 Zhenchu Tang1,2
Zhenchu Tang1,2 Shan Gao3Miao He1,2Qihua Chen1
Shan Gao3Miao He1,2Qihua Chen1 Jia Fang1Yingying Luo1Weiqian Yan1
Jia Fang1Yingying Luo1Weiqian Yan1 Xiaoliu Shi4
Xiaoliu Shi4 Hui Huang4*
Hui Huang4* Jianguang Tang1*
Jianguang Tang1*- 1Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, China
- 2Hunan Key Laboratory of Tumor Models and Individualized Medicine, Second Xiangya Hospital, Central South University, Changsha, China
- 3Department of Gastroenterology, Second Xiangya Hospital, Central South University, Changsha, China
- 4Department of Medical Genetics, Second Xiangya Hospital, Central South University, Changsha, China
Background: Late-onset multiple acyl-CoA dehydrogenase deficiency (LO-MADD) describes a curable autosomal recessive genetic disease caused by ETFDH mutations that result in defects in ETF-ubiquinone oxidoreductase. Almost all patients are responsive to riboflavin. This study describes the clinical presentations and genetic characteristics of five LO-MADD patients.
Methods: From 2018 to 2021, we collected clinical and genetic data on five patients diagnosed with LO-MADD at our hospital and retrospectively analyzed their clinical characteristics, laboratory examination, electromyography, muscle biopsy, genetic analysis, and outcome data.
Results: This study included three males and two females with mean onset age of 37.8 years. Fluctuating exercise intolerance was the most common presentation. Serum creatine kinase (CK) levels were significantly elevated in all patients, and plasma acylcarnitine profiles revealed an increase in long-chain acylcarnitine species in three cases. The urinary organic acid study revealed a high level of hydroxyglutaric acid in all patients. Electrophysiology demonstrated myogenic impairment. Muscle biopsies revealed lipid storage myopathy. Molecular analysis identified nine mutations (three novels and six reported) in ETFDH. Exercise intolerance and muscle weakness were dramatically improved in all patients treated with riboflavin (100 mg) daily following diagnosis.
Conclusions: LO-MADD is caused by ETFDH variants and responds well to riboflavin. Three novel ETFDH pathogenic variants were identified, expanding their spectrum in the Chinese population and facilitating future interpretation and analysis of ETFDH mutations.
Introduction
MADD, also known as glutaric aciduria type II, is an autosomal recessive inherited metabolic disorder caused by imperfection in either electron transfer flavoprotein (ETF, encoded by ETFA and EFTB) or ETF-ubiquinone oxidoreductase (ETF-QO, encoded by ETFDH). Moreover, the defects lead to abnormalities in electron transportation from acyl-CoA dehydrogenases to the respiratory chain (1–3), hindering the oxidation of fatty acids and amino acids, impairing adenosine triphosphate synthesis, and causing abnormal lipid accumulation in muscle (4–6). The clinical manifestations of MADD are incredibly diverse (7) and are generally classified into three types: neonatal onset with congenital anomalies (type I) or without abnormalities (type II), and late-onset MADD (type III). Type I and II MADD are often fatal with patients frequently dying within a few hours or a few days after birth from complications, such as metabolic decompensation or hypertrophic cardiomyopathy (8).
Most MADD patients are of type III, which tends to have a better prognosis. The initial onset age ranges from 6 months (9) to more than 60 years (10). Furthermore, its severity varies considerably (11–13). Clinical manifestations of LO-MADD patients include muscular symptoms and accompany extramuscular symptoms. In the mainland of China, muscle symptoms are the most common presentations, including muscle weakness and exercise intolerance with or without myalgia (14–16). Regarding extramuscular symptoms, they consist of vomiting, diarrhea, dysphagia, fatty liver, palpitation or shortness of breath, and diastolic cardiac dysfunction (14, 16). A high level of serum creatine kinase (CK) is a non-specific phenomenon of LO-MADD. Elevated short-, medium-, and long-chain acylcarnitine species in blood and altered urinary organic acid levels can also be found in some patients during the acute phase (14). Fortunately, most LO-MADD patients exhibit an excellent therapeutic response to riboflavin, indicating why LO-MADD is also referred to as riboflavin response MADD (RR-MADD) (5, 17).
However, without muscle biopsy and molecular study, these patients are frequently misdiagnosed as having polymyositis and treated with glucocorticoids. Although glucocorticoid therapy could partially improve patient's laboratory indicators or clinical symptoms, its overall efficacy is inferior to riboflavin (18). Here, we describe the clinical and genetic characteristics of five LO-MADD patients.
