 Sophia R. L. Vieira
Sophia R. L. Vieira Huw R. Morris
Huw R. Morris
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 04 June 2021
Sec. Dementia and Neurodegenerative Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.679927
Genetics has driven significant discoveries in the field of neurodegenerative diseases (NDDs). An emerging theme in neurodegeneration warrants an urgent and comprehensive update: that carrier status of early-onset autosomal recessive (AR) disease, typically considered benign, is associated with an increased risk of a spectrum of late-onset NDDs. Glucosylceramidase beta (GBA1) gene mutations, responsible for the AR lysosomal storage disorder Gaucher disease, are a prominent example of this principle, having been identified as an important genetic risk factor for Parkinson disease. Genetic analyses have revealed further examples, notably GRN, TREM2, EIF2AK3, and several other LSD and mitochondria function genes. In this Review, we discuss the evidence supporting the strikingly distinct allele-dependent clinical phenotypes observed in carriers of such gene mutations and its impact on the wider field of neurodegeneration.
Autosomal recessive (AR) inheritance is an important cause of paediatric disease, in which parents are carriers of single copies of disease mutations but are unaffected by disease. Their children have a 25% risk of inheriting both disease mutations leading to AR disease with risk to siblings but in general not parents or children of affected subjects. The mutations are usually loss-of-function (deletions, splice site, or nonsense mutations) but sometimes non-synonymous mutations may also result in significant protein dysfunction. AR disease is conventionally defined by bi-allelic (homozygous or compound heterozygous) mutations leading to a disease phenotype, with heterozygous mutation carrier status (which may be associated with haploinsufficiency) typically considered benign. Interestingly, there may be survival benefits associated with heterozygote carrier status, particularly in resistance to infectious disease which has been proposed to underlie the higher-than-expected carrier frequency of AR disease gene alleles observed in some populations (1). Resistance to cholera and typhoid fever in individuals who carry cystic fibrosis heterozygote alleles supports this hypothesis (1). However, an emerging prominent theme has changed our view of the biology of heterozygote carrier status and has important implications for genetic counselling and the development of new therapies. Carrier status of predominantly paediatric-onset AR disease may lead to an increased risk of a spectrum of adult-onset neurodegenerative diseases (NDDs) (2, 3).
Studies of families and large population cohorts have revealed that heterozygous and biallelic mutations in the same gene can lead to clinically distinct diseases (2, 3). The extensively studied glucosylceramidase beta (GBA1) gene exemplifies this principle. Classically associated with Gaucher disease (GD), a rare AR lysosomal storage disorder (LSD), GBA1 mutations are now known to be a common genetic risk factor for Parkinson disease (PD) (2). The notion that GBA1-mutation positive PD clinically, pathologically, and pharmacologically mirrors idiopathic PD triggered a generalised hypothesis of shared genetic and pathophysiological mechanisms behind PD and LSDs (2). Indeed, beyond GBA1 and other LSD gene variants, there is growing recognition of a more widespread association between rare AR diseases and common NDDs. Research to clarify this has recently gathered significant momentum; mutations in GRN, TREM2, EIF2AK3, and several mitochondria function genes (CLN8, LMBRD1, MPI, MRPS34, and MUC1) determine strikingly distinct allele-dependent clinical phenotypes.
We propose that: (i) this phenomenon is likely to occur frequently; (ii) progress with large-scale genome/whole exome sequencing will uncover further examples, spanning numerous diseases and causative genes, and (iii) annotation with respect to recessive disease enables meaningful variant interpretation. Unbiased approaches are required to identify rare gene variants, typically missed in genome-wide association studies (GWAS) which focus on common (occurring with a minor allele frequency of >1 or 5%) variants. Diverse, transethnic study cohorts are ideal to account for variations in carrier frequencies of AR disease gene alleles. Caution is warranted when presuming pathogenicity of rare variants, many of which may have no effect on protein function and disease risk (4).
In this Review, we explore the overlap between AR disease mutations and later onset NDDs, with key genes selected to showcase this genetic convergence. Facilitating the evolution toward personalised medicine, such a paradigm holds the promise of augmenting pre-existing knowledge of NDD pathogenesis, improved patient stratification in clinical trials and boosting the development of targeted disease-modifying therapies. A greater understanding of this link will enable clinicians to offer the most accurate counselling to carriers of such disease genes. This Review presents multiple strands of evidence to confirm this association, including phenotypic assessments of AR disease patients and obligate carriers, overlapping neuropathological features, animal models, and unbiased case-control genetic analyses. Here, we specifically focus on the interplay between AR LSDs, neuroinflammatory and mitochondrial disorders, and NDD risk in risk allele carriers.
The GBA1 gene located on chromosome 1q21-22, includes 11 exons and 10 introns. GBA1 encodes β-glucocerebrosidase (GCase), a lysosomal enzyme involved in sphingolipid metabolism catalysing the hydrolysis of glucosylceramide (GlcCer) into glucose and ceramide (Table 1). Biallelic GBA1 mutations result in GD, characterised by GlcCer-laden macrophages in visceral tissue and clinical presentations of hepatosplenomegaly, pancytopenia, bone disease, and neurological deficits (5). Former GD classifications comprised 3 types, non-neuronopathic (type 1) and neuronopathic (acute, type 2, and chronic, type 3) (5, 6). However, the notion of a spectrum of GD phenotypes, with varying degrees of severity, is now widely accepted (5).
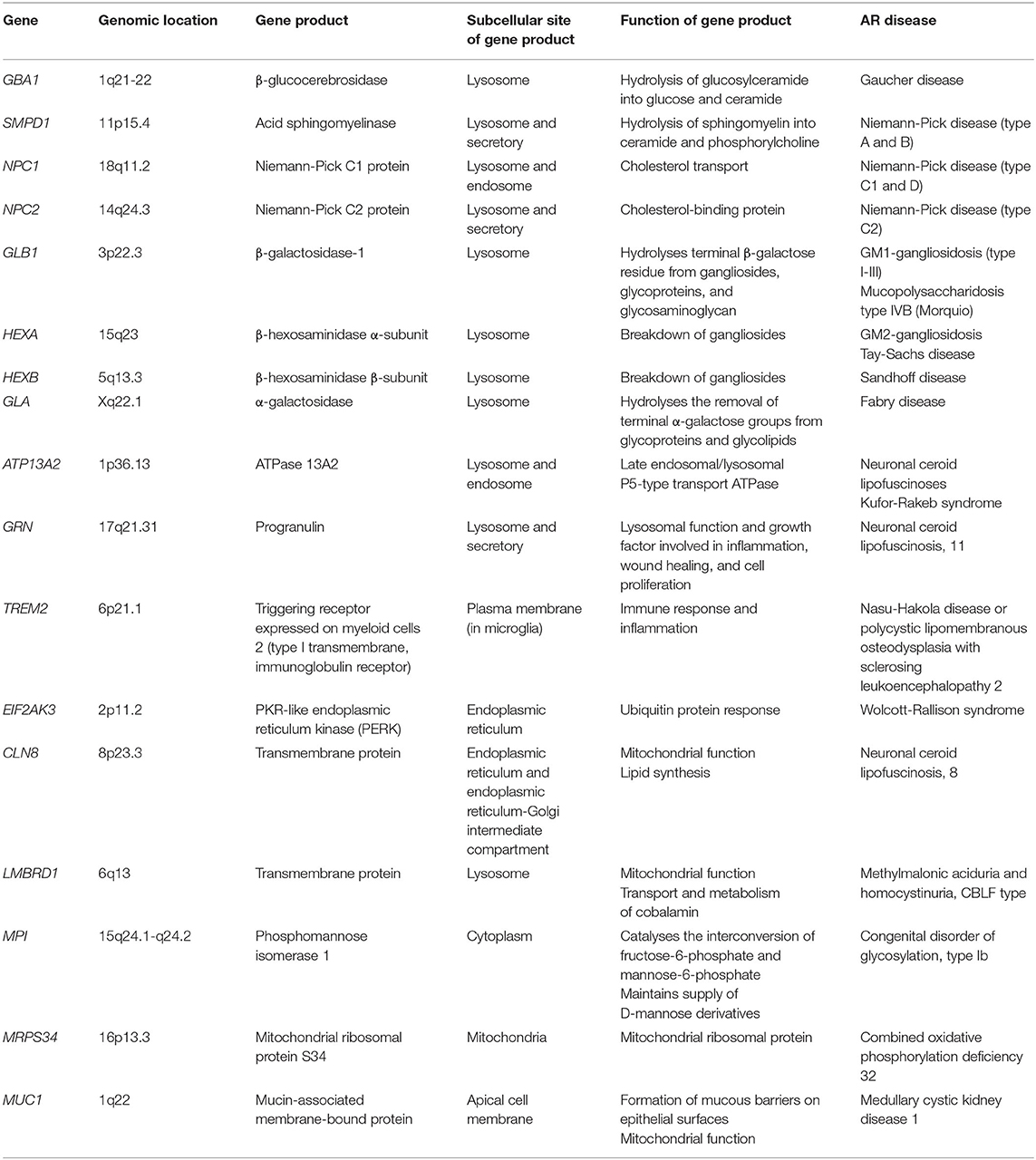
Table 1. Structure and function of genes involved in both AR disease and neurodegeneration.
