 Haitian Nan
Haitian Nan Li Liu
Li Liu Kexin Xie
Kexin Xie Liyong Wu
Liyong Wu- Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
Introduction: Hereditary spastic paraplegias (HSPs) are genetic neurodegenerative diseases. The most common form of pure HSP that is inherited in an autosomal dominant manner is spastic paraplegia type 4 (SPG4), which is caused by mutations in the SPAST gene. Different theories have been proposed as the mechanism underlying SPAST-HSP for different types of genetic mutations, including gain- and loss-of-function mechanisms. To better understand the mutation mechanisms, we performed genetic analysis and investigated a truncating SPAST variant that segregated with disease in one family.
Objectives and methods: We described a pure HSP pedigree with family members across four generations. We performed genetic analysis and investigated a novel frameshift pathogenic variant (c.862_863dupAC, p. H289Lfs*27) in this family. We performed reverse transcription-polymerase chain reaction (RT-PCR), Sanger sequencing, and quantitative RT-PCR using total RNA from an Epstein-Barr virus-induced lymphoblastoid cell line produced from the proband. We also performed Western blotting on cell lysates to investigate if the protein expression of spastin is affected by this variant.
Results: This variant (c.862_863dupAC, p. H289Lfs*27) co-segregated with pure HSP in this family and is not registered in any public database. Measurement of SPAST transcripts in lymphoblasts from the proband demonstrated a reduction of SPAST transcript levels through likely nonsense-mediated mRNA decay. Immunoblot analyses demonstrated a reduction of spastin protein expression levels in lymphoblasts.
Conclusion: We report an SPG4 family with a novel heterozygous frameshift variant p.H289Lfs*27 in SPAST. Our study implies haploinsufficiency as the pathogenic mechanism for this variant and expands the known mutation spectrum of SPAST.
Introduction
Although all related to axonal degeneration in the pyramidal tract, hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous neurodegenerative disorders that cause progressive weakening and spasticity in the lower limbs (1, 2). The age of onset varies from early childhood to 70 years of age, and they can be inherited via autosomal dominant (AD), autosomal recessive (AR), X-linked, or mitochondrial mechanisms (3). Pure HSP is defined as a mostly isolated pyramidal syndrome that mostly affects the lower limbs, either with or without vibration sensation impairment and urine urgency, while complicated HSP has a more complex clinical presentation and additional neurological abnormalities (2).
The most common form of pure HSP is called spastic paraplegia type 4 (SPG4), which is caused by a heterozygous mutation of SPAST and is identified in over 25% of HSP cases (4, 5). Most of the time, SPG4 is regarded as a pure HSP (6). Spastin, a microtubule-severing AAA ATPase that is encoded by SPAST, controls microtubule dynamics and is crucial for cell division and neurogenesis (Figure 1) (7). Spastin transcript is widely expressed in non-neuronal tissues and the nervous system (8), but the disease phenotype is remarkably restricted to corticospinal axons (9). It is still debatable whether the toxic gain-of-function characteristics of the mutant spastin proteins or haploinsufficiency is the cause of the disease (6). We report a novel frameshift pathogenic variant in SPAST that co-segregated with a pure phenotype of HSP in a four-generation family. Our data suggest that the c.862_863dupAC (p. H289Lfs*27) variant exerts its pathogenic effect by haploinsufficiency.
