 Valentina Del Dotto
Valentina Del Dotto Valerio Carelli
Valerio Carelli
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 09 June 2021
Sec. Neuro-Ophthalmology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.681326
This article is part of the Research Topic Hereditary Optic Neuropathies: A New Perspective View all 11 articles
In the year 2000, the discovery of OPA1 mutations as causative for dominant optic atrophy (DOA) was pivotal to rapidly expand the field of mitochondrial dynamics and describe the complex machinery governing this pathway, with a multitude of other genes and encoded proteins involved in neurodegenerative disorders of the optic nerve. OPA1 turned out to be a much more complex protein than initially envisaged, connecting multiple pathways beyond its strict role in mitochondrial fusion, such as sensing of OXPHOS needs and mitochondrial DNA maintenance. As a consequence, an increasing need to investigate OPA1 functions at multiple levels has imposed the development of multiple tools and models that are here reviewed. Translational mitochondrial medicine, with the ultimate objective of translating basic science necessary to understand pathogenic mechanisms into therapeutic strategies, requires disease modeling at multiple levels: from the simplest, like in yeast, to cell models, including the increasing use of reprogrammed stem cells (iPSCs) from patients, to animal models. In the present review, we thoroughly examine and provide the state of the art of all these approaches.
In the year 2000, the human OPA1 gene came to attention, as heterozygous mutations were associated with dominant optic atrophy (DOA) (1, 2), a blinding disorder originally described by the Danish ophthalmologist Paul Kjer in 1959, which usually leads to optic atrophy in the first decade of life (3). This gene encodes a protein for which the role as one of the mitochondrial factors involved in the machinery regulating mitochondrial dynamics, promoting fusion of the inner membrane of mitochondria and being key to mitochondrial DNA (mtDNA) maintenance, was clear by analogy with the orthologous genes MGM1/MSP1 in yeast (4–6). OPA1 was rapidly revealed to have a very complex expression regulation, coming in multiple isoforms due to alternative splicing (7, 8). Only recently have we begun to understand the need for this complexity, revealing how sophisticated and possibly flexible the function of this protein is (9). In fact, besides the role of OPA1 in mitochondrial fusion, by studying OPA1 dysfunction in DOA patients and cell models, its implication in regulating bioenergetics has rapidly emerged (10, 11), contributing to mitochondrial cristae shaping and regulating apoptosis (12, 13), maintaining mtDNA integrity and copy number (14–17), contributing to mitochondrial quality control (18, 19), and ultimately representing a key cross-road for mitochondrial homeostasis, which is central to cell survival and function (20–22).
The identification of OPA1 mutations as causative for selective neurodegeneration of retinal ganglion cells (RGCs) leading to optic atrophy was thus instrumental in opening a very active field of investigation for neurology and neuro-ophthalmology as well as in understanding new mechanistic pathways regulating mitochondria homeostasis and primarily mitochondrial dynamics (23). Congruently, the expanding spectrum of dominant and recessive mutations affecting the OPA1 protein has been reflected into a progressively larger landscape of clinical phenotypes linked to OPA1 dysfunction, including the vast catalog of the so-called DOA plus syndromes (14, 15) dominated by neurodegeneration and multisystem involvement (24), including multiple sclerosis (25, 26), Parkinsonism and dementia (18, 27), infantile Leigh syndrome (28), and cardiomyopathy (17).
Given the constantly growing interest in OPA1 function and dysfunction in relation to human diseases, their modeling is essential to rapidly progress our understanding and possibly provide therapeutic options. Thus, we review the currently available tools for the study of this protein, looking at the future of this rapidly evolving field.
Yeast, as a simple, easy-to-manipulate eukaryotic organism, is an extremely helpful model with which to understand mitochondrial function, providing insight into mitochondrial pathways and their regulation. Indeed, yeast has been instrumental in identifying key functions of MGM1 and MSP1, the orthologous of OPA1 in Saccharomyces cerevisiae and Schizosaccharomyces pombe, respectively (Figure 1). Mgm1 was first identified for its role in mtDNA maintenance (4, 29) and its relative effect on respiratory competence (4, 6). Subsequent reports highlighted the fusion capacity (30–32) and its role for cristae structure stability (32, 33). Similarly, Msp1 has been characterized as a protein involved in mtDNA maintenance (5), in mitochondrial network stability and respiratory function (34), and in the fusion of mitochondria (35). Mgm1 is proteolytically processed in two forms, long and short, by the rhomboid-type protease Pcp1, the homolog of the mammalian PARL (36, 37), whereas Msp1 is cleaved by both rhomboid and m-AAA proteases (38). The presence of both short and long forms of Mgm1 is required to maintain a tubular mitochondrial morphology and proper mtDNA amount (36, 39). The increased ratio of long to short form exerts a dominant negative effect on mitochondrial fusion, and a functional GTPase in the short form, but not in the long form, is necessary to tune fusion (39). Furthermore, one of the transmembrane segments of the dynamin Msp1 is required for mtDNA maintenance but not for the fusion activity (40).
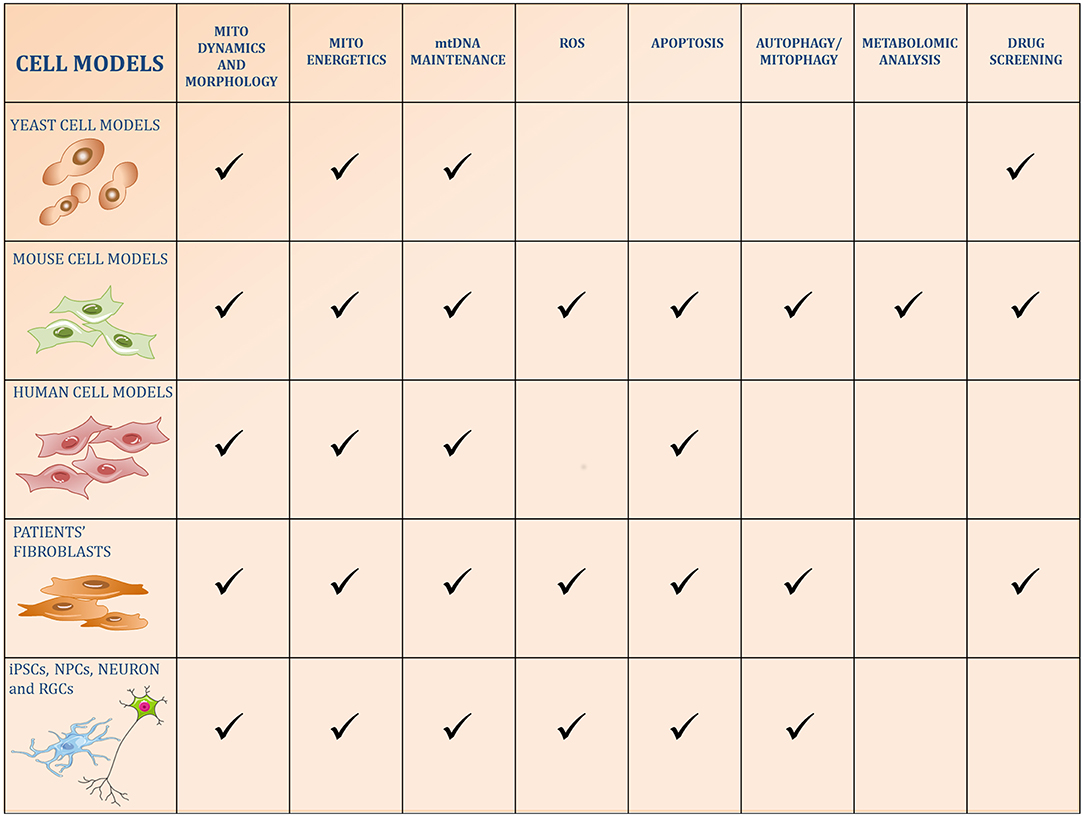
Figure 1. Schematic figure of the five categories of cell models used to study OPA1 functions. The pathways reported in the literature to be regulated by OPA1 or altered by its kd/ko/mutations in the different models are identified by a checkmark.
Yeast may be used as a model of mitochondrial diseases to investigate the effect of pathogenic mutations on mitochondrial function. Due to the weak conservation of amino acid sequences between Mgm1 and OPA1 and the inefficiency of the human GTPase in complementing MGM1 depletion, a chimeric protein composed of the N-terminal region of Mgm1 and the catalytic region of OPA1 (41) has been generated. Our group and other investigators used this chimeric model to dissect the effect of several mutations found in DOA patients, showing that pathogenic mutations caused mtDNA loss and mitochondrial fragmentation, reduced the processing of long to short forms, and impaired the respiratory capacity and oxidative growth in yeast (41–43). In addition to the validation of the pathogenicity, the possibility to compare haploid and diploid strains, expressing the mutant only or in combination with the wild-type chimeric protein, allowed for discrimination between hypomorphic, null, or dominant alleles (42).