To date, at least 495 GBA1 mutations responsible for GD have been identified, encompassing point mutations, insertions, deletions, frameshifts, and recombinant alleles (6). The carrier frequency of GBA1 mutations is significantly higher among the Ashkenazi Jewish (AJ) population (1 in 14–18) compared to the non-AJ population (<1%) (2, 7). Mutation nomenclature has been recently updated to include the 39-amino-acid leader sequence (newer numbering of the mutated amino acid is shown in parentheses). Five mutations are particularly prevalent, namely p.N370S (p.N409S), p.L444P (p.L483P), IVS2+1G>A, c.84GGIns, and RecNciI (the non-homologous recombination between the functional GBA1 gene and its pseudogene, GBAP) (6). Genotype-phenotype correlations have been performed; N370S (p.N409S) and L444P (p.L483P) homozygosity are mainly associated with mild type 1 GD and severe neurological involvement, respectively (5).
Both biallelic and heterozygous GBA1 mutation carrier status, the latter typically considered benign, confer an increased risk for developing PD. Recognition of this association began in the clinic, with case reports describing parkinsonian symptoms in type 1 GD patients (8), and later in obligate and confirmed GBA1 carriers (9, 10). Postmortem examinations revealed prominent α-synuclein-positive inclusions (Lewy bodies, LB) in the brains of GD patients and GBA1 carriers, a pathological hallmark of PD (11, 12). Follow-up large-scale, multicentre analyses confirmed the presence of GBA1 mutations in 4–15% of PD patients (up to 31.3% in PD AJ cohorts), increasing the lifetime risk of developing PD by up to 20-fold (2). There is no significant difference in PD risk between heterozygous and biallelic GBA1 mutation carriers (13). For GD-causing mutations, PD risk correlates with the predicted severity of GD: a recent meta-analysis reports that mild and severe GBA1 mutations have an odds ratio of 2.2 and 10.3, respectively (14). Carriers of severe GBA1 mutations who developed PD have an earlier age-at-onset and accelerated rates of dementia than those harbouring mild mutations (14, 15). Non-pathogenic GBA1 polymorphisms in GD patients, p.E326K (p.E365K) and T369M (p.T408M), are also significantly associated with PD, highlight the complexity of the GD/PD relationship (16, 17).
GBA1-associated PD (GBA1-PD) closely resembles typical sporadic PD. GBA1-PD patients exhibit the classic triad of resting tremor, bradykinesia, and rigidity. Distinct characteristics of GBA1-PD patients include: younger age of disease onset (by 4–5 years on average); reduced rigidity and an increased frequency of non-motor symptoms, such as cognitive impairment and hyposmia, compared to idiopathic PD patients (7, 18, 19). Of interest, a recent 6-year longitudinal study confirmed increased rates of such prodromal non-motor features in GBA1 mutation carriers without PD, reporting a significant deterioration in scores for depression, cognition, olfaction, and rapid eye movement (REM) sleep behaviour disorder (RBD) (20). Such prodromal abnormalities are notably identical to those with idiopathic PD. Further follow-up of healthy GBA1 mutation carriers could enable early diagnoses in those progressing to clinical PD. Recently, unaffected GBA1 carriers have been identified to have microglial activation before the development of overt features of PD (21). The longitudinal clinical and biochemical characterisation of this unique patient cohort may discern biomarkers indicative of PD phenoconversion, prior to significant irreversible neurodegeneration.
A major challenge remains to understand the mechanism by which single GBA1 mutations lead to PD, and how closely this recapitulates the pathogenesis of GD. Several hypotheses have been proposed including: (1) a loss-of-function model characterised by GCase deficiency and its subsequent effect on GlcCer accumulation, lipid homeostasis and α-synuclein degradation, (2) gain-of-function of mutant GCase enhancing α-synuclein aggregation or preventing its degradation via autophagy or the ubiquitin-proteasome pathway, and (3) the GCase/α-synuclein bidirectional positive feedback loop, in which reduced GCase activity leads to an accumulation of α-synuclein and α-synuclein accumulation further contributes to a decrease in GCase activity, leading to a self-propagating disease (Figure 1) (22). Interestingly, trafficking of mutant GCase (typically sequestered in the ER) to the lysosome reduced ER stress and reverse locomotor deficits in Drosophila with mutant GBA1 (23), leading to the subsequent analyses of small molecule chaperones in clinical trials (24). Such work highlights the importance of GCase dysfunction in PD pathogenesis, however all proposed pathogenic mechanisms have significant limitations. Null mutations, c.84GGIns, IVS2+1G>A and p.R359X (p.R398T), do not encode the GCase protein but yet have been reported in PD, a finding conflicting with the gain-of-function theory. Similarly, incomplete penetrance of PD in GBA1 mutation carriers, despite being associated with GCase deficiency, poses a key challenge to the loss-of-function hypothesis. The rate of PD phenoconversion has been reported as 10–30% before age 80, highlighting the notion that GCase deficiency is by no means predictive of PD (25, 26). Further, modifying factors, such as Cathepsin B, play an important role in PD phenoconversion (27). Interestingly, the mutual exclusivity of the aforementioned hypotheses has been challenged. It is possible that the pathogenic pathways may overlap, with evidence indicating that both gain and loss-of-function mechanisms may contribute to PD pathogenesis (28).
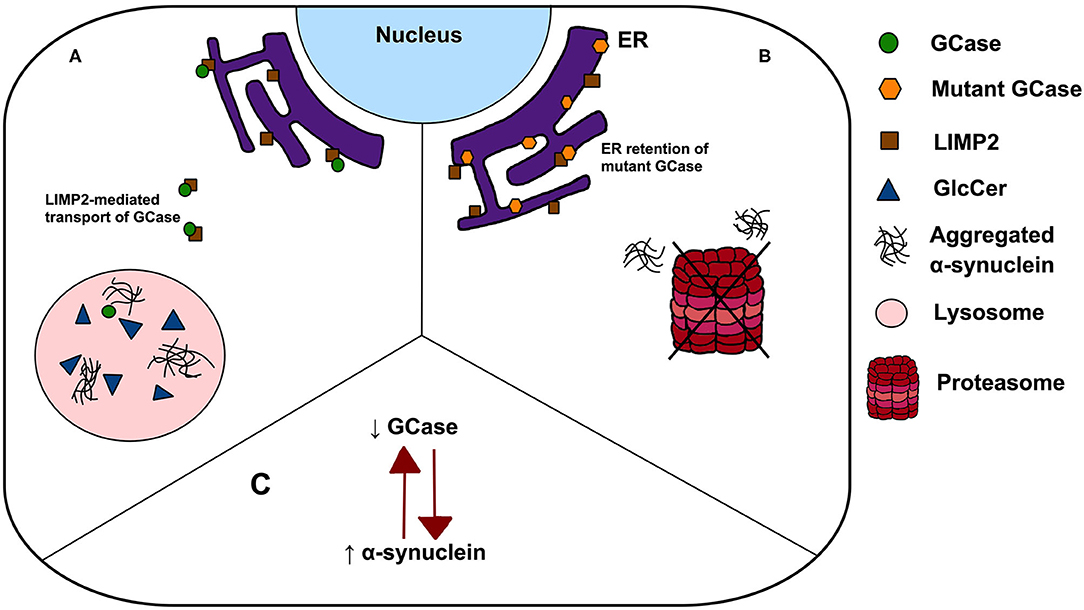
Figure 1. Possible mechanisms linking GBA1 mutations to PD. (A) The loss-of-function theory postulates that In GBA1 mutation-positive patients, deficient GCase results in GlcCer accumulation, lipid dyshomeostasis and α-synuclein aggregation; (B) the gain-of-function model states that ER retention of mutant GCase leads to ER stress, enhancing α-synuclein aggregation and prevent its degradation via autophagy or the ubiquitin-proteasome pathway and (C) a bidirectional positive feedback loop, in which reduced GCase activity leads to an accumulation of α-synuclein and α-synuclein accumulation further contributes to a decrease in GCase activity.
Synucleinopathies are a diverse group of NDDs characterised by the deposition of inclusions composed of aggregated insoluble α-synuclein, in the form of amyloid fibrils, within the central nervous system. PD, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) are the most common synucleinopathies. A higher-than-expected GBA1 mutation burden has been reported among DLB patients compared to control subjects, ranging in frequency from 3 to 34% depending on population heterogeneity, variations in sequencing methods and clinical diagnostic criteria (29, 30). Analyses of neuropathologically confirmed DLB concur with such findings (31). Indeed, a comprehensive, multicentre study revealed an odds ratio of 8.3 for GBA1 carrier status in DLB, suggesting that the role of GBA1 mutations in the genetic aetiology of DLB may even surpass that of PD (32). Analogous to GBA1-PD, GBA1-mutation positive DLB patients demonstrate subtle phenotypic differences compared to their sporadic counterparts, notably an earlier age of onset and greater prevalence of RBD (30). Overall, there is strong support for the notion that GBA1 mutations exert a large effect on susceptibility for a spectrum of synucleinopathies.