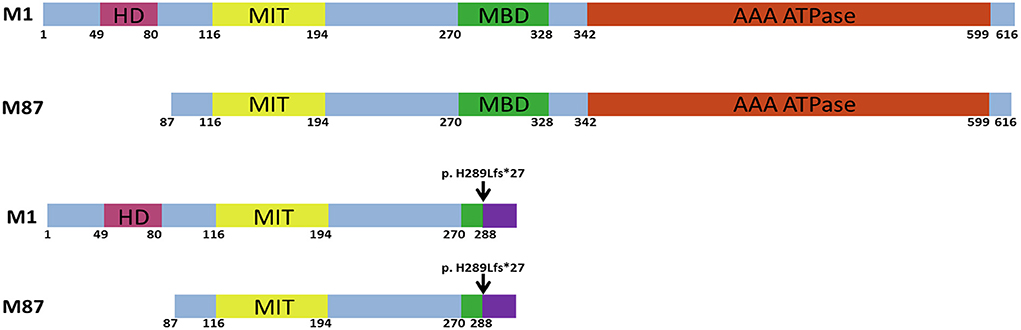
Figure 1. Schematic structure of the human wild-type and mutant spastin proteins. M1 and M87 spastin isoforms are presented. HD, hydrophobic domain; MIT, microtubule interacting and transport domain; MBD, microtubule-binding domain; AAA ATPase, ATPase associated with diverse cellular activities domain. The black arrow indicates the location of the c.862_863dupAC frameshift variant.
Materials and methods
Whole-exome sequencing study
The genomic DNA was isolated from peripheral blood leukocytes (QIAamp DNA Blood Kits, QIAGEN). Exome capture was performed with a SureSelect Human All Exon V6+UTR (89Mb) Kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was carried out on a HiSeq2500 (Illumina, San Diego, CA, USA) using a HiSeq SBS Kit V4 (Illumina), which generated 100-bp reads. The average and minimum sequencing depths were 125× and 20× , respectively. The reference databases utilized included hg38 (GRCh38) (http://genome.ucsc.edu), HGMD (https://portal.biobase-international.com), gnomAD (http://gnomad.broadinstitute.org), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and dbSNP (https://www.ncbi.nlm.nih.gov/SNP).
Cell culture and reverse transcription-polymerase chain reaction (RT-PCR)
We performed RT-PCR, Sanger sequencing, and quantitative RT-PCR using total RNA from an Epstein-Barr virus (EBV)-induced lymphoblastoid cell line (LCL) of the proband. Mononuclear cells from the patient's peripheral blood were immortalized using the concentrated EBV supernatant as previously described (10). Total RNA from cultured lymphoblastoid cells from the proband (Figure 2A, III-5) was extracted using Trizol (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using a Bio-Rad's iScript™ gDNA Clear cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Complementary DNA was then amplified by long-range PCR using a primer set designed to amplify from exon 1 to exon 17 of spastin M87 isoform. Quantitative RT-PCR (qRT-PCR) was performed with SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad). Normalization and relative quantification of the expression levels of the SPAST gene was performed using the ΔCT method, with constitutively expressed gene 18SRNA as an internal control. The primer sequences used in this study were listed in Supplementary Table 1.
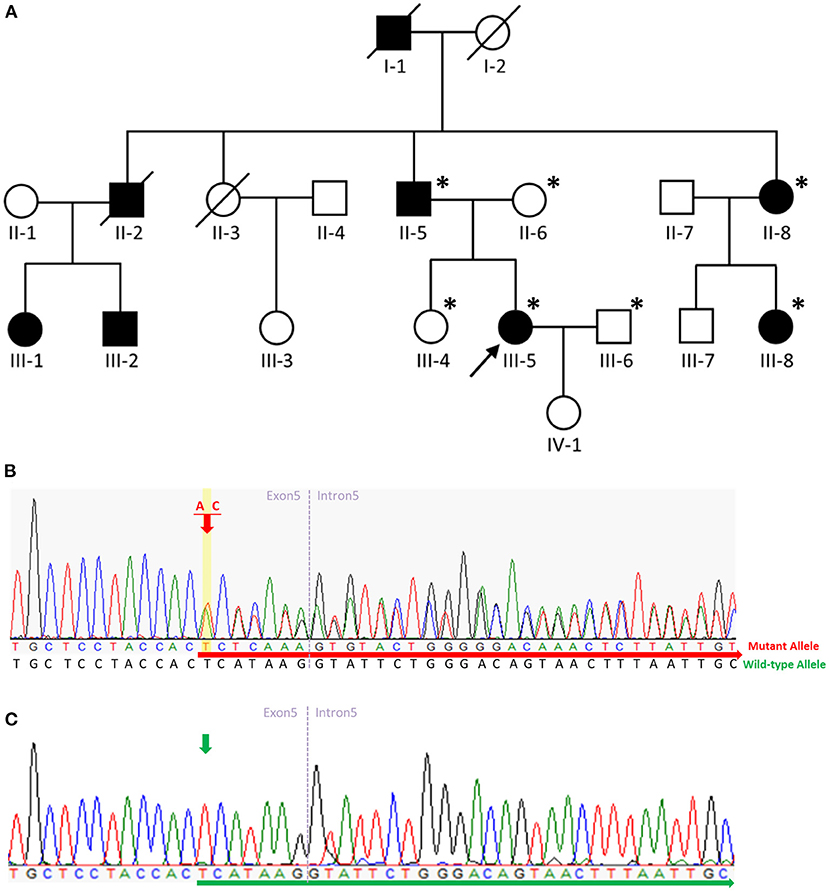
Figure 2. Pedigree of the SPG4 family and sequence analysis. (A) The proband is indicated (arrow). Squares indicate males; circles, females; slashes, deceased individuals; black symbols, individuals with symptoms of HSP; and unshaded symbols, individuals without symptoms of HSP. Patients evaluated clinically and genetically are each denoted by an asterisk. (B) Sequence analysis revealed a c.862_863dupAC frameshift variant in exon 5 of SPAST in the proband, her father, aunt, and aunt's daughter. The red arrow indicates the c.864 nucleotide. (C) Sequence analysis revealed no variant in exon 5 of SPAST in the proband's mother, husband, or sister. The green arrow indicates the c.864 nucleotide.