Yeast is also a valuable system to screen for compounds with therapeutic potential to treat mitochondrial diseases as a “repurposing” strategy. Two research teams applied this approach in the context of OPA1 mutations (44, 45). In the first screening, a collection of 1,600 repurposed drugs has been used and five compounds were able to suppress the lethality in restrictive conditions caused by a GTPase mutation of MSP1 in S. pombe. Two drugs were able to rescue mtDNA depletion, and one of them recovered also mitochondrial network morphology (44). In the second screening, our group used the yeast strains with mutations on MGM1 or on the chimeric Mgm1-OPA1 to evaluate the effect of more than 2,500 drugs from two chemical libraries (45). In this case, out of the 26 drugs able to rescue the oxidative growth phenotype, six of them reduced the mitochondrial DNA instability in yeast and were analyzed also in mammalian cell models (45). Therefore, although some differences are present between Mgm1/Msp1 and OPA1, yeast has proven to be a simple and very useful first-line tool for large-scale OPA1 drug screenings, and, coupled with more physiological models to cross-validate the outcomes, it might speed up the discovery of effective therapies for DOA.
The study of OPA1 in mammalian models, such as mouse and rat cells, in particular the mouse embryonic fibroblasts (MEFs), has been instrumental in deepening our understanding of the complex role played by OPA1 in multiple mitochondrial pathways (Figure 1).
Studies in MEFs led to the identification of the OPA1 cleavage sites S1 and S2, encoded by exon 5 and 5b and cleaved by OMA1 and YME1L, respectively, to generate a balanced mixture of long and short forms (46, 47). Recently, a previously suggested third site (48) has been identified, named S3, located in exon 4b, and cleaved by YME1L (49).
Overexpression or silencing of OPA1 in MEFs highlighted its fundamental role in maintaining a filamentous mitochondrial network and sustaining fusion (50, 51), supporting respiratory efficiency (51), and controlling cristae integrity and apoptosis (13). However, Opa1 knockout (ko) mouse fibroblasts have also been extremely helpful in understanding the DOA pathogenic mechanism given the clear-cut phenotype due to the complete lack of the protein. Indeed, Opa1 null cells have no fusion activity (9, 46, 52, 53), decreased the number of mtDNA and nucleoids (9, 53, 54), and caused profound alterations of cristae structure integrity (9, 53, 55). Furthermore, Opa1 ablation and OPA1 overexpression in mouse fibroblasts clarified the interplay between cristae and OPA1 in respiratory chain supercomplexe (RCS) assembly (56) and identified the ATP synthase as the effector of the OPA1-mediated protection of mitochondrial functions (57). This model also provided details on the relationship between OPA1 and other interactors to sustain cristae architecture, such as the MICOS protein MIC60 (58, 59) and the SLC25A solute carriers family members, which activate the OPA1-mediated cristae remodeling in response to different energetic conditions (60).
Moreover, the complete absence of the protein was also instrumental in dissecting two fundamental and debated questions key to OPA1 complexity: the role of the isoforms and of the long and short forms. Expression of each of the eight isoforms in Opa1 null MEFs disclosed that they are all able to rescue mtDNA content, cristae organization, and energetics (9), whereas the short forms are more efficient in restoring energetic efficiency (9, 53) and enhancing cell survival under oxidative stress (61). Importantly, it has been shown that long forms support mitochondrial fusion (9), whereas both long and short forms are necessary to preserve an interconnected mitochondrial network (9, 46, 53). More recently, it has been reported that the presence of multiple isoforms generating different short forms is necessary to achieve a more filamentous mitochondrial network (49), confirming our seminal study (9).
The model of Opa1 null MEFs has been also employed to study the pathogenic effect of OPA1 missense mutations associated with DOA in an isogenic genetic background, evaluating the capability to restore mitochondrial defects (42, 62). We substantiated the more severe effect of mutations affecting the GTPase domain when compared to those in the dynamin domain or compared to hypomorphic mutations. This was true for all the mitochondrial readouts analyzed, faithfully mirroring the severity of clinical phenotypes (42). Additionally, these cell models underwent metabolomic and lipidomic studies (63–65). Opa1 ko MEFs showed bioenergetic changes with altered metabolism of aspartate, glutamate, nucleotides, and NAD (63), and variations in triglycerides and lipids involved in membrane remodeling and in cell signaling pathways (64), similarly to what has been observed in primary Opa1-depleted cortical neurons (66). An analysis of OPA1 mutated MEFs ranked the allele severity with the metabolic and lipid alterations, highlighting an increased spermine/spermidine ratio and a reduction in hydroxyproline, amino acid pool, and several phospholipids (65). Interestingly, the increase in glutathione in Opa1 null MEFs (63) has been recently confirmed in a second study (67) and reported, together with the increased dependency on cysteine transport, as a metabolic adaptive mechanism to afford protection against oxidative stress in these cells (67). Finally, this cell model has been also used for drug screening on amelioration of mitochondrial readouts with different missense mutations, proving to be a valuable tool in testing new DOA therapeutic interventions (45).
After the discovery that mutations in the OPA1 gene are the prevailing cause of DOA, several studies have been carried out using immortalized or human tumor-derived cells (Figure 1). These models have been instrumental in elucidating the manifold roles of this GTPase in mitochondrial homeostasis, avoiding the individual variations intrinsic to patient-derived primary cells.
Subcellular localization experiments were instrumental in identifying OPA1 as an inner-membrane protein, exposing the C-terminal portion in the IMS (68–71), which is present as a mixture of long and short forms (71, 72), the latter generated by the activity of the proteases YME1L (72, 73) and OMA1 (48). As the major pathogenic mechanism in OPA1-related DOA is haploinsufficiency, OPA1 downregulation has highlighted mitochondrial network fragmentation (12, 70, 73–75) and complete inhibition of mitochondria fusion (74, 76), confirming its pro-fusion role in mitochondrial dynamics. In addition, several studies found dissipation of the mitochondrial membrane potential (12, 76), drastic disorganization of the cristae structure (12, 70, 74, 75, 77), alteration of MICOS assembly (77) and increased sensitivity to apoptosis with the release of cytochrome c (12, 74, 76, 78). The link between OPA1, cristae integrity and apoptosis was confirmed in Hek293 cells by expressing a mutant OPA1 resistant to disassembly of its oligomers, which blocked cytochrome c release and apoptosis (79). Downregulation of OPA1 protein also induced mtDNA depletion (16, 75), bioenergetic defect (75, 80), altered Ca2+ homeostasis (75, 81) and mitophagy (82). Specific silencing of the three alternative splicing exons has been used in the attempt to unravel a precise mitochondrial function of the individual OPA1 isoforms (16, 83).
The pro-fusion role of long forms has been reported in several studies, where overexpression of long OPA1 was more efficient compared with the long/short combination in ameliorating the mitochondrial network morphology in knockdown (Kd)-OPA1 cells (71) and in SH-SY5Y cells after hypoxia and re-oxygenation injury (84). Furthermore, it has been reported that SIRT4 overexpression increased OPA1 long-form promoting fusion, counteracting fission and mitophagy (85).
Human cells have been used also to confirm the new function identified in adipose-like mouse fibroblasts (86), where OPA1 localized in lipid droplets serves as an A-kinase anchoring protein (AKAP) and allows PKA to phosphorylate perilipin 1 to favor lipolytic stimulation (87).
Fibroblasts and lymphoblasts derived from patients are a model extensively used to study the pathophysiology of OPA1 mutations. The majority of the alterations identified in the previous models have been reconfirmed in patients' cells (Figure 1). Numerous studies of fibroblasts and lymphoblasts reported defective mitochondrial network dynamics (11, 18, 27, 42, 78, 88–95), energetic metabolism (11, 42, 88, 91, 93, 94, 96), cristae structure maintenance (11, 27, 93), and increased sensitivity to apoptosis stimuli (11, 96). Depletion of mtDNA copy number in fibroblasts has been reported in a few cases in the presence of missense (42, 96) or compound heterozygous mutations (19, 43).
Recently, increased ROS production (94, 96), low levels of antioxidant enzymes (97), and alteration of calcium uptake (98) have been also reported. Furthermore, direct involvement of OPA1 in quality control has been revealed in fibroblasts (18, 19, 95), where missense mutations induced an increase in both autophagy and mitophagy processes. Interestingly, basal mitophagy was increased in fibroblasts bearing dominant-negative OPA1 mutations, whereas OPA1 haploinsufficiency seems to correlate with a reduction in mitochondrial turnover and autophagy (95).