Conflicting evidence has emerged regarding the association between GBA1 and MSA. Sequencing of GBA1 coding regions and flanking splice sites in 969 MSA patients (in diverse populations–574 Japanese, 223 European, and 172 North American) revealed a GBA1 mutation carrier frequency of 1.75% compared to 0.73% in control subjects (33). Albeit modest, such mutations were reportedly associated with the cerebellar variant of MSA, notably a more prevalent clinical variant in Asian populations (33). Further investigations have yielded negative results, indicating no association at all (34, 35). Such findings highlight a distinct pathogenesis for MSA, a disease in which α-synuclein aggregates in oligodendroglial cytoplasmic inclusions rather than neurones. Moreover, preliminary work failed to show an association between GBA1 mutations and other parkinsonian disorders, progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD), which primarily involve the deposition of tau rather than alpha-synuclein (35). Given the difficulty with genetic power in rare diseases, large-scale multicentre consortia will be required to generate large patient cohorts to fully elucidate the pathogenic contribution of GBA1 mutations.
A reliable and validated biomarker is an urgent unmet need in PD to improve diagnostic accuracy, enable accurate disease monitoring and prognostic predictions. Biomarkers for early detection of PD phenoconversion in GBA1 mutation carriers prior to significant neuronal loss are required for maximal therapeutic benefit. There are inconsistent reports of a decrease in blood GCase activity in GBA1-PD and idiopathic PD patients compared to controls (36–39). Interestingly, Cerri et al. (40) found that plasma exosomal/total α-synuclein ratio inversely correlates with GCase activity and disease severity in idiopathic PD patients, replicated in subsequent studies (41). CSF GCase activity is reduced in PD patients independent of their GBA1 mutation carrier status (42). Further, a combination of multiple markers comprising GCase activity has emerged as a more accurate diagnostic tool for PD. Lysosomal markers (GCase, β-hexosaminidase, cathepsin D), total α-synuclein and AD-associated protein Aβ42 demonstrated adequate diagnostic accuracy (sensitivity 84%, specificity 75%) in samples from the PD BioFIND cohort (42). GCase activity, oligomeric/total α-synuclein ratio and age distinguished PD from neurological controls with a sensitivity of 82% and specificity of 71% (43).
Previously limited to symptomatic therapies enhancing dopaminergic neurotransmission, the discovery of GBA1 mutations as key aetiological players in PD has driven a novel therapeutic approach. Targeting the GCase/α-synuclein pathway could be significant for GBA1-PD patients, asymptomatic GBA1 mutation carriers at risk of PD phenoconversion and even idiopathic PD patients, where GCase deficiency has also been observed (44).
There are a variety of therapeutic approaches to LSDs particularly GD, including enzyme replacement therapy (ERT), substrate reduction therapy (SRT) and chaperone treatment to enable trafficking of misfolded protein. ERTs are in development and licenced for use in LSDs. If the link between LSD carrier status and late onset NDDs relates to partial enzyme deficiency, enzyme replacement would be a logical and effective treatment/ preventative strategy. However, as outlined above it is not clear that there is a direct relationship between partial enzyme deficiency and late onset NDDs but potentially this may be a future therapeutic approach. Indeed, modulating GCase activity is a focal point for current investigations of disease-modifying therapies in PD.
The inhibitory small-molecule chaperone (SMC), Ambroxol, binds to the active site of mutant GCase, mobilising sequestered mutant GCase from the endoplasmic reticulum (ER) to the lysosome and thus facilitating the normal action of GCase in GlcCer hydrolysis. Promising preclinical data has been reported in two clinical trials assessing the efficacy of Ambroxol in PD and PD dementia patients (ClinicalTrials.gov identifiers: NCT02941822 and NCT02914366, respectively). The Phase IIA prospective, open-label AiM-PD trial reported a modulatory effect on cerebrospinal fluid (CSF) GCase and α-synuclein (24). Notably, two further Phase II trials are due to commence aiming to assess the clinical efficacy of ambroxol on the cognitive, neuropsychiatric and functional outcomes in DLB patient cohorts (ClinicalTrials.gov identifier: NCT04405596 and NCT04588285). Non-inhibitory SMC LTI-291, which modulates GCase post-translational folding by binding to sites other than the active site, is being evaluated in GBA1-PD patients by Lysosomal Therapeutic Inc. (Netherlands Trial Register: NTR6960 and NTR7299). Targeting glycosphingolipid accumulation via GlcCer synthase antagonists (substrate reduction), a successful treatment for GD, offers an alternative therapeutic strategy in PD. The GlcCer synthase inhibitor, venglustat (GZ/SAR402671), is safe and well-tolerated in GBA1-PD patients but did not meet its trial's primary endpoint in PD patients and development has been halted as of January 2021 (ClinicalTrials.gov identifier: NCT02906020) (45). The therapeutic potential of GlcCer synthase inhibitors has been the source of much debate with emerging investigations reporting no sphingolipid accumulation in idiopathic PD and GBA1-PD brains (46, 47). Further, GBA1-targeted gene therapy as a means to enhance GCase activity is also under investigation. The ongoing PROPEL study, due to be completed in 2027, is evaluating the therapeutic use of intracisternal administration of gene therapy candidate, PR001A, in GBA1-PD patients (ClinicalTrials.gov identifier: NCT04127578).
The powerful example of GBA1 provided the main rationale for broader studies of LSD gene variants in neurodegeneration. Extensive GD registries, notably the International Collaborative Gaucher Group (ICGG) Gaucher Registry, are an invaluable asset, rendering it relatively easy to perform large genetic studies and gather substantial evidence of the GBA1-PD association. Due to disease rarity, most studies of non-GBA1 LSD gene candidates have used small sample sizes, likely being underpowered to detect rare alleles or those with modest effect sizes. The implementation of aggregate burden association tests for joint analyses significantly improves statistical power (48). Employing this framework, Robak et al. (49) revealed an excessive LSD variant burden in PD patients compared to controls. An examination of 54 LSD genes in two large independent cohorts confirmed associations at the GBA1 and SMPD1 loci, alongside newly implicating ASAH1, SLC17A5 and CTSD as PD susceptibility genes (49). Biallelic ASAH1, SLC17A5, and CTSD mutations cause Farber lipogranulomatosis, Salla disease and neuronal ceroid lipofuscinosis (CLN10), respectively. Acid ceramidase (ASAH1) is involved in ceramide metabolism, specifically the degradation of ceramide into sphingosine. Further, CTSD (Cathepsin D) expression level has been implicated in α-synuclein processing and aggregation (50). Such results recapitulate findings from case reports, reinforcing the importance of lysosomal dysfunction in PD pathogenesis (49).
Multiple investigations of cellular and animal models, as well as neuropathological and phenotypical assessments of patients, point to a genetic parallelism between NDDs and predominantly-AR inherited LSDs. Determining the precise contribution of LSD genes in large population cohorts is crucial. The work by Robak et al. (49) provides evidence for a wide association between LSD genes and PD but more work is needed. Below, we summarise further evidence implicating an association between non-GBA1-associated LSDs and NDD, largely focusing on PD.
Biallelic mutations in either the SMPD1, NPC1, or NPC2 gene, which are key regulators of sphingolipid metabolism, cause the rare LSD Niemann-Pick disease (NPD). Pathological NPD hallmarks include systemic sphingomyelin accumulation. Extensive phenotypic variability is observed, ranging from psychomotor retardation to visceral organ abnormalities (such as cardiovascular disease and hepatosplenomegaly).
The SMPD1 gene, encoding lysosomal enzyme acid sphingomyelinase (ASM), has recently emerged as a PD susceptibility locus (51). Akin to GCase, ASM hydrolyses sphingolipids to ceramide (Figure 2), suggesting a shared pathogenic pathway to PD. Neuropathological examinations of ASM knockout models revealed significant abnormalities, including: neuronal accumulation of distended lysosomes; cerebellar Purkinje cell degeneration; increased α-synuclein levels and deteriorating motor function (52, 53). Further supporting this link, comprehensive genotyping analyses have identified at least 20 candidate, putative damaging SMPD1 risk alleles for PD (49, 51). Individual SMPD1 mutations exhibit differential effects on PD risk, a finding reminiscent of the variable effect of mild vs. severe GBA1 mutations (51). Following the discovery of over 100 SMPD1 mutations, an increasing need exists to define PD risk amongst carriers of such mutations (54).