Immunoblotting
Cultured lymphoblastoid cells from the proband and a 58-year-old wild-type control unrelated to this family were harvested and lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, and protease inhibitors). Equal amounts of protein extracts (20μg/lane) were subjected to SDS-PAGE. After SDS-PAGE, proteins were transferred onto the PVDF membrane and then immunoblotted using an anti-spastin rabbit polyclonal antibody (#PA5-53581), which could recognize both the M1 and M87 spastin isoforms (Immunogen sequence region: amino acids 234-366). To check for loading, blots were re-probed with a mouse monoclonal anti-actin antibody (#A3854). The proteins were visualized with HRP-labeled secondary antibodies using a chemiluminescence detection system (Immobilon Forte, Millipore). Digital images were captured using ImageQuant LAS4000 (GE Healthcare).
Results
Clinical study
We describe a four-generation family with spastic paraplegia that is inherited in an autosomal dominant manner. The proband (Figure 2A, III-5), a 55-year-old female, had walked normally until age 35 when she developed gait unsteadiness and difficulty descending the staircase. Over time, her gait gradually became slow and spastic, and she experienced frequent falls. At age 48, she had to walk with the aid of crutches. A neurological examination at age 55 revealed a scissor gait, lower-limb muscle weakness, ankle clonus, and brisk bilateral knee reflexes. No muscle hypertonia or atrophy was detected, and plantar reflexes were flexor. Her cognitive functioning was normal, and no signs of cerebellar, sensory, or autonomic abnormalities were found. Studies on nerve conduction and magnetic resonance imaging of the brain and spinal cord were unremarkable.
The proband's deceased paternal grandfather (I-1), deceased uncle (II-2), father (II-5), aunt (II-8), and aunt's daughter (III-8) all presented with spastic gait. The proband's uncle's daughter (III-1) presented with spastic gait during childhood, while the proband's uncle's son (III-2) presented with spastic gait in his fifties. The proband's father (II-5) aged 84 presented with spastic gait in his early thirties. All the affected family members showed a pure form of HSP, and they could be ambulatory with crutches. The proband's 59-year-old sister (III-4) was unaffected. The clinical characteristics of the patients with the p.H289Lfs*27 variant in SPAST in this family are summarized in Supplementary Table 2.