Although these cells have been very useful when studying mitochondrial alterations, the presence of the wild-type allele often hides the effect of the mutated one, prompting the need to use galactose media to force oxidative metabolism and reveal the mitochondrial dysfunctions. Also, nuclear and mitochondrial genomes may variably contribute to the great heterogeneity observed in clinical and cellular phenotypes due to the same OPA1 mutation. For example, analysis of mtDNA copy number revealed a depletion only in one of two fibroblasts with the same R445H mutations derived by patients belonging to the same family (42), suggesting that other genetic or environmental factors may contribute. Concordantly, mitochondrial OMI/HTRA2 has been recently reported as a new gene modifier of phenotype variability (99).
Although with these limitations, fibroblasts, if combined with other models, may be functional in identifying new pathways, confirming pathogenic mechanisms, or validating the efficacy of therapeutic molecules, as we recently reported (45).
In the last decade, the establishment of induced pluripotent stem cell (iPSC) technology has provided the opportunity to generate in vitro human models of neurological disorders. The iPSCs are stem cells-like reprogrammed in vitro from patient-derived primary cells, which can be then differentiated into specific somatic cell types (100). The iPSCs and terminally differentiated cells, therefore, allow us to study the exact cell type affected in the human disease to identify the pathological mechanism and to conduct drug screening on a human genetic background (Figure 1).
Human iPSCs lines have been generated from patients' fibroblasts carrying different OPA1 mutations, such as the p.Gln621Ter (101) and the p.Ser545Arg (102) heterozygous mutations, and the compound heterozygous mutations causing Behr syndrome (103). All these iPSCs could differentiate into the three germ layers (endoderm, mesoderm, and ectoderm) (101–103).
Increased apoptosis and inefficient capability to differentiate into a neural rosette and RGCs were reported for the iPSCs derived from two other fibroblasts of patients carrying the intronic mutation c.2496+1G>T, suggesting the impact of apoptosis on RGCs possibly leading to early or congenital optic atrophy (104), as previously proposed by optical coherence tomography clinical studies (105). The addition of the neural induction medium, the secreted signaling molecule noggin, or the estrogen hormone promoted the differentiation into RGCs of the OPA1+/− iPSCs, possibly by inhibiting apoptosis (104).
Dopaminergic neurons carrying an OPA1 mutation causing haploinsufficiency were generated via iPSCs from two patients belonging to the same family that, interestingly, developed different clinical phenotypes: isolated DOA or DOA with syndromic Parkinsonism (106). Both the cell lines showed a reduction in oxygen consumption rate (OCR), complex I levels, and activity, whereas only neurons derived from the patient with Parkinsonism presented mitochondrial fragmentation and an increase of the OPA1 short forms. In this study possible genetic modifiers of the different clinical phenotypes in the two patients, presenting the same OPA1 mutation, were not identified (106).
Neural progenitor cells (NPCs) from two OPA1 missense mutations causing Parkinsonism and dementia (18) presented bioenergetic defect, mitochondrial network fragmentation, increased ROS levels, and alteration in the lysosome pathway (107). Even in this case, the OPA1-mutated NPCs showed a survival deficit, which was rescued by selective inhibition of necroptosis (107). The iPSC-derived neurons bearing these two OPA1 mutations have been further studied in a microfluidic system where different neuronal subtypes were cultured together with a patterned organization of their projections and synaptic terminals (108). Analysis of neuronal projections revealed altered content, distribution, and movement of mitochondria along the axons of the neurons. Moreover, the impairment in synapse formation was followed by a progressive loss of synaptic contacts over time in the mutant neurons, suggesting a depletion of mitochondria in neuronal projections as a cause of loss of neuronal connectivity and neurodegeneration (108).
Recently, human embryonic stem cells (hESCs) with OPA1 haploinsufficiency induced by the CRISPR-Cas9 technology have been characterized and differentiated into NPCs. Interestingly, several genes involved in NPCs differentiation, GABAergic interneuron formation, and retinal development were downregulated in OPA1+/− NPCs due to increased CpG methylation (109). The increased ROS levels and downregulated FOXG1 expression, a factor crucial for GABAergic neuronal formation and retinal development, were confirmed also in NPCs derived from two other patients bearing the common c.2873_2876delTTAG mutation, suggesting that OPA1 haploinsufficiency may result in aberrant nuclear DNA methylation and an altered transcriptional program (109).
Although studying the cells targeted by the disease accelerates our understanding of OPA1-derived dysfunctions, these two-dimensional methods to studying neuronal dysfunction do not completely recapitulate the complexity of a human brain (110). The recently optimized protocols for modeling in vitro brain and retina organogenesis (111) represent a further fundamental step instrumental in disease modeling.
Modeling of DOA has also been carried out in Drosophila melanogaster (Figure 2) and these studies emphasized the role of increased ROS production in leading to the pathologic phenotype, showing how this could be partially counterbalanced by antioxidant therapy (112). This supports the proposal of therapeutic approaches with antioxidants in human patients with DOA. The heterozygous mutant flies showed an age-dependent perturbation of visual functions, heart alterations (113), lifespan reduction, elevated ROS level, and the presence of irregular and dysmorphic mitochondria in the skeletal muscle (114). In summary, these two studies on the Opa1 mutant Drosophila model showed an age-dependent multisystemic disorder, resembling the syndromic forms of DOA “plus” (113, 114).
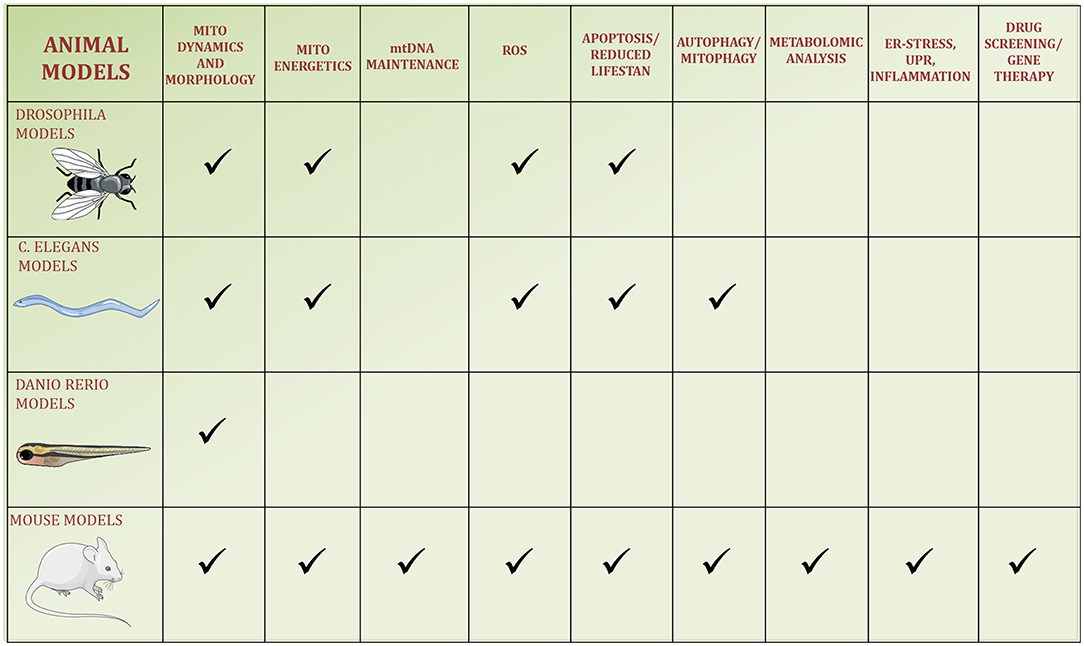
Figure 2. Schematic figure of the four animal models used to study OPA1. The pathways reported in the literature to be regulated by OPA1 or altered by its kd/ko/mutations in the animal models are identified by a checkmark.
The tight interconnection between OPA1 mutations and ROS alteration has been further confirmed in Caenorhabditis elegans (C. elegans) models. Indeed, the mutations in the eat-3 gene, the OPA1 worm orthologous gene, increased susceptibility to damage from free radicals, as shown by increased sensitivity to paraquat and to the loss of the mitochondrial superoxide dismutase sod-2 (115). Transgenic worms carrying loss of function eat-3 mutations presented fragmented mitochondria (115–117) and aberrant cristae architecture (115, 117). The eat-3 mutation caused an age-dependent and progressive deficit in movement, as well as in muscle and neuronal function (117), somehow mirroring the DOA “plus” clinical phenotypes. Interestingly, increased levels of elongated mitochondria have been linked to longevity, as mitochondrial fusion allows the survival of older animals (118), a finding supported by a second study showing that eat-3 mutation reduced median animal lifespan (117), as already reported in the Drosophila model (114).
