 Ji-Hoon Na
Ji-Hoon Na Min Jung Lee
Min Jung Lee Chul Ho Lee
Chul Ho Lee Young-Mock Lee
Young-Mock Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 10 December 2021
Sec. Neurogenetics
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.752467
Background and Purpose: Recent advances in molecular genetic testing have led to a rapid increase in the understanding of the genetics of Leigh syndrome. Several studies have suggested that Leigh syndrome with MT-ND3 mutation is strongly associated with epilepsy. This study focused on the epilepsy-related characteristics of Leigh syndrome with MT-ND3 mutation identified in a single tertiary hospital in South Korea.
Methods: We selected 31 patients with mitochondrial DNA (mtDNA) mutations who were genetically diagnosed with mtDNA-associated Leigh syndrome. Among them, seven patients with MT-ND3 mutations were detected. We reviewed various clinical findings such as laboratory findings, brain images, electroencephalography data, seizure types, seizure frequency, antiepileptic drug use history, and current seizure status.
Results: The nucleotide changes in the seven patients with the Leigh syndrome with MT-ND3 mutation were divided into two groups: m.10191T>C and m.10158T>C. Six of the seven patients were found to have the m.10191T>C mutations. The median value of the mutant load was 82.5%, ranging from 57.9 to 93.6%. No particular tendency was observed for the first symptom or seizure onset or mutant load. The six patients with the m.10191T>C mutation were diagnosed with epilepsy. Three of these patients were diagnosed with Lennox–Gastaut syndrome (LGS).
Conclusion: We reported a very strong association between epilepsy and MT-ND3 mutation in Leigh syndrome, particularly the m.10191T>C mutation. The possibility of an association between the epilepsy phenotype of the m.10191T>C mutation and LGS was noted.
Leigh syndrome (or subacute necrotizing encephalomyelopathy) is the most common clinical syndrome of mitochondrial disease that occurs in the pediatric patients. Leigh syndrome is further divided into mitochondrial DNA (mtDNA) associated and nuclear associated (1–4). Recent advances in molecular genetic testing approaches have led to a rapid increase in the understanding of the genetics of Leigh syndrome. Due to this comprehensive genomic testing, a genotype–phenotype correlation has been actively attempted according to the genetic mutations of Leigh syndrome. In particular, the genotype–phenotype correlation of mtDNA-associated Leigh syndrome is more challenging, as heteroplasmy must be considered and genetic mutation type (2–5).
In total, mutations in 14 mitochondrial genes are known to be associated with Leigh syndrome (MT-ATP6, MT-CO3, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6, MT-T1, MT-TK, MT-TL1, MT-TL2, MT-TV, and MT-TW) (4). Genotype–phenotype studies of each of these mutations are currently underway. Among them, several studies have found that Leigh syndrome with MT-ND3 mutation (hereafter referred as Leigh syndrome with MT-ND3 mutation) is strongly associated with epilepsy (6–8). This knowledge may assist neurologists with the development of treatment plans and prognosis predictions for the patients with Leigh syndrome with the MT-ND3 mutation.
Therefore, in this study, we focused on the epilepsy-related characteristics of the cases of Leigh syndrome with MT-ND3 mutation identified in a single tertiary hospital in Korea.
Patients with mtDNA mutations who were clinically diagnosed with Leigh syndrome between 2006 and 2018 at the Mitochondrial Disease Clinic of Gangnam Severance Hospital in the Republic of Korea were selected. The clinical diagnosis of Leigh syndrome was based on stringent diagnostic criteria described by Rahman et al. (2–4). The criteria is outlined as follows: presentation of a progressive neurological disease with symptoms of motor and intellectual developmental delays, signs and symptoms of brainstem and/or basal ganglia disease, raised lactate concentrations in the blood and/or cerebrospinal fluid (CSF), and bilateral symmetric hyperintense signal abnormality in the brainstem and/or basal ganglia shown on T2-weighted images in MRI of the brain. Sequencing of whole mtDNA was performed in all the patients clinically diagnosed with Leigh syndrome using next-generation sequencing (NGS) technology. Among them, 31 patients were genetically diagnosed with mtDNA-associated Leigh syndrome and among these, 7 patients with MT-ND3 mutations were detected. Each parent received a detailed explanation of this study and signed an informed consent form before their child participated in this study. This study was approved by the Institutional Review Board of Gangnam Severance Hospital, Yonsei University College of Medicine (3-2017-0168).