Materials and Methods
Patient Inclusion
We performed ETFDH mutation screening in 12 patients with pathologically confirmed lipid storage myopathy (LSM) from June 2018 to January 2021 at the Second Xiangya Hospital. LSM was diagnosed by the appearance of excess lipid droplets in muscle fibers detected by oil red O staining. Additionally, lipid storage caused by glucocorticoid or mitochondrial disease was excluded. Five patients with ETFDH mutations were enrolled. All participants provided written informed consent. Additionally, the Ethics Committee of the Second Xiangya Hospital approved this study.
Biochemical and Muscle Biopsy Studies
Before riboflavin administration, blood, and urine samples from five patients were collected for laboratory examinations, including urinary organic acid and blood acyl-carnitine analyses. After the patients' consent, all patients underwent quadriceps femoris muscle biopsy under local anesthesia. Serial frozen slices of fresh muscle were stained using H&E and Oil Red O (ORO) after being frozen in isopentane and chilled in liquid nitrogen. Additionally, modified Gomori trichrome (MGT), periodic acidic Schiff (PAS), adenosine triphosphatase (ATPase), nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), succinate dehydrogenase (SDH), cytochrome c oxidase (COX), and non-specific esterase (NSE) were performed. We fixed these samples sequentially in 2.5% glutaraldehyde and 1% osmium tetroxide and then embedded them in Epon, and finally examined the ultrathin sections using electron microscopy.
Molecular Studies
We extracted genomic DNA (gDNA) from peripheral blood of subjects using a DNeasy Blood & Tissue Kit (MagPure Buffy Coat DNA Midi KF Kit) according to instructions. Following that, gDNA samples were sent to WUHAN BGI MEDICAL TESTING for the next-generation sequencing (NGS) analysis. We compared the sequences with those in other databases, including NCBI dbSNP, Human Gene Mutation Database (HGMD), 1000 human genome data set, and a database of 100 Chinese healthy adults. The sequences were further analyzed for pathogenicity using several bioinformatics programs, such as CADD, GERP, and MutationTaster. All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods.
Results
Clinical Features
This study included two female and three male participants. All patients had a normal birth and developmental milestones. The average onset age was 37.8 years with a range of 26–50 years, and the average duration between disease onset and confirmation was 9 months. All five patients complained about fluctuating muscle weakness and exercise intolerance, aggravated by a high workload, staying up late, or irregular diet with predominant involvement of proximal extremities (5/5), neck (3/5), throat (3/5), and facial (3/5) muscles. Three patients presented with myalgia. However, no patient suffered from muscle atrophy, encephalopathy, hypoglycemia, or seizure. Ultrasound studies revealed a patent foramen ovale in patient 2 and fatty liver in patient 5. Further details are summarized in Table 1.
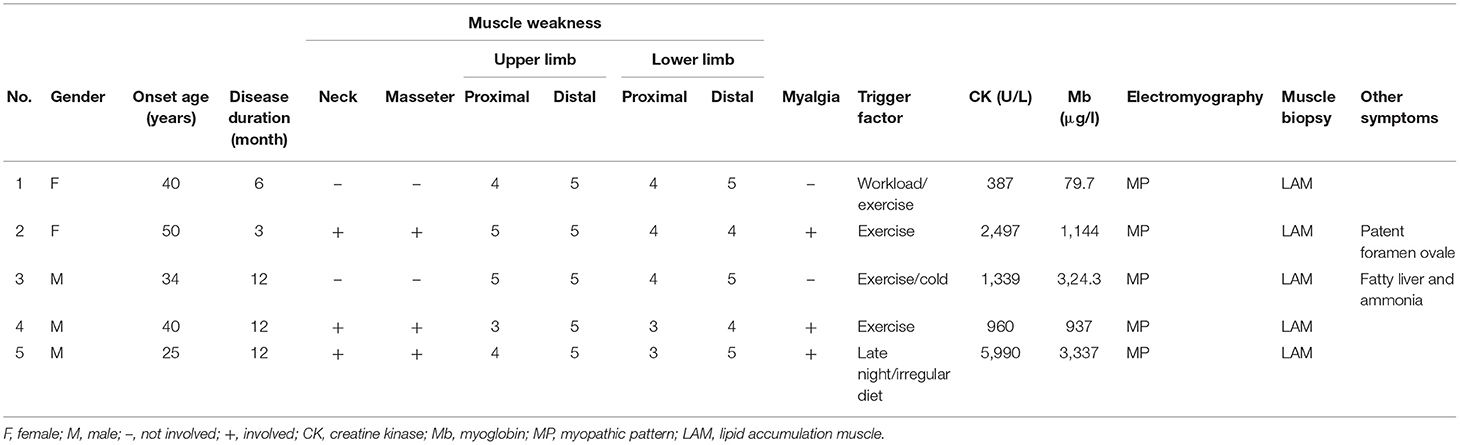
Table 1. Clinical presentations of five patients with LAMADD in our study.