The increased autophagy/mitophagy as a key pathogenic mechanism in DOA has been confirmed also in C. elegans, where the expression of mutated OPA1 in GABAergic axons reduced mitochondrial content in axons, a phenotype that was counteracted by depletion of the ATG8 homolog lgg-2 (119).
The effect of Opa1 depletion in the early development has been studied in a Danio rerio model using the antisense morpholino (120). The Opa1 morphants showed developmental delay, decreased blood circulation velocity, reduction of the eye size and heart rate, defects associated with increased mitochondrial fragmentation in muscle cells, and impaired bioenergetics (120).
The alteration of mitochondrial networks in Opa1 morphants has been reported more recently (121), confirming the main role of OPA1 in mitochondrial morphology.
Three mouse models carrying a heterozygous germline mutant Opa1 allele have been reported to date and have been instrumental in studying the pathogenic mechanisms of DOA (Figure 2) (122–124). Screening an ENU-mutagenized DNA library of mouse DNA led to generating two of the murine models, both recapitulating the human genetic defects that induce haploinsufficiency. The first, B6;C3-Opa1329−355del mutant mouse, leads to a splice error with the skipping of exon10, which ultimately causes an in-frame deletion of 27 amino acid residues in the dynamin GTPase domain (122). The second, B6;C3-Opa1Q285STOP mutant mouse, results in a truncated protein (123). Both models have about a 50% reduction in OPA1 expression and the homozygous condition is embryonically lethal, pointing to the crucial role played by OPA1 during fetal development. The third model is a knock-in mouse carrying the common OPA1 c.2708_2711delTTAG mutation in humans in a C57Bl6/J mouse background (124). In these mice, a 25% reduction in OPA1 protein was recognized in the brain, retina, optic nerve, and glycolytic fibers, whereas the reduction reached 50% in oxidative fibers and heart. Also, in this model, the homozygous condition was embryonically lethal.
In all three models, the heterozygous animals displayed a mild, age-dependent ocular phenotype with well-documented RGCs dysfunction and loss (122, 124–126). At histopathology, degenerative features were observed in the optic nerves including demyelination, various degrees of axonal degeneration, and abnormalities of mitochondria at the ultrastructure level (123–126).
Increased autophagy was reported in the RGCs of the B6;C3-Opa1Q285STOP mutant mouse (127) and in the glycolytic fibers, RGCs, and peripheral neurons of the Opa1delTTAG mutant mouse (124), highlighting the autophagic elimination of mitochondria with impaired fusion. More recently, increased mitophagy has been reported in the B6;C3-Opa1Q285STOP mice (128) and confirmed in mouse RGCs overexpressing a mutated OPA1, showing also that reduced axonal mitochondrial density was linked to increased autophagic mitochondrial degradation in the RGCs soma, in close proximity to axonal hillocks (119). The B6;C3-Opa1329−355del mice were also reported to present an imbalance of redox state possibly increasing mitochondrial ROS, as suggested by the decrease in aconitase activity and induction of antioxidant defenses (97). Metabolic analysis of the optic nerve in female Opa1delTTAG mice identified changes in the concentrations of metabolites involved in neuroprotection and of phospholipids, some of them suggestive of myelin sheath alteration (129). All these dysfunctions may lead most RGCs to death but not the melanopsin-expressing RGCs, which are reported to survive in two DOA mice models (130, 131) according to evidence that also in humans this cell type is relatively resistant to cell death in mitochondrial optic neuropathies (132).
In summary, the phenotype in these mouse models resembles sufficiently the human disease, characterized by loss of RGCs and optic nerve atrophy. In humans, the disease may vary in clinical severity, from severe congenital cases to very mild, subclinical disease disclosed only by accurate ophthalmological investigations (133, 134). Interestingly, systemic examination of these animals revealed mild neuromuscular impairment, including decreased locomotor activity, abnormal clutching reflex, and tremor, in analogy to the continuum clinical spectrum in humans, ranging from DOA to DOA “plus” (134).
Remarkably, interesting results were obtained by a closer investigation of the RGCs synaptic connectivity in the B6;C3-Opa1Q285STOP mutant animals, looking at their dendrites instead of focusing only on axons (135, 136). Counterintuitively, the earliest pathological changes occurred in RGCs dendrites, showing pruning and marked reduction in their synaptic connectivity (135, 136). This dendropathy is, however, congruent with the crucial role played by OPA1 and mitochondrial fusion in maintaining dendrites and their synapses (137).
Furthermore, with aging impaired cardiac function has been reported in all three mice models. Indeed, at onset of blindness, the aged B6;C3-OPA1Q285STOP mice also showed cardiomyopathy, characterized by disruption of mitochondrial organization, mtDNA depletion, bioenergetic defect, and defective cardiac mitochondria (138). Reduction in cardiac adaptation to pressure overload and heart hypertrophy were observed also in the B6;C3-Opa1329−355del mice (139). Similar alterations in mitochondrial calcium handling affected these Opa1 mutant mice (139), observed also in the third Opa1delTTAG mice, together with increased sensitivity to cardiac ischemia/reperfusion injuries (140). Interestingly, cardiac involvement in patients carrying OPA1 mutation has been reported for the first time in two patients harboring a homozygous recessive OPA1 mutation leading to a fatal encephalopathy with progressive hypertrophic cardiomyopathy (17).
A further mouse model with deleted Opa1 in pancreatic beta cells showed glucose intolerance and impaired insulin secretion. It also presented a reduced glucose-stimulated ATP production and developed hyperglycemia. Focusing on mitochondrial function, the beta cells presented a severe alteration of mitochondrial structure and a reduction of subunits' level and activity of Complex IV (141).
Recently, muscle-specific Opa1 ablation mice models have been generated to investigate the tissue-specific role of Opa1 in muscle, highlighting its metabolic role (142–144). Pereira et al. showed that in mutant mice the progressive mitochondrial dysfunction led to an increased metabolic rate, muscle atrophy, and insulin resistance induced by a mechanism involving ER stress and secretion of fibroblast growth factor 21 (FGF21) (142). Increased FGF21 has been observed also in another muscle-specific Opa1-deletion mouse model, where, together with ER stress and activation of the unfolded protein response (UPR), a catabolic program with muscle loss was activated, leading to systemic aging and premature death (143). In a third study, the enhanced FGF21 level and premature death elicited by Opa1 ablation were associated with muscle inflammation characterized by NF-kB activation and increased expression of pro-inflammatory genes; these features were blocked by mtDNA depletion and repression of TLR9 (144). All these features are of great interest, as plasma cytokines, including FGF21, have been recently validated as biomarkers for mitochondrial diseases, especially those with prominent muscle involvement (145, 146).
From a therapeutic point of view, all these mice models are instrumental in testing drugs or therapies. Indeed, the Opa1delTTAG mouse has been already tested for OPA1 isoform 1 gene therapy, proving to mitigate the OPA1-induced RGCs degeneration, encouraging a possible clinical translation in DOA patients (147). Furthermore, the most recent mouse model with RGC-specific overexpression of mutant OPA1 was instrumental in showing that contrasting the excess autophagy by various strategies was effective to avoid RGCs degeneration and restore vision (119).
Over the last decade, to elucidate the OPA1 role in the fusion of the inner mitochondrial membrane, various in vitro assays with purified OPA1 have been developed. Initially, the simpler yeast mgm1 has been studied by purifying short Mgm1 (s-Mgm1), revealing that the protein exhibits GTP activity, self-assembles into low order oligomer, and interacts specifically with negatively charged phospholipids present in the mitochondrial membranes (148). Furthermore, it has been reported that s-Mgm1 oligomerization and its binding to mitochondrial phospholipids strongly stimulates its GTPase activity, and it assembles onto liposomes and boosts liposome interaction, indicating that s-Mgm1 can tether opposing membranes mediating their fusion (149). This hypothesis was supported by cryo-electron microscopy studies and liposome fusion assays, showing that s-Mgm1 self-associates to tether opposing membranes with a gap of 15 nm and the oligomers undergo GTP-dependent structural changes that may induce fusion (150). Together with the ability to cause phospholipid clustering, s-Mgm1 was also reported to trigger local membrane bending and the formation of tubular structures (151). The investigations of both long and short Mgm1 forms in fusion highlighted that both forms are preferentially inserted into liposomes containing the lipid cardiolipin acting together in trans to form a functional unit required for mitochondrial fusion (152).