The clinical manifestations of Leigh syndrome with MT-ND3 mutation included in this study were reviewed. The first symptoms at disease onset and organ involvement at the time of the last follow-up were investigated as general characteristics of the patients. Patients were divided into two groups according to the criteria of early-onset Leigh syndrome: (1) <24 months old and (2) >24 months old when the first symptom occurred. The first symptom at disease onset was used to identify the most dominant symptom in the patients. For organ involvement at the time of the last follow-up, all the organs involved in Leigh syndrome, including the central nervous system (CNS), muscles, respiratory system, gastrointestinal system (e.g., gastroesophageal reflux disease, motility disorders), endocrine system, kidneys (e.g., tubular acidosis), eyes (e.g., optic atrophy, ptosis), ears, and skeletal system, were checked.
The prevalence of epilepsy was high in the patients with Leigh syndrome with MT-ND3 mutation. Therefore, the epilepsy-related characteristics of all the seven patients were examined and have been described in detail including seizure types, seizure frequency, electroencephalography (EEG) findings, antiepileptic drug history, and current seizure status.
The plasma lactate levels of the patients were obtained and defined as mild, moderate, or severe, if they were higher than the normal reference value by at least 2-fold, 3-fold, or 4-fold, respectively (9, 10). These values were evaluated as sequential variables ranging from one to four. Muscle biopsies were also performed on the patients and observed under light and electron microscopes. Light microscopy revealed specific findings for mitochondrial diseases such as ragged red fibers and electron microscopy revealed pleoconia and megaconia. In addition, muscle biopsy samples were processed using routine immunohistochemical staining (9).
Mitochondrial DNA-associated Leigh syndrome is characterized by bilateral symmetric hyperintense signal abnormalities in the brainstem and/or basal ganglia observed on T2-weighted MRIs (4). The MRI findings of the patients were obtained and separated according to the affected area such as the basal ganglia, brainstem, and thalamus. MRS can also be useful in detecting lactate peaks in the brain (11). Therefore, MRS abnormalities were obtained to establish the diagnosis.
Pathogenic mutations, including point mutations and large deletions, can occur in the mitochondrial genome. Therefore, the diagnosis of mtDNA-related Leigh syndrome should include the detection and quantification of sequence changes at any position of the mitochondrial genome (12). Whole mitochondrial gene sequence analysis was performed on all the patients who were clinically diagnosed with Leigh syndrome in this study. NGS technology was used to genetically confirm mtDNA Leigh syndrome and quantify the heteroplasmic mutant load of mtDNA. The sequence results were compared with the human mitochondrial reference (GenBank ID: NC_012920.1) (12–14).
Deoxyribonucleic acid was extracted from peripheral blood leukocytes using the QIAcube System and the QIAamp DNA Blood Mini Extraction Kit (Qiagen, Hilden, Germany, UK) and stored in 10 mM Tris buffer solution at −20°C. mtDNA was amplified using a long-range PCR. PCR reaction conditions were 98°C for 30 s, 30 cycles of 98°C for 10 s, 72°C for 8 min 15 s, and a final extension at 72°C for 10 min. PCR products were run on a 1% agarose gel and the expected 16.5 Kb fragments were excised. DNA was purified using Agencourt AMPure XP (Beckman Coulter, Brea, California, USA). Quantification was performed using the 4,200 TapeStation (Agilent Technologies, Santa Clara, California, USA) (15).
The PCR product was fragmented into 150–200 base pair (bp) segments with a NEBNext dsDNA Fragmentase® (New England Biolabs, Ipswich, Massachusetts, USA), according to the protocol of the manufacturer. The enzyme-fragmented PCR product was used as an input to the Accel-NGS® 2S PCR-free DNA Library Kit, following the protocol of the manufacturer. The final libraries were evaluated on the 4,200 TapeStation (Agilent Technologies, Santa Clara, California, USA) and quantified using Qubit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Libraries were sequenced via synthesis on Miseq for paired 150-bp read lengths using the Illumina MiSeq V3 Kits (Illumina, San Diego, California, USA) (15–17).
The sequenced reads were mapped to the human mitochondria reference (NC_012920) with Burrows-Wheeler Aligner and variants were identified using the Genome Analysis Toolkit. The sequence variants were filtered using various quality parameters. For NGS technology, each template was sequenced individually; therefore, quantitative analysis of the heteroplasmic mutant load was possible by counting the number of mtDNA reads (18, 19).
The genetic and clinical characteristics of the seven patients with Leigh syndrome with MT-ND3 mutation are given in Table 1. Statistical analysis was performed using SPSS version 20.0 for Windows (IBM Corporation, Armonk, New York, USA). Descriptive statistics were used including the median and range. The mutation load of mtDNA in each patient was displayed as a percentage and treated as a continuous variable for statistical analysis. The Pearson correlation coefficients (r) were calculated to see the correlation between two continuous variables. The statistical significance level was set at p < 0.05.
