
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 09 September 2021
Sec. Movement Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.703970
This article is part of the Research TopicGenetics of Paroxysmal Movement DisordersView all 9 articles
 Stephan Lauxmann1,2*
Stephan Lauxmann1,2* Lukas Sonnenberg1,2,3
Lukas Sonnenberg1,2,3 Nils A. Koch2,3
Nils A. Koch2,3 Christian Bosselmann1
Christian Bosselmann1 Natalie Winter1
Natalie Winter1 Niklas Schwarz1
Niklas Schwarz1 Thomas V. Wuttke1,4
Thomas V. Wuttke1,4 Ulrike B. S. Hedrich1
Ulrike B. S. Hedrich1 Yuanyuan Liu1Holger Lerche1
Yuanyuan Liu1Holger Lerche1 Jan Benda2,3
Jan Benda2,3 Josua Kegele1
Josua Kegele1Introduction: Among genetic paroxysmal movement disorders, variants in ion channel coding genes constitute a major subgroup. Loss-of-function (LOF) variants in KCNA1, the gene coding for KV1.1 channels, are associated with episodic ataxia type 1 (EA1), characterized by seconds to minutes-lasting attacks including gait incoordination, limb ataxia, truncal instability, dysarthria, nystagmus, tremor, and occasionally seizures, but also persistent neuromuscular symptoms like myokymia or neuromyotonia. Standard treatment has not yet been developed, and different treatment efforts need to be systematically evaluated.
Objective and Methods: Personalized therapeutic regimens tailored to disease-causing pathophysiological mechanisms may offer the specificity required to overcome limitations in therapy. Toward this aim, we (i) reviewed all available clinical reports on treatment response and functional consequences of KCNA1 variants causing EA1, (ii) examined the potential effects on neuronal excitability of all variants using a single compartment conductance-based model and set out to assess the potential of two sodium channel blockers (SCBs: carbamazepine and riluzole) to restore the identified underlying pathophysiological effects of KV1.1 channels, and (iii) provide a comprehensive review of the literature considering all types of episodic ataxia.
Results: Reviewing the treatment efforts of EA1 patients revealed moderate response to acetazolamide and exhibited the strength of SCBs, especially carbamazepine, in the treatment of EA1 patients. Biophysical dysfunction of KV1.1 channels is typically based on depolarizing shifts of steady-state activation, leading to an LOF of KCNA1 variant channels. Our model predicts a lowered rheobase and an increase of the firing rate on a neuronal level. The estimated concentration dependent effects of carbamazepine and riluzole could partially restore the altered gating properties of dysfunctional variant channels.
Conclusion: These data strengthen the potential of SCBs to contribute to functional compensation of dysfunctional KV1.1 channels. We propose riluzole as a new drug repurposing candidate and highlight the role of personalized approaches to develop standard care for EA1 patients. These results could have implications for clinical practice in future and highlight the need for the development of individualized and targeted therapies for episodic ataxia and genetic paroxysmal disorders in general.
Episodic ataxia type 1 (EA1) is an autosomal dominant ion channel disorder mainly caused by missense variants in the KCNA1 gene on chromosome 12 (1) encoding the α-subunit of the voltage-gated potassium channel KV1.1 (Figure 1), which are expressed in the peripheral and the central nervous system including cerebellum and mostly present as heterotetramers with KV1.2 and KV1.4 subunits (2). It has been shown previously that loss-of-function (LOF) variants cause neuronal hyperexcitability due to disturbed repolarization and subsequently prolonged duration of action potentials [APs; (3)]. Finally, these biophysical changes clinically result in typical frequent and short-lasting attacks (seconds to minutes) which are characterized by gait incoordination, limb ataxia, truncal instability, dysarthria, nystagmus, tremor, occasionally seizures [around 10%; (4, 5); overview in (6)], but also persistent neuromuscular symptoms like myokymia or neuromyotonia (7). Patients often experience multiple attacks per day, some of which may cluster (5) and can have preceding sensory warning symptoms (8, 9). Common triggers for the paroxysmal events are startle reactions, vigorous activity, change in posture (e.g., from sitting to standing), emotion, hunger, alcohol, or intercurrent illness. The age of onset of EA1 attacks is usually in early childhood or adolescence [almost all below 20 years, on average 7.8 years; (7)] and attacks can abate in adulthood (8, 9). Cerebellar function is typically normal between attacks, but persistent cerebellar features have also been described (7) and disease course in general does not seem to follow a progressive mode (10). Brain imaging in patients with EA1 is typically unremarkable but may reveal cerebellar atrophy [around 10% of cases; (7)].
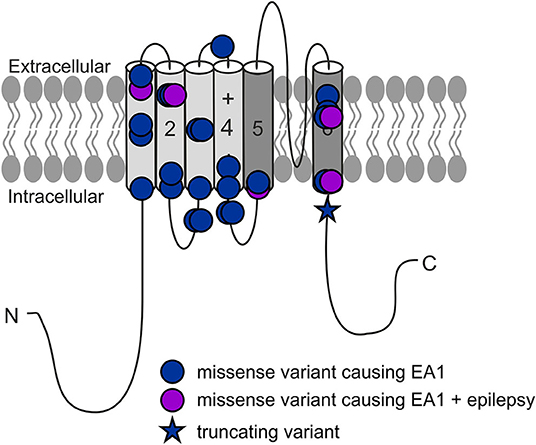
Figure 1. All reported KCNA1 variants with EA1 phenotype.
Already in 1994 “point mutations” in KCNA1 were associated with EA1 (1), but the majority of variants and an ever-growing number of de novo variants have been identified in the last decade by new (and affordable) technologies such as next generation sequencing providing the basis for subsequent pathophysiological studies (summary in Table 1). At the same time, the unfolding complexity of the pathophysiological landscape of genetic paroxysmal movement disorders, such as episodic ataxias, emerges as one likely reason for limited therapeutic success experienced with a variety of therapeutic approaches, demonstrating the need for standards in the care of this increasing group. While a small subset of individuals with EA1 caused by LOF variants in KCNA1 has shown quite good therapeutic response to the commonly used carbonic anhydrase inhibitor acetazolamide (AZA), five of nine attempted treatments were not successful (see Table 1; Figure 2). Although intuitive personalized therapeutic regimens tailored to disease-causing pathophysiological mechanisms may offer the specificity required to overcome intractability, for example, by using an enhancer for voltage-gated potassium channels (11), they are currently not available in clinical standard care. Nevertheless, by following this precision medicine approach, sodium channel blockers (SCBs) seem to constitute a promising alternative providing a biophysical rationale by “narrowing” the otherwise broadened shape of APs (12) through enhancement of the inactivated state (13). In addition, some SCBs show substance-specific and distinct mechanisms, which could contribute to functional compensation of variant channels. Riluzole, for example, displays a reduction of the persistent sodium current (14) and additional direct effects on heterotetramers of voltage-gated potassium channels (15, 16), thereby owning additional potential to restore the loss of KV1.1 channel function. These results could have implications for clinical practice in the future and highlight the need for the development of individualized and targeted therapies for episodic ataxia and genetic paroxysmal disorders in general.
