA, Coulis G, Duarte J, Farahat PK, Mannaa AH, Cauchii J, et al. Immunophenotyping of inclusion body myositis blood T and NK cells. Neurology. (2022). doi: 10.1212/WNL.0000000000200013
70. Pandya JM, Fasth AE, Zong M, Arnardottir S, Dani L, Lindroos E, et al. Expanded T cell receptor Vβ-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum. (2010) 62:3457–66. doi: 10.1002/art.27665
71. Rojana-Udomsart A, Bundell C, James I, Castley A, Martinez P, Christiansen F, et al. Frequency of autoantibodies and correlation with HLA-DRB1 genotype in sporadic inclusion body myositis (s-IBM): a population control study. J Neuroimmunol. (2012) 249:66–70. doi: 10.1016/j.jneuroim.2012.04.007
72. Rothwell S, Cooper RG, Lundberg IE, Gregersen PK, Hanna MG, Machado PM, et al. Immune-Array analysis in sporadic inclusion body myositis reveals hla-drb1 amino acid heterogeneity across the myositis spectrum. Arthritis Rheumatol (Hoboken, NJ). (2017) 69:1090–9. doi: 10.1002/art.40045
73. Mastaglia FL, Needham M, Scott A, James I, Zilko P, Day T, et al. Sporadic inclusion body myositis: HLA-DRB1 allele interactions influence disease risk and clinical phenotype. Neuromuscul Disord. (2009) 19:763–5. doi: 10.1016/j.nmd.2009.07.015
74. Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. (2009) 40:19–31. doi: 10.1002/mus.21386
75. Zanusso G, Vattemi G, Ferrari S, Tabaton M, Pecini E, Cavallaro T, et al. Increased expression of the normal cellular isoform of prion protein in inclusion-body myositis, inflammatory myopathies and denervation atrophy. Brain pathology (Zurich, Switzerland). (2001) 11:182–9. doi: 10.1111/j.1750-3639.2001.tb00390.x
76. Askanas V, Engel WK, Alvarez RB, Glenner GG. beta-Amyloid protein immunoreactivity in muscle of patients with inclusion-body myositis. Lancet. (1992) 339:560–1. doi: 10.1016/0140-6736(92)90388-J
77. Britson KA, Ling JP, Braunstein KE, Montagne JM, Kastenschmidt JM, Wilson A, et al. Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci Transl Med. (2022) 14:eabi9196. doi: 10.1126/scitranslmed.abi9196
78. Catalán-García M, Garrabou G, Morén C, Guitart-Mampel M, Hernando A, Díaz-Ramos À, et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci. (2016) 130:1741–51. doi: 10.1042/CS20160080
79. Johari M, Vihola A, Palmio J, Jokela M, Jonson PH, Sarparanta J, et al. Comprehensive transcriptomic analysis shows disturbed calcium homeostasis and deregulation of T lymphocyte apoptosis in inclusion body myositis. J Neurol. (2022). doi: 10.1101/2021.06.30.450477
80. Nogalska A, Wojcik S, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp Neurol. (2007) 204:610–8. doi: 10.1016/j.expneurol.2006.12.014
81. Ikenaga C, Date H, Kanagawa M, Mitsui J, Ishiura H, Yoshimura J, et al. Muscle transcriptomics shows overexpression of cadherin 1 in inclusion body myositis. Ann Neurol. (2022). doi: 10.1002/ana.26304
82. Askanas V, Engel WK. Inclusion-body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol. (2007) 19:550–9. doi: 10.1097/BOR.0b013e3282efdc7c
83. Snedden AM, Kellett KAB, Lilleker JB, Hooper NM, Chinoy H. The role of protein aggregation in the pathogenesis of inclusion body myositis. Clin Exp Rheumatol. (2022) 40:414–24. doi: 10.55563/clinexprheumatol/pp0oso
84. Belafsky PC, Mouadeb DA, Rees CJ, Pryor JC, Postma GN, Allen J, et al. Validity and reliability of the Eating Assessment Tool (EAT-10). Ann Otol Rhinol Laryngol. (2008) 117:919–24. doi: 10.1177/000348940811701210
85. Taira K, Yamamoto T, Mori-Yoshimura M, Fujita S, Oya Y, Nishino I, et al. Obstruction-related dysphagia in inclusion body myositis: cricopharyngeal bar on videofluoroscopy indicates risk of aspiration. J Neurol Sci. (2020) 413:116764. doi: 10.1016/j.jns.2020.116764
86. Walter MC, Lochmüller H, Toepfer M, Schlotter B, Reilich P, Schröder M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study. J Neurol. (2000) 247:22–8. doi: 10.1007/s004150050005
87. Dobloug C, Walle-Hansen R, Gran JT, Molberg Ø. Long-term follow-up of sporadic inclusion body myositis treated with intravenous immunoglobulin: a retrospective study of 16 patients. Clin Exp Rheumatol. (2012) 30:838–42.
