
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 13 October 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.721108
 Adriana Gata-Garcia1,2Amit Porat3
Adriana Gata-Garcia1,2Amit Porat3 Lior Brimberg1
Lior Brimberg1 Bruce T. Volpe1
Bruce T. Volpe1 Patricio T. Huerta2,4
Patricio T. Huerta2,4 Betty Diamond1,2*
Betty Diamond1,2*Autism Spectrum Disorder (ASD) is a group of neurodevelopmental conditions that is four times more commonly diagnosed in males than females. While susceptibility genes located in the sex chromosomes have been identified in ASD, it is unclear whether they are sufficient to explain the male bias or whether gonadal hormones also play a key role. We evaluated the sex chromosomal and hormonal influences on the male bias in a murine model of ASD, in which mice are exposed in utero to a maternal antibody reactive to contactin-associated protein-like 2 (Caspr2), which was originally cloned from a mother of a child with ASD (termed C6 mice henceforth). In this model, only male mice are affected. We used the four-core-genotypes (FCG) model in which the Sry gene is deleted from the Y chromosome (Y−) and inserted into autosome 3 (TgSry). Thus, by combining the C6 and FCG models, we were able to differentiate the contributions of sex chromosomes and gonadal hormones to the development of fetal brain and adult behavioral phenotypes. We show that the presence of the Y chromosome, or lack of two X chromosomes, irrespective of gonadal sex, increased the susceptibility to C6-induced phenotypes including the abnormal growth of the developing fetal cerebral cortex, as well as a behavioral pattern of decreased open-field exploration in adult mice. Our results indicate that sex chromosomes are the main determinant of the male bias in the maternal C6-induced model of ASD. The less dominant hormonal effect may be due to modulation by sex chromosome genes of factors involved in gonadal hormone pathways in the brain.
Autism Spectrum Disorder (ASD) is a group of neurodevelopmental conditions that manifest early in childhood. ASD is characterized by varying degrees of impairment in social interaction and communication, and by restricted interests and repetitive behaviors (DSM-V). In the U.S., about 1 in 59 children was diagnosed with ASD in 2014 (1). Both genetic and environmental factors contribute to the etiology of ASD [reviewed in (2)]. A recent study estimated the heritability of ASD to be 80% (3). However, environmental factors, especially those present during critical periods of prenatal and perinatal brain development, play an essential role in modulating the risk to develop ASD and may account, in part, for the phenotypic variability observed (2).
ASD is four times more frequently diagnosed in males than in females (1). A strong male preponderance is not unique to ASD; other neuropsychiatric conditions, especially those diagnosed earlier in life, also show a male sex bias [reviewed in (4)]. Conversely, neuropsychiatric conditions with a female predominance, including anorexia nervosa and internalizing disorders such as depression and anxiety, present during puberty or later in life [reviewed in (4–6)]. Identifying factors that contribute to neuropsychiatric syndrome susceptibility in either males or females will increase our understanding of brain development and the pathogenesis of these conditions while also providing a foundation for the discovery of new treatments.
Maternal brain-reactive antibodies and gonadal hormones include some of the environmental factors in utero that may contribute to ASD risk and its sex bias. This study evaluated the role of gonadal hormones and sex chromosomes in the male bias we observed in a maternal antibody-induced mouse model of ASD (7). In this model, mice were exposed in utero to C6, a monoclonal antibody reactive to contactin-associated protein-like 2 (Caspr2) cloned from a mother of a child with ASD, or to an isotype matched control antibody, B1. Caspr2 is a cell-adhesion molecule expressed by neurons (8, 9). Rare and common variants of CNTNAP2, the gene encoding Caspr2, have been linked to an increased risk of ASD or ASD-related phenotypes including language delay and developmental language disorders (10–20). In utero exposure to C6, but not to B1, leads to thinning of the cortical plate (CP), impaired social interactions, and increased repetitive behaviors in male offspring only in C57BL/6 mice (7). Here, we used the four-core-genotypes (FCG) mouse model to isolate gonadal hormone from sex chromosome contributions to the male bias observed (21). The FCG model allows for the dissociation of gonadal development from sex chromosome complement through the deletion of the Sry gene from the Y chromosome (referred to as Y−) and the insertion of the Sry transgene into autosome 3 (TgSry), leading to the development of mice with four genotypes: XY− TgSry, XX TgSry, XY−, and XX (21, 22), with the first two having and the second two lacking male hormone synthesis in utero. Our results demonstrate that the male bias of thinner CP seen during fetal development is driven by sex chromosome differences. Furthermore, we found that, in this strain of mice, in utero exposure to C6 antibody causes increased sustained anxiety-like behavior which may also be sex chromosome dependent.
All animal experimentation was performed in accordance with the National Institutes of Health (NIH) Guidelines, under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Feinstein Institutes for Medical Research.
As described previously (7), human embryonic kidney fibroblast 293T cells (HEK-293T, ATCC CRL 11268TM) were split into culture dishes (100 × 20 mm) in high glucose DMEM (HyClone, GE Healthcare), supplemented with heat inactivated fetal bovine serum (FBS, 10%), glutamine (1%) and penicillin-streptomycin (1%, HyClone, GE Healthcare). After 16–24 h, at 70–80% confluence, the medium was replaced with SFM4Transfx-293 (Hyclone, GE Healthcare) supplemented with 10% FBS, 1% glutamine and 1% penicillin-streptomycin (HyClone, GE Healthcare). Cells were co-transfected 8 h later, using Lipofectamine LTX Reagent (Invitrogen), with IgH and IgL encoding plasmids (5 μg). Supernatants were collected after 7 days, and the antibodies were purified on protein G-sepharose beads (GE Healthcare, Life Technologies). Glycine buffer (0.1 M, pH 3.5) and Tris-HCl (1 M, pH 8) were used for antibody elution and pH neutralization, respectively. Purified antibody was dialyzed in PBS and its concentration was determined by Nanodrop. Antibody integrity was assessed on NuPAGE 4–12% BisTris gels (Invitrogen) stained with Coomassie blue.
C57BL/6 female mice and XY− TgSry male mice (6–10 weeks old) obtained from the Jackson Laboratory were used for timed pregnancies. In detail, two wild type females and one XY− TgSry male mouse were housed together for 15 h starting at the beginning of the dark cycle. The male was removed after 15 h. This mating scheme produced offspring with the genotypes XY− TgSry (termed XYM henceforth), XX TgSry (termed XXM), XY− (termed XYF), and XX (termed XXF) (Figure 1A). Embryonic age was determined by the time of male mouse removal which was defined as embryonic day E0.5. Pregnant females were randomly assigned to receive either C6 (anti-Caspr2 IgG, 200 μg) or B1 (control IgG, 200 μg) by retro-orbital sinus injection under light isoflurane anesthesia on E13.5. Subsequently, E15.5 embryos were harvested, genotyped, and processed for brain histology (Figure 1B). In the fetal analysis, 5–7 litters per antibody were included. No more than two fetuses per genotype were selected from each litter for analysis. When two mice were derived from the same litter, the average score was used. A subgroup of pregnancies was assigned for offspring behavior assessment and allowed to reach full term (Figure 1B). Adult mice from each of the four core genotypes (FCG) were obtained from eight litters for C6 and six litters for B1.
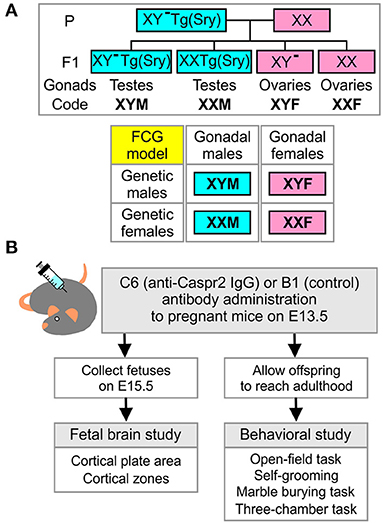
Figure 1. Four-core-genotypes model, combined with in utero exposure to C6, to study maternal antibody-induced model of ASD. (A) Description of the four-core-genotypes (FCG) model in which a C57Bl/6J female mouse (XX) is mated with a XY−(Sry) male mouse (XYM) to generate offspring in which gonadal and chromosomal contributions can be separately studied. (B) Description of the experimental plan for the fetal brain and behavioral studies.
Fetuses were fixed with a 4% paraformaldehyde, 4% sucrose solution for 4 h, at 4°C. They were then cryoprotected at 4°C with a gradual increase in sucrose (45 min in 10% sucrose, 1 h in 20% sucrose, and overnight in 30% sucrose). Fetuses were then submerged in a 1:1 solution of 30% sucrose and OCT compound (Fisher Scientific, Pittsburg, PA, USA) for 1 h at room temperature (RT) and transferred to a cryomold with OCT and frozen on dry ice. Samples were kept at −80°C until cut. Whole embryos were mounted and cut with a Cryostat (Leica, Billerica, MA, USA) and all sections (14 μm) were collected and mounted on gelatin-coated slides. The sampling strategy ensured that sagittal sections 8 and 9, according to the Prenatal Mouse Brain Atlas [(23), p. 295–7], were included. Sections were stored at −80°C until stained.
Sections were thawed for 25 min and rinsed once with 1 × PBS at RT. For antigen retrieval, sections were heated in 10 mM Sodium Citrate Buffer for 10 min at 95°C and cooled to RT with two rinses of 1 × PBS. Blocking was done for 1 h, at RT, with a blocking buffer containing 1 × PBS/0.1% Triton X-100/3% normal goat serum/2.5% bovine serum albumin (BSA). Sections were then incubated with anti-nestin antibody (MAB353, Millipore, Wetzlar, Germany), at 1:400 dilution in 1 × PBS/0.1% Triton X-100/5% BSA, overnight at 4°C. After washing with 1 × PBS/0.1% Tween20, Alexa Fluor 488 goat anti-mouse IgG (A11001, Life Technologies), at 1:400 dilution in 1 × PBS with 0.2% BSA, was used to detect antibody binding (45 min incubation at RT). Secondary antibody was washed off with 1 × PBS/0.1% Tween20, and sections were stained with DAPI (Life Technologies) for 7 min at RT. Sections were washed with 1 × PBS, and coverslipped with Dako Fluorescence Mounting Medium (Dako North America Inc.). Images were obtained with Axio-Imager (Z-1, Zen3.1, Zeiss, Peabody, MA, USA).