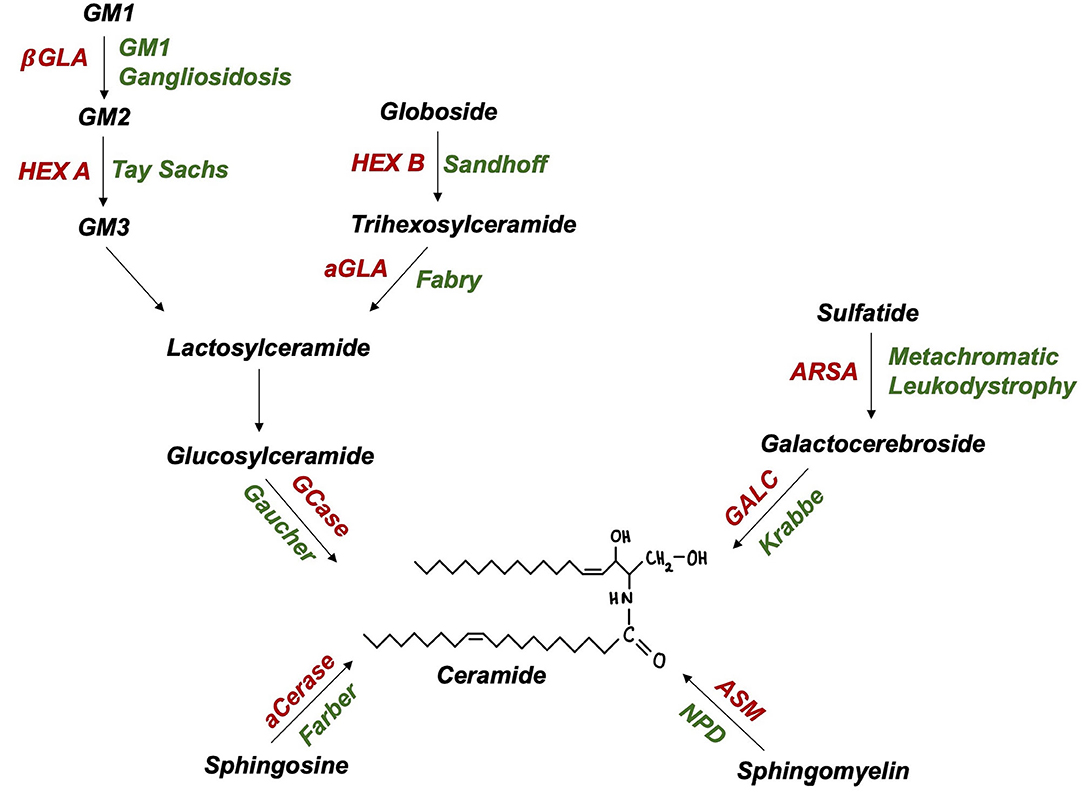
Figure 2. Ceramide and glycolipid metabolism in lysosomes (enzymes in red, autosomal recessive disease in green): aCerase, acid ceramidase; ASM, acid sphingomyelinase; ARSA, arylsulfatase; GALC, galactocerebrosidase; GCase, glucocerebrosidase; aGLA, a-galactosidase; βGLA, β-galactosidase; HEX A/B, hexosaminidase A/B; NPD, Niemann-Pick disease.
NPC heterozygosity may predispose to late-onset neurodegeneration. PD, parkinsonism, atypical parkinsonism (e.g., PSP and CBD), and tremor disorders have been observed in heterozygous NPC1 and NPC2 mutation carriers (55–58). Non-motor features including impaired olfaction, urinary urgency, constipation, impotence, orthostatic hypotension, and sleep disturbance were also present (55). Further studies have either failed to support the link between heterozygous NPC1 mutations and PD (59) or support the notion of its genetic influence beyond movement disorders with reports indicating a pathogenic role in Alzheimer's disease (AD) (60).
Biallelic GLB1 mutations are pathogenic for GM1 gangliosidosis, an AR LSD characterised by β-galactosidase deficiency and ganglioside substrate accumulation within lysosomes (Figure 2). Adult-onset GM1 gangliosidosis patients display increased rates of parkinsonism, ranging from 7.5 to 48% (61, 62). GM1 deficiency was also reported in degenerating dopaminergic neurones of the substantia nigra from idiopathic PD brains (63). The administration of long-term GM1 ganglioside therapy in PD patients resulted in lower rates of symptom progression (64). Indeed, altered GM1-mediated interactions between lipid rafts and α-synuclein may contribute to the formation of pathogenic, oligomeric α-synuclein (65). Further, GM1 ganglioside metabolism imbalances have also been reported in AD and Huntington's disease (66). The rarity of GM1 gangliosidosis and thus, heterozygous GLB1 mutation carriers, poses a critical challenge to researchers attempting to achieve this and may explain recent failures to find a GLB1-PD association in genetic case-control studies (49).
Tay-Sachs and Sandhoff disease (GM2 Gangliosidoses) are LSDs caused by biallelic HEXA and HEXB mutations, encoding lysosomal enzyme β-hexosaminidase α-subunits and β-subunits, respectively (Figure 2). PD-like syndromes, including bradykinesia, tremor, and rigidity have been observed in both GM2 gangliosidosis patients and their relatives (67, 68). Neuropathological examinations revealed enhanced α-synuclein deposition in the brains of GM2 gangliosidosis patients (69). In parallel, PD patients exhibit a reduction in CSF β-hexosaminidase activity (42). A biochemical association between PD and GM2 gangliosidosis is further supported by findings of reduced ubiquitin C-terminal hydrolase (UCH-L1) levels in cultured fibroblasts and brain extracts of Sandhoff disease and disease carrier mouse models (70). Together such pathological evidence highlights the importance of lysosomal function in PD however, investigations of the genetic basis of this association are yet to be conducted.
Recent studies on α-galactosidase A (GLA) deficiency, causing the rare X-linked LSD Fabry disease, suggest there may be an increased risk of developing PD in GLA mutation-positive individuals (71). Reduced GLA activity has been reported in dried blood spots and leukocytes collected from idiopathic PD patients compared to healthy controls (72, 73). Stratified analysis by gender revealed no significant difference in GLA activity in male idiopathic PD patients compared to controls, contradicting findings observed in female PD patients (72). Following case reports of parkinsonism in Fabry patients (74), Wise et al. (71) conducted an online survey and family history questionnaire of 90 Fabry patients, finding that 2 patients had received a PD diagnosis (risk of 11.1% by 70 years of age) and a significant family history of PD was present (7.4% had one first degree relative clinically diagnosed with PD). High prevalence rates of PD in late-onset phenotype of Fabry have been subsequently replicated (75). The mechanism by which reduced GLA activity is associated with abnormal α-synuclein aggregation remains unknown.
The neuronal ceroid lipofuscinoses (NCLs) are a heterogenous group of predominantly-AR inherited disorders, characterised by progressive degeneration of the brain and retina. Clinical manifestations are diverse, encompassing retinopathy, epilepsy, motor, and cognitive deficits. Extensive deposition of autofluorescent storage material lipofuscin pathologically defines this disease. With NCL-pathogenic mutations now identified in 13 different genes, an increased understanding of the molecular basis of NCLs has generated compelling evidence of an overlap between altered NCL gene function and NDDs. Highlighting this concept is the recent discovery that biallelic loss-of-function progranulin (GRN) gene mutations cause NCL whereas heterozygous loss-of-function mutations cause frontotemporal dementia (FTD, discussed later). Rare variants in the NCL gene MFSD8 are also candidate risk factors for FTD (76).
Mutations in ATP13A2 are associated with NCL and Kufor-Rakeb syndrome, a rare juvenile-onset form of PD. Clinical features of parkinsonism are well-documented in the later stages of CLN3-associated juvenile NCL (77), showing improvement post levodopa treatment (78). Functional evidence for nigrostriatal pathology, indicative of PD, revealed reduced striatal dopamine transporter density in the putamen and to a lesser extent in the caudate nucleus of juvenile NCL patients (77). Moreover, Atp13a2−/− mice exhibit overlapping pathological signatures of NCL (intraneuronal lipofuscin accumulation) and PD (aggregated insoluble α-synuclein) in the hippocampus (79).
The GRN gene, situated on chromosome 17q21.31, contains 13 exons and encodes the 88-kDa glycoprotein progranulin (PGRN). Ubiquitously expressed, PGRN is implicated in multiple processes including neuroinflammation, tumorigenesis, and lysosome biology. PGRN rose to prominence in the neuroscience community in 2006 following the emergence of two landmark studies causally linking heterozygous loss-of-function GRN mutations to familial FTD (80, 81). FTD encompasses a clinically and pathologically heterogenous group of conditions associated with selective atrophy of the frontal and temporal lobes of the brain, comprising the second leading cause of early-onset dementia (82). Interestingly, GRN mutations account for 3–26% of familial FTD and perhaps as much as 5% of sporadic cases (82, 83). GRN mutations lead to mis-localised TDP-43 pathology, in common with other genetic and sporadic forms of FTD (84).
Since it was first associated with FTD, over 110 pathogenic GRN mutations have been described (85). The AD&FTD Mutation Database (http://www.molgen.ua.ac.be/FTDmutations) lists most of these mutations (85). GRN haploinsufficiency is thought to underlie GRN-associated FTD pathogenesis, with heterozygous GRN mutations resulting in a loss-of-function, owing to (1) mutant mRNA clearance by nonsense-mediated decay, (2) prevention of mRNA translation due to missense or splice site mutations of the initiator methionine codon and (3) production of non-functional and unstable protein (81, 86). The net effect is a concomitant reduction of ~50% in PGRN levels, with some estimates at 70–80% (87). Epigenetic modifications may contribute to the lower-than-expected PGRN expression in heterozygous GRN mutation carriers (88). GWAS identified lysosomal transmembrane protein 106B (TMEM106B) variants as genetic modifiers of PGRN expression (89). Carriers of the TMEM106B rs1990622 protective minor allele exhibit elevated GRN plasma levels and delayed age of FTD onset (90), although others have contested such findings (91). TMEM106B variants may also account for the age-dependent and incomplete penetrance of GRN-associated FTD (GRN-FTD), estimated as 50–60% at 60 years and ~90% by age 70 years (81, 92).