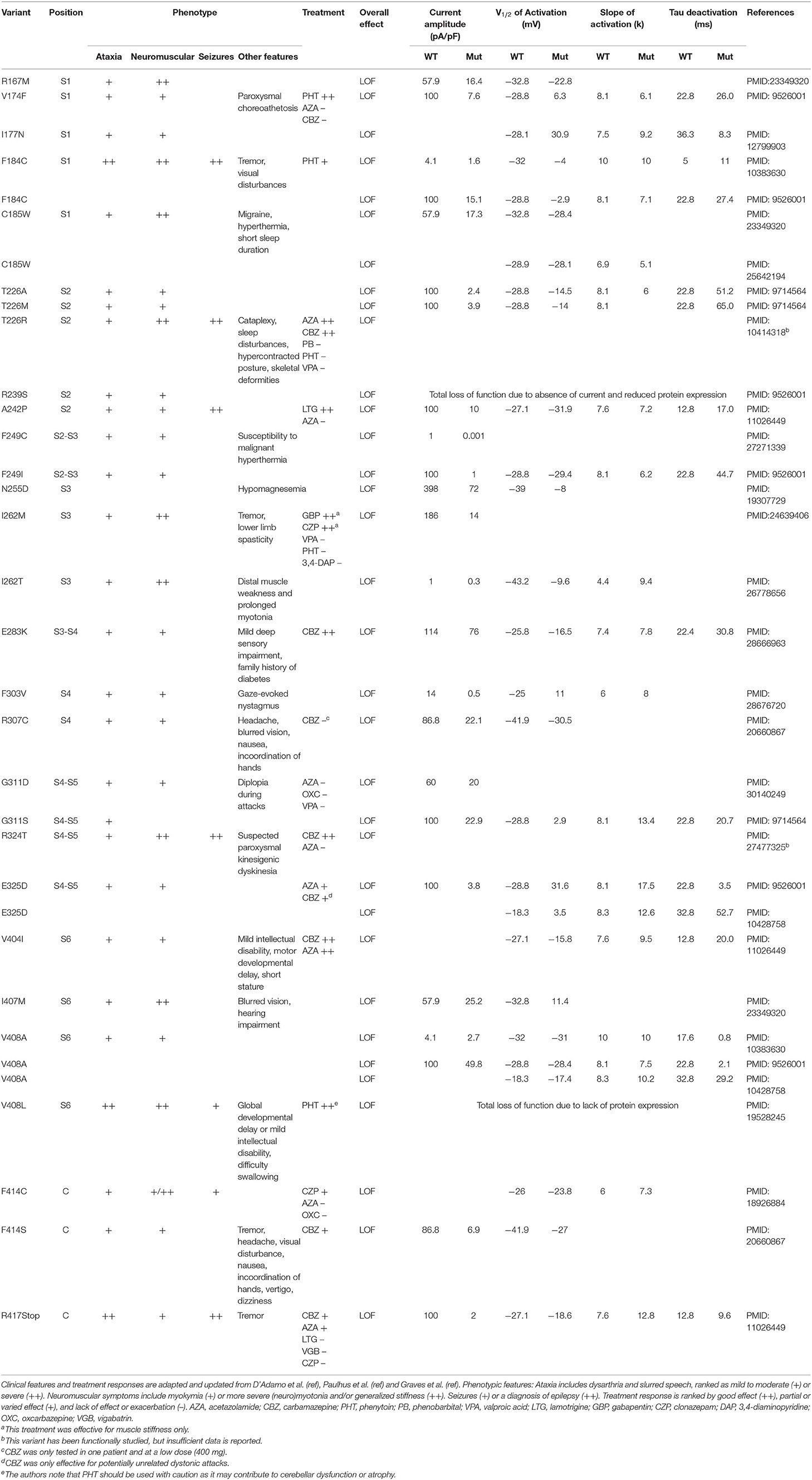
Table 1. Biophysical features and treatment response of all identified variants in KCNA1 patients.
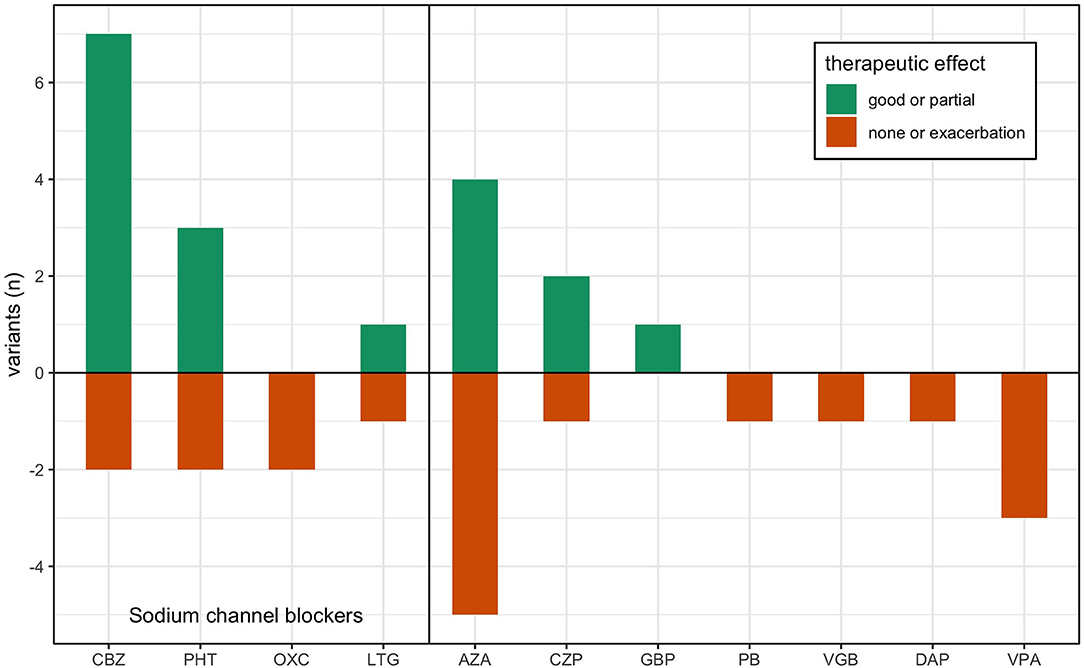
Figure 2. Treatment response in previous reports of KCNA1 variants. Data is adapted from Table 1, where previous single case reports of EA1 are grouped by their respective disease-causing variant. Treatment responses are reported ranked by good effect (dark green), partial or varied effect (light green), and lack of effect or exacerbation (red) per the author's assessment. AZA, acetazolamide; CBZ, carbamazepine; PHT, phenytoin; PB, phenobarbital; VPA, valproic acid; LTG, lamotrigine; GBP, gabapentin; CZP, clonazepam; DAP, 3,4-diaminopyridine; OXC, oxcarbazepine; VGB, vigabatrin.
Toward this aim, we (i) reviewed all available clinical reports on treatment response and functional consequences of all KCNA1 variants causing EA1, (ii) examined the potential effects on neuronal excitability of all variants using a single compartment conductance-based model and set out to assess two SCBs (carbamazepine and riluzole) regarding their potential to restore the identified underlying pathophysiological mechanisms of KCNA1 variant channels, and (iii) provide a comprehensive review of the literature considering all types of episodic ataxia.
To study the potential effects of SCBs on neuronal firing behavior of KCNA1 variants, we simulated the concentration dependent effects of carbamazepine and riluzole.
We simulated neuronal firing behavior in a single-compartment conductance-based model of a pyramidal cell, based on Pospischil et al. (17). A cortical pyramidal neuron model was chosen as it is well-characterized, versatile, and these neurons are known to express KV1.1 channels (18, 19). The model cell has a cylindrical shape with length L and diameter d and its membrane equation
sums up a sodium current, INa = gNam3h [V – ENa], a delayed rectifier potassium current, IK= gKn4 [V – EK], an KV1.1 current, IA = gKV1.1su [V – EK], an M-type potassium current, IM = gMp [V – EK], a leak current, IL = gL[V – EL], the input current, Iinput, and the capacitive current with membrane capacitance C = 1μF/cm2. The dynamics of the gating variables m, h, n, p, s, u of the ionic currents Ii with maximal conductance gi and reversal potential Ei follow
where ẋi is the derivative of the gating parameter xi with respect to time, αi(V) are the opening rates and βi(V) the closing rates. Steady-state activation and inactivation curves as well as time constants are given by x∞, i(V) = αi(V)/[αi(V) + βi(V)] and τi(V) = 1/[αi(V) + βi(V)], respectively.
Parameters for gating variables m, h, and n are taken from Traub and Miles (20); p is taken from Yamada (21); and s and u are taken from Ranjan et al. (22) and fitted to the mean wildtype biophysical parameters in Table 1. All parameters that differ from their original sources are gNa: 56 mS/cm2, gK: 5.4 mS/cm2, gM: 0.075 mS/cm2, gKV1.1: 0.6 mS/cm2, gL: 0.0205 mS/cm2, ENa: 50 mV, EK: −90 mV, EL: −70.3 mV, d = L: 61.4 μm, τmax, p: 608 ms.
To be able to apply changes as reported from electrophysiological characterizations of the kinetics of mutated ionic currents to the activation and inactivation properties, we fitted a sigmoid function
to the respective steady-state curves x∞, i. where ki is the slope factor, V1/2, i is the half-activation voltage, and γ an exponent to improve the accuracy of the fit. A persistent current could be added by changing a. The parameters for the different gates are summarized in Table 2.