The cortical plate (CP) was identified as the highly DAPI-dense band of cells, most distal from the ventricle. This was confirmed on the alternate slides, stained with nestin, which was absent in this region (24). Microscope images (10 × ) of fetal sections stained with DAPI were digitally analyzed using a program that generated orthogonal lines so that a 2-D cortical region was isolated for measurement (Figure 2A). The program allowed the placement of points along the borders of the CP and the ventricle, which were defined using the zoom function (Pyramid, Axiophot2.1, Zen 3.1, Zeiss), and the software joined the points. The CP area was measured as the region contained within the superior and inferior borders of the appropriately joined points and the orthogonal lines (Figure 2A). Next, the region containing the subplate (SP), intermediate zone (IZ), and ventricular zone (VZ) was measured, and defined as the ‘cortical zones' (CZ) area (Figure 2A); the marginal zone was not included in the CZ area. The CP/CZ ratio was calculated by dividing the two areas, followed by repeated sampling (20–30 × ) throughout the length of the defined CP within the section. Additionally, the VZ was defined by the presence of spindle-shaped cells on the edge of the tissue and bright DAPI stain, the IZ was distinguished as the densely-packed DAPI-stained cells (dorsal to VZ), whereas the SP was defined as the sparsely-packed region located dorsal to IZ and ventral to the DAPI-dense CP (Figure 2A). We evaluated 1–3 alternate sections per fetal brain with the CP area, CZ area, CP/CZ ratio, as well as the SP, IZ, and VZ areas being averaged for the entire section. The mean values were used for statistical comparisons across groups. The investigator that performed the CP analysis was blinded to both genotype and antibody exposure. A separate investigator performed the statistical analysis.
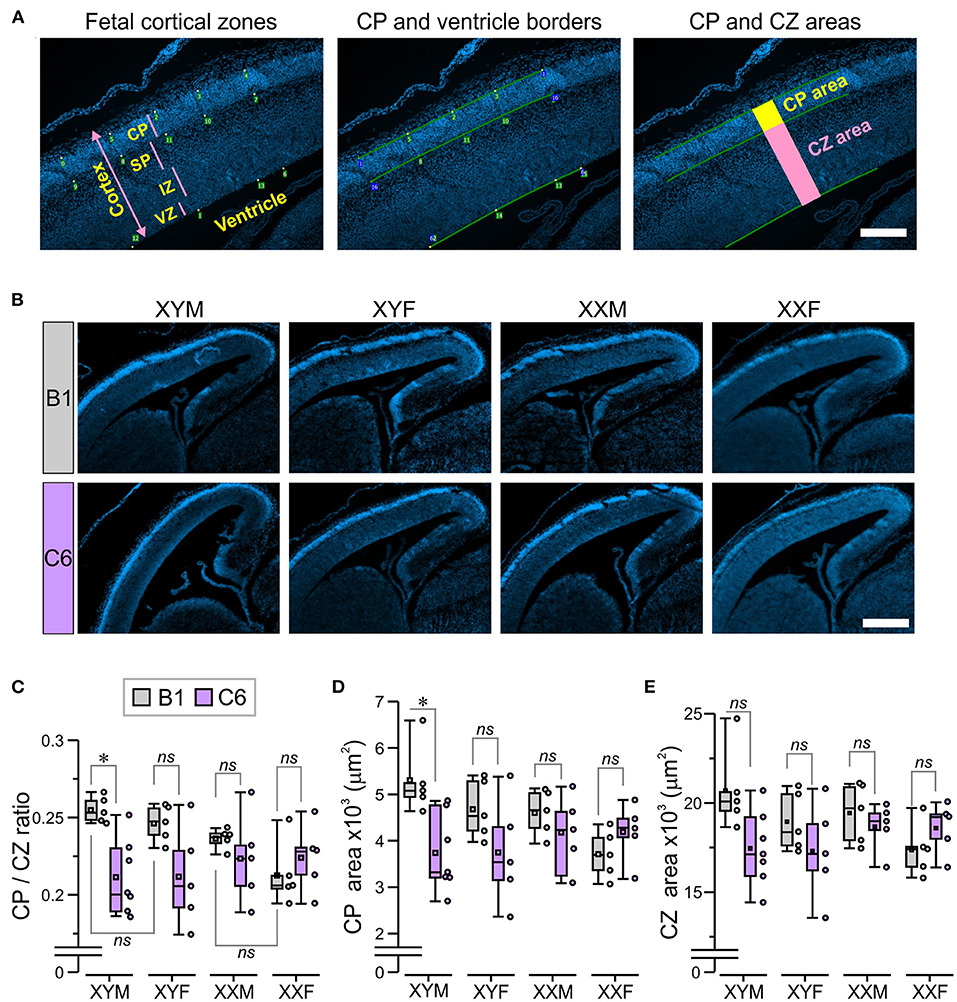
Figure 2. The presence of the Y chromosome predisposes fetuses to develop a smaller cortical plate due to C6 exposure in utero. We established mating pairs between wild type C57Bl/6J female and XYM male mice. This mating scheme produced offspring with four genotypes, whose gonadal development was independent of sex chromosome complement. Offspring from 5 to 7 litters for each antibody were analyzed, with no more than two offspring per genotype selected from each litter. (A) The micrographs describe the technique for measuring the cortical plate (CP) area and the CZ area, which included the subplate (SP), intermediate zone (IZ), and ventricular zone (VZ; as explained in detail in the Methods. Scale = 100 μm. (B) The panels show staining with DAPI, which was used to identify the CP in E15.5 fetuses. Scale = 400 μm. (C–E) Box-and-whisker plots represent median and Q1–Q3 quartiles (whiskers are 10–90 range). Dots represent individual measurements. (C) Quantification of CP/CZ ratio shows that C6-exposed XYM fetuses had a smaller ratio relative to their respective B1-exposed controls. (D) Measurement of the CP area. Compared to their B1 control fetuses, C6-XYM fetuses had a significantly smaller CP area. (E) Measurement of the CZ area. There were no significant differences in the cortical area between C6 and B1 exposed mice irrespective of genotype; 2-way ANOVA with Tukey test was used for statistical comparisons. See Table 1 and Supplementary Table 1 for details of statistical testing; ns, non-significant; *P < 0.05.
FCG mice exposed to C6 or B1 IgG in utero were housed under a reversed dark (9:00–21:00) and light (21:00–9:00) cycle, with ad libitum access to food and water. All manipulations were conducted during the dark phase, at least 1 h after turning lights off, and male and female mice were assessed on different days. Prior to behavioral assessments, mice were handled three times, for 15 min each, on separate days. At 6–11 weeks of age, mice underwent an observational screen, which we have described in detail previously (24), to assess muscle and spinal, spinocerebellar, sensory, neuropsychiatric, and autonomic functions (Table 2). Behavioral assessments were performed at 14–26 weeks of age and included the open-field task (14–15 weeks of age), followed by the marble burying task (17–20 weeks of age), and finally the three-chamber task (22–26 weeks of age). Experiments were conducted and analyzed according to randomly assigned cage numbers which did not indicate mouse genotype or antibody exposure. The investigator was blinded to both genotype and antibody exposure.
This task was used to examine locomotor activity, habituation to a novel chamber, and anxiety-like behavior by placing the mice in the center of a square arena (40 × 40 cm2) with gray walls (35 cm high) and allowing them to freely explore the chamber during two sessions (10 min each) separated by 24 h. The sessions were recorded with a centrally placed video camera directly above the arena which fed the signal to the tracking software (EthoVision XT 14.0, Noldus, Attleboro, MA, USA) used for automated analysis of animal behaviors including distance traveled, velocity, time spent moving, time spent in the center of the arena (18.9 × 18.9 cm2), and self-grooming. We used customized settings, within EthoVision, to detect only grooming bouts >2 s, thus reducing the detection of extra-short bouts.
Repetitive behavior was examined with the marble burying task (25) in which mice were placed in the center of a square arena (40 × 40 cm2, 35-cm wall height) containing 25 black glass marbles (1.2 cm diameter) placed on top of corn cob bedding (5 cm depth) in a 5 × 5 grid pattern arrangement. The mice freely explored the environment for 20 min and at the end of the session, the number of buried marbles (>50% of marble surface area covered by bedding material) was recorded.
This task was used to measure social approach, by placing the subject mouse in an apparatus (60 cm × 40 cm, 40-cm wall height) with clear plexiglass walls, which was subdivided into 3 chambers with sealable doors between chambers. A subject mouse was placed in the center chamber with the left and right chambers sealed off; the doors were opened, and the subject was allowed to explore the empty arena, with access to the 3 chambers, for 10 min. The subject mouse was then gently guided to the center chamber and access to the side chambers was sealed off. An age and sex matched (XYM or XXF) unfamiliar “mouse-stimulus” was placed in one of the side chambers, confined by a cylinder (diameter 9 cm, height 20 cm) with the bottom 5.5 cm perforated with holes (diameter, 0.5 cm). The cylinder was 3D-printed with clear methacrylate resin (FormLabs, Somerville, MA, USA). Each mouse-stimulus was acclimated to the cylinder for 10 min prior to the experiment. An identical but empty methacrylate cylinder was used as a novel object and placed in the second side chamber. The subject mouse was then allowed to explore the arena for 10 min, with access to the three chambers. The chamber used for the mouse-stimulus and the novel object were alternated between trials. The three-chambered apparatus and the methacrylate cylinders were cleaned prior to each trial with 70% ethanol followed by water and wiped dry. Video tracking software (Etovision XT 14.0) was used to obtain the total time spent interacting with the mouse-stimulus and with the novel object. A discrimination ratio was computed by the formula (TMS-TO)/(TMS + TO), in which TMS denotes the time near the mouse-stimulus and TO denotes the time near the novel object.
Most datasets were analyzed with two-way analysis of variance (ANOVA), with “genotype” (XYM, XXM, XYF and XXF) and “antibody” (C6 and B1) as factors, which was followed by post-hoc Tukey correction for multiple comparisons. For the open-field task, time series data were analyzed with 2-way repeated measures ANOVA (RMANOVA) followed by post-hoc Bonferroni correction. For the two sessions of the open-field task, data were analyzed with 3-way ANOVA (with genotype, antibody and sessions as factors) followed by Tukey correction for multiple comparisons. For the three-chamber task, 3-way ANOVA was also used, with genotype, antibody and “stimulus” (mouse-stimulus and object) as factors, followed by Tukey correction for multiple comparisons. The software Origin [Origin Pro 2021b (64-bit) SR2, OriginLab, Northampton, MA] was used for statistical tests. P < 0.05 were considered statistically significant.