Phenotypic characteristics of GRN-FTD are highly variable; intrafamilial heterogeneity in clinical presentation is frequently observed (93). GRN mutation carriers exhibit considerable variation in age of symptom onset, ranging from 35 to 89 years (94). Consecutive generations of the same family display a >10–20 year difference in age of disease onset (94). Neither disease duration nor age of onset correlate with GRN mutation status, type, or location (95). Prominent clinical features of GRN-FTD include behavioural variant FTD subtype (bvFTD), significant apathy and social withdrawal (80, 81). Language dysfunction consistent with progressive non-fluent aphasia may occur in the initial stages of disease (93). The occurrence of extrapyramidal features has been noted in ~40% of GRN-FTD patients, including dystonia, asymmetric parkinsonism, and CBS with limb apraxia, typically unresponsive to levodopa treatment (93). Further, delusions and hallucinations present more frequently, occurring in up to 30% of FTD patients harbouring GRN mutations (96). Characteristic neuroimaging features of GRN-FTD brains consist of early parietal involvement, white matter hyper-intensities in atrophied regions and accelerated rates of asymmetric brain atrophy (93, 97). GRN mutations are pathologically associated with FTLD-TDP subtype A, characterised by neuronal cytoplasmic inclusions and irregular dystrophic neurites in neocortical layer 2 (84). Notably, TDP-43 inclusions are not specific for FTLD, also being present in motor neurones of amyotrophic lateral sclerosis (ALS) patients, consistent with the FTD-ALS spectrum (82).
Illustrating pleiotropy, while heterozygous GRN mutations are pathogenic for FTD, loss of both alleles results in a separate disease entity, NCL with GRN designated as CLN11 (3). Interestingly, GRN mutations were first associated with a late onset NDD and only several years later with their corresponding AR disease. Indeed, the unanticipated 2012 discovery of homozygous c.813_816del GRN mutations in siblings with NCL (3), and later reports of co-occurrence of FTD and NCL within a single family (98), adds further complexity to the GRN-FTD association. Subsequent observations of NCL-like pathobiochemical features in FTD patients with GRN mutations indicates a greater role of lysosomal dysfunction in FTD pathogenesis (99). The involvement of TMEM106B and charged multivesicular body 2B (CHMP2B) in FTD, both with key functions residing in the lysosome, strengthen this association (82).
Targeting PGRN haploinsufficiency represents a potential therapeutic avenue in GRN-FTD. Identifying presymptomatic cohorts suitable for treatment is facilitated by the specificity and sensitivity of PGRN plasma levels as a predictor of GRN-null mutations, and the significant lag (5–10 years) between the onset of neuropathological changes and symptoms (100, 101). Multiple recent trials for FTD disease-modification target PGRN deficiency. Histone deacetylase inhibitor FRM-0334 demonstrated adequate CSF penetration and increased PGRN expression in preclinical models (102). FRM-0334 failed to result in significant changes in PGRN concentrations in 28 subjects with GRN mutations in a recent Phase II trial (103), perhaps due to inadequate FRM-0334 exposure (ClinicalTrials.gov identifier: NCT02149160). Evaluations of the safety, tolerability and efficacy of anti-sortilin monoclonal antibody AL001 in GRN-FTD patients are underway in a Phase II trial (INFRONT-2, ClinicalTrials.gov identifier: NCT03987295), and as of July 2020, a Phase III trial (INFRONT-3, ClinicalTrials.gov identifier: NCT04374136). Further, the use of calcium-channel blocker nimodipine as a treatment for progranulin insufficiency from GRN mutations has been explored (ClinicalTrials.gov identifier: NCT01835665). An 8-week, open-label, dose-finding study revealed no significant alterations in CSF and plasma PGRN concentrations post nimodipine treatment (104). Moreover, alternative approaches under investigation include alkalizing compounds (namely, chloroquine, and amiodarone) and gene therapy to reverse GRN haploinsufficiency (105–107). Multicentre Phase I/II studies are currently investigating the safety, tolerability and efficacy of intra-cisternal adeno-associated viral vector administration of PR006 (PROCLAIM study, ClinicalTrials.gov identifier: NCT04408625) and PBFT02 in GRN-FTD patients (upliFT-D study, ClinicalTrials.gov identifier: NCT04747431). Endpoint outcomes include blood and CSF PGRN measurements, with trial completion expected in 2027.
Heterozygous GRN loss-of-function mutations have been confirmed in clinically diagnosed AD patients (108). Indeed, the T allele of single nucleotide polymorphism (SNP) rs5848 located in the 3′ untranslated region of GRN particularly contributes to an increased AD risk (109), known to reduce GRN mRNA levels in the brain, plasma, and peripheral mononuclear cells (110, 111). Indeed, an inverse correlation exists between AD risk and serum PGRN levels, with AD patients homozygous for the T allele of rs5848 exhibiting the lowest PGRN levels (112). Increased miR-659 binding may underlie such translational inhibition of GRN (112). Further, GRN mutations were also reported in CBD and ALS, albeit evidence for the latter has been rather inconsistent (113, 114). Large-scale studies, pathological verification of diagnosis and meta-analyses are needed to confirm the wider pathogenic role of GRN mutations.
AD is a genetically complex disorder, defined clinically by a progressive memory and cognitive decline, and pathologically by extracellular amyloid (Aβ) plaques and intracellular deposits of neurofibrillary tangles (NFTs). Rare, highly penetrant mutations in presenilin-1/2 (PSEN1/2), amyloid precursor protein (APP) and the ε4 allele of Apolipoprotein E (APOE) impart a notable risk for late-onset AD (115). Recent genomic studies revealed rare heterozygous TREM2 variants as novel genetic risk factors for the disease, offering new keys to decipher AD pathogenesis (115, 116).
TREM2, a gene located on chromosome 6p21.1, comprises 5 exons and encodes a transmembrane receptor required for myeloid (or microglial) inflammatory response. To date, over 60 coding TREM2 variants have been identified, displaying various degrees of population frequency (117). The Alzforum Mutation database provides an up-to-date registry of all potentially pathogenic TREM2 variants (https://www.alzforum.org/mutations). The most well-studied TREM2 variant p.R47H (rs75932628) was found to confer a 2- to 4-fold increase in AD risk, a finding independently replicated in numerous North-American and European populations (118, 119). Albeit the p.R47H-AD association failed to be confirmed in several East Asian and other cohorts, perhaps due to diverse population-specific TREM2 allele frequencies (120, 121). Rare TREM2 variants other than p.R47H are more frequently found in non-Caucasian populations. Reports indicate an increased frequency of novel variants, p.A130V, p.A192T, p.H157Y, and p.S183C in Chinese AD patients (122–124). Allelic heterogeneity was confirmed following the identification of p.W191X and p.L211P variants in African American AD subjects, prompting investigations of different ethnic groups for disease risk variant discovery (120). It is not clear how TREM2 variants increase an individual's AD risk. In vitro studies aiming to elucidate the biochemical effects of such variants report decreased TREM2 stability, altered interactions with lipoprotein ligands and phagocytic functions (125–127). Contrastingly, analyses of AD brains have yielded somewhat inconsistent results, with TREM2 variants demonstrating no alterations in structure, stability, expression, or ligand affinity (127–129). Nevertheless, TREM2 variants lead to an impairment of microglial recruitment and response to Aβ plaques, resulting in exacerbation of AD neuropathology. Two loss-of-function mechanisms have been proposed indicating that TREM2 signalling is required to (1) drive the neuroprotective transformation of microglia (130), and (2) sustain microglial cellular energetic and biosynthetic metabolism, allowing their response to stressors (131). Further investigations of TREM2-associated AD patient-derived induced pluripotent stem cells and animal models expressing such risk variants should clarify the precise mechanism of the TREM2-AD association.
Assessments of the association between particular AD endophenotypes and TREM2 variants are yet to reach definitive conclusions, hindered by the small sample size of TREM2 variant carriers. In general, heterozygous TREM2 p.R47H AD patients typically demonstrate clinical, pathological and neuroimaging features indistinguishable from idiopathic AD (132, 133). An earlier age of AD onset (by ~5 years) and accelerated disease progression have been reported in TREM2 p.R47H carriers, albeit others contest such findings (132, 133). Other variants within or adjacent to the TREM locus, namely TREM1 intronic variants rs6910730 and rs7759295, may augment (134) or reduce (135) the rate of cognitive decline in AD. Clinically, partial support exists for an increased incidence of psychiatric symptoms and parkinsonian signs, particularly evident in initial stages of the disease (136). Moreover, European AD patients harbouring TREM2 p.R47H variants display elevated levels of CSF total and phosphorylated tau protein, whereas Aβ42 levels were left unaffected (137).
