 Shirong Li1,2
Shirong Li1,2 Junyu Lin1
Junyu Lin1 Chunyu Li1
Chunyu Li1 Yongping Chen1
Yongping Chen1 Bei Cao1
Bei Cao1 Tianmi Yang1
Tianmi Yang1 Qianqian Wei1
Qianqian Wei1 Bi Zhao1
Bi Zhao1 Xueping Chen1
Xueping Chen1 Huifang Shang1*
Huifang Shang1*- 1Department of Neurology, Laboratory of Neurodegenerative Disorders, Rare Diseases Center, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Neurology, Guizhou Provincial People's Hospital, Guiyang, China
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by loss of the upper and lower motor neurons from the motor cortex, brainstem, and spinal cord. Most ALS cases are sporadic, with 5–10% having a positive family history. Autosomal dominant polycystic kidney disease (ADPKD) is a heritable renal disease that eventually results in end-stage kidney disease. PKD1 is the most prevalent causative gene for ADPKD, accounting for ~85% of cases. Both diseases are currently considered untreatable. In this study, we report a large family that includes 10 patients with ALS phenotype, 3 asymptomatic SOD1-H47R carriers, and 6 with the ADPKD phenotype. Using whole exome sequencing, we found a novel likely pathogenic variant (p.R2787P) in PKD1 among patients with ADPKD, and a pathogenic variant (p.H47R) in SOD1 among patients with ALS. This study highlights the possibility that two different autosomal dominantly inherited diseases can co-exist independently within the same family. Phenotype—genotype correlations among these patients are also described. This research contributes novel phenotype and genotype characteristics of ALS with SOD1 mutations and ADPKD with PKD1 mutations.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder mainly characterized by degeneration of upper and lower motor neurons, which causes death at a median of 3 years after onset (1). A recent study determined an average global incidence of 1.59 and a prevalence of 4.42 per 100,000 individuals (2). Family ALS (fALS) is mainly caused by mutations of four genes: C9ORF72, SOD1, FUS, and TARDBP. Variants of the copper zinc superoxide dismutase 1 (SOD1) gene are responsible for 10–20% of fALS cases (3).
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary renal disease, with an estimated prevalence of 1 in 2,000–4,000 individuals (4–6). About half of patients with ADPKD develop end-stage kidney disease (ESKD) by age 60 (7). ADPKD presents structural and functional kidney defects and various accompanying extrarenal complications. Polycystic kidney disease (PDK) 1 (PKD1) and 2 (PKD2) have been identified as the genes related to ADPKD (8).
To date, no cases of co-occurrence of these two genetic diseases, PKD and ALS, within a single family have been reported. Herein, we report a large family in which SOD1-ALS and PKD1-ADPKD occurred independently.
Materials and methods
Family members
Proband and family members, who were from the Han Chinese population, were recruited by the Department of Neurology, West China Hospital of Sichuan University. Clinical investigations indicated that there was a total of 73 family members, among which 10 patients with ALS phenotype, 3 asymptomatic SOD1-H47R carriers, and 6 with the ADPKD phenotype (Figure 1). Patients with ALS were examined by at least two senior neurologists and diagnosed according to the revised EI Escorial criteria (9). Deceased patients had presented with the ALS or ADPKD phenotype before death.