We established mating pairs between wild type C57Bl/6J female and XY− TgSry male mice (Figure 1A), which delivered offspring with the genotypes XY− TgSry (termed XYM), XX TgSry (termed XXM), XY− (termed XYF), and XX (termed XXF) (Figure 1A). We administered intravenous C6 or B1 antibody to pregnant mice on embryonic day E13.5 and harvested the fetuses on day E15.5 (7) (Figure 1B). We used the XYM and XXF mice as internal controls to confirm that, as in wild type C57BL/6J mice, in utero exposure to C6 led to ASD-like phenotypes in males but not females in this mouse strain. Offspring from 5 to 7 litters for each antibody were analyzed, with no more than two offspring per genotype selected from each litter. Not all genotypes were present in all litters.
We measured the CP/CZ ratio (Figure 2A) in hormonal and chromosomal males and females exposed to C6 or B1, as we had shown this ratio to be diminished in C57BL/6 males, but not female mice exposed to C6 in utero (7). Crucially, we observed no effect of C6 administration in XXF offspring (Figures 2B,C, P > 0.1, Table 1) and a decrease in the CP/CZ ratio in XYM offspring (Figure 2C), replicating our previous results in C57BL/6 mice (7). Indeed, we found that C6-exposed XYM fetuses had a significantly smaller CP/CZ ratio when compared to B1-XYM control fetuses (Figure 2C; q = 4.89, P = 0.029, see Table 1 and Supplementary Table 1 for details on statistical tests), which was likely due to a decrease in CP area specifically and not to smaller SP, IZ or VZ. Of note, the C6-treated fetuses displayed high variability in the CP/CZ ratio and, predictably, two-way ANOVA revealed a significant effect for the “antibody” factor [C6 vs. B1 groups, F(1, 41) = 9.1, P = 0.0048] and the antibody × genotype interaction [F(3, 41) = 3.29, P = 0.03], while the post-hoc Tukey correction confirmed the C6 vs. B1 effect (q = 4.41, P = 0.004). However, the comparisons of the interactions for all the other groups failed to reach statistical significance (Supplementary Table 1).
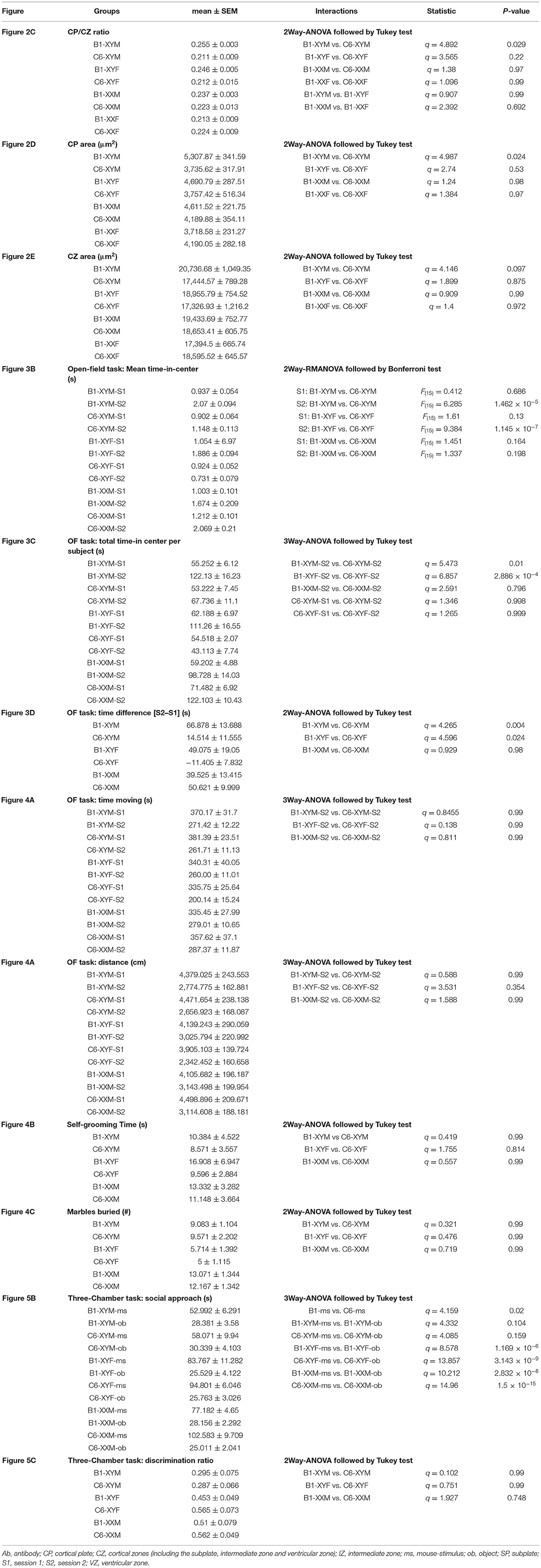
Table 1. Statistical analysis for the parameters presented in the figures.
Importantly, the CP/CZ ratio of B1-XYM and B1-XYF offspring did not differ (Figure 2C, P > 0.1, Table 1) suggesting that sex hormones do not determine the CP size in XY mice. Also, B1-XXM and B1-XXF mice had an equivalent CP/CZ ratio (Figure 2C, P > 0.1, Table 1), suggesting again that sex hormones do not regulate cortical structure in XX mice.
Additionally, when compared to their respective control group, the CP area was significantly smaller in C6-XYM fetuses (Figure 2D, P < 0.05, Table 1). As in the case of the CP/CZ ratio, two-way ANOVA showed significant effects for antibody [F(1, 41) = 6.74, P = 0.001] and antibody × genotype interaction [F(3, 41) = 3.42, P = 0.027], whereas the Tukey test confirmed the C6 vs. B1 effect (q = 3.82, P = 0.001), a significant C6-XYM vs. B1-XYM interaction (q = 4.98, P = 0.024) and lack of significance for all other interactions (Supplementary Table 1). Moreover, in utero exposure to C6 maternal antibody did not alter the CZ area in any of the groups (Figure 2E, P > 0.1, Table 1), which was verified by two-way ANOVA followed by Tukey test (Supplementary Table 1). To further confirm this point, we performed separate measurements of the areas of the subplate, intermediate zone, and ventricular zone (Figure 2A) and, again, found no differences across the groups (P > 0.5, Supplementary Table 1). Hormones were not identified as a contributing factor to the male bias of a C6-induced decrease in CP area at the E15.5 stage of development because neither gonadal female nor gonadal male XX fetuses were significantly affected by antibody exposure.
To evaluate the role of sex chromosomes and gonadal hormones on the male bias of C6-induced behavioral deficits, we conducted a series of behavioral studies on adult FCG mice that were exposed to C6 or B1 in utero on E13.5. We include 3 groups in these studies (XYM, XYF and XXM) as our previous studies in wildtype C57BL/6 mice revealed no effect of C6 exposure on female offspring and there was no structural phenotype associated with C6 exposure in XXF offspring (Figures 2B,C). Initially, we performed an observational screen and surveyed several variables to assess muscle, spinal, spinocerebellar, sensory, neuropsychiatric, and autonomic functions (Table 2). We found that, for each genotype, there were no significant differences in the scores obtained for C6 and B1 mice (Table 2).
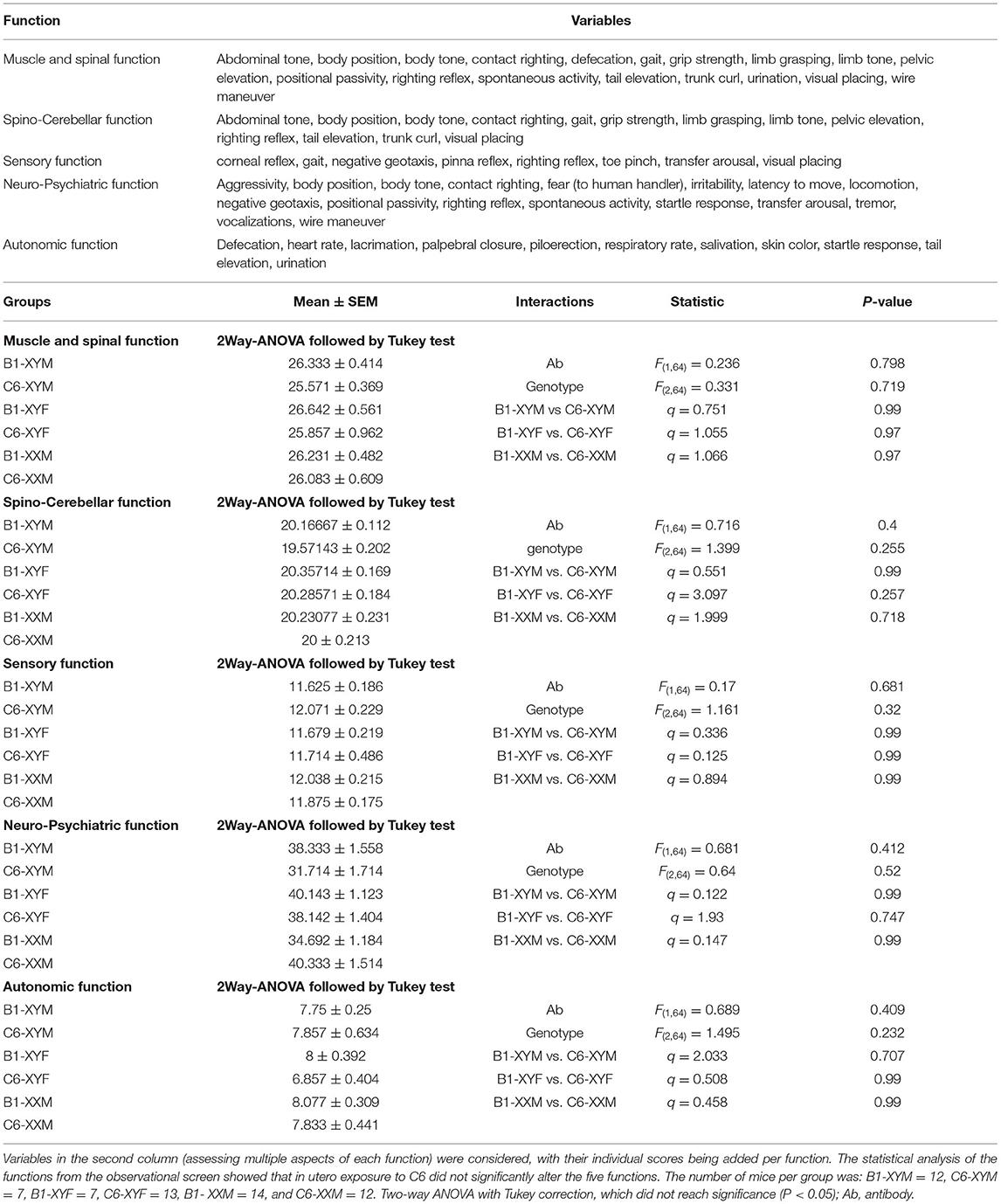
Table 2. Five functions were evaluated in the observational screen.