Genetic study
We carried out WES of genomic DNA from the proband (III-5). We examined variants of 168 genes known to be involved in Charcot-Marie-Tooth disease or HSP (Supplementary Table 3). Through this analysis, we identified a heterozygous frameshift variant (c.862_863dupAC, p. H289Lfs*27 [NM_014946]) in exon 5 of the SPAST gene in the patient and did not find any additional causal genetic variants in other genes. We then examined exon 5 of the SPAST gene in the proband (III-5), the proband's father (II-5), mother (II-6), sister (III-4), husband (III-6), aunt (II-8), and aunt's daughter (III-8) by Sanger sequencing. On Sanger sequencing, we found the c.862_863dupAC variant in SPAST in a heterozygous state in the proband, her father, aunt, and aunt's daughter (Figure 2B). This variant was not detected in the proband's mother, husband, or sister without symptoms (Figure 2C), indicating the variant co-segregated with pure HSP in this family. This variant was not present in HGMD (https://portal.biobase-international.com; Last Access Date: 2022-10-22), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; Last Access Date: 2022-10-22), or GnomAD (http://gnomad.broadinstitute.org; Last Access Date: 2022-10-22), thus we considered it a novel molecular variant causing SPG4. Bioinformatic analyses using Mutation Taster (http://www.mutationtaster.org) predicted that this variant was disease-causing. This variant is designated as pathogenic in accordance with the criteria of the American College of Medical Genetics and Genomics (ACMG) (PVS1 + PS4 + PM2 + PP1 + PP3 + PP4) (11). It is noteworthy that the c.866_870del (p.His289fs) and c.867_868del (p.His289fs) variants were reported on ClinVar. The c.867_868del (p.His289fs) variant was also reported in the literature with one patient presenting a pure form of HSP and another SPG4 pedigree complicated with dysarthria (12). Including the variant we report here, all three truncating variants arising from the same amino acid (His289) seem to be pathogenic and causal for SPG4. Moreover, the c.843_846dup (p.Gly283fs) and c.883dup (p.Thr295fs) variants which are located nearby were also reported on ClinVar. While these two variants have not been reported in the literature, they were both classified as pathogenic on ClinVar.
Transcription analysis
To analyze the mRNA expression affected by the variant (c.862_863dupAC, p. H289Lfs*27) in SPAST, we performed RT-PCR using total RNA from an EBV-induced lymphoblastoid cell line produced from peripheral blood mononuclear cells of the proband (10, 13). Agarose gel electrophoresis of the long-range RT-PCR products revealed only the 1,494 bp band expected in both the patient and the control, but no aberrant bands (Figure 3A). Sanger sequencing from both directions of the RT-PCR products identified only canonical transcripts in the patient (Figure 3B). The same results were obtained with a different set of primers targeting a shorter fragment (exon3-exon8). Agarose gel electrophoresis of the shorter RT-PCR products revealed only the 603 bp band expected in both the patient and control (Figure 3C). Sanger sequencing of the RT-PCR products in both directions identified only canonical transcripts in the patient (Figure 3D). Therefore, no additional spastin isoforms were found, making alternative splicing less likely. Moreover, the truncated transcript was not detected in the patient. We then examined mRNA levels in the proband's lymphoblasts by qRT-PCR. The results showed that the expression of SPAST transcripts was decreased by approximately 0.5-fold in the patient compared to the six wild-type controls when normalized to the 18SRNA levels (Figure 4A).