Biochemical Studies and Muscle Biopsy
Serum CK levels were significantly elevated in all patients, ranging from 2 to 20 times the upper limit of normal (310 μ/l). Meanwhile, lactate dehydrogenase and myoglobin were increased to some extent. At first, two patients were misdiagnosed with polymyositis and treated with glucocorticoids (1.0 mg/kg weight per day). After glucocorticoid treatment, the 50-year-old female (patient 2) had no change in symptoms or laboratory testing. Meanwhile, after steroid treatment, the 26-year-old male (patient 5) with fatty liver and increased liver enzymes exhibited significant decreases in CK, aspartate aminotransferase, lactate dehydrogenase, and blood ammonia. However, the weak muscle strength remained unchanged. For these two patients, glucocorticoid treatment was discontinued once muscle biopsy and molecular analysis revealed MADD. Riboflavin 100 mg per day was administered once following LO-MADD diagnosis.
Urine organic acid analysis revealed excessive levels of 2-hydroxyglutaric acid-3 and 3-hydroxyglutaric-3 in all patients (5/5), whereas some other C5-C10 dicarboxylic acids were increased in three patients (3/5). Blood acylcarnitine analysis revealed that one patient (1/5) had a decreased degree of free carnitine, and three patients (3/5) had a high concentration of long-chain acylcarnitine.
Before riboflavin treatment, electromyography was performed in all patients (5/5), the results revealed a myopathic pattern without nerve conduction deterioration.