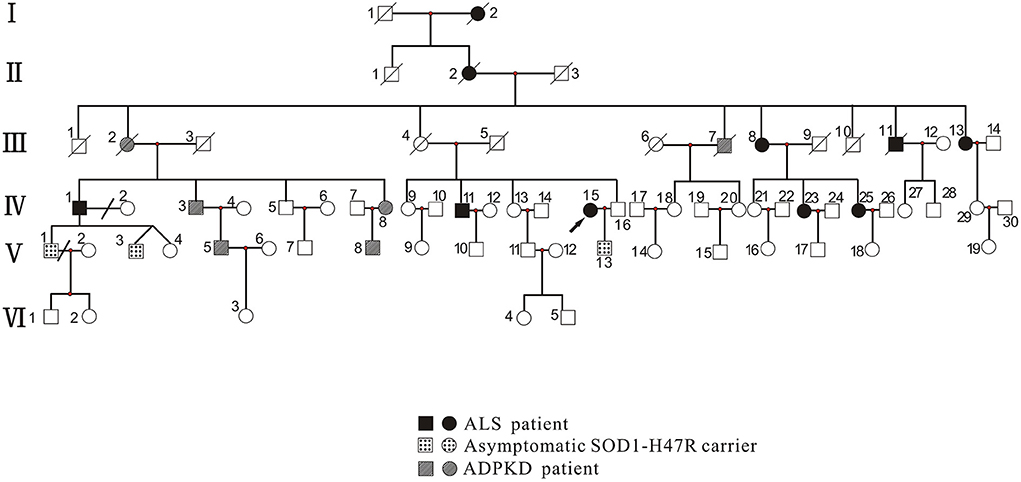
Figure 1. Pedigree of family with ALS and ADPKD. Patients with ALS included those with the ALS phenotype (III-13, IV-1, IV-11, IV-15, IV-23, and IV-25) who carried the SOD1 p.H47R mutation (note that I-2, II-2, III-8, and III-11 did not undergo genetic testing). Asymptomatic SOD1-H47R carriers were those who had no ALS phenotype but carried the SOD1 p.H47R mutation (V-1, V-3, and V-13). Patients with ADPKD had the ADPKD phenotype (IV-3, IV-8, V-5, and V-8) and carried the PKD1 p.R2787P variant (note that III-2 and III-7 did not undergo genetic testing). Circles indicate women. Squares indicate men. Arrow indicates proband. Diagonal lines indicate deceased individuals.
The ADPKD diagnostic criteria were: aged < 30 years with >2 renal cysts (on one side or 1 on each side); age 30–59 years with ≥2 cysts (on each side); or aged ≥60 years with ≥4 cysts (on each side). However, the diagnostic criteria can be modified if the patient has a family history, extrarenal manifestations, or positive genetic tests for diseases such as polycystic liver, intracranial aneurysms, or cardiac valvular abnormalities (10). The following clinical data were collected: sex, age, education, height, weight, age of onset, site of symptoms onset, presence of respiratory failure, and wheelchair dependency. Body mass index (BMI) was calculated as body weight (kg) divided by height squared (m2). Survival time was defined as the duration from disease onset to death. Detailed neurological examinations were conducted to assess upper and lower motor neuron signs. Disease severity was evaluated using the ALS functional rating scale-revised (ALSFRS-R). Frontal lobe function was assessed using the Frontal Assessment Battery (FAB) scale. Cognitive impairment was assessed using the Montreal Cognitive Assessment (MoCA) scale. We also collected blood and cerebrospinal fluid (CSF) samples of the proband members. Electromyography (EMG), abdominal Doppler ultrasound (DU), and magnetic resonance imaging (MRI) of the head or abdomen were also conducted on patients. This research was approved by the Ethics Committee of the West China Hospital, Sichuan University.
Whole exome sequencing and multiplex ligation-dependent probe amplification
A total of 19 DNA samples were available. Genomic DNA from blood samples was extracted using standard procedures (QIAGEN, Valencia, CA). Using a GenCap capture kit (MyGenostics GenCap Enrichment Technologies, Beijing, China). Enrichment libraries were sequenced using a DNBSEQ (DNBSEQ-T7) (MGI, Shenzhen, China) sequence for paired-end reads of 150 bp. After sequencing, raw data were saved in FASTQ format. Quality control filters were applied to remove low-quality reads. Reads were aligned to the human reference genome (UCSC Genome Browser build hg19) using Burrows-Wheeler Aligner20. Single-nucleotide polymorphism (SNP) and insertion/deletion variants were detected by GATK HaplotypeCaller. Data were then transformed to variant call format and variants further annotated by ANNOVAR and associated with multiple databases, including 1000 Genomes, ESP6500, dbSNP, and ExAC, and predicted by SIFT, PolyPhen-2, Mutation Taster, and GERP++. Pathogenicity of variation loci was also analyzed according to the American College of Medical Genetics and Genomics (ACMG) genetic variation classification criteria and guidelines (11, 12). Sanger sequencing was used to confirm identified mutations. MPLA was also conducted to exclude spinal muscular atrophy (SMA).