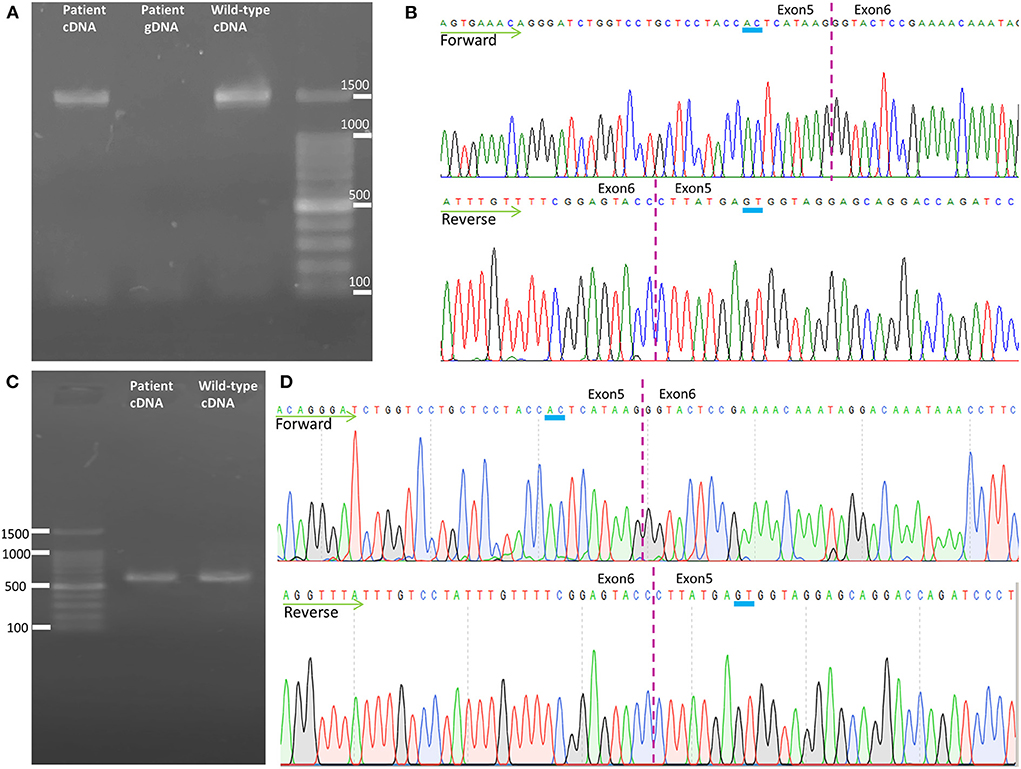
Figure 3. Transcription analysis of spastin in lymphoblast cell lines. (A) Agarose gel electrophoresis of the long-range RT-PCR products revealed only the cDNA fragment of 1,494 bp corresponding to the predicted canonical transcripts in both the patient and the control. (B) Sanger sequencing of long-range RT-PCR products revealed only the canonical transcript in the proband, which was reconfirmed on sequencing with a reverse primer. The transcript with the frameshift variant was not detected. (C) Agarose gel electrophoresis of the shorter RT-PCR products revealed only the 603 bp band expected in both the patient and control. (D) Sanger sequencing of the shorter RT-PCR products in both directions identified only canonical transcripts in the patient. cDNA, complementary DNA; gDNA, genomic DNA.
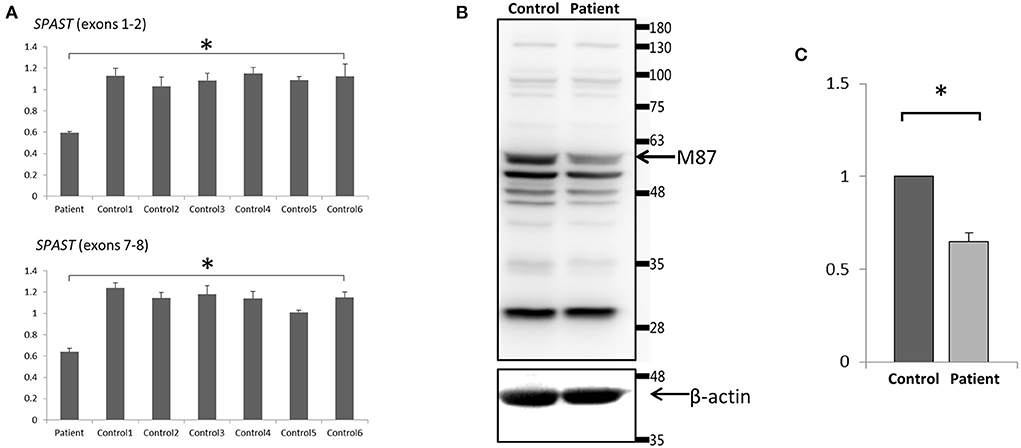
Figure 4. Expression analysis of spastin in lymphoblast cell lines. (A) Quantitative PCR of SPAST cRNA showed a ~50% decrease standardized against 18SRNA in the proband compared with 6 wild-type controls. Data were the mean (±SD) of three independent experiments, Mann-Whitney U test; *p < 0.01. (B) Western Blotting analysis revealed a decreased level of the spastin protein in the patient compared to the control. Blots are representative images from 3 independent experiments. (C) Densitometry analysis of the peak density normalized over β-actin showed a ~40% decrease of spastin protein in the patient compared with the control. Data were the mean (±SD) of three independent experiments, *P < 0.05. The Mann-Whitney test was used to compare the two groups.