88. Jørgensen AN, Jensen KY, Nielsen JL, Frandsen U, Hvid LG, Bjørnshauge M, et al. Effects of blood-flow restricted resistance training on mechanical muscle function and thigh lean mass in sIBM patients. Scand J Med Sci Sports. (2022) 32:359–71. doi: 10.1111/sms.14079
89. Jørgensen AN, Aagaard P, Frandsen U, Boyle E, Diederichsen LP. Blood-flow restricted resistance training in patients with sporadic inclusion body myositis: a randomized controlled trial. Scand J Rheumatol. (2018) 47:400–9. doi: 10.1080/03009742.2017.1423109
90. Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nature reviews Rheumatology. (2019) 15:257–72. doi: 10.1038/s41584-019-0186-x
91. Snitow ME, Bhansali RS, Klein PS. Lithium and therapeutic targeting of GSK-3. Cells. (2021) 10:255. doi: 10.3390/cells10020255
92. Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM, et al. pilot randomized trial of oxandrolone in inclusion body myositis. Neurology. (2002) 58:1081–7. doi: 10.1212/WNL.58.7.1081
93. Mendell JR, Sahenk Z, Al-Zaidy S, Rodino-Klapac LR, Lowes LP, Alfano LN, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther. (2017) 25:870–9. doi: 10.1016/j.ymthe.2017.02.015
94. Hanna MG, Badrising UA, Benveniste O, Lloyd TE, Needham M, Chinoy H, et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. (2019) 18:834–44. doi: 10.1016/S1474-4422(19)30200-5
95. Hargitai J, Lewis H, Boros I, Rácz T, Fiser A, Kurucz I, et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Commun. (2003) 307:689–95. doi: 10.1016/S0006-291X(03)01254-3
96. Ahmed M, Machado PM, Miller A, Spicer C, Herbelin L, He J, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med. (2016) 8:331ra41. doi: 10.1126/scitranslmed.aad4583
97. Kim J, Kundu M, Viollet B, Guan KL, AMPK. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
98. Mammen AL. Inclusion body myositis: autoimmune or myodegenerative disease? Neurology. (2022) 98:521–2. doi: 10.1212/WNL.0000000000200188
99. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
100. Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per Med. (2011) 8:161–73. doi: 10.2217/pme.11.7
101. Stunnenberg BC, Berends J, Griggs RC, Statland J, Drost G, Nikles J, et al. N-of-1 trials in neurology: a systematic review. Neurology. (2022) 98:e174–e85. doi: 10.1212/WNL.0000000000012998
102. Weihl CC, Miller SE, Hanson PI, Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet. (2007) 16:919–28. doi: 10.1093/hmg/ddm037
103. Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. (2010) 19:1741–55. doi: 10.1093/hmg/ddq050
Keywords: inclusion body myositis, idiopathic inflammatory myopathies, neurodegenerative diseases, aging, individualized medicine
Citation: Naddaf E (2022) Inclusion body myositis: Update on the diagnostic and therapeutic landscape. Front. Neurol. 13:1020113. doi: 10.3389/fneur.2022.1020113
Received: 15 August 2022; Accepted: 12 September 2022;
Published: 27 September 2022.
Edited by:
Mohamed Kazamel, University of Alabama at Birmingham, United StatesReviewed by:
Kenkichi Nozaki, University of Alabama at Birmingham, United StatesCopyright © 2022 Naddaf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elie Naddaf, bmFkZGFmLmVsaWVAbWF5by5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.