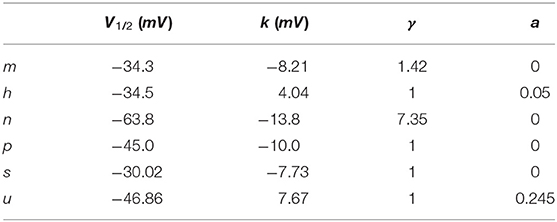
Table 2. Gating parameters.
The model was integrated with the Euler forward method and time steps Δt = 0.01 ms. All simulations and analysis were done with custom Python 3.8 software.
We simulated membrane responses to injected step currents ranging from 0 to 0.75 nA for 2 s and analyzed the voltage traces for their firing rate. The resulting f-I curve was then analyzed for its rheobase and its area under the curve (AUC) computed for the first 200 pA of steady-state firing.
The observed effects of each KCNA1 variant on the biophysical properties of IKV1.1 activation (V1/2, slope, and current amplitude; Table 1) were implemented in the model KV1.1 current, and the effects of KCNA1 variants on the firing behavior were simulated.
The effect of each of KCNA1 variant i on the rheobase and AUC of the f-I curve was quantified relative to the unchanged wildtype model with
The effects of SCBs carbamazepine and riluzole were modeled with alterations of current properties affected by the respective SCBs. For carbamazepine (CBZ), concentration dependent decreases in INa amplitude [1/(1 + [CBZ]/227 μM); (23)] and hyperpolarizing shifts in INa inactivation [−20.5 mV/(1 + 116.4 μM/[CBZ]); (24)] are modeled. These effects are in line with the effects seen at 100 μM in human hippocampal slices (25). The effects of 0, 5, 10, 15, 20, 25, 37.5, 50, 75, and 100 μM carbamazepine were simulated for each KCNA1 variant.
Concentration dependent riluzole (RLZ) induced decreases in persistent INa amplitude [1 − 0.99365487/(1 + (2 μM/[RLZ])1.36); (14)], INa amplitude [1 − [RLZ]1.05/(51 + [RLZ]1.05); (26)], IK amplitude [(1 − [RLZ]/(88 μM + [RLZ]); (26)] as well as concentration-dependent hyperpolarizing shifts in INa [; (14)] are simulated for each KCNA1 variant from 0 to 30 μM in 5 μM increments.
For each KCNA1 variant and the carbamazepine or riluzole concentration, firing behavior was quantified and compared to wildtype model firing with percentage change in AUC and Δrheobase.
MEDLINE, the Cochrane Library, conference abstracts, thesis and dissertations, preprint servers (bioRxiv, medRxiv), internet websites, and reference lists were searched in April 2021 to identify articles for inclusion. Only articles in English were considered. Keywords have been: “episodic ataxia” and all subtypes (EA 1-9), “KCNA1,” “KV1.1,” “CACNA1A,” “CACNB4,” “EAAT1,” “SLC1A3,” “UBR4,” “NaV1.2,” “SCN2A,” “ATP1A3,” “PRRT2,” “SLC2A1,” “TBC1D24,” “KCNA2,” “CEP290,” “FGF14,” “NALCN,” “paroxysmal movement disorders,” “functional consequences,” and “biophysical features.” Four reviewers independently screened literature search results and abstracted data from included studies. Descriptive analysis was conducted.
In this study, we found 34 reports about 28 different variants in the KCNA1 gene including electrophysiological data about their functional consequences. In 15 patients, 36 treatment efforts with 12 different drugs were described (see Table 1; Figure 2): 9 trials each with acetazolamide (AZA) and carbamazepine (CBZ), 5 trials with phenytoin (PHT), 3 trials with valproic acid (VPA), 2 trials with oxcarbazepine (OXC), lamotrigine (LTG), and clonazepam (CZP), and 1 trial with gabapentin (GBP), 3.4-diaminopyridine (DAP), vigabatrin (VGB), and phenobarbital (PB). Altogether, most positive results were seen for CBZ with seven reports of beneficial effects. One patient benefitted only in respect of potentially unrelated dystonic attacks and another non-responding patient was tested only with a low dose (400 mg/d). Effects of another SCB, PHT, were also remarkable, since three of five patients showed substantial clinical improvement. AZA showed mixed results with four patients exhibiting amelioration and five patients without positive effects. For all other drugs, only one or two reports are available, partially with unspecific positive clinical effects on muscle stiffness (see Table 1).
All variants with described ionic current kinetics showed distinct LOF behavior considering isolated KV1.1 function, which usually leads to hyperexcitability in a neuronal network. Common mechanisms of almost all of the 21 variants are reduced current amplitudes and depolarizing shifts of the steady-state activation curve (see Table 1; Figure 3). The only patient with a positive response to LTG (Figure 3A, green dot) had negative shift of the KCNA1 activation curve. Positive responders to CBZ (Figure 3A, orange dots) showed a medium shift, and PHT responders (Figure 3A, red dots) showed a pronounced shift of the half activation voltage.
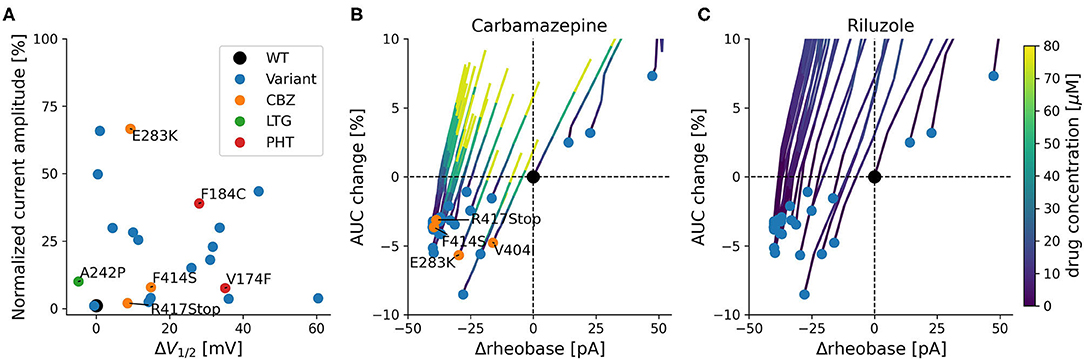
Figure 3. Functional effects of KCNA1 variants with episodic ataxia 1 phenotype. (A) Current amplitudes and shifts of the variant's steady-state activation curves in relation to wildtype channels for all functionally characterized EA1 variants with available data. Positive treatment effects to SCBs are indicated (CBZ: orange, PHT: red, LTG: green). Selected variants are labeled with their respective mutation. (B,C) Simulated effects of variants on firing rheobase and change in AUC. Rheobase is measured relative to the wildtype (Δrheobase), and AUC change represents the percentage change relative to wildtype of the area under the curve of the fI-curve for the first 200 pA of steady-state firing. Blue dots depict available KCNA1 variants. Orange dots represent patients that responded positively to CBZ (in B). Colored lines depict concentration dependent simulated changes in rheobase and AUC caused by CBZ (B) or RLZ (C).
Simulation-based estimates of KCNA1 variant effects predict decreases in rheobase and relative mild decreases in firing rate quantified as the area under the curve of the f-I curve (AUC). These predictions were associated with increased neuronal excitability and were present in all but three KCNA1 variants (Figures 3B,C).
The SCBs carbamazepine (CBZ) and riluzole (RLZ) both increased rheobase, consequently reducing neuronal excitability. In addition, they both counteracted the reduction in firing rate by increasing AUC (Figures 3B,C). The simulated responses to the drugs seem to be consistent over the different variants and have comparable slopes between CBZ and RLZ simulations, with RLZ being more sensitive to the respective drug concentration. The most effective drug in EA1 patients, carbamazepine, demonstrated positive treatment effects in KCNA1 variants that have moderate changes in AUC and rheobase (F414S, V404I, R417Stop, and E283K). R307C variant channels exhibited also moderate AUC and rheobase changes, but the clinical reports did not show positive treatment effects with only low-dose carbamazepine (400 mg/day) in a limited dataset.