A set of behavioral assessments was focused on key ASD-like phenotypes, such as the presence of anxiety, repetitive behaviors, and social impairments. We studied adult mice from eight litters exposed to C6 and six litters exposed to B1. Not all the genotypes were represented in each litter. Anxiety is often a comorbid psychiatric condition in patients with ASD. Therefore, we evaluated C6-exposed FCG mice for increased anxiety-like behavior in an open-field task in which the animals were allowed to freely explore a well-lit square arena during two sessions (S1 and S2), lasting 10 min each, and separated by 24 h (Figure 3A). In the context of the open-field task, anxiety-like behavior is defined as avoidance to explore the center of the apparatus, which some claim occurs more prominently at the early stage of the task. However, it is clear that initial exposure to the chamber triggers habituation, a type of non-associative learning, in which the mice adapt their behavioral responses due to the continual exposure to the novel environment. Therefore, in this study, we decided to define sustained anxiety-like behavior as a mouse spending significantly less time in the center of the arena during S2, after the initial S1 habituation session. We, thus, calculated ‘time-in-center' scores for consecutive 10-s intervals in S1 and S2, which we used to build time series graphs (Figure 3B). During S1, we found that all groups (regardless of antibody exposure or genotype) displayed similar time-in-center scores (Figure 3B, P > 0.1, Table 1). During S2, C6-XYM mice and C6-XYF mice did not increase their time-in-center scores while the other groups spent significantly more time in the center area than they had during S1 (Figure 3B, P < 0.01, Table 1, Supplementary Table 2).
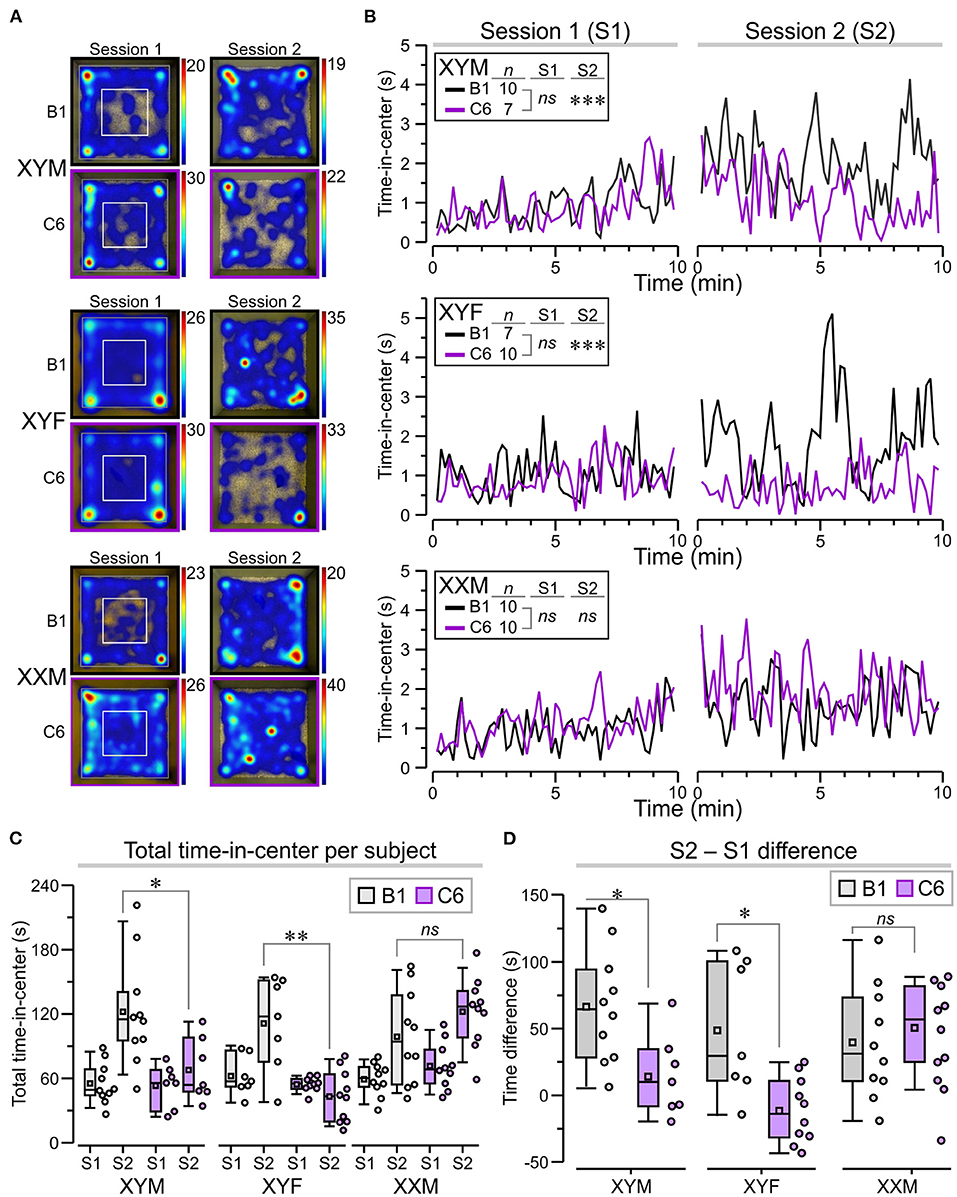
Figure 3. Sustained anxiety-like behavior in C6-exposed XYM and XYF mice during the open-field task. (A) Top-view heatmaps of the open-field task for representative XYM, XYF, and XXM mice that were exposed in utero to anti-Caspr2 antibody (C6) or control antibody (B1). The color scale at the right of each heatmap represents seconds. (B) Graphs show the time spent at the center of the arena, taken over regular 10-s intervals during session 1 (S1) and session 2 (S2), for all groups. RMANOVA with Bonferroni post-hoc tests reveal significantly lower time-in-center, during S2, for C6-XYM mice compared to B1-XYM mice as well as C6-XYF mice compared to B1-XYF mice. (C) Box-and-whisker plots for time-in-center show mean (small square), median, Q1–Q3 quartiles (box), and 10–90 range (whiskers) for S1 and S2. Dots represent individual mice; 3-way ANOVA, followed by Tukey test, was used for statistical comparisons. (D) Difference for time-in-center between S2 and S1 show statistical differences for the XYM and XYF cohorts; 2-way ANOVA with Tukey test was used for statistical comparisons. See Table 1 and Supplementary Table 2 for details of statistical testing; ns = non-significant, *P < 0.05, **P < 0.01, ***P < 0.001.
To further demonstrate the anxiety-like effect in C6-XYM and C6-XYF groups, we computed the overall time-in-center for each subject during S1 and S2 (Figure 3C) and found that neither C6-XYM nor C6-XYF mice increased the amount of time spent in the center during S2 (P = 0.99 for both groups, Table 1, Supplementary Table 2). In contrast, C6-XXM mice significantly increased their time-in-center during S2 (P < 0.05, Table 1, Supplementary Table 2). Also, B1-XYM mice had higher time-in-center during S2 (Figure 3C, P < 0.05, Table 1, Supplementary Table 2). Furthermore, a comparison of B1 vs C6 groups during S2 confirmed that C6-XYM and C6-XYF mice spent significantly less time-in-center than their B1 counterparts (Figure 3C, P < 0.05 for both groups, Table 1, Supplementary Table 2), whereas C6-XXM and B1-XXM animals spent a similar amount time in the center of the arena (Figure 3C, P = 0.796, Table 1, Supplementary Table 2). Finally, we computed the time difference for time-in-center during the two sessions (S2–S1) and found that the C6-XYM and C6-XYF groups had significantly lower scores than B1-XYM and B1-XYF groups, respectively (Figure 3D, P < 0.05 for both groups, Table 1, Supplementary Table 2). Taken together, these data show a sustained anxiety-like phenotype associated with sex chromosome complement.
We also assessed locomotor activity in the open-field task by measuring the total time moving and the total distance traveled during each period to ensure that this was not a confounding variable. We focused on S2 as it was in S2 that we observed differences related to genotype and antibody exposure. During S2, all the groups showed significantly less time moving when compared to S1 (Figure 4A, P < 0.05, Table 1, Supplementary Table 2); nevertheless, the C6-exposed offspring had similar time-moving scores when compared to B1 control mice (Figure 4A, P > 0.5, Table 1, Supplementary Table 2). Additionally, there were no significant differences in distance traveled during S2 for C6-exposed mice compared to their respective B1-exposed controls (Figure 4A, P > 0.05, Table 1, Supplementary Table 2).