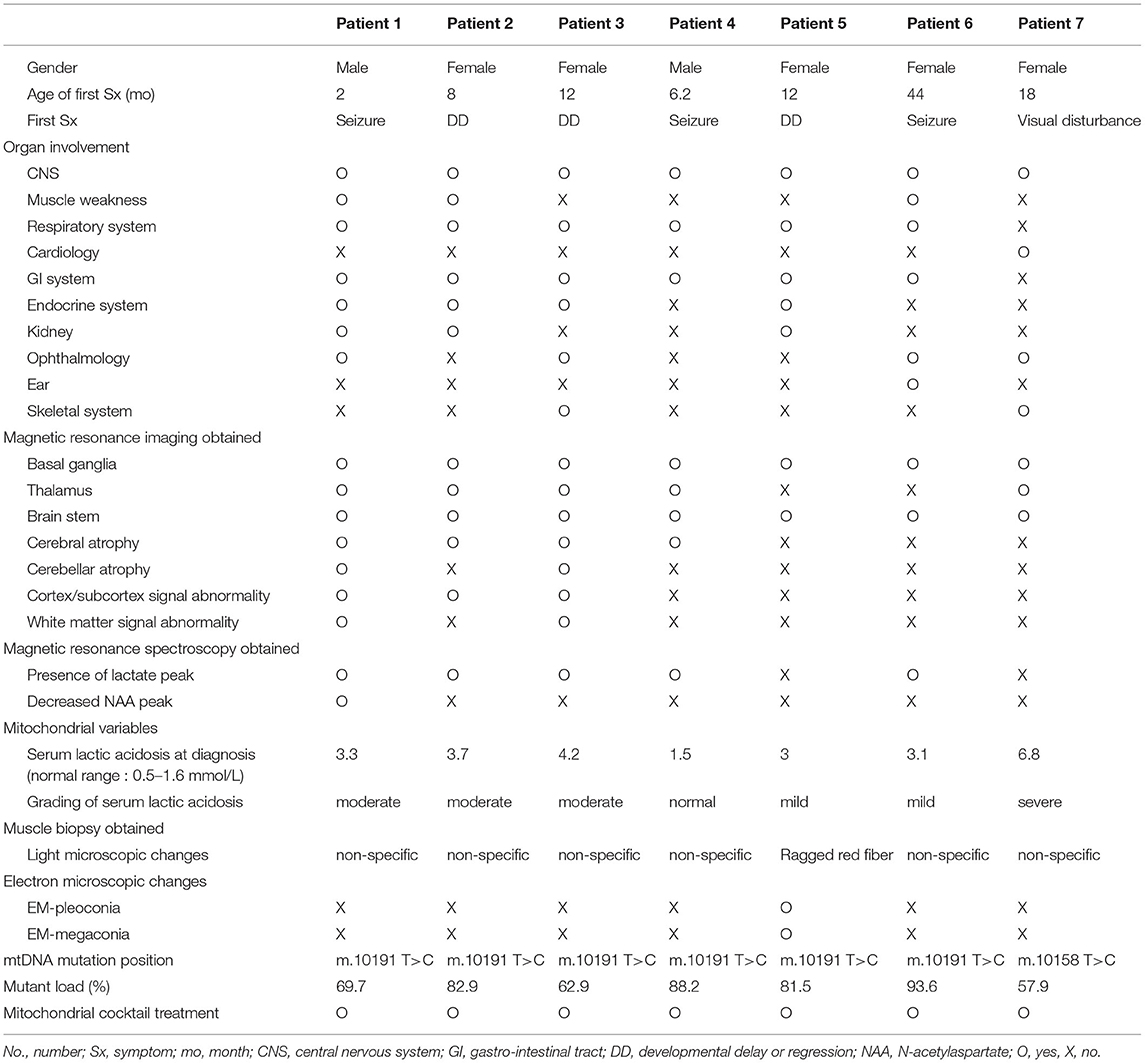
Table 1. General characteristics of Leigh syndrome with MT-ND3 mutation (Total N = 7).
Among the seven patients with Leigh syndrome with MT-ND3 mutation, the male-to-female ratio was 2:5. The median age at first symptom onset was 12 months and ranged from 12 to 44 months. Six of the seven patients experienced early-onset Leigh syndrome and, therefore, developed symptoms before 24 months of age. The first symptoms of the patients were primarily developmental delay/regression and seizures (three patients each). The organ involvement in the patients at the time of the last follow-up was identified. CNS symptoms were observed in 100% of the patients, followed by symptoms related to the gastrointestinal (GI) tract (n = 6, 85.7%) and the respiratory system (n = 6, 85.7%) and ophthalmological symptoms (n = 5, 71.4%). Normotonic or hypertonic muscle tone was observed in all the patients. Three patients had symptoms of dystonia. All the patients had gait disorder and three patients with muscle weakness could not walk at all. Among the patients, six patients were receiving enteral tube feeding, two patients underwent gastrostomy, and four patients were fed through temporary enteral tube feeding (nasogastric tube). The median age at the time of Leigh syndrome diagnosis was 22 months (range, 5–70 months). The median interval time between the development of the first symptom and Leigh syndrome diagnosis was 12 months (range, 1–44 months) (Table 1).