Results
Clinical and genetic features of the 10 patients with ALS and 3 asymptomatic SOD1-H47R carriers
The proband (Figure 1, IV-15) was a 57-year-old female with an 11 year history of progressive muscle weakness. The patient first noticed right lower limb weakness at age 46, followed by weakness and atrophy of all four limbs with proximal to distal progression. Neurological examination revealed severe muscle weakness in upper [Medical Research Council (MRC) 3/5] and lower (MRC 2/5) limbs, marked atrophy of all four limbs, hyporeflexia of all four limbs, and decreased muscle tone. No other abnormal neurological signs were found. She became wheelchair-dependent 6 years after onset. At diagnosis, her ALSFRS-R score was 27, the MoCA score was 27 (with 6 years of education), and she had a BMI of 24.0 kg/m2. Neurofilament light chain (NFL) in CSF was significantly elevated (1,349.27 pg/ml, normal: < 830.00 pg/ml). EMG revealed acute and chronic denervation in muscles of all limbs and rectus abdominis. Head MRI and abdominal DU showed no abnormalities. MLPA detected no abnormality in SMN1/SMN2 genes. WES revealed a pathogenic (PS3+PS4+PM1+PM2_Supporting+PM5) variant (c.140A>G p.H47R) in the SOD1 gene (Figure 2A). Verified SOD1 genes of other family members are shown in Supplementary Table 1. Unfortunately, the proband's parents were deceased and their clinical data and genetic evaluations were unavailable.
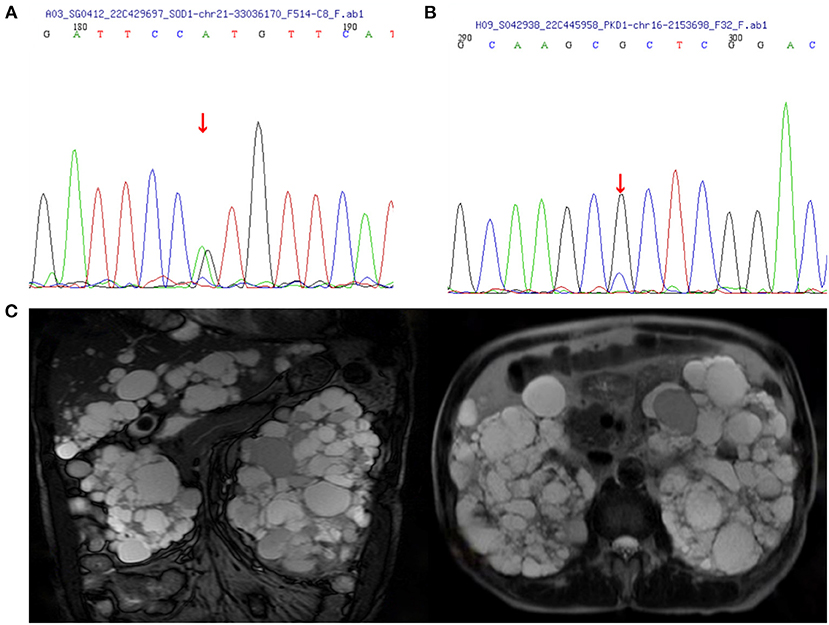
Figure 2. Images of polycystic kidney disease and sanger sequence. (A) SOD1:c.140(exon2)A>G. (B) PKD1:c.8360(exon23)G>C. (C) MRI images of the case IV-3 associated with polycystic liver disease and polycystic kidney disease.