EA type 2 is not only the best-characterized but also represents the most common form of EAs, with an estimated prevalence of about 1/100,000 (10). Typical paroxysmal attacks are commonly longer than in EA1 (lasting hours to days) and are characterized by recurrent debilitating spells of unsteadiness, incoordination, vertigo, and dysarthria. Most patients with EA2 exhibit disease onset in early childhood; however, an overall wide range between 2 and up to 32 years has been reported. Frequency of episodes may vary from once or twice a year to several per week and may occur spontaneously or triggered by physical stress (frequently with first attacks while playing sports), fatigue, emotional distress, exercise, or drinks (i.e., alcohol and coffee). Interictal signs are common and may include slowly progressive ataxia and nystagmus [i.e., gaze-evoked, rebound, or primary position downbeat nystagmus, (27)]. Brain imaging may reveal cerebellar atrophy of the vermis. Acetazolamide is considered the drug of choice and may significantly reduce both frequency and severity of ataxic attacks (28). Additionally, 4-aminopyridine, a selective blocker for voltage-gated potassium channels (KV1 family) and FDA/EMA approved for symptomatic treatment of multiple sclerosis, has also been demonstrated to be effective in adolescent and adult individuals with EA2 (29).
EA2 is genetically determined by heterozygous variants in the CACNA1A gene, coding for the α1A subunit of P/Q type voltage-dependent calcium channels [Figure 4; (30)]. Electrophysiological studies mainly postulated LOF mechanisms underlying EA2 (31, 32), while in patients with familial hemiplegic migraine (FHM1) gain-of-function (GOF) variants have been identified. In contrast, mixed biophysical features (LOF and GOF) may account for epilepsy phenotypes (33). Of note, spino-cerebellar ataxia type 6 (SCA6) is caused by CAG triplet expansion in the same gene (34).
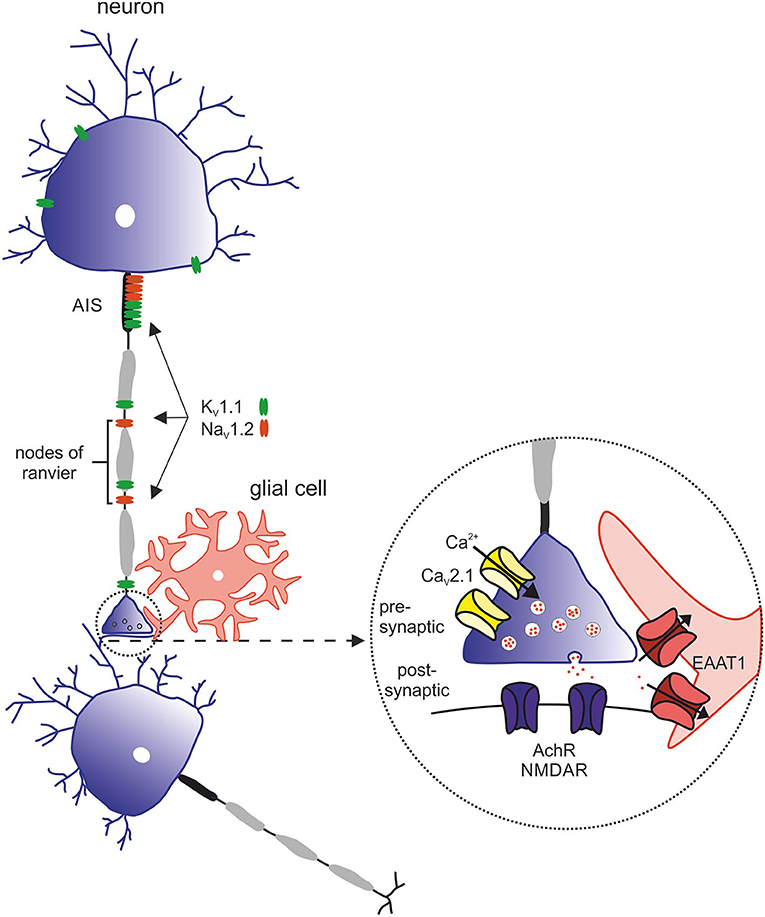
Figure 4. Overview of relevant ion channels and transporters in pathophysiology of episodic ataxia subtypes. Morphology of the neuron with its structural compartments—soma and dendrites (purple), axon initial segment (AIS, gray), and nodes of ranvier (black). Relevant ion channels of episodic ataxia subtypes: KV1.1 (green) → EA1, expressed in soma, AIS and nodes of ranvier. NaV1.2 (red) → EA9, predominantly expressed in the AIS and nodes of ranvier. Inset with representative synapse: CaV2.1 (yellow) → EA2, presynaptic. EAAT1 (red) → EA6, postsynaptic. AchR and NMDAR (purple), postsynaptic.
EA5 is associated with mutations in CACNB4 (35), which encodes the voltage-dependent L-type calcium channel subunit β4 (Figure 4). The phenotypic spectrum cannot be determined well, since only very few individuals with EA5 have been described so far: By screening pedigrees with familial epilepsy and ataxia, Escayg et al. (35) identified the p.(482X) variant in an individual with juvenile myoclonic epilepsy without episodic ataxia and p.(Cys104Phe) in a family with episodic ataxia and another family with generalized epilepsy (GTCS and atypical absences) and “praxis-induced” seizures. In 2019, two further individuals (father and daughter) have been published with the same missense mutation [p.(Cys104Phe)], who suffered from mild adult onset episodic ataxia (36). Recently, two siblings have been reported with a complex neurodevelopmental disorder carrying a homozygous variant p.(Leu126Pro), though without episodic ataxia (37). Compared to EA2 patients, patients suffer from similar symptoms, but onset of manifestation is later in EA5. Long-term follow-up in one adult EA5 case has shown permanent ataxia. Brain imaging is typically unremarkable. Acetazolamide seems to be effective. Functional studies of the variants revealed alterations in the calcium channel function, such as decreasing time constants of variant channels leading to a decrease of net inward flow of calcium ions into activation. However, the authors state that functional alterations in the oocyte expression system are rather subtle and the pedigrees are too small to provide strong evidence for linkage (35). Identification of additional patients with mutations in CACNB4 and EA phenotype failed, and designation remains contentious.
The first description of episodic Ataxia type 6 (EA6) was in 2005 in a 10-year-old boy with symptoms of EA as well as progressive ataxia, seizures, and migraine headaches with prolonged alternating hemiplegia triggered by mild head trauma and fever (38, 39). The phenotype could be linked to a de novo heterogenous missense mutation in SLC1A3, encoding the glial glutamate transporter EAAT1 (excitatory amino acid transporter 1, see Supplementary Figure 1). This mutation leads to an exchange of the highly preserved proline to arginine at position 290 [p.(Pro290Arg)]. Next to this mutation, four other mutations in SLC1A3 could be associated with EA6: p.(Met128Arg) (40), p.(CysC186Ser) (41), p.(Val393Iso) (42), and p.(Arg499Glu) (43). All listed mutations are associated with clinical symptoms of EA as well as additional complaints such as migraine, tremor or slurred speech. Onset of disease ranged from 10 years [p.(Pro290Arg)] to 55 years [p.(Val393Iso)] (42). Symptoms partially responded to treatment with acetazolamide (39, 42). In addition, the mutation p.(Thr387Pro) was described in a case of a 22-year-old man with severe migraine headache accompanied by transient neurological deficits, including visual disturbances, prominent dysphasia, and unilateral sensory and motor deficits (44). The main purpose of the five EAAT subtypes is the reuptake of the excitatory neurotransmitter glutamate in the brain. In addition, they can work as anion channels (45, 46). In the latter function, the main glial glutamate transporter in the cerebellum, EAAT1, seems to play a crucial role in astrocytic anion homeostasis and volume regulation, especially in Bergmann Glia cells during glial maturation (47). In heterologous expression systems could be demonstrated that the mutation p.(Pro290Arg) impairs the glutamate transport rate, while increasing channel activity leading to an increased anion efflux (46, 48). This could be confirmed in a transgenic mouse model by Kovermann and others (46). The glutamate-activated chloride efflux in Bergmann glia triggered the apoptosis of these cells during infancy. The loss of Bergmann glia resulted in reduced glutamate uptake and impaired neural network formation in the cerebellar cortex (46). The migraine associated mutation p.(Thr387Pro) on the other hand diminishes hEAAT1 glutamate uptake rates by an altered ligand-binding to the transporter and reduces the number of hEAAT1 in the surface membrane (44). Functional studies of the other mutations mentioned are missing to date.