Magnetic resonance imaging was performed for all the patients. Involvement of the basal ganglia was identified as 100%, followed by the involvement of the brainstem or thalamus. MRS was performed for six of the patients presenting the m.10191T> C mutation and lactate peaks were detected in five of the six patients (Table 1 and Figure 1).
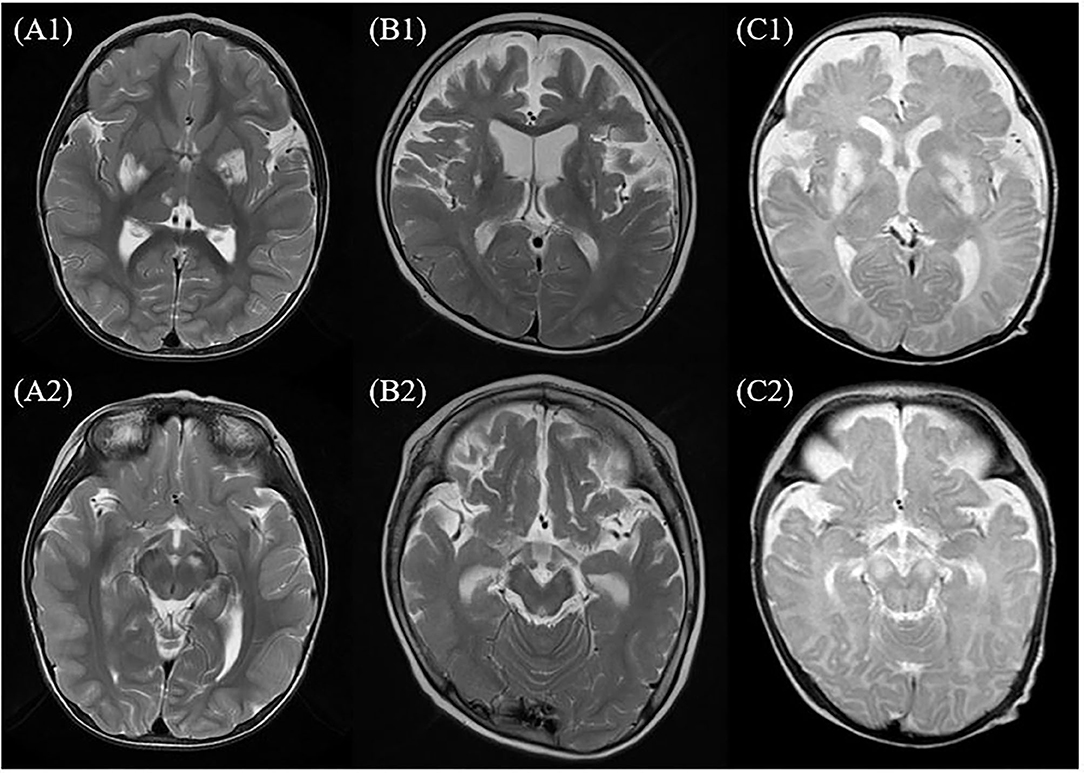
Figure 1. MRI of the brain findings in the patients with Leigh syndrome with MT-ND3 mutation. This figure shows some findings from T2-weighted axial images of MRI of the brain in the patients with Leigh syndrome with MT-ND3 mutation. (A1,A2) Hyperintense signal abnormalities are observed in both the basal ganglia, brainstem, and thalamus (patient 7). (B1,B2) Hyperintense signal abnormalities in both the basal ganglia and diffuse brain atrophy prominently at bilateral frontotemporal lobes with secondary ventricular dilatation are observed (patient 2). (C1,C2) Diffuse abnormal signal changes at both the basal ganglia, thalami, and brainstem are observed and diffuse brain atrophy is also observed (patient 1).