Genetic testing revealed a c.140A>G (p.H47R) variant in the SOD1 gene in members of III-13, IV-1, IV-11, IV-23, IV-25, V-1, V-3, and V-13, in addition to the IV-15 proband (Supplementary Table 1). At enrollment, 3 family members, including V-1 (age 40 years), V-3 (age 19 years), and V-13 (age 30 years) had no symptoms of muscle weakness or atrophy or abnormal neurological signs. Among all affected members, I-2, II-2, III-8, and III-11 did not receive genetic screening for SOD1 as they were deceased, or DNA samples were unavailable. Detailed clinical characteristics are shown in Table 1. All patients, including I-2, II-2, III-8, III-11, III-13, IV-1, IV-11, IV-23, and IV-25, in addition to the proband, developed lower limb weakness as their initial symptom, which progressed to other limbs, with no bulbar onset. Aside from IV-11, those who had 2 years of education had lower MoCA and FAB scores (17 and 14, respectively), and no patient had cognitive impairment. I-2 and III-11 had long survival times (20 and 30 years, respectively), though II-2 had asphyxia during the final two survival years.
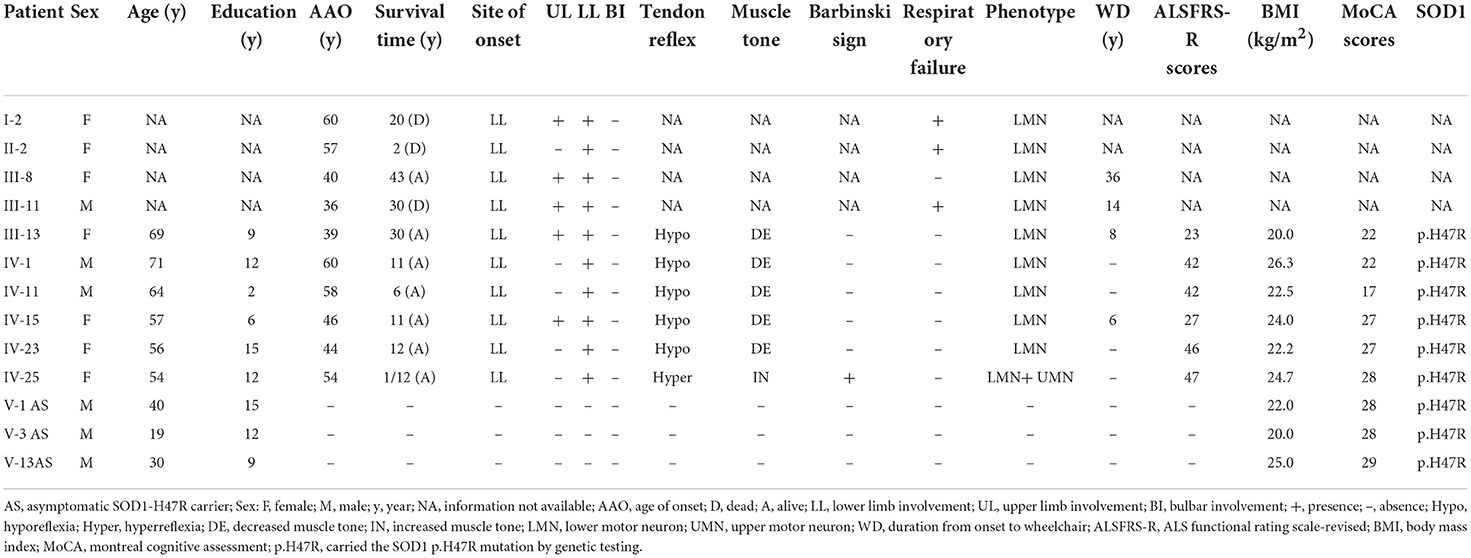
Table 1. Clinical and genetic features of 10 patients with ALS and 3 asymptomatic SOD1-H47R carriers.