The mechanism by which biallelic loss-of-function mutations in the same gene TREM2, are pathogenic for the very rare Nasu-Hakola disease (NHD), or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, poses an interesting conundrum. NHD is a rare condition associated with spontaneous fractures, multifocal bone cysts and early-onset dementia. NHD can also be caused by biallelic TYROBP mutations, with reports speculating a role for TYROBP mutations in AD (138). Notably, the TREM2 R47H variant is associated with AD but not a causative NHD allele, striking similarities with the GBA1 E326K gene and highlighting mechanistic divergence. Evaluations of several NHD patients by neuroimaging and functional nuclear imaging (99mTc-ECD SPECT) tests revealed key abnormalities including: marked cortical hypometabolism; Aβ deposition in the grey matter of the inferior frontal and occipital lobes, and hypoperfusion of the basal ganglia with concomitant visuospatial memory deficits (139, 140). Immunohistochemistry analyses were less supportive of AD pathology in NHD brains; reports of almost undetectable Aβ plaques and only a small number of NFT-bearing neurones imply that loss-of-function of TREM2 may not exacerbate AD pathology in NHD (141).
Further work is required to determine how TREM2 variants influence AD risk and cause NHD. The clinical implications of such variants are potentially significant. Intriguing findings of CSF soluble TREM2 levels as a dynamic marker of microglial activity and thus AD progression, indicate the need to validate its use as a biomarker (142). Additionally, TREM2 may represent a potential therapeutic target in AD. In vivo elevation of TREM2 expression confers a rescuing effect, notably promoting microglial survival, reducing amyloid accumulation, and ameliorating memory deficits in AD mouse models (143, 144).
Heterozygous and biallelic TREM2 variants have been found in FTD cases, particularly bvFTD with atypical presentations, namely seizures (145, 146). A greater severity of TREM2 dysfunction is associated with TREM2 variants pathogenic for both NHD and FTD: nonsense p.Q33X variant introduces a premature stop codon, halting TREM2 expression; p.T66M and p.Y38C variants, residing in the Ig fold, lead to deficits in TREM2 protein folding, maturation, and expression (145, 146). Initial case-control studies of the association of heterozygous TREM2 variants as genetic risk factors for FTD in the general population highlighted T96K, L211P, and R47H as significant risk variants (147). Subsequent analyses failed to replicate such findings, prompting the notion that TREM2 only significantly influences FTD risk at the gene, not variant, level (137).
Findings of TREM2 variants in other NDDs have been less definitive. The TREM2 p.R47H variant appears to confer susceptibility to ALS and PD (148, 149). Of note, underlying differences in minor allele frequency for TREM2 p.R47H in study cohorts across populations may account for the discrepant results generated in attempts to confirm this reported association (150). Further investigations of TREM2 variant burden in PSP, MSA, and LBD patients have been conducted, yielding inconclusive findings (151–153).
Emerging findings have implicated numerous genes in AR disease and neurodegeneration (see Table 2 for summary). Additional GWAS of 1,114 PSP cases and 3,247 controls revealed heterozygous EIF2AK3 rs7571971 common variants, located at the 2p11.2 locus, as significant genetic risk factors for PSP (162). In 2000, mutations in EIF2AK3 were found to be pathogenic for the rare AR Wolcott-Rallison syndrome, a form of monogenic neonatal diabetes mellitus with concomitant skeletal dysplasia and recurrent hepatitis (163). This discovery highlights intriguing insights into PSP pathogenesis. Encoding PERK, a key regulator of the ubiquitin protein response (UPR), it has been proposed that PERK dysfunction increases ER stress-mediated neuronal damage, ultimately leading to PSP. Indeed, multiple lines of evidence implicate UPR perturbation as a common pathway in NDDs. EIF2AK3 p.R240H carrier status confers increased AD susceptibility (161), especially in APOE ε4-positive subjects (164). Upregulated UPR has also been observed in PD, FTD, and ALS; intriguingly, pharmacological inhibition of PERK signalling is neuroprotective in animal models of such diseases (165–167). Such work supports the need to explore EIF2AK3 variant burden in a broad range of NDDs.
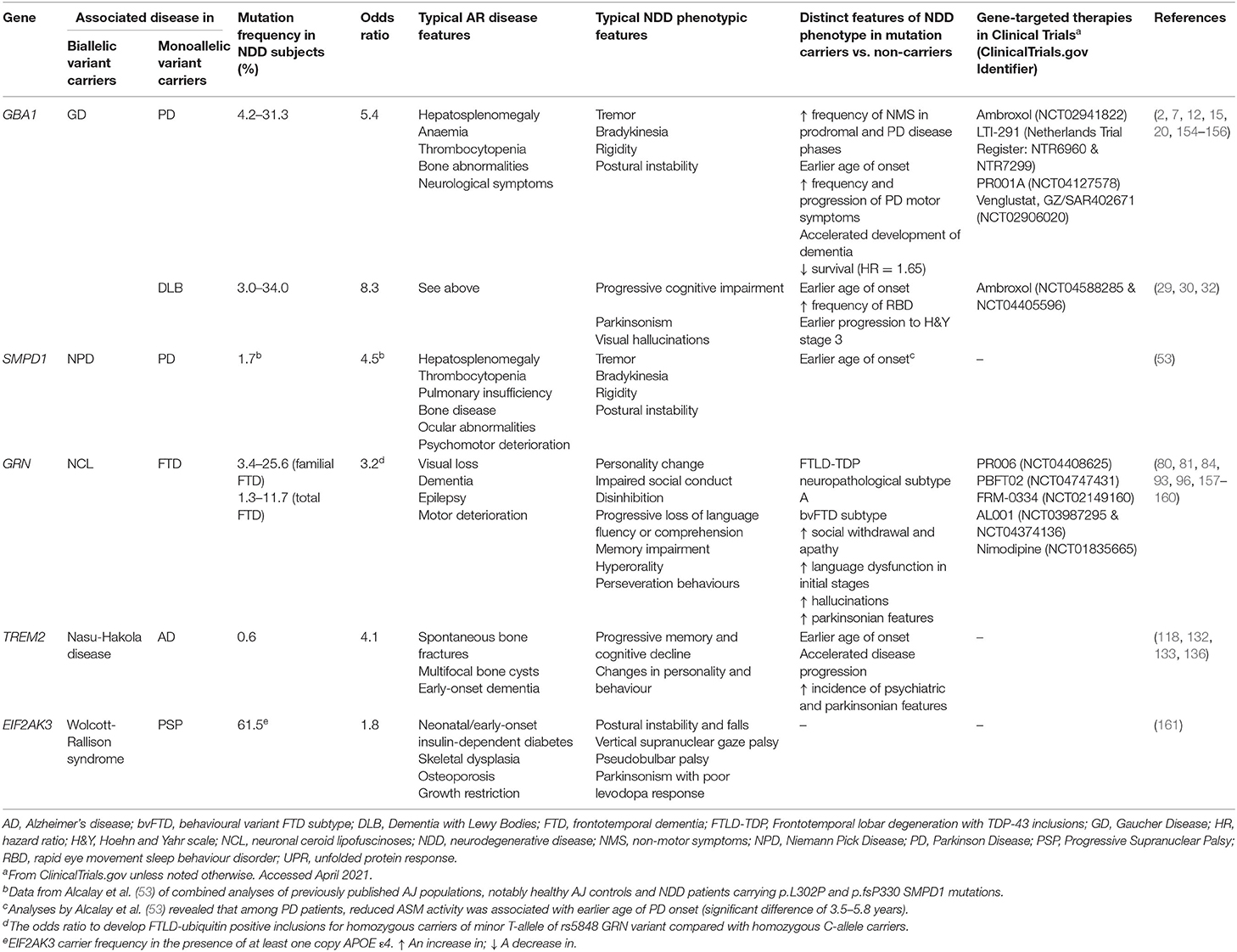
Table 2. Summary table of the main genes associated with AR disease and NDDs (genes with significant evidence supporting association selected).
More comprehensive understanding of the genetics of mitochondrial disease revealed new heterozygous gene variants, involved in mitochondria function, to be associated with PD risk and later age of disease onset (168). Robust evidence implicates the genes CLN8, LMBRD1, MPI, MRPS34, and MUC1, pathogenic for the following AR diseases, NCL8, Methylmalonic aciduria and homocystinuria, congenital disorder of glycosylation type Ib, Combined oxidative phosphorylation deficiency 32 and Medullary cystic kidney disease 1, respectively (168). It is likely further unbiased analyses may yield more genomic associations between NDDs and rare AR diseases; therapeutics targeting mitochondrial processes may be beneficial in the initial stages of PD.