The median value of serum lactic acidosis at diagnosis was 3.3 and ranged from 1.5 to 6.8. The serum lactic acidosis of six of the seven patients was mildly to severely increased. No specific findings were identified from muscle biopsies using light and electron microscopy. The seven patients with Leigh syndrome with MT-ND3 mutation were divided into the m.10191T>C and the m.10158T>C groups, according to the respective nucleotide change. The majority of the patients (n = 6) were in the m.10191T>C group. The median value of the mutant load was 82.5%, ranging from 57.9 to 93.6%. All the patients received mitochondrial cocktail treatment such as coenzyme, L-carnitine, and multivitamins from the time of diagnosis. No particular tendency was observed for the first symptom, first seizure onset, or mutant load. The Pearson correlation coefficient and significance level are r = 0.470, p = 0.347 in Figure 2A; r = 0.523, p = 0.287 in Figure 2B; and r = 0.374, p = 0.465 in Figure 2C, respectively (Table 1 and Figure 2).
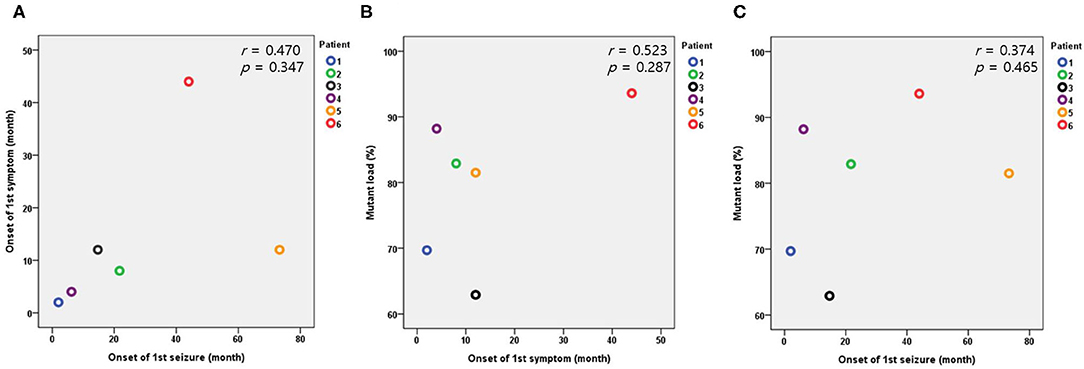
Figure 2. Relationships between the onset of the first symptom, first seizure, and mutant load in the patients with the m.10191T>C mutation. (A) Relationship between the onset of the first symptom and first seizure. (B) Relationship between the onset of the first symptom and mutant load. (C) Relationship between the onset of the first seizure and mutant load.
The prevalence of epilepsy was high in Leigh syndrome with MT-ND3 mutation (n = 6). In particular, patients 1, 2, 3, 4, 5, and 6 (m.10191T>C mutation) were diagnosed with epilepsy and treated accordingly. The median age at first seizure was 18.2 months and ranged from 2 to 73.3 months. The seizure types consisted of two generalized tonic seizures, two focal seizures, one focal-to-secondary generalized seizure, and one mixed-type seizure. Three of the six patients with epilepsy had a history of status epilepticus. In the EEG findings at the last follow-up, the EEG patterns of Lennox–Gastaut syndrome (LGS), such as multifocal sharp, generalized sharp and slow wave (GSSW) discharge, and generalized paroxysmal fast activity (GPFA), were observed in three of the six patients with epilepsy (50% of the m.10191T>C mutation group, patient numbers 1, 2, and 3). These three patients had no history of infantile spasm. Five of the six patients with epilepsy were taking multiple antiseizure medications (ASMs) and two patients had attempted the ketogenic diet (KD) (lipid: nonlipid ratio: 4:1). No patients experienced serious side effects due to the ASMs, but the KD was discontinued early due to poor oral intake and GI problems in the two patients. The current seizure status compared to the initial seizure frequency was investigated and the two seizure-free patients were identified. One patient experienced > 90% reduction in seizure frequency a second patient experienced 50–90% reduction in seizure frequency, a third patient experienced < 50% reduction in seizure frequency, and a fourth patient experienced no change. The current status of deterioration was also measured and progressive deterioration was observed in four of the seven patients of Leigh syndrome with MT-ND3 mutation and the remaining three patients had a static status and no longer experienced progressive deterioration (Table 2).

Table 2. Epilepsy characteristics of Leigh syndrome with MT-ND3 mutation (Total N = 7).