Similar results have been obtained by studying the molecular mechanisms of OPA1-mediated fusion. Indeed, the association with liposomes containing negative phospholipids, such as cardiolipin, enhanced the GTPase activity of short forms of OPA1, promoted their assembly into oligomers, and led to membrane tubulation, activities that were selectively impaired by DOA mutations (62). Using an in vitro purified mitochondria system, it has been shown that mitochondrial fusion is dependent on proteolytic processing of long forms (153). To disentangle the debated issue of the fusogenic capability of OPA1 forms, Ban et al. purified and analyzed short and long forms (154). The in vitro membrane fusion assays revealed that long forms and cardiolipin on opposite sides cooperate in mitochondrial inner membrane fusion, a process that is efficiently accelerated by a defined amount of short form and lowered by its excess level. On the other hand, a cardiolipin-independent interaction of long forms located on opposite membranes was reported to promote membrane tethering, thus sustaining the cristae structure (154). Concordantly, an in vitro reconstitution system, able to distinguish the sequential steps in membrane fusion, disclosed that the short form mediates membrane tethering, the long form is sufficient for membrane docking, hemifusion and low levels of content release, and the short form cooperates with the long form to mediate efficient and fast membrane pore opening (155). Importantly, as seen in other studies, the excess levels of short form inhibited fusion activity (155). Cryo-electron microscopic structures revealed that the short form presents the classic dynamin-like structure, can bind to membranes, induces membrane tubulation by forming a helical array, and GTPgS binding promotes changes in S-OPA1 assembly from a “closed” to an “open” conformation (156).
New mechanistic insight into how OPA1/Mgm1 mediates the membrane fusion has been provided by the recently solved crystal structures of short Mgm1 from Chaetomium thermophilum (157) and Saccharomyces cerevisiae (158). The fungus short Mgm1 was reported to form stalk-mediated tetramers and assemble on positively or negatively curved membranes (157), whereas the yeast short Mgm1 forms a concave membrane-associated head-to-tail trimeric structure built by intermolecular interactions (158). The crystal structure of the minimal GTPase domain (MGD) of OPA1 and biochemical analysis, instead, revealed that it can form nucleotide-dependent and -independent dimers, which may combine to form higher-order oligomers (159).
All these in vitro findings resolve some conflicting results obtained with cellular studies and helped to highlight the complexity of OPA1 activities, fundamental to maintain and support the inner membrane structure and fusion.
The investigation of OPA1 function has witnessed a tremendous deal of progress in the last decade thanks to a multitude of new models and approaches that we here reviewed. This progress was instrumental in refining the role of mitochondria in RGCs survival and to deepen our understanding of DOA pathogenesis to possibly establish effective therapies for these patients, currently an unmet need that urges a research effort. A few proof of principle studies already provided interesting results, such as by pharmacologically or genetically limiting the overactive autophagy in RGCs (119), or refining strategies based on gene therapy approaches (147). The multifaceted role of OPA1 in mitochondrial function and, more generally, in cell metabolism continues to surprise, and the possible therapeutic role of slight OPA1 overexpression has also been explored to counteract an array of different conditions (160–162), which is well-beyond the specific field of inherited optic neuropathies.
VD and VC contributed to the writing of the manuscript. Both authors contributed to the article and approved the submitted version.
This work was supported by the Italian Ministry of Health through the Ricerca Corrente funding to the Istituto di Ricovero e Cura a Carattere Scientifico Istituto delle Scienze Neurologiche di Bologna; and VC was supported by the Italian Ministry of Health (RF-2018-12366703 REORION grant) and by the Italian Ministry of University and Research (MUR) (PRIN-2017 20172T2MHH grant).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. (2000) 26:207–10. doi: 10.1038/79936
2. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. (2000) 26:211–5. doi: 10.1038/79944
3. Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. (1959) 164:1–147.
4. Guan K, Farh L, Marshall TK, Deschenes RJ. Normal mitochondrial structure and genome maintenance in yeast requires the dynamin-like product of the MGM1 gene. Curr Genet. (1993) 24:141–8. doi: 10.1007/BF00324678
5. Pelloquin L, Belenguer P, Menon Y, Ducommun B. Identification of a fission yeast dynamin-related protein involved in mitochondrial DNA maintenance. Biochem Biophys Res Commun. (1998) 251:720–6. doi: 10.1006/bbrc.1998.9539
6. Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol. (2000) 151:341–52. doi: 10.1083/jcb.151.2.341
7. Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. (2001) 109:584–91. doi: 10.1007/s00439-001-0633-y
8. Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C. Eight human OPA1 isoforms, long and short: what are they for? Biochim Biophys Acta Bioenerg. (2018) 1859:263–9. doi: 10.1016/j.bbabio.2018.01.005
9. Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, et al. OPA1 Isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. (2017) 19:2557–71. doi: 10.1016/j.celrep.2017.05.073
10. Lodi R, Tonon C, Valentino ML, Iotti S, Clementi V, Malucelli E, et al. Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Ann Neurol. (2004) 56:719–23. doi: 10.1002/ana.20278
11. Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. (2008) 131:352–67. doi: 10.1093/brain/awm335
12. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. (2003) 278:7743–6. doi: 10.1074/jbc.C200677200
13. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. (2006) 126:177–89. doi: 10.1016/j.cell.2006.06.025
14. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain. (2008) 131:338–51. doi: 10.1093/brain/awm298
15. Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. (2008) 131:329–37. doi: 10.1093/brain/awm272
16. Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. (2011) 21:12–20. doi: 10.1101/gr.108696.110
17. Spiegel R, Saada A, Flannery PJ, Burté F, Soiferman D, Khayat M, et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J Med Genet. (2016) 53:127–31. doi: 10.1136/jmedgenet-2015-103361
18. Carelli V, Musumeci O, Caporali L, Zanna C, La Morgia C, Del Dotto V, et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol. (2015) 78:21–38. doi: 10.1002/ana.24410
19. Liao C, Ashley N, Diot A, Morten K, Phadwal K, Williams A, et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology. (2017) 88:131–42. doi: 10.1212/WNL.0000000000003491
20. Del Dotto V, Fogazza M, Lenaers G, Rugolo M, Carelli V, Zanna C. OPA1: how much do we know to approach therapy? Pharmacol Res. (2018) 131:199–210. doi: 10.1016/j.phrs.2018.02.018
21. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
22. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
23. Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. (2015) 11:11–24. doi: 10.1038/nrneurol.2014.228
24. Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. (2010) 133:771–86. doi: 10.1093/brain/awq007
25. Verny C, Loiseau D, Scherer C, Lejeune P, Chevrollier A, Gueguen N, et al. Multiple sclerosis-like disorder in OPA1-related autosomal dominant optic atrophy. Neurology. (2008) 70:1152–3. doi: 10.1212/01.wnl.0000289194.89359.a1
26. Yu-Wai-Man P, Spyropoulos A, Duncan HJ, Guadagno JV, Chinnery PF. A multiple sclerosis-like disorder in patients with OPA1 mutations. Ann Clin Transl Neurol. (2016) 3:723–9. doi: 10.1002/acn3.323
27. Lynch DS, Loh SHY, Harley J, Noyce AJ, Martins LM, Wood NW, et al. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to OPA1 mutations. Neurol Genet. (2017) 3:e188. doi: 10.1212/NXG.0000000000000188
28. Rubegni A, Pisano T, Bacci G, Tessa A, Battini R, Procopio E, et al. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur J Paediatr Neurol. (2017) 21:671–7. doi: 10.1016/j.ejpn.2017.04.004
29. Jones BA, Fangman WL. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev. (1992) 6:380–9. doi: 10.1101/gad.6.3.380
30. Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, et al. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. (2003) 160:303–11. doi: 10.1083/jcb.200209015
31. Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. (2003) 14:2342–56. doi: 10.1091/mbc.e02-12-0788
32. Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. (2006) 127:383–95. doi: 10.1016/j.cell.2006.09.021
33. Amutha B, Gordon DM, Gu Y, Pain D. A novel role of Mgm1p, a dynamin-related GTPase, in ATP synthase assembly and cristae formation/maintenance. Biochem J. (2004) 381:19–23. doi: 10.1042/BJ20040566
34. Pelloquin L, Belenguer P, Menon Y, Gas N, Ducommun B. Fission yeast Msp1 is a mitochondrial dynamin-related protein. J Cell Sci. (1999) 112:4151–61.