Quadriceps femoris muscle biopsy the presence of intracytoplasmic vacuoles on H&E in all five patients as well as a large number of cytoplasmic vacuoles stained with ORO, indicating lipid storage myopathy. In contrast, other stains exhibited no aberration. Under electron microscope, the number of lipids deposited in muscle fibers increased significantly. Here, we used patients 2 and 3 as representative samples in Figure 1.
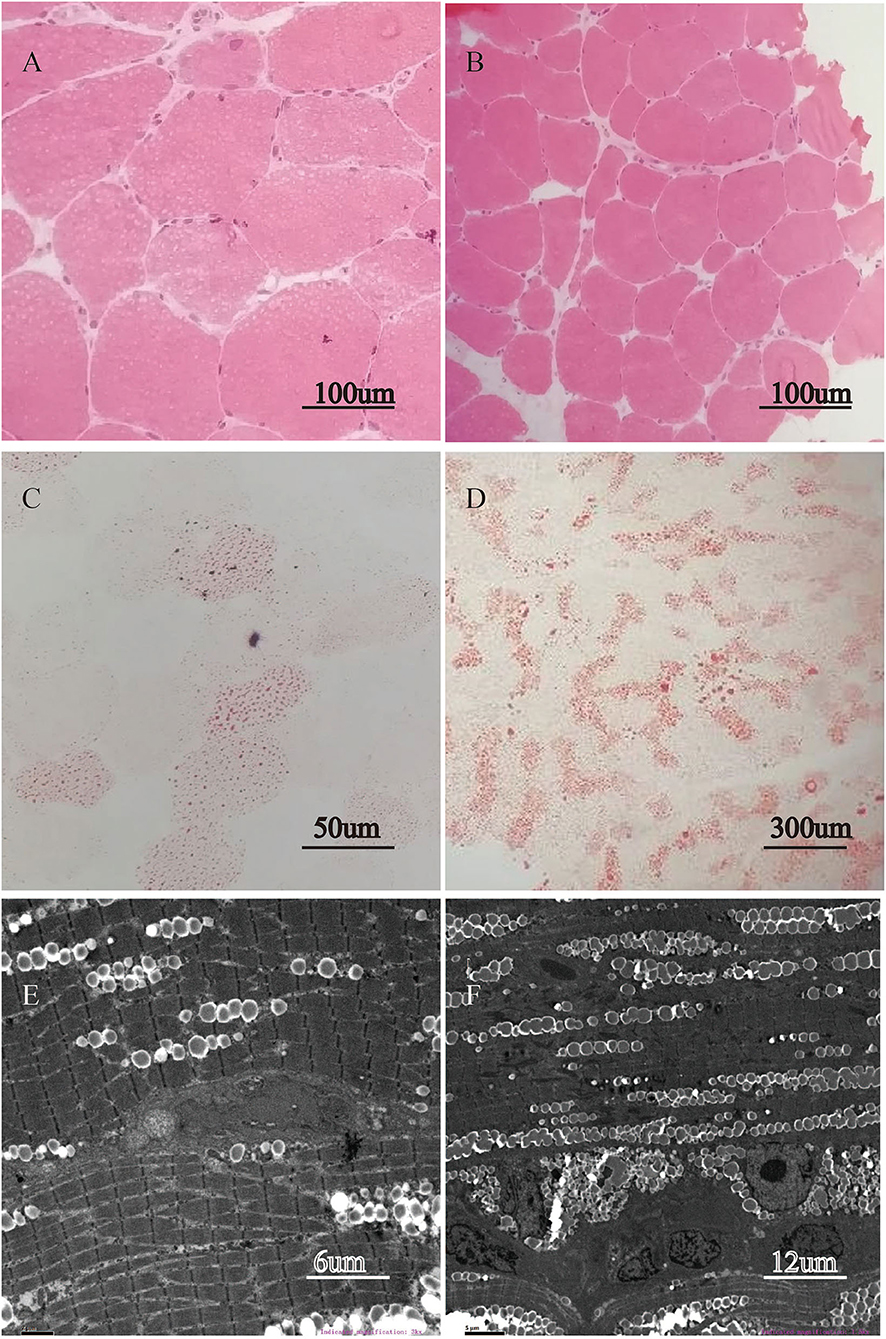
Figure 1. Lipid storage in quadriceps femoris muscle biopsy. H&E stain revealed intracytoplasmic vacuoles appearing in most fibers of patient 2 (A, magnification × 400) and patient 3 (B, magnification × 200). ORO stain demonstrated abnormal accumulation of lipid droplets in muscle tissue of patient 2 (C, magnification × 400), and of patient 3 (D, magnification × 60). The electron microscopic ultrastructural muscle fiber study reveals the substantial deposition of lipid droplets between myofibrils in patient 2 (E, magnification × 3,000) and in patient 3 (F, magnification × 1,500).
Molecular Studies
Nine different ETFDH mutations were identified. One patient presented a homozygous mutation (c.770A>G), whereas four exhibited compound heterozygous mutations (c.236C>G and c.1448C>T, c.152G>A and c.524G>T, c.295C>T and c.582_583 ins TA, c.920C>T and c.257G>A) (details in Table 2). The c.152G>A, c.236C>G, c.295C>T, c.524G>T, c.770A>G, and c.1448C>T mutations were reported previously (5, 14, 19–22). Furthermore, c.770A>G was one of the most popular pathogenicity mutations in China (14, 16, 23). Three novel mutations were identified, namely, c.257G>A, c.582-583insTA, and c.920C>T, which are not included in the Human Gene Mutation Database (HGMD) or ClinVar.
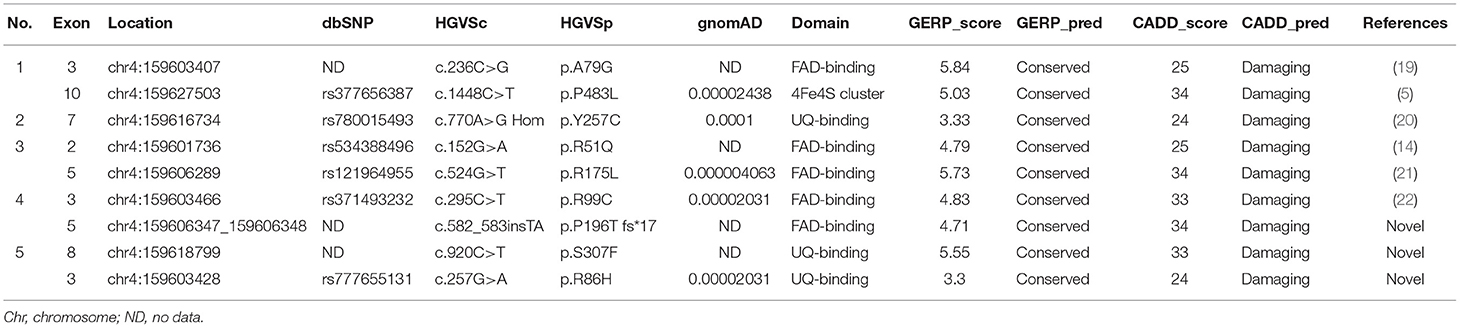
Table 2. ETFDH gene mutation in five patients in this study.