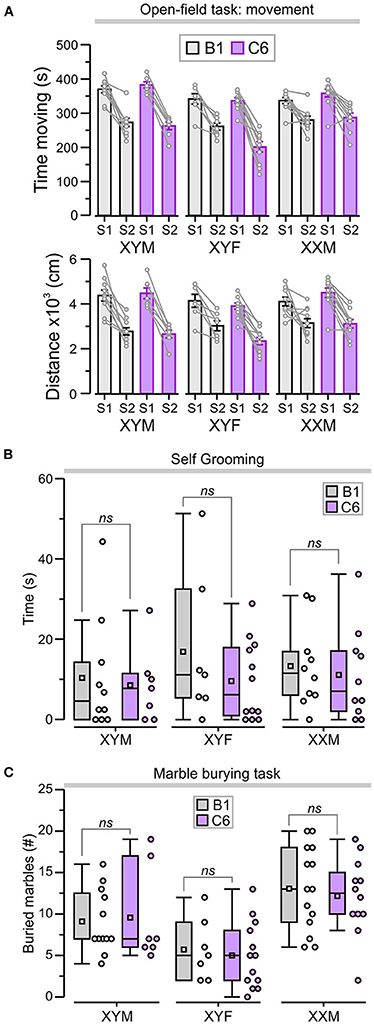
Figure 4. C6 exposure in utero did not lead to an increase in repetitive behaviors. (A) Top, graph shows the total time (mean ± SEM) moving during sessions 1 and 2 (S1 and S2). Bottom, total distance traveled (mean ± SEM) as a measure of locomotor activity in the open-field task. The lines linking each S1 and S2 represent the scores for individual animals. (B) Box-and-whisker plots for cumulative time self-grooming show median and Q1–Q3 quartiles (whiskers are 10–90 range). Dots represent individual animals. C6-exposed mice did not exhibit increased self-grooming compared to B1 control groups, irrespective of genotype. (C) Box-and-whisker plots show the number of marbles buried (more than 50% of the surface area covered by bedding) during the marble-burying task. The number of marbles buried was not significantly different between B1- and C6-exposed mice, irrespective of genotype; 2-way ANOVA with Tukey test was used for statistical comparisons. See Table 1 and Supplementary Table 2 for details of statistical testing; ns = non-significant.
We evaluated the C6-exposed FCG mice for stereotypic and compulsive behaviors using cumulative time self-grooming and number of marbles buried. We found that XYM, XYF, and XXM mice exposed to C6 in utero did not display an increase in repetitive behaviors when compared to their respective B1 controls in either self-grooming (Figure 4B, P > 0.4, Table 1, Supplementary Table 2) or the marble burying task (Figure 4C, P > 0.6, Table 1, Supplementary Table 2).
Lastly, we used the three-chamber task [adapted from Yang et al. (26)] to explore the influence of sex chromosomes and gonadal hormones on the decreased social interactions produced by in utero exposure to C6 in male mice (7) (Figure 5A). We defined social approach as the total amount of time spent interacting with an unfamiliar mouse (referred to as “mouse-stimulus”) compared to the amount of time spent interacting with a novel object. Normal social approach was defined as spending more time near the mouse-stimulus and less time near the object. We found that XYF and XXM mice, regardless of C6 or B1 exposure, preferred the mouse-stimulus compared to the object (Figures 5A,B, P < 0.01, Table 1, Supplementary Table 2). Interestingly, C6-XYM mice did not show significantly higher exploration of the mouse-stimulus but, paradoxically, B1-XYM mice failed to show a preference for the mouse-stimulus (Figures 5A,B, P > 0.1, Table 1, Supplementary Table 2). Additionally, we computed social discrimination ratios (for which positive values reflect a predilection for the mouse-stimulus), which further demonstrated that B1- and C6-exposed mice of all groups preferred the mouse-stimulus instead of the object (Figure 5C, Table 1, Supplementary Table 2). These results indicate that XYM, XYF and XXM mice exposed to C6 displayed normal social approach. We were unable to replicate the phenotype seen in C6 exposed male wild type mice (7) in the FCG model.
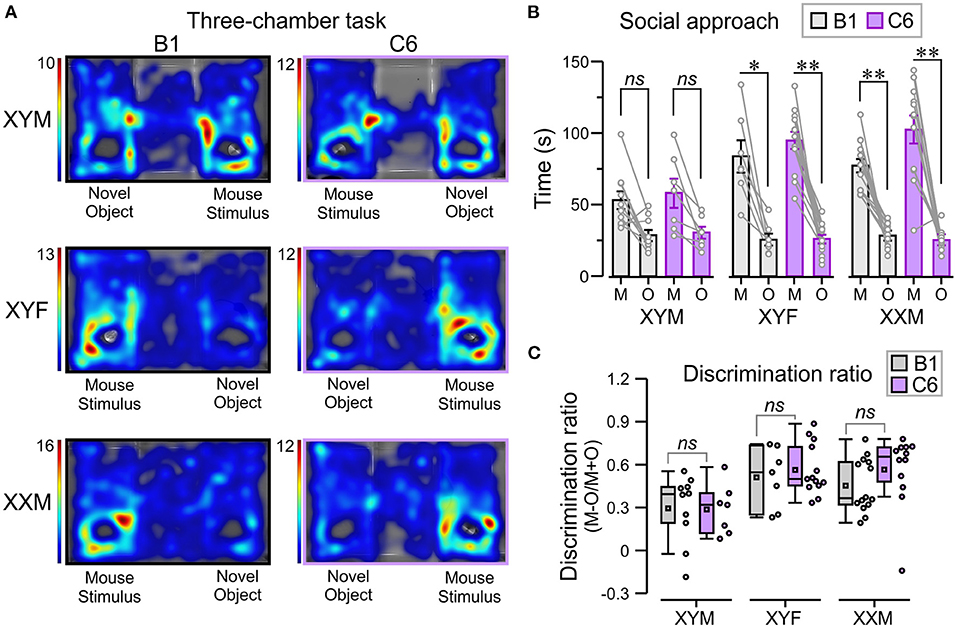
Figure 5. In utero C6 exposure did not affect social approach. We used the three-chamber task to evaluate social approach. The number of mice per group was: B1-XYM = 10, C6-XYM = 7, B1-XYF = 7, C6-XYF = 13, B1-XXM = 14, and C6-XXM = 12. (A) Representative trials of the social approach session displayed as top-view heatmaps for individual B1- and C6-exposed XYM, XYF, and XXM mice. (B) Time (mean ± SEM) spent interacting with the novel object (O) and the “mouse-stimulus” (M). The lines linking each mouse-stimulus and object represent the scores for individual subjects. All the groups tested showed social preference for the mouse-stimulus compared to the novel object; 3-way ANOVA with Tukey test was used for these comparisons. (C) Box-and-whisker plots show the discrimination ratio for social interaction. C6- and B1-exposed mice displayed similar discrimination between mouse-stimulus and novel object, irrespective of genotype; 2-way ANOVA with Tukey test was used for these comparisons. See Table 1 and Supplementary Table 2 for details of statistical testing; ns, non-significant, *P < 0.05, **P < 0.001.
In this study we used the FCG mouse model to distinguish the contributions of sex chromosomes and gonadal hormones to abnormal cortical development and adult ASD-like phenotypes in a model of maternally-induced ASD by the anti-Caspr2 antibody, C6. Analysis of the fetal brains showed that C6-exposed XYM fetuses had significantly smaller CP/CZ ratio and CP area, when compared to B1-XYM control fetuses. Remarkably, the B1-XYM and B1-XYF fetal cortices did not differ in their CP/CZ ratio and CP area, and this was also the case for the B1-XXM and B1-XXF groups, which strongly suggest that sex hormones were not responsible for determining the CP size in either XY or XX mice. Moreover, behavioral assessment of adult offspring revealed a behavioral pattern of sustained anxiety-like behavior in C6-XYM and C6-YXF mice, in which they spent significantly less time in the center of an open-field arena during a second exposure after the initial habituation session. Additionally, adult C6-exposed XYM, XYF and XXM mice did not seem to exhibit abnormalities in locomotion, self-grooming, the marble-burying task and the three-chamber social approach task.
C6 antibody exposure in our model occurs throughout a limited window during in utero development, from time of antibody administration on E13.5 to IgG exclusion by the blood brain barrier between E16.5 and E17.5. Thus, we propose that the sex-biased effects of the C6 antibody depend on the roles that sex chromosome genes and gonadal hormones have on the critical brain developmental processes ongoing at the time of exposure. Our findings support a predominant influence of sex chromosome complement on the susceptibility to C6-induced ASD-like phenotypes in mice exposed mid-gestation.
Changes in cortical thickness, including thinning (27–32) and thickening (33–35), have been observed in ASD. We have previously shown that in utero exposure to C6 antibody leads to thinning of the CP in male mice (7). Using the FCG mouse model, we found that both XYM and XYF but not XXM or XXF mice had a significantly smaller CP/CZ ratio when exposed to C6. Thus, presence of the Y chromosome or lack of two copies of the X chromosome is key in determining the susceptibility to cortical thinning, irrespective of gonadal hormones. The mechanism for the thinned cortical plate has not yet been determined. Sex chromosome complement has previously been identified to be important for brain development and the establishment of brain sex differences (21, 36–40). In congruence with our findings, the analysis of MR brain images from individuals with complete androgen insensitivity syndrome (CAIS) and Klinefelter syndrome suggests a sex chromosome gene-dosage effect on the thickness of the motor cortex (39) and the temporal, orbitofrontal and lingual cortices (41), respectively. Savic and Arver (41) propose that sex differences in motor cortex development are predominantly established by X-chromosome genes that escape inactivation and lack a Y-chromosome homolog whereas in the superior temporal cortex these differences are likely influenced by X-chromosome escapee genes with Y-chromosome homologs. Similar potential sex chromosome gene dosage relationships have been proposed to account for the brain structural differences identified in individuals with sex chromosome aneuploidies (42–44). Indeed, Vawter et al. (45) identified fourteen X-chromosome genes that are differentially expressed in XXY compared to XY individuals, twelve of which were significantly correlated with measure of verbal cognition. Furthermore, Good et al. (42) propose that haploinsufficiency of a subset of X-linked genes, including the gene encoding monoamine oxidase B, contribute to the brain structural abnormalities and neurocognitive deficits observed in Turner syndrome. The increased prevalence of ASD in individuals with sex chromosome aneuploidies [reviewed in (46–51)] supports the hypothesis that sex chromosome gene dosage contributes to ASD susceptibility and suggests that it may contribute to the sex bias in our model.
While sex chromosomes are essential for brain differentiation and sex specific behaviors (21, 37, 52–59), gonadal hormones also modulate developmental processes and contribute to sex differences in brain anatomy. Indeed, both human and animal studies have found a mixed contribution of gonadal hormones and sex chromosomes to sex differences in brain structure, with the predominant factor being region specific (38, 39, 41, 43, 6041; 39). In particular, testosterone exposure has been implicated in determining parietal and occipital cortical thickness in individuals with CAIS and sex chromosome aneuploidies (39, 41). Additionally, data from the FCG mouse model also suggest a predominant gonadal hormone contribution to differences in cortical thickness and volume in the adult brain as we found in the comparison of B1-exposed XXF and XXM mice in the fetal brain (38, 60). Based on these data, gonadal hormones might have been predicted to modulate the effects of C6 on fetal cortical thickness, however, they mainly address hormone exposure during puberty (61, 62). Furthermore, gonadal hormone and signaling deficiencies observed in individuals with CAIS and sex chromosome aneuploidies may act as confounders when studying the association between sex chromosomes and brain sexual differentiation.