35. Guillou E, Bousquet C, Daloyau M, Emorine LJ, Belenguer P. Msp1p is an intermembrane space dynamin-related protein that mediates mitochondrial fusion in a Dnm1p-dependent manner in S. pombe. FEBS Lett. (2005) 579:1109–16. doi: 10.1016/j.febslet.2004.12.083
36. Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem. (2003) 278:27781–8. doi: 10.1074/jbc.M211311200
37. McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature. (2003) 423:537–41. doi: 10.1038/nature01633
38. Leroy I, Khosrobakhsh F, Diot A, Daloyau M, Arnauné-Pelloquin L, Cavelier C, et al. Processing of the dynamin Msp1p in S. pombe reveals an evolutionary switch between its orthologs Mgm1p in S. cerevisiae and OPA1 in mammals. FEBS Lett. (2010) 584:3153–7. doi: 10.1016/j.febslet.2010.05.060
39. Zick M, Duvezin-Caubet S, Schäfer A, Vogel F, Neupert W, Reichert AS. Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett. (2009) 583:2237–43. doi: 10.1016/j.febslet.2009.05.053
40. Diot A, Guillou E, Daloyau M, Arnauné-Pelloquin L, Emorine LJ, Belenguer P. Transmembrane segments of the dynamin Msp1p uncouple its functions in the control of mitochondrial morphology and genome maintenance. J Cell Sci. (2009) 122:2632–9. doi: 10.1242/jcs.040139
41. Nolli C, Goffrini P, Lazzaretti M, Zanna C, Vitale R, Lodi T, et al. Validation of a MGM1/OPA1 chimeric gene for functional analysis in yeast of mutations associated with dominant optic atrophy. Mitochondrion. (2015) 25:38–48. doi: 10.1016/j.mito.2015.10.002
42. Del Dotto V, Fogazza M, Musiani F, Maresca A, Aleo SJ, Caporali L, et al. Deciphering OPA1 mutations pathogenicity by combined analysis of human, mouse and yeast cell models. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:3496–514. doi: 10.1016/j.bbadis.2018.08.004
43. Nasca A, Rizza T, Doimo M, Legati A, Ciolfi A, Diodato D, et al. Not only dominant, not only optic atrophy: expanding the clinical spectrum associated with OPA1 mutations. Orphanet J Rare Dis. (2017) 12:89. doi: 10.1186/s13023-017-0641-1
44. Delerue T, Tribouillard-Tanvier D, Daloyau M, Khosrobakhsh F, Emorine LJ, Friocourt G, et al. A yeast-based screening assay identifies repurposed drugs that suppress mitochondrial fusion and mtDNA maintenance defects. Dis Model Mech. (2019) 12:dmm036558. doi: 10.1242/dmm.036558
45. Aleo SJ, Del Dotto V, Fogazza M, Maresca A, Lodi T, Goffrini P, et al. Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum Mol Genet. (2021) 29:3631–45. doi: 10.1093/hmg/ddaa244
46. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. (2007) 178:749–55. doi: 10.1083/jcb.200704110
47. Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. (2009) 187:1023–36. doi: 10.1083/jcb.200906084
48. Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. (2009) 187:959–66. doi: 10.1083/jcb.200906083
49. Wang R, Mishra P, Garbis SD, Moradian A, Sweredoski MJ, Chan DC. Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol Biol Cell. (2021) 32:157–68. doi: 10.1091/mbc.E20-09-0605
50. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
51. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. (2005) 280:26185–92. doi: 10.1074/jbc.M503062200
52. Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. (2007) 130:548–62. doi: 10.1016/j.cell.2007.06.026
53. Lee H, Smith SB, Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem. (2017) 292:7115–30. doi: 10.1074/jbc.M116.762567
54. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. (2010) 141:280–9. doi: 10.1016/j.cell.2010.02.026
55. Hu C, Shu L, Huang X, Yu J, Li L, Gong L, et al. OPA1 and MICOS regulate mitochondrial crista dynamics and formation. Cell Death Dis. (2020) 11:940. doi: 10.1038/s41419-020-03152-y
56. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
57. Quintana-Cabrera R, Quirin C, Glytsou C, Corrado M, Urbani A, Pellattiero A, et al. The cristae modulator optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun. (2018) 9:3399. doi: 10.1038/s41467-018-05655-x
58. Barrera M, Koob S, Dikov D, Vogel F, Reichert AS. OPA1 functionally interacts with MIC60 but is dispensable for crista junction formation. FEBS Lett. (2016) 590:3309–22. doi: 10.1002/1873-3468.12384
59. Glytsou C, Calvo E, Cogliati S, Mehrotra A, Anastasia I, Rigoni G, et al. Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. (2016) 17:3024–34. doi: 10.1016/j.celrep.2016.11.049
60. Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. (2014) 33:2676–91. doi: 10.15252/embj.201488349
61. Lee H, Smith SB, Sheu S-S, Yoon Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J Biol Chem. (2020) 295:6543–60. doi: 10.1074/jbc.RA119.010983
62. Ban T, Heymann JAW, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. (2010) 19:2113–22. doi: 10.1093/hmg/ddq088
63. Bocca C, Kane MS, Veyrat-Durebex C, Chupin S, Alban J, Kouassi Nzoughet J, et al. The metabolomic bioenergetic signature of Opa1-disrupted mouse embryonic fibroblasts highlights aspartate deficiency. Sci Rep. (2018) 8:11528. doi: 10.1038/s41598-018-29972-9
64. Bocca C, Kane MS, Veyrat-Durebex C, Nzoughet JK, Chao de la Barca JM, Chupin S, et al. Lipidomics reveals triacylglycerol accumulation due to impaired fatty acid flux in Opa1-disrupted fibroblasts. J Proteome Res. (2019) 18:2779–90. doi: 10.1021/acs.jproteome.9b00081
65. Chao de la Barca JM, Fogazza M, Rugolo M, Chupin S, Del Dotto V, Ghelli AM, et al. Metabolomics hallmarks OPA1 variants correlating with their in vitro phenotype and predicting clinical severity. Hum Mol Genet. (2020) 29:1319–29. doi: 10.1093/hmg/ddaa047
66. Chao de la Barca JM, Arrázola MS, Bocca C, Arnauné-Pelloquin L, Iuliano O, Tcherkez G, et al. The metabolomic signature of Opa1 deficiency in rat primary cortical neurons shows aspartate/glutamate depletion and phospholipids remodeling. Sci Rep. (2019) 9:6107. doi: 10.1038/s41598-019-42554-7
67. Patten DA, McGuirk S, Anilkumar U, Antoun G, Gandhi K, Parmar G, et al. Altered mitochondrial fusion drives defensive glutathione synthesis in cells able to switch to glycolytic ATP production. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118854. doi: 10.1016/j.bbamcr.2020.118854
68. Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. (2002) 523:171–6. doi: 10.1016/s0014-5793(02)02985-x
69. Satoh M, Hamamoto T, Seo N, Kagawa Y, Endo H. Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem Biophys Res Commun. (2003) 300:482–93. doi: 10.1016/s0006-291x(02)02874-7
70. Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. (2004) 279:18792–8. doi: 10.1074/jbc.M400920200
71. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. (2006) 25:2966–77. doi: 10.1038/sj.emboj.7601184
72. Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombès A, et al. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell. (2008) 100:315–25. doi: 10.1042/BC20070110
73. Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. (2007) 178:757–64. doi: 10.1083/jcb.200704112
74. Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. (2005) 280:35742–50. doi: 10.1074/jbc.M505970200
75. Kushnareva YE, Gerencser AA, Bossy B, Ju WK, White AD, Waggoner J, et al. Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. (2013) 20:353–65. doi: 10.1038/cdd.2012.128
76. Lee Y, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. (2004) 15:5001–11. doi: 10.1091/mbc.e04-04-0294
77. Stephan T, Brüser C, Deckers M, Steyer AM, Balzarotti F, Barbot M, et al. MICOS assembly controls mitochondrial inner membrane remodeling and crista junction redistribution to mediate cristae formation. EMBO J. (2020) 39:e104105. doi: 10.15252/embj.2019104105
78. Olichon A, Landes T, Arnauné-Pelloquin L, Emorine LJ, Mils V, Guichet A, et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. (2007) 211:423–30. doi: 10.1002/jcp.20950
79. Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, et al. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. (2008) 31:557–69. doi: 10.1016/j.molcel.2008.07.010
80. Jang S, Javadov S. OPA1 regulates respiratory supercomplexes assembly: the role of mitochondrial swelling. Mitochondrion. (2020) 51:30–9. doi: 10.1016/j.mito.2019.11.006
81. Fülöp L, Szanda G, Enyedi B, Várnai P, Spät A. The effect of OPA1 on mitochondrial Ca2+ signaling. PLoS ONE. (2011) 6:e25199. doi: 10.1371/journal.pone.0025199
82. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. (2016) 12:689–702. doi: 10.1080/15548627.2016.1151580
83. Olichon A, Elachouri G, Baricault L, Delettre C, Belenguer P, Lenaers G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. (2007) 14:682–92. doi: 10.1038/sj.cdd.4402048
84. Sun Y, Xue W, Song Z, Huang K, Zheng L. Restoration of Opa1-long isoform inhibits retinal injury-induced neurodegeneration. J Mol Med (Berl). (2016) 94:335–46. doi: 10.1007/s00109-015-1359-y
85. Lang A, Anand R, Altinoluk-Hambüchen S, Ezzahoini H, Stefanski A, Iram A, et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (Albany NY). (2017) 9:2163–89. doi: 10.18632/aging.101307
86. Pidoux G, Witczak O, Jarnæss E, Myrvold L, Urlaub H, Stokka AJ, et al. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. (2011) 30:4371–86. doi: 10.1038/emboj.2011.365
87. Rogne M, Chu D-T, Küntziger TM, Mylonakou M-N, Collas P, Tasken K. OPA1-anchored PKA phosphorylates perilipin 1 on S522 and S497 in adipocytes differentiated from human adipose stem cells. Mol Biol Cell. (2018) 29:1487–501. doi: 10.1091/mbc.E17-09-0538
88. Amati-Bonneau P, Guichet A, Olichon A, Chevrollier A, Viala F, Miot S, et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann Neurol. (2005) 58:958–63. doi: 10.1002/ana.20681
89. Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferré M, Guillet V, et al. OPA1-associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol. (2009) 41:1855–65. doi: 10.1016/j.biocel.2009.04.012
90. Spinazzi M, Cazzola S, Bortolozzi M, Baracca A, Loro E, Casarin A, et al. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum Mol Genet. (2008) 17:3291–302. doi: 10.1093/hmg/ddn225
91. Nochez Y, Arsene S, Gueguen N, Chevrollier A, Ferré M, Guillet V, et al. Acute and late-onset optic atrophy due to a novel OPA1 mutation leading to a mitochondrial coupling defect. Mol Vis. (2009) 15:598–608.