Protein expression analysis
To investigate the protein expression of spastin affected by this variant, Western blot analysis of spastin was performed on cultured lymphoblastoid cell lysates from the proband and wild-type control. The band that would correspond to the wild-type spastin M87 protein (60 kD) [NP_055761] was detected, however, the spastin protein level was decreased in the patient (~40%) compared to the control (Figures 4B,C). Therefore, we may conclude that the mutant SPAST transcript harboring the premature termination codon (p.H289Lfs*27) was subjected to nonsense-mediated mRNA decay pathway and degraded. The band that would correspond to the wild-type spastin M1 protein (68 kDa) was not detected (Figure 4B), nor was it detected by immunoblotting using a spastin antibody raised specifically against the N-terminal region of the M1 spastin isoform (data not shown).
Discussion
We report an SPG4 family with a novel heterozygous frameshift pathogenic variant in SPAST. No complicated HSP-related clinical signs were noticed in any affected family members, and the disease in this pedigree was apparently autosomal dominant uncomplicated HSP. All the affected family members presented with a relatively mild phenotype, but they showed somehow different ages of onset, from childhood to over fifty years old.
All types of mutations have been described in SPAST. Missense mutations are primarily found in the AAA domain, but nonsense, splice-site, and insertion/deletion mutations can be found throughout the gene in various regions (Figure 1) (14). Together with the variant we report, three truncating variants arising from the same amino acid (His289) have been reported, indicating a possible mutation hotspot in SPAST. However, functional studies were not performed on c.866_870del (p.His289fs) and c.867_868del (p.His289fs) variants identified in SPG4 patients. Our study may provide preliminary functional evidence that all three SPAST gene variants are probably pathogenic.
Two start codons found in SPAST mRNA control the synthesis of two different spastin isoforms, M1 (68 kDa) and M87 (60 kDa) (15). Both isoforms also have low levels of splicing variants (16). In our study, only wild-type M87 (60 kDa) spastin isoform was detected by Western blot analysis. The M1 isoform was not detectable. Our results are consistent with previous studies that M87 predominates in all tissues and at all developmental stages, whereas M1 is mostly detectable in nerve tissue (15, 17). Although we cannot be certain that the observed decrease in spastin protein in lymphoblasts is representative of the brain or spinal cord where the M1 isoform of spastin is more abundant than in other tissues, our study may reveal the transcription and translation characteristics of the p.H289Lfs*27 variant in SPAST in this patient.