Treatment and Follow Up
Since MADD diagnosis was confirmed, riboflavin alone (100 mg daily) was administered, dramatically improving exercise intolerance and muscle weakness within the first week. They later recovered completely following 2–3 months of riboflavin treatment. Furthermore, no further MADD attacks were detected after 2 months to 2 years of follow-up visits.
Discussion
This study described five LO-MADD patients from the Hunan province of China, exhibiting exercise intolerance and muscle weakness with or without myalgia. The favored sites of involvement include proximal limb, cervical, facial, and throat muscles. Muscle biopsy affirmed abnormal lipid accumulation in myofibers, and electromyography demonstrated myogenic impairment without nerve damage. Anomalies were discovered in blood and urine metabolite tests. Two patients misdiagnosed with polymyositis received glucocorticoids. One of them gained no benefit, and the other showed improvement in laboratory parameters without clinical improvement. Within a few weeks, high-dose riboflavin treatment offered noticeable relief for the five patients.
Consistent with another study (24) and a cohort study published in a Chinese journal about MADD patients from Hunan province, and unlike the coastal areas, two of the three frequent ETFDH mutations in China, c.250G>A and c.1227A>C (14, 16, 25) were not prevalent in Hunan. Our result indicates that the mutational spectrum may be heterogeneous in disparate areas. Perhaps the prevalence of c.250G>A mutation is confined to the coastal regions of China, Thailand, and Singapore. As a result, additional epidemiological surveys need to conducted. In addition, three novel mutations in ETFDH, including two missense mutations (c.257G>A and c.920C>T) and one frameshift mutation (c.582-583insTA), were identified in merely five cases, indicating that the mutational spectrum of LO-MADD might be broader than that expected in Mainland China.
Similar to previously reported MADD patients, the main clinical manifestations of our patients were exercise intolerance and muscle weakness, which are easily provoked by a high workload, starvation, or infection. ETF-QO protein, a component of the mitochondrial respiratory chain, is responsible for transferring electrons from ETF in the mitochondrial matrix to the ubiquinone pool located in the inner mitochondrial membrane (26, 27), which is critical for fatty acid β-oxidation (FAO). However, RR-MADD–associated mutant ETF-QO, a protein with a gentle structural defect that partly but not wholly disrupts FAD-binding to flavoprotein (1, 2), was unstable and easily degraded by the ubiquitin-proteasome pathway (28). Therefore, the impaired FAO resulted from ETFDH mutations impacts energy supply, resulting in lipid accumulation in muscle and induces an increase of its upstream substrates (short-, medium-, and long-chain acylcarnitine) and a decrease in free carnitine, which binds to acylcarnitine. Patients often complain about muscle weakness and exercise intolerance without efficient energy supplements, explaining the male-dominant tendency in some studies (14–16). Due to the remaining function of ETF-QO, RR-MADD patients often present with mild symptoms. By acting as a molecular chaperone to mediate folding defects, the binding of FAD improves the conformational stability of the variant ETF-QO protein (29, 30). As a result, supplementation with riboflavin, the precursor of FAD, upregulates the amount of ETF-QO holoenzyme by producing more FAD that facilitates binding to apoenzymes (31, 32) improving FAO. In addition, glucocorticoids could treat some RR-MADD patients by alleviating oxidative stress associated with MADD (33).
In our study, patient 4 with compound heterozygous variants, a frameshift mutation c.582_583insTA (p.P196T fs*17) and a missense variant c.295C>T (p.R99C) had a relatively average age of onset and typical clinical picture of LOMADD. Nevertheless, previous reports reveal that patients with heterozygous frameshift and missense mutations appear to have an earlier age of onset than those with heterozygous missense mutations (32, 34–37). Additionally, they frequently exhibit episodes of vomiting, drowsiness, appetite loss, asthenia, and acetonemic breath (32, 34–37), none of which are prevalent in LOMADD. A high dose of riboflavin (100 mg/day or more) often resulted in excellent improvements for the suffers mentioned previously. In contrast, patients with heterozygous frameshift mutations or homozygosity for null mutations are usually diagnosed with type I or II MADD (38) and are almost unresponsive to riboflavin (8). Interestingly, a 6-month-old girl with a heterozygous frameshift and synonymous mutations, which led to exon 5 skipping and truncated protein production, did not exhibit consistent type I or II MADD symptoms. Instead, the child was diagnosed with LOMADD at the age of 6 months and better responded to riboflavin (9). Therefore, we hypothesize that frameshift mutations are more likely to form truncated proteins, impairing ETF-QO protein function and increasing the likelihood of younger onset age. Nevertheless, the clinical manifestations of frameshift and missense ETFDH mutations and their responsiveness to riboflavin remain investigated.