92. Bonifert T, Karle KN, Tonagel F, Batra M, Wilhelm C, Theurer Y, et al. Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain. (2014) 137:2164–77. doi: 10.1093/brain/awu165
93. Agier V, Oliviero P, Lainé J, L'Hermitte-Stead C, Girard S, Fillaut S, et al. Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta. (2012) 1822:1570–80. doi: 10.1016/j.bbadis.2012.07.002
94. Kao S-H, Yen M-Y, Wang A-G, Yeh Y-L, Lin A-L. Changes in mitochondrial morphology and bioenergetics in human lymphoblastoid cells with four novel OPA1 mutations. Invest Ophthalmol Vis Sci. (2015) 56:2269–78. doi: 10.1167/iovs.14-16288
95. Kane MS, Alban J, Desquiret-Dumas V, Gueguen N, Ishak L, Ferre M, et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J Cell Mol Med. (2017) 21:2284–97. doi: 10.1111/jcmm.13149
96. Zhang J, Liu X, Liang X, Lu Y, Zhu L, Fu R, et al. A novel ADOA-associated OPA1 mutation alters the mitochondrial function, membrane potential, ROS production and apoptosis. Sci Rep. (2017) 7:5704. doi: 10.1038/s41598-017-05571-y
97. Millet AMC, Bertholet AM, Daloyau M, Reynier P, Galinier A, Devin A, et al. Loss of functional OPA1 unbalances redox state: implications in dominant optic atrophy pathogenesis. Ann Clin Transl Neurol. (2016) 3:408–21. doi: 10.1002/acn3.305
98. Fülöp L, Rajki A, Maka E, Molnár MJ, Spät A. Mitochondrial Ca2+ uptake correlates with the severity of the symptoms in autosomal dominant optic atrophy. Cell Calcium. (2015) 57:49–55. doi: 10.1016/j.ceca.2014.11.008
99. Napolitano F, Terracciano C, Bruno G, Nesti C, Barillari MR, Barillari U, et al. Intrafamilial “DOA-plus” phenotype variability related to different OMI/HTRA2 expression. Am J Med Genet A. (2020) 182:176–82. doi: 10.1002/ajmg.a.61381
100. Rowe RG, Daley GQ. Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet. (2019) 20:377–88. doi: 10.1038/s41576-019-0100-z
101. Galera-Monge T, Zurita-Díaz F, Moreno-Izquierdo A, Fraga MF, Fernández AF, Ayuso C, et al. Generation of a human iPSC line from a patient with an optic atrophy “plus” phenotype due to a mutation in the OPA1 gene. Stem Cell Res. (2016) 16:673–6. doi: 10.1016/j.scr.2016.03.011
102. Zurita-Díaz F, Galera-Monge T, Moreno-Izquierdo A, Corton M, Ayuso C, Garesse R, et al. Establishment of a human DOA “plus” iPSC line, IISHDOi003-A, with the mutation in the OPA1 gene: c.1635C>A; p.Ser545Arg. Stem Cell Res. (2017) 24:81–4. doi: 10.1016/j.scr.2017.08.017
103. Hauser S, Schuster S, Theurer Y, Synofzik M, Schöls L. Generation of optic atrophy 1 patient-derived induced pluripotent stem cells (iPS-OPA1-BEHR) for disease modeling of complex optic atrophy syndromes (Behr syndrome). Stem Cell Res. (2016) 17:426–9. doi: 10.1016/j.scr.2016.09.012
104. Chen J, Riazifar H, Guan M-X, Huang T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res Ther. (2016) 7:2. doi: 10.1186/s13287-015-0264-1
105. Barboni P, Carbonelli M, Savini G, Foscarini B, Parisi V, Valentino ML, et al. OPA1 mutations associated with dominant optic atrophy influence optic nerve head size. Ophthalmology. (2010) 117:1547–53. doi: 10.1016/j.ophtha.2009.12.042
106. Jonikas M, Madill M, Mathy A, Zekoll T, Zois CE, Wigfield S, et al. Stem cell modeling of mitochondrial Parkinsonism reveals key functions of OPA1. Ann Neurol. (2018) 83:915–25. doi: 10.1002/ana.25221
107. Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson's disease models. Cell Rep. (2018) 22:2066–79. doi: 10.1016/j.celrep.2018.01.089
108. Iannielli A, Ugolini GS, Cordiglieri C, Bido S, Rubio A, Colasante G, et al. Reconstitution of the human nigro-striatal pathway on-a-chip reveals OPA1-dependent mitochondrial defects and loss of dopaminergic synapses. Cell Rep. (2019) 29:4646–56.e4. doi: 10.1016/j.celrep.2019.11.111
109. Caglayan S, Hashim A, Cieslar-Pobuda A, Jensen V, Behringer S, Talug B, et al. Optic atrophy 1 controls human neuronal development by preventing aberrant nuclear DNA methylation. iScience. (2020) 23:101154. doi: 10.1016/j.isci.2020.101154
110. Chiaradia I, Lancaster MA. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat Neurosci. (2020) 23:1496–508. doi: 10.1038/s41593-020-00730-3
111. Cowan CS, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, et al. Cell types of the human retina and its organoids at single-cell resolution. Cell. (2020) 182:1623–40.e34. doi: 10.1016/j.cell.2020.08.013
112. Yarosh W, Monserrate J, Tong JJ, Tse S, Le PK, Nguyen K, et al. The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet. (2008) 4:e6. doi: 10.1371/journal.pgen.0040006
113. Shahrestani P, Leung H-T, Le PK, Pak WL, Tse S, Ocorr K, et al. Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific manner. PLoS ONE. (2009) 4:e6867. doi: 10.1371/journal.pone.0006867
114. Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS ONE. (2009) 4:e4492. doi: 10.1371/journal.pone.0004492
115. Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, et al. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. (2008) 4:e1000022. doi: 10.1371/journal.pgen.1000022
116. Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. (2009) 186:525–40. doi: 10.1083/jcb.200905070
117. Byrne JJ, Soh MS, Chandhok G, Vijayaraghavan T, Teoh JS, Crawford S, et al. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol Life Sci. (2019) 76:1967–85. doi: 10.1007/s00018-019-03024-5
118. Chaudhari SN, Kipreos ET. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun. (2017) 8:182. doi: 10.1038/s41467-017-00274-4
119. Zaninello M, Palikaras K, Naon D, Iwata K, Herkenne S, Quintana-Cabrera R, et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat Commun. (2020) 11:4029. doi: 10.1038/s41467-020-17821-1
120. Rahn JJ, Stackley KD, Chan SSL. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE. (2013) 8:e59218. doi: 10.1371/journal.pone.0059218
121. Eijkenboom I, Vanoevelen JM, Hoeijmakers JGJ, Wijnen I, Gerards M, Faber CG, et al. A zebrafish model to study small-fiber neuropathy reveals a potential role for GDAP1. Mitochondrion. (2019) 47:273–81. doi: 10.1016/j.mito.2019.01.002
122. Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. (2007) 130:1029–42. doi: 10.1093/brain/awm005
123. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
124. Sarzi E, Angebault C, Seveno M, Gueguen N, Chaix B, Bielicki G, et al. The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain. (2012) 135:3599–613. doi: 10.1093/brain/aws303
125. Heiduschka P, Schnichels S, Fuhrmann N, Hofmeister S, Schraermeyer U, Wissinger B, et al. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. (2010) 51:1424–31. doi: 10.1167/iovs.09-3606
126. Nguyen D, Alavi MV, Kim KY, Kang T, Scott RT, Noh YH, et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. (2011) 2:e240. doi: 10.1038/cddis.2011.117
127. White KE, Davies VJ, Hogan VE, Piechota MJ, Nichols PP, Turnbull DM, et al. OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest Ophthalmol Vis Sci. (2009) 50:2567–71. doi: 10.1167/iovs.08-2913