There has been a large non-consanguineous Irish family described whose members suffer from episodic ataxia beginning in the second year of life (49). Typical ictal features were unsteadiness, weakness, and dysarthria triggered by stress or tiredness. Compared to EA1-7, the onset of the EA in this family happens to be much earlier, muscular hypertrophy is absent, and there is no vertigo, tinnitus, limb, or neck jerking during the attacks. Minimal gaze-evoked nystagmus has been reported but no prominent eye movement disturbances. Interestingly, the affected individuals responded well to clonazepam, but acetazolamide was not effective. The disease has been linked to the genes UBR4 [p.(Arg5091His)], which encodes an ubiquitine-protein ligand, and HSPG2, which encodes the structural protein perlecan. The authors suggested UBR4 to be the more likely causative gene but did not exclude that HSPG2 may also play a critical role in disease mechanism. The exact mechanism on how UBR4 mutation may lead to the phenotype is not well-understood yet, although UBR4 seems to play a role in calcium signaling (50). In a Korean study, where whole exome sequencing was performed in 39 individuals with episodic ataxia, four new possible or probable pathogenic variants in UBR4 [p.(Tyr4877Cys), p.(Arg4111His), p.(Ala5042Val), and p.(Ala2581Val)] have been identified. Two of them [p.(Tyr4877Cys) and p.(Arg41111His)] co-occurred with variants in CACNA1A which is why the authors suggested that UBR4 variants may have a modifier effect to CACNA1A (42).
Episodic ataxia type 9 (EA9) is caused by pathogenic variants in SCN2A, which encodes the α-subunit of the voltage-gated sodium channel NaV1.2. EA9 is characterized by an onset of ataxic episodes in the first years of life, which are usually preceded by tonic or generalized tonic-clonic seizures. The ataxic episodes have a broad phenotypic spectrum including walking difficulties, headache, vomiting, pain, slurred speech, and “dizziness” (51, 52). Symptoms manifest between the first months and 14 years of life and last from minutes to several weeks. The frequency varies between daily and one episode a year. The development of the patients ranges from slight retardation or mild autistic features to normal psychomotor development. Standard pharmaceutical treatment with acetazolamide was effective in half of the patients (53).
So far, two mutation hotspots have been identified. One of these is the mutation p.(Ala263Val), while the other includes the S4 segment and its cytoplasmic loop in domain IV of the channel (54). Electrophysiological studies showed a profound gain-of-function with a 3-fold increased persistent sodium current by the p.(Ala263Val) variant (51). Further investigations also revealed GOF effects by a hyperpolarizing shift of the activation curve [p.(Arg1882Gly)] or by increased current density along with a depolarizing shift of the steady-state inactivation curve [p.(Arg1882Gly) in combination with p.(Gly1522Ala)] (54). All these findings are consistent with neuronal hyperexcitability.
In contrast to the aforementioned types of episodic ataxia the following genes are associated with episodic ataxia, but not OMIM listed as distinct groups: ATP1A3 (55), PRRT2 (56), SLC2A1 (57), TBC1D24 (58), KCNA2 (59), CEP290 (60), FGF14 (61), and NALCN (62).
Episodic ataxia types 3, 4, and 7 have not yet been linked to specific genes or variants. Most descriptions are based on single families with phenotypes that differ from EA1 and EA2 in conjunction with unremarkable screening of variants in KCNA1 and CACNA1A.
Following the descriptions of EA1 and EA2, Steckley et al. (63) broadened the phenotypic spectrum of EAs by reporting a Canadian family of Mennonite heritage with vestibular ataxia, vertigo, tinnitus, and interictal myokymia. Interictal nystagmus was not present and episodes were shorter distinguishing this family from EA2 patients. Although sharing some common features with EA4 such as vertigo and tinnitus, the absence of other features (see section Episodic Ataxia Type 4) justifies this entity: Patients did respond to treatment with acetazolamide. Linkage analysis mapped the disease locus to a 4-cM region on 1q42; a causative mutation has not yet been identified (64).
EA4, also known as periodic vestibulocerebellar ataxia (PATX) or North Carolina autosomal dominant ataxia, is an autosomal dominant disorder described in two families from North Carolina (65) and characterized by recurrent episodes of vertigo and ataxia associated with ocular abnormalities (diplopia, deficient smooth pursuit, gaze-evoked nystagmus). Attacks typically last for hours and acetazolamide is not effective. The onset varies from the third to the sixth decade. Slowly progressive cerebellar ataxia has been recorded in some affected individuals. Linkage analysis excluded the loci for EA1 and EA2 (66). Although sharing some common features with EA3 and designation of EA4 was suggested at the time of the mapping of EA3 (64), some other features justify this entity: abnormal eye movements, including abnormal smooth pursuit, nystagmus, and abnormal vestibuloocular reflex. Interictal myokymia was absent and patients did respond to treatment with acetazolamide.
EA7 has been reported in one American 4-generation family with autosomal dominant inheritance. The seven affected members presented with exercise- and excitement-triggered episodes including EA, weakness, and slurred speech (67) lasting hours to days. Two affected family members reported vertigo during attacks. EA onset was before the age of 20 years and attack frequency ranged from monthly to yearly and tended to decrease with age. There were no interictal findings on neurologic examination. By genomewide linkage and haplotype analysis this distinct type could be linked to chromosome 19q13 (67).
Since this article has a focus on genetic forms of episodic ataxias, acquired EAs are only mentioned briefly. Some authors proposed that adult onset and short episodes are the most specific features for distinguishing “symptomatic” (i.e., in the context of an acquired neurological disease) from primary EAs (68). First descriptions come from Multiple Sclerosis patients. Hereby, it is postulated that midbrain lesions may lead to interruption of the cerebello–thalamo–cortical pathway with subsequent induction of parietal diaschisis manifesting paroxysmal movement disorders, such as EA (69). Other causing diseases are Bickerstaff's-like brainstem encephalitis (70), stroke (71), Behcet's disease (72), paraneoplastic limbic encephalitis (e.g., Anti-Hu), and anti–contactin associated protein-like 2 (CASPR2) antibody-related autoimmune limbic encephalitis presenting with amnesia and seizures (73, 74). Anti-Hu-associated paraneoplastic limbic encephalitis presenting with EA and behavioral changes evolving to intractable epilepsy has been reported (75).
Reviewing treatment efforts of EA1 patients show that less than half of the reported patients responded to acetazolamide (AZA) treatment but also revealed that the majority of patients responded to different SCBs, in particular carbamazepine (CBZ) and phenytoin (PHT; Table 1; Figure 2). For all other drugs, no reasonable statements can be made due to low numbers (≤3 treatment trials). Unfortunately, the small size of reported treatment responses is a major limitation of this study, which emphasizes even more the necessity to review the known facts and develop effective treatment approaches. While for PHT only five patient reports were available of which three showed beneficial effects, seven responses to CBZ and two reports without clinical improvement could be found including one patient treated with only a minimum dose (400 mg/d). It is worth pointing out that the therapeutic response is variable even among individuals with the same genotype. Whether symptoms will be improved by a given drug can therefore not be fully predicted based on the expressed variant and associated biophysical alterations of channel function as genetic background or environmental factors may play important roles. Interestingly, a study of monozygotic twins with EA1 demonstrated inhomogeneous disease severity among the siblings thereby highlighting the impact of non-genetic factors (76).
In this study, we predict decreased rheobases and AUC in all but 3 KCNA1 variants (I177N, V408A, and F414C) suggesting that LOF KCNA1 variants generally induce an overall increase in neuronal excitability. The three variants with increased rheobases and AUC lack a complete electrophysiological characterization of the biophysical properties of the KV1.1 current (Table 1) and thus the simulated effects of these variants on neuronal firing must be interpreted with caution. Patients with these variants were not treated with CBZ and as such conclusions as to the benefit or potential harm of treatment of these patients with CBZ cannot be made.
Remarkably, biophysical dysfunction of KV1.1 channels in EA1 patients is almost exclusively based on depolarizing shifts of steady-state activation (Figure 3A), leading to LOF of KCNA1 variant channels. Recently, Zhao et al. (77) predicted in silico that these depolarizing shifts broaden APs and interfere negatively with high frequency AP firing, suggesting that SCBs would be deleterious in EA1. However, the observed increase in neuronal excitability predicted by our models and the increased firing rates and decreased rheobases observed with KV1.1 channel block in sensory neurons (78), as well as clinical data reflecting positive outcomes of SCBs in EA1 patients (Table 1; Figure 2) provide a rational basis for the suggestion of the use of SCBs in EA1.