To date, different mechanistic hypotheses for the etiology of truncating variants in SPAST-HSP have been proposed. We have summarized the pathogenic truncating variants that have had transcription or protein expression analysis performed in previous studies (Supplementary Table 4) (16, 18–26). Haploinsufficiency has been the explanation for truncating SPAST mutations in the majority of the literature. The majority of mRNAs with premature termination codons in SPAST degrade rapidly. Meanwhile, western blot analysis of lymphoblastoid cell lines, fibroblasts, olfactory neurosphere-derived cells, and pluripotent stem cells produced from fibroblasts of the SPG4 patients revealed a decrease in the wild-type spastin protein and the absence of detectable quantities of truncated spastins (16, 19, 21–26). However, some truncating variants (c.550dupT, c.734C > G, and c.985dupA) escape degradation and are stable. Their truncated mutant proteins could accumulate to a higher level than their wild-type counterparts, indicating possible gain-of-function mechanisms (18, 20). The authors concluded that premature stop codons caused by SPAST mutations do not always result in haploinsufficiency (18). They hypothesized that the position of the premature termination codon from an exon-exon junction in SPAST may be crucial to preventing nonsense-mediated mRNA decay. Moreover, the less stable mRNAs with premature termination codons might be translated into truncated spastin proteins with greater stability (18, 20). Therefore, it would be important to perform transcription and protein expression analysis for different truncating SPAST pathogenic variants, as these studies would provide insight into the mechanisms underlying their pathogenicity.
Conclusion
We report a novel frameshift pathogenic variant (p. H289Lfs*27) in SPAST from a four-generation family with a phenotype of pure HSP. The truncating variant in SPAST resulted in mRNA instability and reduced protein expression levels. Our study implies haploinsufficiency as the pathogenic mechanism for this variant and expands the known mutation spectrum of SPAST.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: [GenBank: BankIt2579948 Seq1 ON453993], https://www.ncbi.nlm.nih.gov/nuccore/ON453993.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Capital Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HN conceived the present idea, collected and analyzed the data, and wrote the manuscript. MC, LL, and KX participated in data collection. LW contributed to the conception and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Beijing Municipal Science and Technology Committee (No.7202060) and National Natural Science Foundation of China (No.81971011).
Acknowledgments
The authors would like to thank the patients who participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1005544/full#supplementary-material
References
1. Meyyazhagan A, Orlacchio A. Hereditary spastic paraplegia: an update. Int J Mol Sci. (2022) 23:1697. doi: 10.3390/ijms23031697
2. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. (1983) 1:1151–5. doi: 10.1016/S0140-6736(83)92879-9
3. Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. (2012) 318:1–18. doi: 10.1016/j.jns.2012.03.025
4. Erfanian Omidvar M, Torkamandi S, Rezaei S, Alipoor B, Omrani MD, Darvish H, et al. Genotype-phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol. (2021) 268:2065–82. doi: 10.1007/s00415-019-09633-1
5. Rattay TW, Boldt A, Volker M, Wiethoff S, Hengel H, Schule R, et al. Non-motor symptoms are relevant and possibly treatable in hereditary spastic paraplegia type 4 (Spg4). J Neurol. (2020) 267:369–79. doi: 10.1007/s00415-019-09573-w
6. Solowska JM, Baas PW. Hereditary spastic paraplegia Spg4: what is known and not known about the disease. Brain. (2015) 138:2471–84. doi: 10.1093/brain/awv178
7. Sakoe K, Shioda N, Matsuura T. A newly identified nes sequence present in spastin regulates its subcellular localization and microtubule severing activity. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118862. doi: 10.1016/j.bbamcr.2020.118862
8. Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, et al. Spastin, a new aaa protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. (1999) 23:296–303. doi: 10.1038/15472