128. Diot A, Agnew T, Sanderson J, Liao C, Carver J, Neves RP das, et al. Validating the RedMIT/GFP-LC3 mouse model by studying mitophagy in autosomal dominant optic atrophy due to the OPA1Q285STOP mutation. Front Cell Dev Biol. (2018) 6:103. doi: 10.3389/fcell.2018.00103
129. Chao de la Barca JM, Simard G, Sarzi E, Chaumette T, Rousseau G, Chupin S, et al. Targeted metabolomics reveals early dominant optic atrophy signature in optic nerves of Opa1delTTAG/+ mice. Invest Ophthalmol Vis Sci. (2017) 58:812–20. doi: 10.1167/iovs.16-21116
130. Perganta G, Barnard AR, Katti C, Vachtsevanos A, Douglas RH, MacLaren RE, et al. Non-image-forming light driven functions are preserved in a mouse model of autosomal dominant optic atrophy. PLoS ONE. (2013) 8:e56350. doi: 10.1371/journal.pone.0056350
131. González-Menéndez I, Reinhard K, Tolivia J, Wissinger B, Münch TA. Influence of Opa1 mutation on survival and function of retinal ganglion cells. Invest Ophthalmol Vis Sci. (2015) 56:4835–45. doi: 10.1167/iovs.15-16743
132. La Morgia C, Ross-Cisneros FN, Sadun AA, Hannibal J, Munarini A, Mantovani V, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain. (2010) 133:2426–8. doi: 10.1093/brain/awq155
133. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. (2004) 23:53–89. doi: 10.1016/j.preteyeres.2003.10.003
134. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies–disease mechanisms and therapeutic strategies. Prog Retin Eye Res. (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
135. Williams PA, Morgan JE, Votruba M. Opa1 deficiency in a mouse model of dominant optic atrophy leads to retinal ganglion cell dendropathy. Brain. (2010) 133:2942–51. doi: 10.1093/brain/awq218
136. Williams PA, Piechota M, von Ruhland C, Taylor E, Morgan JE, Votruba M. Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain. (2012) 135:493–505. doi: 10.1093/brain/awr330
137. Li Z, Okamoto K-I, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. (2004) 119:873–87. doi: 10.1016/j.cell.2004.11.003
138. Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, et al. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc. (2012) 1:e003012. doi: 10.1161/JAHA.112.003012
139. Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. (2012) 94:408–17. doi: 10.1093/cvr/cvs117
140. Le Page S, Niro M, Fauconnier J, Cellier L, Tamareille S, Gharib A, et al. Increase in cardiac ischemia-reperfusion injuries in Opa1+/− mouse model. PLoS ONE. (2016) 11:e0164066. doi: 10.1371/journal.pone.0164066
141. Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. (2011) 22:2235–45. doi: 10.1091/mbc.E10-12-0933
142. Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. (2017) 36:2126–45. doi: 10.15252/embj.201696179
143. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. (2017) 25:1374–89.e6. doi: 10.1016/j.cmet.2017.04.021
144. Rodríguez-Nuevo A, Díaz-Ramos A, Noguera E, Díaz-Sáez F, Duran X, Muñoz JP, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. (2018) 37:e96553. doi: 10.15252/embj.201796553
145. Lehtonen JM Forsström S Bottani E Viscomi C Baris OR Isoniemi H . FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology. (2016) 87:2290–9. doi: 10.1212/WNL.0000000000003374
146. Maresca A, Del Dotto V, Romagnoli M, La Morgia C, Di Vito L, Capristo M, et al. Expanding and validating the biomarkers for mitochondrial diseases. J Mol Med (Berl). (2020) 98:1467–78. doi: 10.1007/s00109-020-01967-y
147. Sarzi E, Seveno M, Piro-Mégy C, Elzière L, Quilès M, Péquignot M, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. Sci Rep. (2018) 8:2468. doi: 10.1038/s41598-018-20838-8
148. Meglei G, McQuibban GA. The dynamin-related protein Mgm1p assembles into oligomers and hydrolyzes GTP to function in mitochondrial membrane fusion. Biochemistry. (2009) 48:1774–84. doi: 10.1021/bi801723d
149. Rujiviphat J, Meglei G, Rubinstein JL, McQuibban GA. Phospholipid association is essential for dynamin-related protein Mgm1 to function in mitochondrial membrane fusion. J Biol Chem. (2009) 284:28682–6. doi: 10.1074/jbc.M109.044933
150. Abutbul-Ionita I, Rujiviphat J, Nir I, McQuibban GA, Danino D. Membrane tethering and nucleotide-dependent conformational changes drive mitochondrial genome maintenance (Mgm1) protein-mediated membrane fusion. J Biol Chem. (2012) 287:36634–8. doi: 10.1074/jbc.C112.406769
151. Rujiviphat J, Wong MK, Won A, Shih Y-L, Yip CM, McQuibban GA. Mitochondrial genome maintenance 1 (Mgm1) protein alters membrane topology and promotes local membrane bending. J Mol Biol. (2015) 427:2599–609. doi: 10.1016/j.jmb.2015.03.006
152. DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. (2009) 186:793–803. doi: 10.1083/jcb.200906098
153. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. (2014) 19:630–41. doi: 10.1016/j.cmet.2014.03.011
154. Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. (2017) 19:856–63. doi: 10.1038/ncb3560
155. Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife. (2020) 9:e50973. doi: 10.7554/eLife.50973
156. Zhang D, Zhang Y, Ma J, Zhu C, Niu T, Chen W, et al. Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. Elife. (2020) 9:e50294. doi: 10.7554/eLife.50294
157. Faelber K, Dietrich L, Noel JK, Wollweber F, Pfitzner A-K, Mühleip A, et al. Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature. (2019) 571:429–33. doi: 10.1038/s41586-019-1372-3
158. Yan L, Qi Y, Ricketson D, Li L, Subramanian K, Zhao J, et al. Structural analysis of a trimeric assembly of the mitochondrial dynamin-like GTPase Mgm1. Proc Natl Acad Sci USA. (2020) 117:4061–70. doi: 10.1073/pnas.1919116117
159. Yu C, Zhao J, Yan L, Qi Y, Guo X, Lou Z, et al. Structural insights into G domain dimerization and pathogenic mutation of OPA1. J Cell Biol. (2020) 219:e201907098. doi: 10.1083/jcb.201907098
160. Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, et al. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. (2015) 21:845–54. doi: 10.1016/j.cmet.2015.04.016
161. Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. (2015) 21:834–44. doi: 10.1016/j.cmet.2015.05.007
Keywords: OPA1 mutations, dominant optic atrophy, OPA1, mitochondria, cell models, mouse models, iPSCs, retinal ganglion cells
Citation: Del Dotto V and Carelli V (2021) Dominant Optic Atrophy (DOA): Modeling the Kaleidoscopic Roles of OPA1 in Mitochondrial Homeostasis. Front. Neurol. 12:681326. doi: 10.3389/fneur.2021.681326
Received: 16 March 2021; Accepted: 22 April 2021;
Published: 09 June 2021.
Edited by:
Victoria Susan Pelak, University of Colorado, United StatesReviewed by:
M. Esther Gallardo, Research Institute Hospital 12 de Octubre, SpainCopyright © 2021 Del Dotto and Carelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Del Dotto, dmFsZW50aW5hLmRlbGRvdHRvMkB1bmliby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.