Neurological examination of all affected and available members, except for IV-25 showed lower motor neuron involvement. IV-25 had mild lower limb weakness and predominate upper motor neuron affected signs including increased muscle tone, hyperreflexia, and positive Hoffmann and Babinski signs. All patient EMGs showed neurogenic changes in limb and paraspinal muscles.
Clinical and genetic features of 6 patients with ADPKD
Within this family, case IV-3 was a 70-year-old man who developed gross hematuria and hypertension, and his abdominal MRI (Figure 2C) showed multiple liver cysts and renal cysts when he was age 40 years. He was diagnosed with PKD and later developed ESKD at age 60 when he began hemodialysis therapy. His most recent (April 18, 2022) blood biochemical indices were urea 17.4 mmo/l, creatinine 1,176 umol/l and glomerular filtration rate (GFR) < 15 ml/min. WES revealed a novel heterozygous missense variant (c.8360G>C p.R2787P) on exon 23 of the PKD1 gene (Figure 2B). The validated PKD1 genes of other family members are shown in Supplementary Table 1. The integrative genomics viewer (IGV) screenshot shows the number of reads supporting each allele (Supplementary Figure 4). The sequencing depth of chr16:2153698 (PKD1 p.R2787P) is 438 and the “G” variant was 208. This variant is located in the mutation hot spot area (Supplementary Figures 3A,B) and was absent from both the 1000 Genomes Project and the Human Genetic Variation Database. It was predicted as harmful by multiple software analyses (SIFT, PolyPhen-2, Mutation Taster, and GERP++). The homology comparison of the amino acid sequences of PKD1 between homo sapiens and other animals is shown in Supplementary Figures 2A,B. The mutated amino acid variant was located in the conserved region, indicating that the mutation might affect the gene's protein function. Cases III-2 and III-7 had a history of PKD but did not undergo genetic screening as they were deceased. Sanger sequencing identified a mutation of the PKD1 gene (c.8360G>C p.R2787P) in cases IV-8, V-5, and V-8. Abdominal DU showed multiple renal and liver cysts (Supplementary Figure 1). The detailed characteristics of these patients with PKD are outlined in Table 2. This variant was thus co-segregated in the family and considered a likely pathogenic variant (PM1+PM2_Supporting+PP1+PP2+PP3) according to the ACMG guidelines.
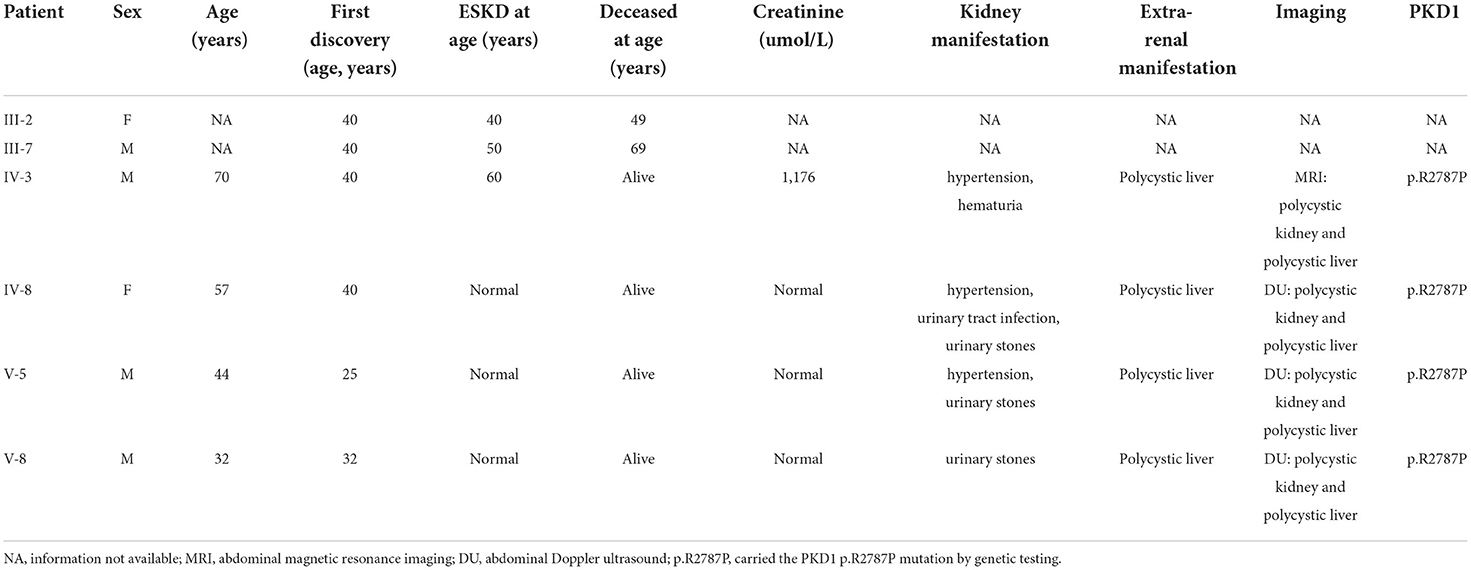
Table 2. Clinical and genetic features of six ADPKD patients.
Discussion
The present study has described a large Chinese family with 10 patients with ALS phenotype-−3 with asymptomatic SOD1 p.H47R variant carriers and 6 with the ADPKD phenotype—in whom we performed WES, MLPA, and Sanger sequencing. The ADPKD phenotype was caused by a novel, likely pathogenic mutation (c.8360G>C, p.R2787P) in PKD1, and the ALS phenotype was caused by a pathogenic variant (c.140A>G, p.H47R) in SOD1. This is the first reported family to have simultaneously co-occurring ADPKD and ALS.