RLZ has been shown to inhibit neuronal firing across the CNS (23, 79–84), as well as inhibiting axonal firing (85). In a computational Purkinje cell model of ataxia exhibiting increased excitability, firing is returned to wildtype when multiple RLZ effects are taken into account (86), suggesting that the pleiotropy of RLZ is key to its efficacy. Additionally, RLZ decreases induced neuronal hyperexcitability in a number of animal disease models (87–89) with Hassani et al. (89) observing firing decreases in lesioned but not in unlesioned rats. The neuroprotective ability of RLZ to decrease hyperexcitability provides a further rationale for its potential in the treatment of EA1.
In our single compartment-based modeling presented here, predicted drug effects for riluzole are based on known mechanisms including reduction of persistent sodium channels, a notable shift of the steady-state inactivation and a reduced sodium conductance. However, distinctively to other SCBs in this study, RLZ also has the potential to reduce potassium conductance by dramatically slowing down the inactivation of KV1.4 channels within heterotetramers (16). In contrast to these reflected effects, this study neglects further additional mechanisms, such as an increase of calcium-activated potassium currents (90), an antagonism on NMDA receptors leading to reduced excitatory postsynaptic currents (80), and any effects of RLZ at the network level. In particular, RLZ partially inhibits high-voltage activated (HVA) calcium currents by up to 20% (91); however, this is specific to N and P/Q-type calcium channels (92), suggesting that RLZ inhibits calcium influx at presynaptic terminals. Although our single neuron simulations predict increased firing frequencies (AUC) as a result of RLZ, this may be mitigated in part at the network level through decreased synaptic transmission. Taking these and further additional mechanisms into account which are not reflected in our model, RLZ holds even greater potential in the treatment of EA1.
Our modeling-based findings for RLZ demonstrate similar effects on KCNA1 variant firing to CBZ with increases in rheobase and AUC albeit with lower concentrations. Given the effectiveness of CBZ in an EA1 patient subset and the similarity of the effects of RLZ on firing rate, further investigation into the treatment of EA1 with RLZ is warranted. These findings would benefit from clinical data. Whereas, treatment reports of EA1 patients with RLZ are missing, RLZ has shown promise in a double-blind placebo-controlled trial in cerebellar ataxia patients with diverse ataxia etiologies in decreasing ataxia symptom severity with only mild adverse events reported (93). In a different double-blind placebo-controlled trial of RLZ in patients with spinocerebellar or Friedrich's ataxia, RLZ was also effective in treatment of the ataxias without severe adverse effects (94). Large sample size trials of RLZ in homogeneous forms of ataxia are needed to better understand the benefits of RLZ in ataxia and any potential ataxia-type specific efficacy (95, 96). However, the benefits of RLZ across diverse non-episodic ataxias suggest that treatment of common pathophysiological ataxia mechanisms such as altered neuronal firing may be beneficial in episodic ataxias.
Altogether, RLZ holds in our opinion great potential in the treatment of EA1, which needs certainly corroborated by further studies in heterologous expression systems and neurons.
The original contributions presented in the study are included in the Supplementary Material, further inquiries can be directed to the corresponding author.
SL, LS, and NK: designed and conceptualized study, acquisition of data, interpreted the data, and drafted and revised the manuscript for intellectual content. CB, UH, and YL: acquisition of data, interpreted the data, and drafted and revised the manuscript for intellectual content. NW, NS, and TW: interpreted the data and drafted and revised the manuscript for intellectual content. HL: interpreted the data, drafted and revised the manuscript for intellectual content, and study supervision. JB and JK: designed and conceptualized study, drafted and revised the manuscript for intellectual content, and study supervision. All authors approved the submitted version.
This study was supported by the German Research Foundation in the frame of the Research Unit FOR-2715 (Grants Ko4877/3-1, Le1030/15-1, and He8155/1-1) and the German Federal Ministry for Education and Research (TreatION 01GM1907A). TW was supported by an intramural Clinician Scientist Fellowship granted by the Faculty of Medicine, University of Tübingen (419-0-0). We acknowledge support by Open Access Publishing Fund of University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.703970/full#supplementary-material
1. Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. (1994) 8:136–40. doi: 10.1038/ng1094-136
2. Veh RW, Lichtinghagen R, Sewing S, Wunder F, Grumbach IM, Pongs O. Immunohistochemical localization of five members of the KV1 channel subunits: contrasting subcellular locations and neuron-specific co-localizations in rat brain. Eur J Neurosci. (1995) 7:2189–205. doi: 10.1111/j.1460-9568.1995.tb00641.x
3. Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, et al. A mouse model of episodic ataxia type-1. Nat Neurosci. (2003) 6:378–83. doi: 10.1038/nn1025
4. Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, et al. A novel mutation in the human voltage-gated potassium channel gene (KV1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. (1999) 122:817–25. doi: 10.1093/brain/122.5.817
5. Rajakulendran S, Schorge S, Kullmann DM, Hanna MG. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics. (2007) 4:258–66. doi: 10.1016/j.nurt.2007.01.010
6. de Gusmao CM, Garcia L, Mikati MA, Su S, Silveira-Moriyama L. Paroxysmal genetic movement disorders and epilepsy. Front Neurol. (2021) 12:648031. doi: 10.3389/fneur.2021.648031
7. Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. (2014) 137:1009–18. doi: 10.1093/brain/awu012
8. VanDyke DH, Griggs RC, Murphy MJ, Goldstein MN. Hereditary myokymia and periodic ataxia. J Neurol Sci. (1975) 25:109–18. doi: 10.1016/0022-510X(75)90191-4
9. Brunt ER, van Weerden TW. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain. (1990) 113:1361–82. doi: 10.1093/brain/113.5.1361
10. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. (2007) 130:2484–93. doi: 10.1093/brain/awm126
11. Orhan G, Wuttke TV, Nies AT, Schwab M, Lerche H. Retigabine/ezogabine, a KCNQ/K(V)7 channel opener: pharmacological and clinical data. Expert Opin Pharmacother. (2012) 13:1807–16. doi: 10.1517/14656566.2012.706278
12. Begum R, Bakiri Y, Volynski KE, Kullmann DM. Action potential broadening in a presynaptic channelopathy. Nat Commun. (2016) 7:12102. doi: 10.1038/ncomms12102
13. Bayraktar E, Liu Y, Hedrich UB, Sara Y, Lerche H, Wuttke TV, et al. In vitro effects of S-Licarbazepine as a potential precision therapy on SCN8A variants causing neuropsychiatric disorders. bioRxiv. (2021). doi: 10.1101/2021.04.24.441205
14. Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. (2000) 12:3567–74. doi: 10.1046/j.1460-9568.2000.00242.x
15. Du J, Vegh V, Reutens DC. Persistent sodium current blockers can suppress seizures caused by loss of low-threshold D-type potassium currents: predictions from an in silico study of KV1 channel disorders. Epilepsia Open. (2020) 5:86–96. doi: 10.1002/epi4.12379
16. Xu L, Enyeart JA, Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of KV1.4 potassium channels by a voltage-dependent oxidative mechanism. J Pharmacol Exp Ther. (2001) 299:227–37.