While sex hormones act on the brain throughout post-partum life, for gonadal hormones to modulate the susceptibility to C6-induced phenotypes, both the hormones and their receptors must be expressed at the time of antibody exposure. Androgen receptor (AR) mRNA has been detected in the fetal mouse brain as early as E11, with expression in the neocortex, hippocampal cortex, and hypothalamus peaking on E15–16 (63). Estrogen receptor (ER) α protein has been detected as early as E12–14 in the mouse embryonic ventricular and subventricular zones (64) whereas mRNA has been detected as early as E16.5 in the mouse fetal brain (65) and follows a similar pattern in the ventricular zone and cerebral cortex in rats, with the earliest expression on E16 (66). ERβ protein has been detected as early as E12.5 in the mouse brain and was first appreciated in the deep layers of the cerebral cortex on E15.5–16.5 (67). Signaling through AR and ERs has been found to regulate developmental processes in the cortex. Zhang et al. (68) observed an effect of testosterone and estrogen on the differentiation but not proliferation of cortical neurons isolated from rat fetuses on E14. They proposed that these effects are mediated by signaling through the AR and not ERα as its expression was low. In mice, Wang et al. (69) proposed that estrogen regulates neuronal survival and migration through ERβ and that decreased signaling leads to cortical thinning likely secondary to impairments in these processes. While the beginning of the period during which decreased signaling through ERβ in the mouse fetal brain leads to cortical thinning was narrowed down to E14.5 at the earliest, thus overlapping with the period of antibody exposure in our model, no sex bias was observed. This lack of sex bias in the effects of altered signaling through ERβ on cortical structure could account for the insignificant influence of gonadal hormones in our ASD model as one would expect C6 to interact with processes that when altered have different outcomes in males compared to females.
Both testosterone and estrogens are present in mouse fetal circulation at the time of C6 exposure. Fetal Leydig cells in mice arise on E12.5 [reviewed in (70)] and proteins required for testosterone synthesis are first detected between E12.5 and E13.5, coinciding with the earliest reported testosterone production (71). Given that fetal rodent ovaries are thought to produce minimal amounts of estrogen (72), estrogens in the fetal peripheral circulation likely originate from maternal and placental sources. However, of these hormones, only peripherally derived testosterone crosses the blood brain barrier in a form that can be actively used. Estrogens in the peripheral circulation are bound by alpha-fetoprotein, rendering them inaccessible for cell signaling in the brain (73). Thus, alpha-fetoprotein protects the female rodent brain from masculinization and defeminization by estrogen (73) while testosterone aromatization to estrogen masculinizes and defeminizes the male rodent brain. While alpha-fetoprotein may sequester peripheral estrogens, other sources of estrogen including aromatization of testosterone and de novo synthesis from cholesterol have been identified in the developing rodent brain (74–83). Moreover, Martínez-Cerdeño et al. (64) detected aromatase expression in the mouse ventricular and subventricular zones as early as E9. Interestingly, alpha-fetoprotein is also strongly expressed in the ventricular zone from E12 (64). Hence, while there may be differences in the level of estrogen in the brain during development between males and females, C6 may alter brain developmental processes in regions where estrogen signaling is diminished by locally produced alpha-fetoprotein. Furthermore, sex chromosome genes may modulate aromatase and gonadal hormone receptor expression in the brain (84, 85), potentially accounting for the observed dominant genetic effect on C6 susceptibility. In accordance with this hypothesis, decreased expression of aromatase has been detected in individuals with ASD (86, 87).
The behavioral assessment of adult mice revealed that the time spent in the center of an open-field arena did not significantly increase upon repeat exposure in C6-XYM and C6-XYF mice. Furthermore, C6-XYM and C6-XYF mice spent significantly less time in the center of the arena during repeat exposure compared to the control B1 counterparts. We have termed this response sustained anxiety-like behavior, as anxiety has been defined by others as avoidance of the center of an arena in the first exposure. Together, these observations indicate an abnormal behavioral pattern due to in utero C6 exposure in XYM and XYF mice. Conversely, XXM mice showed a significant difference in time spent in the center between the first and repeated exposures independent of C6 or B1 exposure, suggesting that the anxiety phenotypes are driven by sex chromosome complement. Indeed, individuals with sex chromosome aneuploidies, including Klinefelter and Turner Syndromes, have been reported to have an increased prevalence of anxiety (88–90). Additional tests including the elevated plus maze and light-dark box tests may be useful to further characterize the anxiogenic effects of C6 and the relationship to sex chromosome complement.
In this study we identified sex chromosome complement to be essential for the effects of in utero exposure to C6 maternal antibody on fetal brain cortical development and adult behavior. Differences in gene dosage is one potential mechanism through which sex chromosome complement may be increasing the susceptibility of XY mice to C6. Indeed, Xu et al. (59) identified a subset of genes outside the pseudo-autosomal region of the sex chromosomes whose level of expression in the brain shows a sex bias. Furthermore, while X chromosome inactivation is a compensatory mechanism for differences in gene dosage between XX and XY complements (91), an estimated 10–15% of the X chromosome genes outside the pseudo-autosomal region escape inactivation in humans and are therefore more highly expressed in XX compared to XY individuals (92, 93). Finally, imprinting can affect expression levels of X chromosome genes in the brain (94–96) and has been associated with impairments in social behavior in Turner Syndrome (97) and cognitive function in a mouse model of this condition (94). Of note, the mouse Y chromosome encodes for 10 × more genes than the human Y chromosome (98). Consequently, if Y chromosome genes account for the male bias observed in our mouse model of maternal antibody induced ASD, our findings may not completely translate to the pathogenesis of ASD in humans exposed in utero to anti-Caspr2 antibody.
While the establishment of sexual dimorphisms involves both gonadal hormones and sex chromosomes, both of which are operative not just during the window of fetal brain exposure to maternal antibody our data suggest that gonadal hormones have a limited role in determining the susceptibility to C6-induced phenotypes, at least in these genetically manipulated mice. It should be noted that sex hormone levels in these mice are not the same as in the C57BL/6 strain in which the C6 model was established. Nevertheless, C6 likely impairs development processes that are regulated by sex chromosome genes. It is possible that these sex chromosome genes modulate gonadal hormone signaling pathways. For example, sex chromosome complement determines sex differences in aromatase and ERβ expression levels in the developing mouse amygdala (84, 85). Given the proposed relationship between cortical thickness and sociability (30) and symptom severity (99) in ASD, further inquiry into the exact mechanism by which sex chromosome complement influences the risk to develop ASD-like phenotypes due to C6 exposure in utero is important. Deciphering the exact mechanisms through which sex chromosome genes compensate for or exacerbate the effects of in utero C6 exposure in females and males, respectively, will be key for expanding our knowledge of brain development and identifying potential therapeutic targets for ASD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC) of the Feinstein Institutes for Medical Research.
AG-G, AP, LB, BV, PH, and BD designed the experiments. AG-G, AP, BV, and PH performed experiments and analyzed the data. AG-G and PH made the final figures. AG-G, PH, and BD wrote the manuscript. All authors approved the manuscript.