In addition, there is a subset of patients with mutations in riboflavin transporter genes or FAD synthase gene, whose clinical presentation was nearly identical to that of RR-MADD, and dramatic improvement was observed after riboflavin treatment (39). However, genetic analyses revealed no mutation in ETFDH, ETFA, or ETFB. In this situation, we should consider mutations of SLC52A1, SLC52A2, SLC52A3, SLC25A32 (40–43), and FLAD1 (44) that cause FAD homeostasis disturbance.
Several limitations should be mentioned: (1) We did not measure riboflavin and coenzyme Q10 levels, which might provide some treatment supply indicators, such as whether patients with compound heterozygous frameshift and missense mutations require a higher concentration of riboflavin in blood to recover compared with patients with heterozygous missense mutations. (2) Five cases appear insufficient to present the entire clinical presentation and characteristics of LO-MADD.
In conclusion, the primary clinical presentations of LO-MADD are muscle weakness and exercise intolerance with or without myalgia, and patients better respond to riboflavin. Based on clinical manifestations alone, it is difficult to differentiate the disease from polymyositis and fatty acid oxidation disorders. A final diagnosis requires a combination of blood and urine metabolite analyses, muscle biopsy, and genetic diagnosis. Three novel pathogenic variants of ETFDH were identified, enriching their spectrum in the Chinese population and facilitating future genetic data interpretation and analysis.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Xiangya Hospital, Central South University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ZT conducted the case series and drafted the manuscript. SG, MH, JF, and YL participated in the collection and collation of clinical data. QC and WY conducted the muscle biopsy and pathological analysis. HH and XS were responsible for data analysis of genetic testing and Sanger sequencing. JT and HH were involved in revising the manuscript critically and have given final approval of the version to be published. All authors have read and approved the manuscript.
Funding
This work was supported by grants Youth Program of the National Natural Science Foundation of China (81901306, MH, 81600996, YL, and 81801123, WY), Huxiang High-Level Talent Gathering Project of Hunan Province (2019RS1011, MH), and Natural Science Foundation of Hunan (2021JJ40857, YL and 2020JJ5810, HH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab. (2002) 77:86–90. doi: 10.1016/s1096-7192(02)00138-5
2. Schiff M, Froissart R, Olsen RK, Acquaviva C, Vianey-Saban C. Electron transfer flavoprotein deficiency: functional and molecular aspects. Mol Genet Metab. (2006) 88:153–8. doi: 10.1016/j.ymgme.2006.01.009
3. Watmough NJ, Frerman FE. The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta. (2010) 1797:1910–6. doi: 10.1016/j.bbabio.2010.10.007
4. Missaglia S, Coleman RA, Mordente A, Tavian D. Neutral lipid storage diseases as cellular model to study lipid droplet function. Cells. (2019) 8:187. doi: 10.3390/cells8020187
5. Olsen RK, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. (2007) 130(Pt 8):2045–54. doi: 10.1093/brain/awm135
6. Yotsumoto Y, Hasegawa Y, Fukuda S, Kobayashi H, Endo M, Fukao T, et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab. (2008) 94:61–7. doi: 10.1016/j.ymgme.2008.01.002
7. Frerman FE, Goodman SI. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci USA. (1985) 82:4517–20. doi: 10.1073/pnas.82.13.4517
8. Olsen RK, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat. (2003) 22:12–23. doi: 10.1002/humu.10226
9. Hu G, Zeng J, Wang C, Zhou W, Jia Z, Yang J, et al. A synonymous variant c.579A>G in the ETFDH gene caused exon skipping in a patient with late-onset multiple acyl-CoA dehydrogenase deficiency: a case report. Front Pediatr. (2020) 8:118. doi: 10.3389/fped.2020.00118