17. Pospischil M, Toledo-Rodriguez M, Monier C, Piwkowska Z, Bal T, Fregnac Y, et al. Minimal Hodgkin-Huxley type models for different classes of cortical and thalamic neurons. Biol Cybern. (2008) 99:427–41. doi: 10.1007/s00422-008-0263-8
18. Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of KV1.1 and KV1.2. two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. (1994) 14:4588–99. doi: 10.1523/JNEUROSCI.14-08-04588.1994
19. Tsaur ML, Sheng M, Lowenstein DH, Jan YN, Jan LY. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron. (1992) 8:1055–67. doi: 10.1016/0896-6273(92)90127-Y
20. Traub RD, Miles R. Multiple modes of neuronal population activity emerge after modifying specific synapses in a model of the CA3 region of the hippocampus. Ann N Y Acad Sci. (1991) 627:277–90. doi: 10.1111/j.1749-6632.1991.tb25931.x
21. Yamada Y. A study of the process of self-concept formation in adolescence: an investigation of self-descriptions using the Twenty Statement Test (TST). Shinrigaku Kenkyu. (1989) 60:245–52. doi: 10.4992/jjpsy.60.245
22. Ranjan R, Logette E, Marani M, Herzog M, Tâche V, Scantamburlo E, et al. A kinetic map of the homomeric voltage-gated potassium channel (Kv) family. Front Cell Neurosci. (2019) 13:358. doi: 10.3389/fncel.2019.00358
23. Kuo JJ, Lee RH, Zhang L, Heckman CJ. Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J Physiol. (2006) 574:819–34. doi: 10.1113/jphysiol.2006.107094
24. Qiao X, Sun G, Clare JJ, Werkman TR, Wadman WJ. Properties of human brain sodium channel α-subunits expressed in HEK293 cells and their modulation by carbamazepine, phenytoin and lamotrigine. Br J Pharmacol. (2014) 171:1054–67. doi: 10.1111/bph.12534
25. Doeser A, Dickhof G, Reitze M, Uebachs M, Schaub C, Pires NM, et al. Targeting pharmacoresistant epilepsy and epileptogenesis with a dual-purpose antiepileptic drug. Brain. (2015) 138:371–87. doi: 10.1093/brain/awu339
26. Zona C, Siniscalchi A, Mercuri NB, Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. (1998) 85:931–8. doi: 10.1016/S0306-4522(97)00604-0
27. Imbrici P, Eunson LH, Graves TD, Bhatia KP, Wadia NH, Kullmann DM, et al. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology. (2005) 65:944–6. doi: 10.1212/01.wnl.0000176069.64200.28
28. Orsucci D, Raglione LM, Mazzoni M, Vista M. Therapy of episodic ataxias: case report and review of the literature. Drugs Context. (2019) 8:212576. doi: 10.7573/dic.212576
29. Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology. (2011) 77:269–75. doi: 10.1212/WNL.0b013e318225ab07
30. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. (2004) 62:17–22. doi: 10.1212/01.WNL.0000101675.61074.50
31. Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. (2010) 588:1905–13. doi: 10.1113/jphysiol.2009.186437
32. Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, Caimi S, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci. (2010) 291:30–6. doi: 10.1016/j.jns.2010.01.010
33. Jiang X, Raju PK, D'Avanzo N, Lachance M, Pepin J, Dubeau F, et al. Both gain-of-function and loss-of-function de novo CACNA 1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. (2019) 60:1881–94. doi: 10.1111/epi.16316
34. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. (1997) 15:62–9. doi: 10.1038/ng0197-62
35. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. (2000) 66:1531–9. doi: 10.1086/302909
36. Gonzalez Sanchez M, Izquierdo S, Alvarez S, Bautista Alonso RE, Berciano J, Gazulla J. Clinical manifestations of episodic ataxia type 5. Neurol Clin Pract. (2019) 9:503–4. doi: 10.1212/CPJ.0000000000000697
37. Coste de Bagneaux P, von Elsner L, Bierhals T, Campiglio M, Johannsen J, Obermair GJ, et al. A homozygous missense variant in CACNB4 encoding the auxiliary calcium channel beta4 subunit causes a severe neurodevelopmental disorder and impairs channel and nonchannel functions. PLoS Genet. (2020) 16:e1008625. doi: 10.1371/journal.pgen.1008625
38. Jen JC. Hereditary episodic ataxias. Ann N Y Acad Sci. (2008) 1142:250–53 doi: 10.1196/annals.1444.016
39. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. (2005) 65:529–34. doi: 10.1212/01.wnl.0000172638.58172.5a
40. Iwama K, Iwata A, Shiina M, Mitsuhashi S, Miyatake S, Takata A, et al. A novel mutation in SLC1A3 causes episodic ataxia. J Hum Genet. (2018) 63:207–11. doi: 10.1038/s10038-017-0365-z
41. de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. (2009) 66:97–101. doi: 10.1001/archneurol.2008.535
42. Choi KD, Kim JS, Kim HJ, Jung I, Jeong SH, Lee SH, et al. Genetic variants associated with episodic ataxia in Korea. Sci Rep. (2017) 7:13855. doi: 10.1038/s41598-017-14254-7
43. Pyle A, Griffin H, Keogh MJ, Horvath R, Chinnery PF. Reply: evaluation of exome sequencing variation in undiagnosed ataxias. Brain J Neurol. (2015) 138:e384. doi: 10.1093/brain/awv088
44. Kovermann P, Hessel M, Kortzak D, Jen JC, Koch J, Fahlke C, et al. Impaired K+ binding to glial glutamate transporter EAAT1 in migraine. Sci Rep. (2017) 7:13913. doi: 10.1038/s41598-017-14176-4
45. Danbolt NC. Glutamate uptake. Prog Neurobiol. (2001) 65:1–105. doi: 10.1016/S0301-0082(00)00067-8
46. Kovermann P, Untiet V, Kolobkova Y, Engels M, Baader S, Schilling K, et al. Increased glutamate transporter-associated anion currents cause glial apoptosis in episodic ataxia 6. Brain Commun. (2020) 2:fcaa022. doi: 10.1093/braincomms/fcaa022
47. Untiet V, Kovermann P, Gerkau NJ, Gensch T, Rose CR, Fahlke C. Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia. (2017) 65:388–400. doi: 10.1002/glia.23098
48. Winter N, Kovermann P, Fahlke C. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain J Neurol. (2012) 135:3416–25. doi: 10.1093/brain/aws255
49. Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia: UBR4 the likely candidate. Eur J Hum Genet. (2014) 22:505–10. doi: 10.1038/ejhg.2013.173
50. Zecevic N, Milosevic A, Ehrlich BE. Calcium signaling molecules in human cerebellum at midgestation and in ataxia. Early Hum Dev. (1999) 54:103–16. doi: 10.1016/S0378-3782(98)00090-5
51. Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. (2010) 75:1454–8. doi: 10.1212/WNL.0b013e3181f8812e
52. Gorman KM, King MD. SCN2A p.Ala263Val variant a phenotype of neonatal seizures followed by paroxysmal ataxia in toddlers. Pediatr Neurol. (2017) 67:111–2. doi: 10.1016/j.pediatrneurol.2016.11.008
53. Schwarz N, Bast T, Gaily E, Golla G, Gorman KM, Griffiths LR, et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur J Paediatr Neurol. (2019) 23:438–47. doi: 10.1016/j.ejpn.2019.03.001
54. Schwarz N, Hahn A, Bast T, Muller S, Loffler H, Maljevic S, et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol. (2016) 263:334–43. doi: 10.1007/s00415-015-7984-0
55. Maas RP, Schieving JH, Schouten M, Kamsteeg EJ, van de Warrenburg BP. The genetic homogeneity of CAPOS syndrome: four new patients with the c.2452G>A (p.Glu818Lys) mutation in the ATP1A3 gene. Pediatr Neurol. (2016) 59:71–5.e1. doi: 10.1016/j.pediatrneurol.2016.02.010
56. Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology. (2012) 79:2115–21. doi: 10.1212/WNL.0b013e3182752c5a
57. Ohshiro-Sasaki A, Shimbo H, Takano K, Wada T, Osaka H. A three-year-old boy with glucose transporter type 1 deficiency syndrome presenting with episodic ataxia. Pediatr Neurol. (2014) 50:99–100. doi: 10.1016/j.pediatrneurol.2013.09.002
58. Zimmern V, Riant F, Roze E, Ranza E, Lehmann-Horn F, de Bellescize J, et al. Infantile-onset paroxysmal movement disorder and episodic ataxia associated with a TBC1D24 mutation. Neuropediatrics. (2019) 50:308–12. doi: 10.1055/s-0039-1688410
59. Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology. (2016) 87:1975–84. doi: 10.1212/WNL.0000000000003309
60. Hamed M, Shetty A, Dzwiniel T, Buller M, Koskinen L, Suchowersky O. Episodic ataxia secondary to CEP290 compound heterozygous mutations: a case report. Mov Disord Clin Pract. (2019) 7:104–6. doi: 10.1002/mdc3.12872