This work was supported by the National Institutes of Health (NIH) grant 5P01AI102852 and NIH grant 5P01AI073693 to BD. It was also supported by the Nancy Lurie Marks Family Foundation (BD). PH was supported by DOD Impact Award W81XWH1910759.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Andrea La Bella for help with experiments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.721108/full#supplementary-material
1. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. (2018) 67:1–23. doi: 10.15585/mmwr.ss6706a1
2. Gata-Garcia A, Diamond B. Maternal antibody and ASD: clinical data and animal models. Front Immunol. (2019) 10:1129. doi: 10.3389/fimmu.2019.01129
3. Bai D, Yip BHK, Windham GC, Sourander A, Francis R, Yoffe R, et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry. (2019) 76:1035–43. doi: 10.1001/jamapsychiatry.2019.1411
4. Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. (2011) 9:e1001081. doi: 10.1371/journal.pbio.1001081
5. Nolen-Hoeksema S, Girgus JS. The emergence of gender differences in depression during adolescence. Psychol Bull. (1994) 115:424–43. doi: 10.1037/0033-2909.115.3.424
6. Rutter M, Caspi A, Moffitt TE. Using sex differences in psychopathology to study causal mechanisms: unifying issues and research strategies. J Child Psychol Psychiatry. (2003) 44:1092–115. doi: 10.1111/1469-7610.00194
7. Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK, et al. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Mol Psychiatry. (2016) 21:1663–71. doi: 10.1038/mp.2016.165
8. Bel C, Oguievetskaia K, Pitaval C, Goutebroze L, Faivre-Sarrailh C. Axonal targeting of Caspr2 in hippocampal neurons via selective somatodendritic endocytosis. J Cell Sci. (2009) 122 :3403–13. doi: 10.1242/jcs.050526
9. Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. (1999) 24:1037–47. doi: 10.1016/S0896-6273(00)81049-1
10. Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. (2008) 82:150–9. doi: 10.1016/j.ajhg.2007.09.005
11. Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. (2008) 82:160–4. doi: 10.1016/j.ajhg.2007.09.015
12. Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. (2008) 82:165–73. doi: 10.1016/j.ajhg.2007.09.017
13. Newbury DF, Paracchini S, Scerri TS, Winchester L, Addis L, Richardson AJ, et al. Investigation of dyslexia and SLI risk variants in reading- and language-impaired subjects. Behav Genet. (2011) 41:90–104. doi: 10.1007/s10519-010-9424-3
14. Nord AS, Roeb W, Dickel DE, Walsh T, Kusenda M, O'Connor KL, et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet. (2011) 19:727–31. doi: 10.1038/ejhg.2011.24
15. O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. (2011) 43:585–9. doi: 10.1038/ng.835
16. Poot M, Beyer V, Schwaab I, Damatova N, Van't Slot R, Prothero J, et al. Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics. (2010) 11:81–9. doi: 10.1007/s10048-009-0205-1
17. Sehested LT, Møller RS, Bache I, Andersen NB, Ullmann R, Tommerup N, et al. Deletion of 7q34-q36.2 in two siblings with mental retardation, language delay, primary amenorrhea, dysmorphic features. Am J Med Genet A. (2010) 152A:3115–9. doi: 10.1002/ajmg.a.33476
18. Steer CD, Golding J, Bolton PF. Traits contributing to the autistic spectrum. PLoS ONE. (2010) 5:e12633. doi: 10.1371/journal.pone.0012633
19. Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. (2006) 354:1370–7. doi: 10.1056/NEJMoa052773
20. Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. (2008) 359:2337–45. doi: 10.1056/NEJMoa0802828
21. De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, et al. A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci. (2002) 22:9005–14. doi: 10.1523/JNEUROSCI.22-20-09005.2002
22. Itoh Y, Mackie R, Kampf K, Domadia S, Brown JD, O'Neill R, et al. Four core genotypes mouse model: localization of the sry transgene and bioassay for testicular hormone levels. BMC Res Notes. (2015) 8:69. doi: 10.1186/s13104-015-0986-2
23. Schambra UB. Prenatal Mouse Brain Atlas. 1st edn. Boston, MA: Springer (2008). doi: 10.1007/978-0-387-47093-1
24. Lee JY, Huerta PT, Zhang J, Kowal C, Bertini E, Volpe BT, et al. Neurotoxic autoantibodies mediate congenital cortical impairment of offspring in maternal lupus. Nat Med. (2009) 15:91–6. doi: 10.1038/nm.1892
25. Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology. (2009) 204:361–73. doi: 10.1007/s00213-009-1466-y
26. Yang M, Silverman JL, Crawley JN. Automated three-chambered social approach task for mice. Curr Protoc Neurosci. (2011) Chapter 8:Unit 8.26. doi: 10.1002/0471142301.ns0826s56
27. Chung MK, Robbins SM, Dalton KM, Davidson RJ, Alexander AL, Evans AC. Cortical thickness analysis in autism with heat kernel smoothing. Neuroimage. (2005) 25:1256–65. doi: 10.1016/j.neuroimage.2004.12.052
28. Hadjikhani N, Joseph RM, Snyder J, Tager-Flusberg H. Anatomical differences in the mirror neuron system and social cognition network in autism. Cereb Cortex. (2006) 16:1276–82. doi: 10.1093/cercor/bhj069
29. Hyde KL, Samson F, Evans AC, Mottron L. Neuroanatomical differences in brain areas implicated in perceptual and other core features of autism revealed by cortical thickness analysis and voxel-based morphometry. Hum Brain Mapp. (2010) 31:556–66. doi: 10.1002/hbm.20887
30. Richter J, Henze R, Vomstein K, Stieltjes B, Parzer P, Haffner J, et al. Reduced cortical thickness and its association with social reactivity in children with autism spectrum disorder. Psychiatry Res. (2015) 234:15–24. doi: 10.1016/j.pscychresns.2015.06.011
31. Scheel C, Rotarska-Jagiela A, Schilbach L, Lehnhardt FG, Krug B, Vogeley K, et al. Imaging derived cortical thickness reduction in high-functioning autism: key regions and temporal slope. Neuroimage. (2011) 58:391–400. doi: 10.1016/j.neuroimage.2011.06.040
32. Wallace GL, Dankner N, Kenworthy L, Giedd JN, Martin A. Age-related temporal and parietal cortical thinning in autism spectrum disorders. Brain. (2010) 133 :3745–54. doi: 10.1093/brain/awq279
33. Hardan AY, Muddasani S, Vemulapalli M, Keshavan MS, Minshew NJ. An MRI study of increased cortical thickness in autism. Am J Psychiatry. (2006) 163:1290–2. doi: 10.1176/ajp.2006.163.7.1290
34. Jou RJ, Minshew NJ, Keshavan MS, Vitale MP, Hardan AY. Enlarged right superior temporal gyrus in children and adolescents with autism. Brain Res. (2010) 1360:205–12. doi: 10.1016/j.brainres.2010.09.005
35. Khundrakpam BS, Lewis JD, Kostopoulos P, Carbonell F, Evans AC. Cortical thickness abnormalities in autism spectrum disorders through late childhood, adolescence, and adulthood: a large-scale MRI study. Cereb Cortex. (2017) 27:1721–31. doi: 10.1093/cercor/bhx038
36. Abel JM, Witt DM, Rissman EF. Sex differences in the cerebellum and frontal cortex: roles of estrogen receptor alpha and sex chromosome genes. Neuroendocrinology. (2011) 93:230–40. doi: 10.1159/000324402
37. Carruth LL, Reisert I, Arnold AP. Sex chromosome genes directly affect brain sexual differentiation. Nat Neurosci. (2002) 5:933–4. doi: 10.1038/nn922
38. Corre C, Friedel M, Vousden DA, Metcalf A, Spring S, Qiu LR, et al. Separate effects of sex hormones and sex chromosomes on brain structure and function revealed by high-resolution magnetic resonance imaging and spatial navigation assessment of the four core genotype mouse model. Brain Struct Funct. (2016) 221:997–1016. doi: 10.1007/s00429-014-0952-0
39. Savic I, Frisen L, Manzouri A, Nordenstrom A, Lindén Hirschberg A. Role of testosterone and Y chromosome genes for the masculinization of the human brain. Hum Brain Mapp. (2017) 38:1801–14. doi: 10.1002/hbm.23483
40. Seney ML, Ekong KI, Ding Y, Tseng GC, Sibille E. Sex chromosome complement regulates expression of mood-related genes. Biol Sex Differ. (2013) 4:20. doi: 10.1186/2042-6410-4-20
41. Savic I, Arver S. Sex differences in cortical thickness and their possible genetic and sex hormonal underpinnings. Cereb Cortex. (2014) 24:3246–57. doi: 10.1093/cercor/bht180
42. Good CD, Lawrence K, Thomas NS, Price CJ, Ashburner J, Friston KJ, et al. Dosage-sensitive X-linked locus influences the development of amygdala and orbitofrontal cortex, and fear recognition in humans. Brain. (2003) 126 :2431–46. doi: 10.1093/brain/awg242
43. Lentini E, Kasahara M, Arver S, Savic I. Sex differences in the human brain and the impact of sex chromosomes and sex hormones. Cereb Cortex. (2013) 23:2322–36. doi: 10.1093/cercor/bhs222
44. Lepage JF, Hong DS, Mazaika PK, Raman M, Sheau K, Marzelli MJ, et al. Genomic imprinting effects of the X chromosome on brain morphology. J Neurosci. (2013) 33:8567–74. doi: 10.1523/JNEUROSCI.5810-12.2013
45. Vawter MP, Harvey PD, DeLisi LE. Dysregulation of X-linked gene expression in Klinefelter's syndrome and association with verbal cognition. Am J Med Genet B Neuropsychiatr Genet. (2007) 144B:728–34. doi: 10.1002/ajmg.b.30454
46. Green T, Flash S, Reiss AL. Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology. (2019) 44:9–21. doi: 10.1038/s41386-018-0153-2
47. Bishop DV, Jacobs PA, Lachlan K, Wellesley D, Barnicoat A, Boyd PA, et al. Autism, language and communication in children with sex chromosome trisomies. Arch Dis Child. (2011) 96:954–9. doi: 10.1136/adc.2009.179747
48. Bruining H, Swaab H, Kas M, van Engeland H. Psychiatric characteristics in a self-selected sample of boys with Klinefelter syndrome. Pediatrics. (2009) 123:e865–70. doi: 10.1542/peds.2008-1954
49. Tartaglia N, Davis S, Hench A, Nimishakavi S, Beauregard R, Reynolds A, et al. A new look at XXYY syndrome: medical and psychological features. Am J Med Genet A. (2008) 146A:1509–22. doi: 10.1002/ajmg.a.32366
50. Tartaglia NR, Wilson R, Miller JS, Rafalko J, Cordeiro L, Davis S, et al. Autism spectrum disorder in males with sex chromosome aneuploidy: XXY/klinefelter syndrome, XYY, and XXYY. J Dev Behav Pediatr. (2017) 38:197–207. doi: 10.1097/DBP.0000000000000429
51. van Rijn S, Swaab H. Vulnerability for psychopathology in Klinefelter syndrome: age-specific and cognitive-specific risk profiles. Acta Paediatr. (2011) 100:908–16. doi: 10.1111/j.1651-2227.2011.02289.x
52. Cox KH, Rissman EF. Sex differences in juvenile mouse social behavior are influenced by sex chromosomes and social context. Genes Brain Behav. (2011) 10:465–72. doi: 10.1111/j.1601-183X.2011.00688.x