10. Kuo YC, Hsueh HW, Hsueh SJ, Lee NC, Hsieh MJ, Chao CC, et al. A systematic review of late-onset and very-late-onset multiple acyl-coenzyme A dehydrogenase deficiency: cohort analysis and patient report from Taiwan. Neuromuscul Disord. (2021) 31:218–25. doi: 10.1016/j.nmd.2021.01.006
11. Beresford MW, Pourfarzam M, Turnbull DM, Davidson JE. So doctor, what exactly is wrong with my muscles? Glutaric aciduria type II presenting in a teenager. Neuromuscul Disord. (2006) 16:269–73. doi: 10.1016/j.nmd.2006.01.001
12. Angelini C, Pennisi E, Missaglia S, Tavian D. Metabolic lipid muscle disorders: biomarkers and treatment. Ther Adv Neurol Disord. (2019) 12:1756286419843359. doi: 10.1177/1756286419843359
13. Macchione F, Salviati L, Bordugo A, Vincenzi M, Camilot M, Teofoli F, et al. Multiple acyl-COA dehydrogenase deficiency in elderly carriers. J Neurol. (2020) 267:1414–9. doi: 10.1007/s00415-020-09729-z
14. Xi J, Wen B, Lin J, Zhu W, Luo S, Zhao C, et al. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. (2014) 37:399–404. doi: 10.1007/s10545-013-9671-6
15. Zhu M, Zhu X, Qi X, Weijiang D, Yu Y, Wan H, et al. Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients. J Hum Genet. (2014) 59:256–61. doi: 10.1038/jhg.2014.10
16. Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J Mol Med. (2011) 89:569–76. doi: 10.1007/s00109-011-0725-7
17. Cornelius N, Byron C, Hargreaves I, Guerra PF, Furdek AK, Land J, et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum Mol Genet. (2013) 22:3819–27. doi: 10.1093/hmg/ddt232
18. Liu XY, Wang ZQ, Wang DN, Lin MT, Wang N. A historical cohort study on the efficacy of glucocorticoids and riboflavin among patients with late-onset multiple acyl-CoA dehydrogenase deficiency. Chin Med J. (2016) 129:142–6. doi: 10.4103/0366-6999.173438
19. Wen B, Li D, Shan J, Liu S, Li W, Zhao Y, et al. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol Genet Metab. (2013) 109:154–60. doi: 10.1016/j.ymgme.2013.04.007
20. Wen B, Dai T, Li W, Zhao Y, Liu S, Zhang C, et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry. (2010) 81:231–6. doi: 10.1136/jnnp.2009.176404
21. Liang WC, Ohkuma A, Hayashi YK, Lopez LC, Hirano M, Nonaka I, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. (2009) 19:212–6. doi: 10.1016/j.nmd.2009.01.008
22. Chien YH, Lee NC, Chao MC, Chen LC, Chen LH, Chien CC, et al. Fatty acid oxidation disorders in a Chinese population in Taiwan. JIMD Rep. (2013) 11:165–72. doi: 10.1007/8904_2013_236
23. Zhao YW, Liu XJ, Zhang W, Wang ZX, Yuan Y. Muscle magnetic resonance imaging for the differentiation of multiple acyl-CoA dehydrogenase deficiency and immune-mediated necrotizing myopathy. Chin Med J. (2018) 131:144–50. doi: 10.4103/0366-6999.222323
24. Huang K, Duan HQ, Li QX, Luo YB, Yang H. Investigation of adult-onset multiple acyl-CoA dehydrogenase deficiency associated with peripheral neuropathy. Neuropathology. (2020) 40:531–9. doi: 10.1111/neup.12667
25. Lan MY, Fu MH, Liu YF, Huang CC, Chang YY, Liu JS, et al. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet. (2010) 78:565–9. doi: 10.1111/j.1399-0004.2010.01421.x
26. Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur J Biochem. (2004) 271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x
27. Missaglia S, Tavian D, Angelini C. ETF dehydrogenase advances in molecular genetics and impact on treatment. Crit Rev Biochem Mol Biol. (2021) 56:360–72. doi: 10.1080/10409238.2021.1908952
28. Liu XY, Chen XJ, Zhao M, Wang ZQ, Chen HZ, Li HF, et al. CHIP control degradation of mutant ETF:QO through ubiquitylation in late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. (2021) 44:450–68. doi: 10.1002/jimd.12361
29. Cornelius N, Frerman FE, Corydon TJ, Palmfeldt J, Bross P, Gregersen N, et al. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum Mol Genet. (2012) 21:3435–48. doi: 10.1093/hmg/dds175