61. Piarroux J, Riant F, Humbertclaude V, Remerand G, Hadjadj J, Rejou F, et al. FGF14-related episodic ataxia: delineating the phenotype of Episodic Ataxia type 9. Ann Clin Transl Neurol. (2020) 7:565–72. doi: 10.1002/acn3.51005
62. Aoyagi K, Rossignol E, Hamdan FF, Mulcahy B, Xie L, Nagamatsu S, et al. A gain-of-function mutation in NALCN in a child with intellectual disability, ataxia, and arthrogryposis. Hum Mutat. (2015) 36:753–7. doi: 10.1002/humu.22797
63. Steckley JL, Ebers GC, Cader MZ, McLachlan RS. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. (2001) 57:1499–502. doi: 10.1212/WNL.57.8.1499
64. Cader MZ, Steckley JL, Dyment DA, McLachlan RS, Ebers GC. A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia. Neurology. (2005) 65:156–8. doi: 10.1212/01.wnl.0000167186.05465.7c
65. Farmer TW, Mustian VM. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol. (1963) 8:471–80. doi: 10.1001/archneur.1963.00460050021002
66. Damji KF, Allingham RR, Pollock SC, Small K, Lewis KE, Stajich JM, et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol. (1996) 53:338–44. doi: 10.1001/archneur.1996.00550040074016
67. Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. (2007) 64:749–52. doi: 10.1001/archneur.64.5.749
68. Marcel C, Anheim M, Flamand-Rouviere C, Heran F, Masnou P, Boulay C, et al. Symptomatic paroxysmal dysarthria-ataxia in demyelinating diseases. J Neurol. (2010) 257:1369–72. doi: 10.1007/s00415-010-5534-3
69. Li Y, Zeng C, Luo T. Paroxysmal dysarthria and ataxia in multiple sclerosis and corresponding magnetic resonance imaging findings. J Neurol. (2011) 258:273–6. doi: 10.1007/s00415-010-5748-4
70. Piffer S, Turri G, Acler M, Richelli S, Cerini R, Fiaschi A, et al. Paroxysmal dysarthria-ataxia in remitting-relapsing Bickerstaff's-like encephalitis. J Neurol Sci. (2014) 341:85–7. doi: 10.1016/j.jns.2014.04.008
71. Matsui M, Tomimoto H, Sano K, Hashikawa K, Fukuyama H, Shibasaki H. Paroxysmal dysarthria and ataxia after midbrain infarction. Neurology. (2004) 63:345–7. doi: 10.1212/01.WNL.0000130252.69304.D2
72. Akman-Demir FG, Eraksoy M, Gürvit IH, Saruhan-Direskeneli G, Aral O. Paroxysmal dysarthria and ataxia in a patient with Behçet's disease. J Neurol. (1995) 242:344–7. doi: 10.1007/BF00878879
73. López Chiriboga AS, Pittock S. Episodic ataxia in CASPR2 autoimmunity. Neurol Neuroimmunol Neuroinflam. (2018) 6:e536. doi: 10.1212/NXI.0000000000000536
74. Joubert B, Gobert F, Thomas L, Saint-Martin M, Desestret V, Convers P, et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflam. (2017) 4:e371. doi: 10.1212/NXI.0000000000000371
75. Langer JE, Lopes MB, Fountain NB, Pranzatelli MR, Thiele EA, Rust RS, et al. An unusual presentation of anti-Hu-associated paraneoplastic limbic encephalitis. Dev Med Child Neurol. (2012) 54:863–6. doi: 10.1111/j.1469-8749.2012.04221.x
76. Graves TD, Rajakulendran S, Zuberi SM, Morris HR, Schorge S, Hanna MG, et al. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology. (2010) 75:367–72. doi: 10.1212/WNL.0b013e3181ea9ee3
77. Zhao J, Petitjean D, Haddad GA, Batulan Z, Blunck R. A common kinetic property of mutations linked to episodic ataxia type 1 studied in the shaker Kv channel. Int J Mol Sci. (2020) 21:7602. doi: 10.3390/ijms21207602
78. Chi XX, Nicol GD. Manipulation of the potassium channel KV1. 1 and its effect on neuronal excitability in rat sensory neurons. J Neurophysiol. (2007) 98:2683–92. doi: 10.1152/jn.00437.2007
79. Cho JH, Choi IS, Lee SH, Lee MG, Jang IS. Contribution of persistent sodium currents to the excitability of tonic firing substantia gelatinosa neurons of the rat. Neurosci Lett. (2015) 591:192–6. doi: 10.1016/j.neulet.2015.02.039
80. Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. (2008) 27:2501–14. doi: 10.1111/j.1460-9568.2008.06211.x
81. MacIver MB, Amagasu SM, Mikulec AA, Monroe FA. Riluzole anesthesia: use-dependent block of presynaptic glutamate fibers. Anesthesiology. (1996) 85:626–34. doi: 10.1097/00000542-199609000-00023
82. Schuster JE, Fu R, Siddique T, Heckman CJ. Effect of prolonged riluzole exposure on cultured motoneurons in a mouse model of ALS. J Neurophysiol. (2012) 107:484–92. doi: 10.1152/jn.00714.2011
83. Siniscalchi A, Bonci A, Mercuri NB, Bernardi G. Effects of riluzole on rat cortical neurones: an in vitro electrophysiological study. Br J Pharmacol. (1997) 120:225–30. doi: 10.1038/sj.bjp.0700905
84. Stutzmann JM, Böhme GA, Gandolfo G, Gottesmann C, Lafforgue J, Blanchard JC, et al. Riluzole prevents hyperexcitability produced by the mast cell degranulating peptide and dendrotoxin I in the rat. Eur J Pharmacol. (1991) 193:223–9. doi: 10.1016/0014-2999(91)90040-W
85. Xie RG, Zheng DW, Xing JL, Zhang XJ, Song Y, Xie YB, et al. Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS ONE. (2011) 6:e18681. doi: 10.1371/journal.pone.0018681
86. Abbasi S, Edrisi M, Mahnam A, Janahmadi M. Computational insights into the neuroprotective action of riluzole on 3-acetylpyridine-induced ataxia in rats. Cell J. (2013) 15:98–107.
87. Boraud T, Bezard E, Stutzmann JM, Bioulac B, Gross CE. Effects of riluzole on the electrophysiological activity of pallidal neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkey. Neurosci Lett. (2000) 281:75–8. doi: 10.1016/S0304-3940(00)00780-1
88. Han GY, Li CY, Shi HB, Wang JP, Su KM, Yin XL, et al. Riluzole is a promising pharmacological inhibitor of bilirubin-induced excitotoxicity in the ventral cochlear nucleus. CNS Neurosci Therap. (2015) 21:262–70. doi: 10.1111/cns.12355
89. Hassani OK, Mouroux M, Bohme GA, Stutzmann JM, Féger J. Riluzole reduces hyperactivity of subthalamic neurons induced by unilateral 6-OHDA lesion in the rat brain. Mov Disord. (2001) 16:1110–4. doi: 10.1002/mds.1219
90. Wang YJ, Lin MW, Lin AA, Wu SN. Riluzole-induced block of voltage-gated Na+ current and activation of BKCa channels in cultured differentiated human skeletal muscle cells. Life Sci. (2008) 82:11–20. doi: 10.1016/j.lfs.2007.10.015
91. Stefani A, Spadoni F, Bernardi G. Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp Neurol. (1997) 147:115–22. doi: 10.1006/exnr.1997.6554
92. Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. (1997) 282:1280–90.
93. Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, et al. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. (2010) 74:839–45. doi: 10.1212/WNL.0b013e3181d31e23
94. Romano S, Coarelli G, Marcotulli C, Leonardi L, Piccolo F, Spadaro M, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2015) 14:985–91. doi: 10.1016/S1474-4422(15)00201-X
95. Romano S, Coarelli G, Vanacore N, Salvetti M, Ristori G. Riluzole in patients with hereditary cerebellar ataxia - authors' reply. Lancet Neurol. (2016) 15:789. doi: 10.1016/S1474-4422(16)00117-4
Keywords: episodic ataxia, sodium channel blockers, KV1.1, conduction-based model, voltage-gated potassium channels, riluzole, precision medicine, KCNA1
Citation: Lauxmann S, Sonnenberg L, Koch NA, Bosselmann C, Winter N, Schwarz N, Wuttke TV, Hedrich UBS, Liu Y, Lerche H, Benda J and Kegele J (2021) Therapeutic Potential of Sodium Channel Blockers as a Targeted Therapy Approach in KCNA1-Associated Episodic Ataxia and a Comprehensive Review of the Literature. Front. Neurol. 12:703970. doi: 10.3389/fneur.2021.703970
Received: 01 May 2021; Accepted: 23 July 2021;
Published: 09 September 2021.
Edited by:
Kishore Raj Kumar, Garvan Institute of Medical Research, AustraliaReviewed by:
Neven Maksemous, Queensland University of Technology, AustraliaCopyright © 2021 Lauxmann, Sonnenberg, Koch, Bosselmann, Winter, Schwarz, Wuttke, Hedrich, Liu, Lerche, Benda and Kegele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Lauxmann, U3RlcGhhbi5MYXV4bWFubkB1bmktdHVlYmluZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.