SOD1, which is on chromosome 21q22.11 and encodes Cu/Zn superoxide dismutase, was identified as the first ALS causative gene in 1993 (13). The SOD1 protein acts as an antioxidant enzyme by converting two superoxide anions, which are byproducts of cellular respiration with oxidant action, into hydrogen peroxide and oxygen (14). Mutant SOD1 induces numerous toxic effects associated with pathological misfolding and aggregation of mutant SOD1 species. To date, more than 238 SOD1 mutations have been reported (http://www.hgmd.cf.ac/uk/). In our previous research, we found that SOD1 is the most common causative gene, followed by FUS, TARDBP, VCP, and OPTN and that it accounts for 21.88% of fALS in Southwest China (15, 16). Analyzing this large family indicated that the patients with ALS carried SOD1 p.H47R variant (also coded as p.H46R), which has been reported to be the most frequent variant in Chinese ALS patients with SOD1 mutations (17). All our patients with the SOD1 p.H47R variant presented with a predominated lower motor neuron deficit phenotype, rare bulbar or cognitive involvement, and considerably slow progression with longer survival time, similar to previous reports of patients with ALS with the same SOD1 mutation (18, 19). Thus, molecular diagnostics may be needed to distinguish this phenotype of ALS from pure lower motor neuron syndrome, such as SMA.
Herein, only the proband underwent CSF NFL assessment, which was significantly increased, consistent with previous reports (20, 21). Of particular note, reduced levels of toxic variants appear to be a promising therapeutic strategy for SOD1-related ALS (14). Further investigations of NFL changes and their relation to ALS progression in asymptomatic carriers are needed and will provide potentially significant future directions for research.
In this family, both IV-1 and his grandmother (II-2) suffered from ALS, while his mother (III-2) suffered from ADPKD. Therefore, we conclude that III-2 was a likely carrier of the SOD1 variant, though she died of ESKD at age 49, likely prior to ALS symptom onset. III-4 was also a likely carrier of the pathogenic SOD1 variant, as III-7 is affected by ADPKD. III-13 recalled that her father (II-3) suffered from transient hematuria at age 50, had no further consultation or diagnosis, and died of suicide at age 55. We thus suspect that the PKD1 variant was inherited by II-3 and not by II-2 (co-occurrence in the same patient). However, one study limitation is that detailed PKD information from the offspring and other family members of II-3 could not be obtained since they had either lost contact or died. Short tandem repeat (STR) analysis was not performed since many third-generation family members had died and blood samples were unavailable.