Emerging evidence presented in this Review highlights a recurring theme of pleiotropic effects of biallelic and heterozygous gene mutations. Carrier status of several AR diseases, hitherto perceived as benign, are now known to confer increased NDD risk. The advent of widespread implementation of high throughput sequencing will certainly unravel further examples. Caution is required when implicating pathogenicity to the presence of de novo gene variants identified via this approach; certain variants may indeed be neutral in the general population. It is important that future case control studies in NDD incorporate our progress in understanding rare AR diseases. Well-designed case control studies will include careful matching of cases and control populations (given the variation in underlying rare allele mutation frequency) and the variants will be annotated with respect to their known AR disease biology. Annotation of rare disease gene variants is an important aspect of evaluating carrier status, clinical counselling, and attributing functional impact to variants. These may be distinct, so for the case of GBA1, two non-GD causing variants, p.E326K (p.E365K) and T369M (p.T408M), are associated with PD. It may be difficult to interpret or predict the effect of rare non-synonymous variants, as seen in GD, knowledge of their effects in AR disease may be helpful and clinical variant databases such as ClinVar may be of use here. Transethnic studies may also be important with different alleles in different populations being associated with disease.
Improved understanding of the genetic basis of NDDs creates opportunities to better elucidate disease pathogenesis and develop targeted therapeutics for genetically, and thus molecularly, defined subsets of patients. In particular, further research is required to ascertain whether the search for treatments for AR disease will benefit carriers of AR disease genes presenting with adult-onset NDDs. Substrate reduction via GlcCer synthase antagonists is one such example, already having been established as a successful therapy for GD (45). The known biology and biochemistry of AR diseases provides new insights into late onset NDD. However, the spectrum and effect of variants may be different in AR disease and NDD, as seen in GBA1 and TREM2 variants with NDD associated alleles may not be pathogenic for AR disease, highlighting mechanistic divergence. Moreover, insights gained in exploring the association between AR disease and NDDs adds greater complexity to genetic counselling, enabling heterozygote carriers of AR disease genes to be fully informed of their risk for adult-onset NDDs. We hope that research in this area will benefit both adults and children suffering from these progressive degenerative conditions.
SV: conception, organization, and execution of the research project and writing of first draft, review, and critique of the manuscript preparation. HM: conception and organization of the research project and review and critique of the manuscript preparation. Both authors contributed to the article and approved the submitted version.
This work was supported by Medical Research Council (UK)–G0700943 and G1100643, Parkinson's UK (PUK-K-1501).
HM is employed by UCL. In the last 24 months he reports paid consultancy from Biogen, Biohaven, Lundbeck; lecture fees/honoraria from Wellcome Trust, Movement Disorders Society. Research Grants from Parkinson's UK, Cure Parkinson's Trust, PSP Association, CBD Solutions, Drake Foundation, Medical Research Council, Michael J Fox Foundation. HM is a co-applicant on a patent application related to C9ORF72—Method for diagnosing a neurodegenerative disease (PCT/GB2012/052140).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Poolman EM, Galvani AP. Evaluating candidate agents of selective pressure for cystic fibrosis. J R Soc Interface. (2007) 4:91–8. doi: 10.1098/rsif.2006.0154
2. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
3. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. (2012) 90:1102–7. doi: 10.1016/j.ajhg.2012.04.021
4. Gambin T, Jhangiani SN, Below JE, Campbell IM, Wiszniewski W, Muzny DM, et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. (2015) 7:54. doi: 10.1186/s13073-015-0171-1
5. Cassinerio E, Graziadei G, Poggiali E. Gaucher disease: a diagnostic challenge for internists. Eur J Intern Med. (2014) 25:117–24. doi: 10.1016/j.ejim.2013.09.006
6. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. (2008) 29:567–83. doi: 10.1002/humu.20676
7. Beavan MS, Schapira AH. Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann Med. (2013) 45:511–21. doi: 10.3109/07853890.2013.849003
8. Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM. (1996) 89:691–4. doi: 10.1093/qjmed/89.9.691
9. Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. (2004) 41:937–40. doi: 10.1136/jmg.2004.024455
10. Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis. (2006) 36:426–8. doi: 10.1016/j.bcmd.2006.02.004
11. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. (2004) 82:192–207. doi: 10.1016/j.ymgme.2004.04.011
12. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. (2009) 132:1783–94. doi: 10.1093/brain/awp044
13. McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH. A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry. (2012) 83:853–4. doi: 10.1136/jnnp-2012-302402
14. Gan-Or Z Amshalom I Kilarski LL Bar-Shira A Gana-Weisz M Mirelman A . Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
15. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
16. Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. (2011) 20:202–10. doi: 10.1093/hmg/ddq454
17. den Heijer JM, Cullen VC, Quadri M, Schmitz A, Hilt DC, Lansbury P, et al. A large-Scale full GBA1 gene screening in Parkinson's disease in the Netherlands. Mov Disord. (2020) 35:1667–74. doi: 10.1002/mds.28112
18. Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-Related Parkinson's disease: dissection of genotype-phenotype correlates in a large Italian Cohort. Mov Disord. (2020) 35:2106–11. doi: 10.1002/mds.28200
19. Olszewska DA, McCarthy A, Soto-Beasley AI, Walton RL, Magennis B, McLaughlin RL, et al. Association between glucocerebrosidase mutations and Parkinson's disease in Ireland. Front Neurol. (2020) 11:527. doi: 10.3389/fneur.2020.00527
20. Avenali M, Toffoli M, Mullin S, McNeil A, Hughes DA, Mehta A, et al. Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J Neurol Neurosurg Psychiatry. (2019) 90:1091–7. doi: 10.1136/jnnp-2019-320394
21. Mullin S, Stokholm MG, Hughes D, Mehta A, Parbo P, Hinz R, et al. Brain microglial activation increased in Glucocerebrosidase (GBA) mutation carriers without Parkinson's disease. Mov Disord. (2021) 36:774–9. doi: 10.1002/mds.28375
22. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. (2012) 11:986–98. doi: 10.1016/S1474-4422(12)70190-4
23. Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep. (2016) 6:31380. doi: 10.1038/srep31380
24. Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. (2020) 77:427–34. doi: 10.1001/jamaneurol.2019.4611
25. Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol Dis. (2011) 46:95–102. doi: 10.1016/j.bcmd.2010.10.006
26. Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. (2012) 78:417–20. doi: 10.1212/WNL.0b013e318245f476
27. Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain. (2020) 143:234–48. doi: 10.1093/brain/awz350
28. Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. (2011) 69:940–53. doi: 10.1002/ana.22400
29. Farrer MJ, Williams LN, Algom AA, Kachergus J, Hulihan MM, Ross OA, et al. Glucosidase-beta variations and Lewy body disorders. Parkinsonism Relat Disord. (2009) 15:414–6. doi: 10.1016/j.parkreldis.2008.08.004
30. Lerche S, Machetanz G, Wurster I, Roeben B, Zimmermann M, Pilotto A, et al. Dementia with lewy bodies: GBA1 mutations are associated with cerebrospinal fluid alpha-synuclein profile. Mov Disord. (2019) 34:1069–73. doi: 10.1002/mds.27731
31. Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VM, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. (2006) 67:908–10. doi: 10.1212/01.wnl.0000230215.41296.18
32. Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. (2013) 70:727–35. doi: 10.1001/jamaneurol.2013.1925
33. Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. (2015) 2:417–26. doi: 10.1002/acn3.185
34. Pihlstrom L, Schottlaender L, Chelban V, Consortium MSAE, Meissner WG, Federoff M, et al. Lysosomal storage disorder gene variants in multiple system atrophy. Brain. (2018) 141:e53. doi: 10.1093/brain/awy124
35. Jamrozik Z, Lugowska A, Slawek J, Kwiecinski H. Glucocerebrosidase mutations p.L444P and p.N370S are not associated with multisystem atrophy, progressive supranuclear palsy and corticobasal degeneration in Polish patients. J Neurol. (2010) 257:459–60. doi: 10.1007/s00415-009-5363-4
36. Papagiannakis N, Xilouri M, Koros C, Stamelou M, Antonelou R, Maniati M, et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson's disease patients. Mov Disord. (2015) 30:1830–4. doi: 10.1002/mds.26433
37. Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson's disease. Sci Rep. (2018) 8:15446. doi: 10.1038/s41598-018-33921-x
38. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. (2015) 138:2648–58. doi: 10.1093/brain/awv179
39. Pchelina S, Emelyanov A, Baydakova G, Andoskin P, Senkevich K, Nikolaev M, et al. Oligomeric alpha-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson's disease. Neurosci Lett. (2017) 636:70–6. doi: 10.1016/j.neulet.2016.10.039
40. Cerri S, Ghezzi C, Sampieri M, Siani F, Avenali M, Dornini G, et al. The exosomal/total alpha-synuclein ratio in plasma is associated with glucocerebrosidase activity and correlates with measures of disease severity in PD patients. Front Cell Neurosci. (2018) 12:125. doi: 10.3389/fncel.2018.00125
41. Xicoy H, Penuelas N, Vila M, Laguna A. Autophagic- and lysosomal-related biomarkers for parkinson's disease: lights and shadows. Cells. (2019) 8:1317. doi: 10.3390/cells8111317
42. Parnetti L, Paciotti S, Eusebi P, Dardis A, Zampieri S, Chiasserini D, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson's disease patients. Mov Disord. (2017) 32:1423–31. doi: 10.1002/mds.27136
43. Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson's disease. Mov Disord. (2014) 29:1019–27. doi: 10.1002/mds.25772
44. Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. (2012) 72:455–63. doi: 10.1002/ana.23614
45. Peterschmitt MJ, Gasser T, Isaacson S, Kulisevsky J, Mir P, Simuni T, et al. Safety, tolerability and pharmacokinetics of oral venglustat in Parkinson disease patients with a GBA mutation. Mol Genetics Metab. (2019) 126:S117. doi: 10.1016/j.ymgme.2018.12.298
46. Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AH. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov Disord. (2015) 30:1085–9. doi: 10.1002/mds.26278
47. Boutin M, Sun Y, Shacka JJ, Auray-Blais C. Tandem mass spectrometry multiplex analysis of glucosylceramide and galactosylceramide isoforms in brain tissues at different stages of Parkinson disease. Anal Chem. (2016) 88:1856–63. doi: 10.1021/acs.analchem.5b04227
48. Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA. (2014) 111:E455–64. doi: 10.1073/pnas.1322563111
49. Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. (2017) 140:3191–203. doi: 10.1093/brain/awx285
50. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. (2009) 2:5. doi: 10.1186/1756-6606-2-5
51. Gan-Or Z Orr-Urtreger A Alcalay RN Bressman S Giladi N Rouleau GA. The emerging role of SMPD1 mutations in Parkinson's disease: implications for future studies. Parkinsonism Relat Disord. (2015) 21:1294–5. doi: 10.1016/j.parkreldis.2015.08.018
52. Macauley SL, Sidman RL, Schuchman EH, Taksir T, Stewart GR. Neuropathology of the acid sphingomyelinase knockout mouse model of Niemann-Pick A disease including structure-function studies associated with cerebellar Purkinje cell degeneration. Exp Neurol. (2008) 214:181–92. doi: 10.1016/j.expneurol.2008.07.026
53. Alcalay RN, Mallett V, Vanderperre B, Tavassoly O, Dauvilliers Y, Wu RYJ, et al. SMPD1 mutations, activity, and alpha-synuclein accumulation in Parkinson's disease. Mov Disord. (2019) 34:526–35. doi: 10.1002/mds.27642
54. Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. (2007) 30:654–63. doi: 10.1007/s10545-007-0632-9
55. Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for Niemann-Pick C1. J Neurol Sci. (2013) 335:219–20. doi: 10.1016/j.jns.2013.08.033
56. Zech M, Nubling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B, et al. Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS ONE. (2013) 8:e82879. doi: 10.1371/journal.pone.0082879
57. Castro-Fernandez C-S, Rodriguez-Sureda V, Martinez-Regueiro R, Aguiar P, Blanco-Arias P, Perez-Sousa C, et al. A heterozygous splicing variant in NPC2 in a patient with PSP [abstract]. In: Proceedings of 20th International Congreess of Parkinson's Disease and Movement Disorders. Berlin (2016).