Epilepsy is a major phenotype of mitochondrial diseases (20). In particular, Leigh syndrome is the most common mitochondrial disease in childhood and high epilepsy morbidity has been reported (21). Among the various presentations of mtDNA-associated Leigh syndrome, several studies have reported a specific gene related to epilepsy. Some studies have found that m.10191T>C, a major nucleotide change in MT-ND3 gene, is strongly related to epilepsy (7, 21). In this study, the six patients with the m.10191T>C mutation were associated with epilepsy, suggesting that this mutation is important in Leigh syndrome with MT-ND3 mutation. Additionally, the results of this study suggested that even the same MT-ND3 mutation may have different phenotypes depending on the location of the point mutation. These results are supported by two important studies. Nesbitt et al. reviewed the clinical spectra of 16 patients with the m.10191T>C mutation from 14 references. This study does not describe a close relationship between epilepsy and m.10191T>C. However, after reviewing the 14 case reports used in this study, seizures were identified in 12 of the 16 (75%) patients with the m.10191T>C mutation. Their EEG findings were unknown, but most were found to be strongly associated with epilepsy. Rebecca et al. added several cases to those reviewed by Nesbitt et al. and modified the clinical features of the patients with the m.10191T>C mutation. In this study, seizures were identified in 15 out of 22 (68.2%) patients with the m.10191T>C mutation (6, 8). In addition, a recent study by Li et al. reviewed 28 patients with the m.10191T>C mutation from 23 references. Among them, 24 (85.7%) patients with the m.10191T>C mutation had epilepsy as their phenotype. Therefore, this study states that the m.10191T>C mutation may be strongly associated with epilepsy and these findings are consistent with those of this study (7). The value of this study lies in the fact that its findings are consistent with those of the previous studies and that it undertakes a more focused analysis of the characteristics of epilepsy in Leigh syndrome with the m.10191T>C mutations.
Lennox–Gastaut syndrome is rare and is one of the most severe forms of epilepsy with childhood onset. Epileptic discharges, known as GSSW complexes and GPFA in EEG, multiple types of seizures, and severe intellectual impairments are the diagnostic triad of LGS. LGS is one of the most complex epileptic disorders to manage, both for the pediatric neurologists and epilepsy specialists (22, 23). In a study by Lee et al., of the 372 patients with a mitochondrial disease, 40 patients were diagnosed with LGS, which represented about 10% of their study cohort (23). To the best of our knowledge, there are no previous studies on the relationship between LGS and mitochondrial disease or Leigh syndrome. Therefore, genotype–phenotype correlations are necessary to establish a cohort and analyze these relationships. In this study, 50% of the six patients with the m.10191T>C mutation and epilepsy had LGS. Although this study included a small cohort, considering that Leigh syndrome with the m.10191T>C mutation and with LGS are both the rare diseases, LGS may be frequently associated with the epilepsy phenotype in the m.10191T>C mutation. If the m.10191T>C mutation has features that are often accompanied by LGS, it may be possible to control the disease by providing intensive LGS treatment to these patients. To the best of our knowledge, no previous studies support this, but we will continue to explore the relationship between LGS and the m.10191T>C mutation in future studies.
Heteroplasmy is an important feature of mtDNA-associated Leigh syndrome. Heteroplasmy rarely affects the type and severity of phenotypes. Within the same mutation, heteroplasmy is expected to be associated with disease severity, reflected by the specific phenotypic presentation or the time of symptom onset (4, 24). However, in this study, no correlation was identified between heteroplasmy and the onset of the first symptom or seizure. In addition, among the patients with MT-ND3 mutation, no significant difference in heteroplasmy was observed between patients with or without LGS. This may be because the total number of patients in this study was relatively small. In addition, the possibility of other factors and of heteroplasmy acting upon the onset of symptoms can also be considered. Also, although not measured in this study, heteroplasmy in brain may be different than in the blood leukocytes measured and might show a closer relationship to the neurologic presentations (7).
Two patients started the KD; one had LGS and the other had not LGS. However, both the patients discontinued the KD after 3 months due to poor oral intake and severe GI problems. This suggested possible intolerance for KDs in the patients with the m.10191T>C mutation, but a larger cohort is necessary to establish a firm conclusion. Kang et al. reported the safety and effective application of the KD in mitochondrial respiratory chain complex defects. In this study, four patients had mitochondrial disease with LGS. The KD was effective for seizure reduction in all the patients. However, the maintenance duration of the KD varied, with one patient discontinuing the KD after 3 months due to severe hypoglycemia and the other three patients maintained the KD for 6, 12, and 24 months. Two patients became seizure free (25). However, this treatment was not conducted through gene-based targeted therapy. Therefore, with the results of this study, the effect and stability of the KD may not be suitable for patients with the m.10191T>C mutation. At present, there are no studies with respect to the relationship between the KD and MT-ND3 or the m.10191T>C mutations. Genotype-based KD studies should be conducted in the future (26).