ADPKD, the fourth leading cause of ESKD worldwide, remains untreatable (22). It is characterized by the development of fluid-filled renal cysts, causing progressive loss of kidney function and culminating in the need for renal replacement therapy or kidney transplant (23). The typical clinical phenotypes also include extrarenal manifestation with polycystic liver and arachnoid cysts. PKD1 and PKD2 account for ~85 and 15% of patients with ADPKD, respectively (24). PKD1 encodes polycystin1, which contains a large N-terminal extracellular region, multiple transmembrane domains, and a cytoplasmic C-tail (25), and plays a role in renal tubular development. To date, more than 2,691 PKD1 variants have been reported (http://www.hgmd.cf.ac.uk/). Herein, a novel heterozygous missense variant c.8360G>C (p.R2787P) in the PKD1 was identified among the family members with ADPKD. These patients also presented with typical ADPKD phenotypes, consistent with previous reports (7).
To our knowledge, this is the first report of the coexistence of important hereditary diseases, ADPKD and ALS, in the same family. Notably, the occurrence of these disorders in the same individual may complicate their clinical presentation or lead to diagnostic delay. Although no member of this family had both diseases, possibly due to the rarity of the two genes on different chromosomes being transmitted simultaneously, this possibility cannot be completely eliminated. The study limitation is that detailed PKD information could not be obtained from other members of II-3's family, though we will continue to follow this family. Genetic counseling for members of such families should consider the overall family context.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Hospital, Sichuan University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HS designed and conceptualized the study. SL, JL, CL, YC, BC, TY, QW, BZ, XC, and HS contributed patient material and clinical data. SL wrote the first draft of the manuscript. HS, JL, and CL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Sichuan Science and Technology Program (Grant No. 2022ZDZX0023).
Acknowledgments
The authors thank the patients and their family members for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1004909/full#supplementary-material
References
1. Bernard E, Pegat A, Svahn J, Bouhour F, Leblanc P, Millecamps S, et al. Clinical and molecular landscape of ALS patients with SOD1 Mutations: novel pathogenic variants and novel phenotypes. A single ALS center study. Int J Mol Sci. (2020) 21:6807. doi: 10.3390/ijms21186807
2. Goutman SA, Hardiman O, Al-Chalabi A, Chio A, Savelieff MG, Kiernan MC, et al. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. (2022) 21:480–93. doi: 10.1016/S1474-4422(21)00465-8
3. Hayashi Y, Homma K, Ichijo H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul. (2016) 60:95–104. doi: 10.1016/j.jbior.2015.10.006
4. Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol. (2018) 29:2593–600. doi: 10.1681/ASN.2018050493
5. Willey C, Kamat S, Stellhorn R, Blais J. Analysis of nationwide data to determine the incidence and diagnosed prevalence of autosomal dominant polycystic kidney disease in the USA: 2013-2015. Kidney Dis. (2019) 5:107–17. doi: 10.1159/000494923
6. Solazzo A, Testa F, Giovanella S, Busutti M, Furci L, Carrera P, et al. The prevalence of autosomal dominant polycystic kidney disease (ADPKD): a meta-analysis of European literature and prevalence evaluation in the Italian province of Modena suggest that ADPKD is a rare and underdiagnosed condition. PLoS ONE. (2018) 13:e0190430. doi: 10.1371/journal.pone.0190430