30. Er TK, Chen CC, Liu YY, Chang HC, Chien YH, Chang JG, et al. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct Biol. (2011) 11:43. doi: 10.1186/1472-6807-11-43
31. Xu J, Li D, Lv J, Xu X, Wen B, Lin P, et al. ETFDH mutations and flavin adenine dinucleotide homeostasis disturbance are essential for developing riboflavin-responsive multiple acyl-coenzyme A dehydrogenation deficiency. Ann Neurol. (2018) 84:659–73. doi: 10.1002/ana.25338
32. Missaglia S, Tavian D, Moro L, Angelini C. Characterization of two ETFDH mutations in a novel case of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Lipids Health Dis. (2018) 17:254. doi: 10.1186/s12944-018-0903-5
33. Olsen RK, Cornelius N, Gregersen N. Genetic and cellular modifiers of oxidative stress: what can we learn from fatty acid oxidation defects? Mol Genet Metab. (2013) 110(Suppl.):S31–9. doi: 10.1016/j.ymgme.2013.10.007
34. Ou M, Zhu L, Zhang Y, Zhang Y, Zhou J, Zhang Y, et al. A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: a case report of a Chinese family. BMC Med Genet. (2020) 21:98. doi: 10.1186/s12881-020-00995-2
35. Hong D, Yu Y, Wang Y, Xu Y, Zhang J. Acute-onset multiple acyl-CoA dehydrogenase deficiency mimicking Guillain-Barre syndrome: two cases report. BMC Neurol. (2018) 18:219. doi: 10.1186/s12883-018-1221-2
36. Law LK, Tang NL, Hui J, Fung SL, Ruiter J, Wanders RJ, et al. Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin Chim Acta. (2009) 404:95–9. doi: 10.1016/j.cca.2009.02.015
37. Purevjav E, Kimura M, Takusa Y, Ohura T, Tsuchiya M, Hara N, et al. Molecular study of electron transfer flavoprotein alpha-subunit deficiency in two Japanese children with different phenotypes of glutaric acidemia type II. Eur J Clin Invest. (2002) 32:707–12. doi: 10.1046/j.1365-2362.2002.01045.x
38. Grunert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis. (2014) 9:117. doi: 10.1186/s13023-014-0117-5
39. Vergani L, Barile M, Angelini C, Burlina AB, Nijtmans L, Freda MP, et al. Riboflavin therapy. Biochemical heterogeneity in two adult lipid storage myopathies. Brain. (1999) 122(Pt 12):2401–11. doi: 10.1093/brain/122.12.2401
40. Bosch AM, Abeling NG, Ijlst L, Knoester H, van der Pol WL, Stroomer AE, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. (2011) 34:159–64. doi: 10.1007/s10545-010-9242-z
41. Ho G, Yonezawa A, Masuda S, Inui K, Sim KG, Carpenter K, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. (2011) 32:E1976–84. doi: 10.1002/humu.21399
42. Haack TB, Makowski C, Yao Y, Graf E, Hempel M, Wieland T, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J Inherit Metab Dis. (2012) 35:943–8. doi: 10.1007/s10545-012-9513-y
43. Schiff M, Veauville-Merllie A, Su CH, Tzagoloff A, Rak M, Ogier de Baulny H, et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N Engl J Med. (2016) 374:795–7. doi: 10.1056/NEJMc1513610
44. Olsen RKJ, Konarikova E, Giancaspero TA, Mosegaard S, Boczonadi V, Matakovic L, et al. Riboflavin-responsive and -non-responsive mutations in fad synthase cause multiple acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. (2016) 98:1130–45. doi: 10.1016/j.ajhg.2016.04.006
Keywords: ETFDH, riboflavin, mutation, late-onset MADD, molecular analysis
Citation: Tang Z, Gao S, He M, Chen Q, Fang J, Luo Y, Yan W, Shi X, Huang H and Tang J (2021) Clinical Presentations and Genetic Characteristics of Late-Onset MADD Due to ETFDH Mutations in Five Patients: A Case Series. Front. Neurol. 12:747360. doi: 10.3389/fneur.2021.747360
Received: 26 July 2021; Accepted: 05 October 2021;
Published: 08 November 2021.
Edited by:
Corrado Italo Angelini, University of Padua, ItalyReviewed by:
Filippo M. Santorelli, Fondazione Stella Maris (IRCCS), ItalyDaniela Tavian, Catholic University of the Sacred Heart, Italy
Copyright © 2021 Tang, Gao, He, Chen, Fang, Luo, Yan, Shi, Huang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianguang Tang, amlhbmd1YW5nX3RhbmdAY3N1LmVkdS5jbg==; Hui Huang, aHVpaHVhbmcwOTE2QGNzdS5lZHUuY24=