Valproic acid (VPA) is a broad-spectrum antiepileptic drug that is widely used as a first-line treatment for most types of epilepsy. In particular, patients with LGS are considered for first-line treatment (27). However, because VPA can induce mitochondrial toxicity through the inhibition and subsequent decreased activity of mitochondrial complexes I and IV, it is contraindicated for patients with a mitochondrial disease (28–30). However, in this study, VPA was used in three of the six patients with the m.10191T>C mutation and an improvement in seizure frequency was observed. Two of the three patients were diagnosed with LGS. VPA therapy may be effective in treating epilepsy depending on the clinical situation and phenotype and may not cause a contraindication. In particular, patients with the m.10191T>C mutation have mtDNA-associated Leigh syndrome with a high morbidity of epilepsy that may be accompanied by refractory epilepsy such as LGS. In such situations, mitochondrial dysfunction may be diagnosed later as the etiology of epilepsy. Therefore, the careful application of VPA may assist in controlling adverse effects or mitochondrial toxicity.
In conclusion, the results of this study indicate a strong association between epilepsy and Leigh syndrome with MT-ND3 mutation, particularly the m.10191T>C mutation. Additionally, there is a possibility of an association between the epilepsy phenotype of the m.10191T>C mutation and LGS. Due to the characteristics of rare disease, there is a limitation in that this study could not be conducted on a large homogeneous patient group. Therefore, a follow-up study or meta-analysis with a large number of patients is needed in the future. This study explored the importance of specific point mutations such as the m.10191T>C mutation, although MT-ND3 mutation is important for genotype–phenotype correlation. Understanding the characteristics of Leigh syndrome with MT-ND3 mutation may assist in predicting disease progression and improving patient survival through timely treatment. This understanding could be the first step in finding biomarkers for the development of new treatments for Leigh syndrome (31).
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
This study was approved by the Institutional Review Board of Gangnam Severance Hospital, Yonsei University College of Medicine (3-2017-0168). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Y-ML conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed and revised the manuscript. J-HN designed the data collection instruments, collected data, carried out the initial analyses, drafted the initial manuscript and revised the manuscript. ML and CL coordinated and supervised data collection. All authors approved the final manuscript as submitted and agree to be accountable for the content of the work.
This study was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Family Welfare, Republic of Korea (Grant Numbers: 2018-31-0425/HI18C1166020018, 2018-31-1061/HI18C1166020019, and 2019-31-1183/HI18C1166020020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to all the staff members, doctors, and statistical consultants who were involved in this study.
1. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. (2003) 348:2656–68. doi: 10.1056/NEJMra022567
2. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. (2016) 79:190–203. doi: 10.1002/ana.24551
3. Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. (1996) 39:343–51. doi: 10.1002/ana.410390311
4. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle (1993–2019).
5. Sofou K, de Coo IFM, Ostergaard E, Isohanni P, Naess K, De Meirleir L, et al. Phenotype-genotype correlations in Leigh syndrome: new insights from a multicentre study of 96 patients. J Med Genet. (2018) 55:21–7. doi: 10.1136/jmedgenet-2017-104891
6. Nesbitt V, Morrison PJ, Crushell E, Donnelly DE, Alston CL, He L, et al. The clinical spectrum of the m.10191T>C mutation in complex I-deficient Leigh syndrome. Dev Med Child Neurol. (2012) 54:500–6. doi: 10.1111/j.1469-8749.2012.04224.x
7. Li TR, Wang Q, Liu MM, Lv RJ. A Chinese family with adult-onset leigh-like syndrome caused by the heteroplasmic m.10191t>c mutation in the mitochondrial MTND3 gene. Front Neurol. (2019) 10:347. doi: 10.3389/fneur.2019.00347
8. Levy RJ, Ríos PG, Akman HO, Sciacco M, Vivo DC, DiMauro S. Long survival in patients with leigh syndrome and the m.10191T>C mutation in MT-ND3: a case report and review of the literature. J Child Neurol. (2014) 29:NP105-110. doi: 10.1177/0883073813506783
9. Eom S, Lee HN, Lee S, Kang HC, Lee JS, Kim HD, et al. Cause of death in children with mitochondrial diseases. Pediatr Neurol. (2017) 66:82–88. doi: 10.1016/j.pediatrneurol.2016.10.006