7. Fukunaga S, Kamei F, Sonoda H, Oba M, Kawanishi M, Egawa M, et al. Detection of autosomal dominant polycystic kidney disease by medical checkup at an early stage. Cureus. (2021) 13:e18595. doi: 10.7759/cureus.18595
8. Oh YK, Park HC, Ryu H, Kim YC, Oh KH. Clinical and genetic characteristics of Korean autosomal dominant polycystic kidney disease patients. Korean J Intern Med. (2021) 36:767–79. doi: 10.3904/kjim.2021.176
9. Brooks BR, Miller RG, Swash M, Munsat TL. World federation of neurology research group on motor neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
10. Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. (2009) 20:205–12. doi: 10.1681/ASN.2008050507
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. (2018) 39:1593–613. doi: 10.1002/humu.23630
13. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. (1993) 364:362. doi: 10.1038/364362c0
14. Abati E, Bresolin N, Comi G, Corti S. Silence superoxide dismutase 1 (SOD1): a promising therapeutic target for amyotrophic lateral sclerosis (ALS). Expert Opin Ther Targets. (2020) 24:295–310. doi: 10.1080/14728222.2020.1738390
15. Chen YP, Yu SH, Wei QQ, Cao B, Gu XJ, Chen XP, et al. Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population. J Med Genet. (2022) 59:840–9. doi: 10.1136/jmedgenet-2021-107965
16. Wei Q, Zhou Q, Chen Y, Ou R, Cao B, Xu Y, et al. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: a case-control study and literature review. Sci Rep. (2017) 7:44606. doi: 10.1038/srep44606
17. Liu ZJ, Lin HX, Wei Q, Zhang QJ, Chen CX, Tao QQ, et al. Genetic spectrum and variability in chinese patients with amyotrophic lateral sclerosis. Aging Dis. (2019) 10:1199–206. doi: 10.14336/AD.2019.0215
18. Zou ZY, Liu MS, Li XG, Cui LY. H46R SOD1 mutation is consistently associated with a relatively benign form of amyotrophic lateral sclerosis with slow progression. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:610–3. doi: 10.1080/21678421.2016.1199698
19. Yamashita S, Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener. (2015) 4:13. doi: 10.1186/s40035-015-0036-y
20. Schreiber S, Spotorno N, Schreiber F, Acosta-Cabronero J, Kaufmann J, Machts J, et al. Significance of CSF NfL and tau in ALS. J Neurol. (2018) 265:2633–45. doi: 10.1007/s00415-018-9043-0
21. Lu CH, Macdonald-Wallis C, Gray E, Pearce N, Petzold A, Norgren N, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. (2015) 84:2247–57. doi: 10.1212/WNL.0000000000001642
22. Ong ACM, Devuyst O, Knebelmann B, Walz G. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. (2015) 385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2
23. Benson KA, Murray SL, Senum SR, Elhassan E, Conlon ET, Kennedy C, et al. The genetic landscape of polycystic kidney disease in Ireland. Eur J Hum Genet. (2021) 29:827–38. doi: 10.1038/s41431-020-00806-5
24. Ma M. Cilia and polycystic kidney disease. Semin Cell Dev Biol. (2021) 110:139–48. doi: 10.1016/j.semcdb.2020.05.003
Keywords: amyotrophic lateral sclerosis, SOD1, autosomal dominant polycystic kidney disease (ADPKD), PKD1, Chinese
Citation: Li S, Lin J, Li C, Chen Y, Cao B, Yang T, Wei Q, Zhao B, Chen X and Shang H (2022) Clinical and genetic study of a Chinese family affected by both amyotrophic lateral sclerosis and autosomal dominant polycystic kidney disease. Front. Neurol. 13:1004909. doi: 10.3389/fneur.2022.1004909
Received: 27 July 2022; Accepted: 16 September 2022;
Published: 20 October 2022.
Edited by:
Xin-Ming Shen, Mayo Clinic, United StatesReviewed by:
Dario Ronchi, University of Milan, ItalyMelissa Nel, University of Cape Town, South Africa
Copyright © 2022 Li, Lin, Li, Chen, Cao, Yang, Wei, Zhao, Chen and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huifang Shang, aGZzaGFuZzIwMDJAMTI2LmNvbQ==