9. Errico A, Ballabio A, Rugarli EI. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet. (2002) 11:153–63. doi: 10.1093/hmg/11.2.153
10. Hui-Yuen J, McAllister S, Koganti S, Hill E, Bhaduri-McIntosh S. Establishment of epstein-barr virus growth-transformed lymphoblastoid cell lines. J Vis Exp. (2011) 57:3321. doi: 10.3791/3321
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Elert-Dobkowska E, Stepniak I, Krysa W, Rajkiewicz M, Rakowicz M, Sobanska A, et al. Molecular spectrum of the spast, Atl1 and Reep1 gene mutations associated with the most common hereditary spastic paraplegias in a group of polish patients. J Neurol Sci. (2015) 359:35–9. doi: 10.1016/j.jns.2015.10.030
13. Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. (2006) 7:118. doi: 10.1186/1471-2164-7-118
14. Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV, et al. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of spast mutations in hereditary spastic paraplegia. Eur J Hum Genet. (2009) 17:187–94. doi: 10.1038/ejhg.2008.147
15. Claudiani P, Riano E, Errico A, Andolfi G, Rugarli EI. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp Cell Res. (2005) 309:358–69. doi: 10.1016/j.yexcr.2005.06.009
16. Svenson IK, Ashley-Koch AE, Gaskell PC, Riney TJ, Cumming WJ, Kingston HM, et al. Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am J Hum Genet. (2001) 68:1077–85. doi: 10.1086/320111
17. Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, et al. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. (2008) 28:2147–57. doi: 10.1523/JNEUROSCI.3159-07.2008
18. Chen R, Du S, Yao Y, Zhang L, Luo J, Shen Y, et al. A novel spast mutation results in spastin accumulation and defects in microtubule dynamics. Mov Disord. (2022) 37:598–607. doi: 10.1002/mds.28885
19. Riano E, Martignoni M, Mancuso G, Cartelli D, Crippa F, Toldo I, et al. Pleiotropic effects of spastin on neurite growth depending on expression levels. J Neurochem. (2009) 108:1277–88. doi: 10.1111/j.1471-4159.2009.05875.x
20. Solowska JM, Rao AN, Baas PW. Truncating mutations of spast associated with hereditary spastic paraplegia indicate greater accumulation and toxicity of the M1 isoform of spastin. Mol Biol Cell. (2017) 28:1728–37. doi: 10.1091/mbc.e17-01-0047
21. Burger J, Fonknechten N, Hoeltzenbein M, Neumann L, Bratanoff E, Hazan J, et al. Hereditary spastic paraplegia caused by mutations in the Spg4 gene. Eur J Hum Genet. (2000) 8:771–6. doi: 10.1038/sj.ejhg.5200528
22. Kawarai T, Montecchiani C, Miyamoto R, Gaudiello F, Caltagirone C, Izumi Y, et al. Spastic paraplegia type 4: a novel spast splice site donor mutation and expansion of the phenotype variability. J Neurol Sci. (2017) 380:92–7. doi: 10.1016/j.jns.2017.07.011
23. Newton T, Allison R, Edgar JR, Lumb JH, Rodger CE, Manna PT, et al. Mechanistic basis of an epistatic interaction reducing age at onset in hereditary spastic paraplegia. Brain. (2018) 141:1286–99. doi: 10.1093/brain/awy034
24. Abrahamsen G, Fan Y, Matigian N, Wali G, Bellette B, Sutharsan R, et al. A patient-derived stem cell model of hereditary spastic paraplegia with spast mutations. Dis Model Mech. (2013) 6:489–502. doi: 10.1242/dmm.010884
25. Charvin D, Cifuentes-Diaz C, Fonknechten N, Joshi V, Hazan J, Melki J, et al. Mutations of Spg4 are responsible for a loss of function of spastin, an abundant neuronal protein localized in the nucleus. Hum Mol Genet. (2003) 12:71–8. doi: 10.1093/hmg/ddg004
Keywords: hereditary spastic paraplegias, SPG4, spastin, SPAST, haploinsufficiency
Citation: Nan H, Chu M, Liu L, Xie K and Wu L (2022) A novel truncating variant of SPAST associated with hereditary spastic paraplegia indicates a haploinsufficiency pathogenic mechanism. Front. Neurol. 13:1005544. doi: 10.3389/fneur.2022.1005544
Received: 28 July 2022; Accepted: 27 October 2022;
Published: 14 November 2022.
Edited by:
Manabu Funayama, Juntendo University, JapanReviewed by:
Angelica D'Amore, Boston Children's Hospital and Harvard Medical School, United StatesQueenie Tan, Duke University Medical Center, United States
Copyright © 2022 Nan, Chu, Liu, Xie and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyong Wu, d215d2x5QGhvdG1haWwuY29t