53. Dewing P, Chiang CW, Sinchak K, Sim H, Fernagut PO, Kelly S, et al. Direct regulation of adult brain function by the male-specific factor SRY. Curr Biol. (2006) 16:415–20. doi: 10.1016/j.cub.2006.01.017
54. Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, et al. Sex chromosome complement and gonadal sex influence aggressive and parental behaviors in mice. J Neurosci. (2006) 26:2335–42. doi: 10.1523/JNEUROSCI.3743-05.2006
55. Kopsida E, Lynn PM, Humby T, Wilkinson LS, Davies W. Dissociable effects of sry and sex chromosome complement on activity, feeding and anxiety-related behaviours in mice. PLoS ONE. (2013) 8:e73699. doi: 10.1371/journal.pone.0073699
56. Maxson SC, Didier-Erickson A, Ogawa S. The Y chromosome, social signals, and offense in mice. Behav Neural Biol. (1989) 52:251–9. doi: 10.1016/S0163-1047(89)90369-5
57. McPhie-Lalmansingh AA, Tejada LD, Weaver JL, Rissman EF. Sex chromosome complement affects social interactions in mice. Horm Behav. (2008) 54:565–70. doi: 10.1016/j.yhbeh.2008.05.016
58. Sluyter F, Bohus B, Beldhuis HJ, van Oortmerssen GA. Autosomal and Y chromosomal effects on the stereotyped response to apomorphine in wild house mice. Pharmacol Biochem Behav. (1995) 52:17–22. doi: 10.1016/0091-3057(95)00092-B
59. Xu J, Burgoyne PS, Arnold AP. Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet. (2002) 11:1409–19. doi: 10.1093/hmg/11.12.1409
60. Markham JA, Jurgens HA, Auger CJ, De Vries GJ, Arnold AP, Juraska JM. Sex differences in mouse cortical thickness are independent of the complement of sex chromosomes. Neuroscience. (2003) 116:71–5. doi: 10.1016/S0306-4522(02)00554-7
61. Bramen JE, Hranilovich JA, Dahl RE, Chen J, Rosso C, Forbes EE, et al. Sex matters during adolescence: testosterone-related cortical thickness maturation differs between boys and girls. PLoS ONE. (2012) 7:e33850. doi: 10.1371/journal.pone.0033850
62. Nguyen TV, McCracken J, Ducharme S, Botteron KN, Mahabir M, Johnson W, et al. Testosterone-related cortical maturation across childhood and adolescence. Cereb Cortex. (2013) 23:1424–32. doi: 10.1093/cercor/bhs125
63. Young WJ, Chang C. Ontogeny and autoregulation of androgen receptor mRNA expression in the nervous system. Endocrine. (1998) 9:79–88. doi: 10.1385/ENDO:9:1:79
64. Martínez-Cerdeño V, Noctor SC, Kriegstein AR. Estradiol stimulates progenitor cell division in the ventricular and subventricular zones of the embryonic neocortex. Eur J Neurosci. (2006) 24:3475–88. doi: 10.1111/j.1460-9568.2006.05239.x
65. Lemmen JG, Broekhof JL, Kuiper GG, Gustafsson JA, van der Saag PT, van der Burg B. Expression of estrogen receptor alpha and beta during mouse embryogenesis. Mech Dev. (1999) 81:163–7. doi: 10.1016/S0925-4773(98)00223-8
66. Miranda RC, Toran-Allerand CD. Developmental expression of estrogen receptor mRNA in the rat cerebral cortex: a nonisotopic in situ hybridization histochemistry study. Cereb Cortex. (1992) 2:1–15. doi: 10.1093/cercor/2.1.1
67. Fan X, Warner M, Gustafsson JA. Estrogen receptor beta expression in the embryonic brain regulates development of calretinin-immunoreactive GABAergic interneurons. Proc Natl Acad Sci USA. (2006) 103:19338–43. doi: 10.1073/pnas.0609663103
68. Zhang L, Chang YH, Barker JL, Hu Q, Maric D, Li BS, et al. Testosterone and estrogen affect neuronal differentiation but not proliferation in early embryonic cortex of the rat: the possible roles of androgen and estrogen receptors. Neurosci Lett. (2000) 281:57–60. doi: 10.1016/S0304-3940(99)00942-8
69. Wang L, Andersson S, Warner M, Gustafsson JA. Estrogen receptor (ER)beta knockout mice reveal a role for ERbeta in migration of cortical neurons in the developing brain. Proc Natl Acad Sci USA. (2003) 100:703–8. doi: 10.1073/pnas.242735799
70. O'Shaughnessy PJ, Baker PJ, Johnston H. The foetal leydig cell– differentiation, function and regulation. Int J Androl. (2006) 29:90–5; discussion 105–108. doi: 10.1111/j.1365-2605.2005.00555.x
71. Büdefeld T, Jezek D, Rozman D, Majdic G. Initiation of steroidogenesis precedes expression of cholesterologenic enzymes in the fetal mouse testes. Anat Histol Embryol. (2009) 38:461–6. doi: 10.1111/j.1439-0264.2009.00975.x
72. Lamprecht SA, Kohen F, Ausher J, Zor U, Lindner HR. Hormonal stimulation of oestradiol-17 beta release from the rat ovary during early postnatal development. J Endocrinol. (1976) 68:343–4. doi: 10.1677/joe.0.0680343
73. Bakker J, De Mees C, Douhard Q, Balthazart J, Gabant P, Szpirer J, et al. Alpha-fetoprotein protects the developing female mouse brain from masculinization and defeminization by estrogens. Nat Neurosci. (2006) 9:220–6. doi: 10.1038/nn1624
74. Amateau SK, Alt JJ, Stamps CL, McCarthy MM. Brain estradiol content in newborn rats: sex differences, regional heterogeneity, and possible de novo synthesis by the female telencephalon. Endocrinology. (2004) 145:2906–17. doi: 10.1210/en.2003-1363
75. Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, et al. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci USA. (2004) 101:865–70. doi: 10.1073/pnas.2630225100
76. Kimoto T, Tsurugizawa T, Ohta Y, Makino J, Tamura H.o, et al. Neurosteroid synthesis by cytochrome p450-containing systems localized in the rat brain hippocampal neurons: N-methyl-D-aspartate and calcium-dependent synthesis. Endocrinology. (2001) 142:3578–89. doi: 10.1210/endo.142.8.8327
77. MacLusky NJ, Walters MJ, Clark AS, Toran-Allerand CD. Aromatase in the cerebral cortex, hippocampus, and mid-brain: ontogeny and developmental implications. Mol Cell Neurosci. (1994) 5:691–8. doi: 10.1006/mcne.1994.1083
78. McEwen BS, Lieberburg I, Chaptal C, Krey LC. Aromatization: important for sexual differentiation of the neonatal rat brain. Horm Behav. (1977) 9:249–63. doi: 10.1016/0018-506X(77)90060-5
79. Naftolin F, Ryan KJ, Davies IJ, Reddy VV, Flores F, Petro Z, et al. The formation of estrogens by central neuroendocrine tissues. Recent Prog Horm Res. (1975) 31:295–319. doi: 10.1016/B978-0-12-571131-9.50012-8
80. Prange-Kiel J, Wehrenberg U, Jarry H, Rune GM. Para/autocrine regulation of estrogen receptors in hippocampal neurons. Hippocampus. (2003) 13:226–34. doi: 10.1002/hipo.10075
81. Rhoda J, Corbier P, Roffi J. Gonadal steroid concentrations in serum and hypothalamus of the rat at birth: aromatization of testosterone to 17 beta-estradiol. Endocrinology. (1984) 114:1754–60. doi: 10.1210/endo-114-5-1754
82. Rune GM, Frotscher M. Neurosteroid synthesis in the hippocampus: role in synaptic plasticity. Neuroscience. (2005) 136:833–42. doi: 10.1016/j.neuroscience.2005.03.056
83. Vreeburg JT, van der Vaart PD, van der Schoot P. Prevention of central defeminization but not masculinization in male rats by inhibition neonatally of oestrogen biosynthesis. J Endocrinol. (1977) 74:375–82. doi: 10.1677/joe.0.0740375
84. Cisternas CD, Tome K, Caeiro XE, Dadam FM, Garcia-Segura LM, Cambiasso MJ. Sex chromosome complement determines sex differences in aromatase expression and regulation in the stria terminalis and anterior amygdala of the developing mouse brain. Mol Cell Endocrinol. (2015) 414:99–110. doi: 10.1016/j.mce.2015.07.027
85. Cisternas CD, Cabrera Zapata LE, Arevalo MA, Garcia-Segura LM, Cambiasso MJ. Regulation of aromatase expression in the anterior amygdala of the developing mouse brain depends on ERβ and sex chromosome complement. Sci Rep. (2017) 7:5320. doi: 10.1038/s41598-017-05658-6
86. Crider A, Thakkar R, Ahmed AO, Pillai A. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol Autism. (2014) 5:46. doi: 10.1186/2040-2392-5-46
87. Sarachana T, Xu M, Wu RC, Hu VW. Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA. A novel candidate gene for autism. PLoS ONE. (2011) 6:e17116. doi: 10.1371/journal.pone.0017116
88. Kili, ç BG, Ergür AT, Ocal G. Depression, levels of anxiety and self-concept in girls with Turner's syndrome. J Pediatr Endocrinol Metab. (2005) 18:1111–7. doi: 10.1515/JPEM.2005.18.11.1111
89. Schmidt PJ, Cardoso GM, Ross JL, Haq N, Rubinow DR, Bondy CA. Shyness, social anxiety, and impaired self-esteem in Turner syndrome and premature ovarian failure. JAMA. (2006) 295:1374–6. doi: 10.1001/jama.295.12.1374
90. Tartaglia N, Cordeiro L, Howell S, Wilson R, Janusz J. The spectrum of the behavioral phenotype in boys and adolescents 47,XXY (Klinefelter syndrome). Pediatr Endocrinol Rev. (2010) 8 (Suppl. 1):151–9.
91. Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. (1961) 190:372–3. doi: 10.1038/190372a0
92. Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci USA. (1999) 96:14440–4. doi: 10.1073/pnas.96.25.14440
93. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. (2005) 434:400–4. doi: 10.1038/nature03479
94. Davies W, Isles A, Smith R, Karunadasa D, Burrmann D, Humby T, et al. Xlr3b is a new imprinted candidate for X-linked parent-of-origin effects on cognitive function in mice. Nat Genet. (2005) 37:625–9. doi: 10.1038/ng1577
95. Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. (2010) 329:682–5. doi: 10.1126/science.1190831
96. Raefski AS, O'Neill MJ. Identification of a cluster of X-linked imprinted genes in mice. Nat Genet. (2005) 37:620–4. doi: 10.1038/ng1567
97. Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper G, et al. Evidence from turner's syndrome of an imprinted X-linked locus affecting cognitive function. Nature. (1997) 387:705–8. doi: 10.1038/42706
98. Hughes JF, Page DC. The biology and evolution of mammalian Y chromosomes. Annu Rev Genet. (2015) 49:507–27. doi: 10.1146/annurev-genet-112414-055311
Keywords: autism spectrum disorder, male bias, sex chromosome, gonadal hormones, four core genotypes, SRY gene, maternal antibody
Citation: Gata-Garcia A, Porat A, Brimberg L, Volpe BT, Huerta PT and Diamond B (2021) Contributions of Sex Chromosomes and Gonadal Hormones to the Male Bias in a Maternal Antibody-Induced Model of Autism Spectrum Disorder. Front. Neurol. 12:721108. doi: 10.3389/fneur.2021.721108
Received: 06 June 2021; Accepted: 14 September 2021;
Published: 13 October 2021.
Edited by:
Serge Nataf, Université Claude Bernard Lyon 1, FranceReviewed by:
Vincent Russel Harley, Hudson Institute of Medical Research, AustraliaCopyright © 2021 Gata-Garcia, Porat, Brimberg, Volpe, Huerta and Diamond. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Betty Diamond, YmRpYW1vbmRAbm9ydGh3ZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.