10. Eom S, Lee YM. Long-term developmental trends of pediatric mitochondrial diseases: the five stages of developmental decline. Front Neurol. (2017) 17:208. doi: 10.3389/fneur.2017.00208
11. Ganetzky RD, Stendel C, McCormick EM, Zolkipli-Cunningham Z, Goldstein AC, Klopstock T, et al. MT-ATP6 mitochondrial disease variants: phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum Mutat. (2019) 40:499–515. doi: 10.1002/humu.23723
12. Wong LJ. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion. (2013) 13:379–87. doi: 10.1016/j.mito.2013.02.001
13. Ma YY, Wu TF, Liu YP, Wang Q, Li XY, Song JQ, et al. Heterogeneity of six children and their mothers with mitochondrial DNA 3243 A>G mutation. Mitochondrial DNA. (2013) 24:297–302. doi: 10.3109/19401736.2012.760071
14. Ma YY, Wu TF, Liu YP, Wang Q, Song JQ, Li XY, et al. Genetic and biochemical findings in Chinese children with Leigh syndrome. J Clin Neurosci. (2013) 20:1591–4. doi: 10.1016/j.jocn.2013.03.034
15. Gould MP, Bosworth CM, McMahon S, Grandhi S, Grimberg BT, LaFramboise T. PCR-free enrichment of mitochondrial DNA from human blood and cell lines for highquality next-generation DNA sequencing. PLoS ONE. (2015) 10:e0139253. doi: 10.1371/journal.pone.0139253
16. Craig DW, Nasser S, Corbett R, Chan SK, Murray L, Legendre C, et al. A somatic reference standard for cancer genome sequencing. Sci Rep. (2016) 6:24607. doi: 10.1038/srep24607
17. Huptas C, Scherer S, Wenning M. Optimized Illumina PCR-free library preparation for bacterial whole genome sequencing and analysis of factors influencing de novo assembly. BMC Res Notes. (2016) 9:269. doi: 10.1186/s13104-016-2072-9
18. Palculict ME, Zhang VW, Wong LJ, Wang J. Comprehensive mitochondrial genome analysis by massively parallel sequencing. Methods Mol Biol. (2016) 1351:3–17. doi: 10.1007/978-1-4939-3040-1_1
19. Preston JL, Royall AE, Randel MA, Sikkink KL, Phillips PC, Johnson EA. High-specificity detection of rare alleles with paired-end low error sequencing (PELE-Seq). BMC Genomics. (2016) 17:464. doi: 10.1186/s12864-016-2669-3
20. Khurana D, Salganicoff L, Melvin J, Hobdell E, Valencia I, Hardison H, et al. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics. (2008) 39:8–13. doi: 10.1055/s-2008-1076737
21. Lee S, Na JH, Lee YM. Epilepsy in Leigh syndrome with mitochondrial DNA mutations. Front Neurol. (2019) 10:496. doi: 10.3389/fneur.2019.00496
22. Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, Feucht M, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. (2009) 8:82–93. doi: 10.1016/S1474-4422(08)70292-8
23. Lee S, Baek MS, Lee YM. Lennox-Gastaut syndrome in mitochondrial disease. Yonsei Med J. (2019) 60:106–4. doi: 10.3349/ymj.2019.60.1.106
24. White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, DiMauro S, et al. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. (1999) 65:474–82. doi: 10.1086/302488
25. Kang HC, Lee YM, Kim HD, Lee JS, Slama A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. (2007) 48:82–8. doi: 10.1111/j.1528-1167.2006.00906.x
26. Na JH, Kim HD, Lee YM. Effective and safe diet therapies for Lennox-Gastaut syndrome with mitochondrial dysfunction. Ther Adv Neurol Disord. (2020) 13:1756286419897813. doi: 10.1177/1756286419897813
27. Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox-Gastaut syndrome: treatment algorithms and practical considerations. Front Neurol. (2017) 8:505. doi: 10.3389/fneur.2017.00505
28. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. (2013) 46:1323–38. doi: 10.1016/j.clinbiochem.2013.06.012
29. Finsterer J, Segall L. Drugs interfering with mitochondrial disorders. Drug Chem Toxicol. (2010) 33:138–51. doi: 10.3109/01480540903207076
30. Finsterer J, Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. (2012) 8:71–9. doi: 10.1517/17425255.2012.644535
Keywords: mitochondrial DNA-associated Leigh syndrome, MT-ND3, m10191T>C, epilepsy, Lennox-Gastaut syndrome
Citation: Na J-H, Lee MJ, Lee CH and Lee Y-M (2021) Association Between Epilepsy and Leigh Syndrome With MT-ND3 Mutation, Particularly the m.10191T>C Point Mutation. Front. Neurol. 12:752467. doi: 10.3389/fneur.2021.752467
Received: 03 August 2021; Accepted: 12 November 2021;
Published: 10 December 2021.
Edited by:
Jing Zhang, Peking University First Hospital, ChinaReviewed by:
Gerald Pfeffer, University of Calgary, CanadaCopyright © 2021 Na, Lee, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young-Mock Lee, eW1sZWVtZEB5dWhzLmFj
†ORCID: Ji-Hoon Na orcid.org/0000-0002-3051-2010
Min Jung Lee orcid.org/0000-0002-0693-4811
Chul Ho Lee orcid.org/0000-0001-8045-4368
Young-Mock Lee orcid.org/0000-0002-5838-249X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.