58. Josephs KA, Matsumoto JY, Lindor NM. Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology. (2004) 63:2189–90. doi: 10.1212/01.WNL.0000145710.25588.2F
59. Ouled Amar Bencheikh B, Senkevich K, Rudakou U, Yu E, Mufti K, Ruskey JA, et al. Variants in the Niemann-Pick type C gene NPC1 are not associated with Parkinson's disease. Neurobiol Aging. (2020) 93:143e1–4. doi: 10.1016/j.neurobiolaging.2020.03.021
60. Kresojevic N, Dobricic V, Svetel M, Kostic V. Mutations in Niemann Pick type C gene are risk factor for Alzheimer's disease. Med Hypotheses. (2014) 83:559–62. doi: 10.1016/j.mehy.2014.08.025
61. Roze E, Paschke E, Lopez N, Eck T, Yoshida K, Maurel-Ollivier A, et al. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord. (2005) 20:1366–9. doi: 10.1002/mds.20593
62. Muthane U, Chickabasaviah Y, Kaneski C, Shankar SK, Narayanappa G, Christopher R, et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord. (2004) 19:1334–41. doi: 10.1002/mds.20193
63. Wu G, Lu ZH, Kulkarni N, Ledeen RW. Deficiency of ganglioside GM1 correlates with Parkinson's disease in mice and humans. J Neurosci Res. (2012) 90:1997–2008. doi: 10.1002/jnr.23090
64. Schneider JS, Gollomp SM, Sendek S, Colcher A, Cambi F, Du W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson's disease patients. J Neurol Sci. (2013) 324:140–8. doi: 10.1016/j.jns.2012.10.024
65. Martinez Z, Zhu M, Han S, Fink AL. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry. (2007) 46:1868–77. doi: 10.1021/bi061749a
66. Magistretti PJ, Geisler FH, Schneider JS, Li PA, Fiumelli H, Sipione S. Gangliosides: treatment avenues in neurodegenerative disease. Front Neurol. (2019) 10:859. doi: 10.3389/fneur.2019.00859
67. Argov Z, Navon R. Clinical and genetic variations in the syndrome of adult GM2 gangliosidosis resulting from hexosaminidase A deficiency. Ann Neurol. (1984) 16:14–20. doi: 10.1002/ana.410160105
68. Inzelberg R, Korczyn AD. Parkinsonism in adult-onset GM2 gangliosidosis. Mov Disord. (1994) 9:375–7. doi: 10.1002/mds.870090325
69. Suzuki K, Iseki E, Togo T, Yamaguchi A, Katsuse O, Katsuyama K, et al. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. (2007) 114:481–9. doi: 10.1007/s00401-007-0264-z
70. Bifsha P, Landry K, Ashmarina L, Durand S, Seyrantepe V, Trudel S, et al. Altered gene expression in cells from patients with lysosomal storage disorders suggests impairment of the ubiquitin pathway. Cell Death Differ. (2007) 14:511–23. doi: 10.1038/sj.cdd.4402013
71. Wise AH, Yang A, Naik H, Stauffer C, Zeid N, Liong C, et al. Parkinson's disease prevalence in Fabry disease: a survey study. Mol Genet Metab Rep. (2018) 14:27–30. doi: 10.1016/j.ymgmr.2017.10.013
72. Alcalay RN, Wolf P, Levy OA, Kang UJ, Waters C, Fahn S, et al. Alpha galactosidase A activity in Parkinson's disease. Neurobiol Dis. (2018) 112:85–90. doi: 10.1016/j.nbd.2018.01.012
73. Wu G, Yan B, Wang X, Feng X, Zhang A, Xu X, et al. Decreased activities of lysosomal acid alpha-D-galactosidase A in the leukocytes of sporadic Parkinson's disease. J Neurol Sci. (2008) 271:168–73. doi: 10.1016/j.jns.2008.04.011
74. Buechner S, De Cristofaro MT, Ramat S, Borsini W. Parkinsonism and Anderson Fabry's disease: a case report. Mov Disord. (2006) 21:103–7. doi: 10.1002/mds.20675
75. Gago MF, Azevedo O, Guimaraes A, Teresa Vide A, Lamas NJ, Oliveira TG, et al. Parkinson's disease and Fabry disease: clinical, biochemical and neuroimaging analysis of three pedigrees. J Parkinsons Dis. (2020) 10:141–52. doi: 10.3233/JPD-191704
76. Geier EG, Bourdenx M, Storm NJ, Cochran JN, Sirkis DW, Hwang JH, et al. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. (2019) 137:71–88. doi: 10.1007/s00401-018-1925-9
77. Aberg L, Liewendahl K, Nikkinen P, Autti T, Rinne JO, Santavuori P. Decreased striatal dopamine transporter density in JNCL patients with parkinsonian symptoms. Neurology. (2000) 54:1069–74. doi: 10.1212/WNL.54.5.1069
78. Aberg LE, Rinne JO, Rajantie I, Santavuori P. A favorable response to antiparkinsonian treatment in juvenile neuronal ceroid lipofuscinosis. Neurology. (2001) 56:1236–9. doi: 10.1212/WNL.56.9.1236
79. Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. (2013) 22:2067–82. doi: 10.1093/hmg/ddt057
80. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
81. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
82. Van Mossevelde S, Engelborghs S, van der Zee J, Van Broeckhoven C. Genotype-phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol. (2018) 14:363–78. doi: 10.1038/s41582-018-0009-8
83. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. (2012) 8:423–34. doi: 10.1038/nrneurol.2012.117
84. Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. (2006) 129:3081–90. doi: 10.1093/brain/awl271
85. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. (2019) 266:2075–86. doi: 10.1007/s00415-019-09363-4
86. van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. (2007) 28:416. doi: 10.1002/humu.9484
87. Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. (2009) 132:583–91. doi: 10.1093/brain/awn352
88. Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, et al. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. (2012) 2:789–98. doi: 10.1016/j.celrep.2012.09.007
89. Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. (2010) 42:234–9. doi: 10.1038/ng.536
90. Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. (2011) 76:467–74. doi: 10.1212/WNL.0b013e31820a0e3b
91. Meeter LH, Patzke H, Loewen G, Dopper EG, Pijnenburg YA, van Minkelen R, et al. Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra. (2016) 6:330–40. doi: 10.1159/000447738
92. Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. (2006) 15:2988–3001. doi: 10.1093/hmg/ddl241
93. Le Ber, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. (2008) 131:732–46. doi: 10.1093/brain/awn012
94. van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. (2008) 7:965–74. doi: 10.1016/S1474-4422(08)70194-7
95. Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. (2015) 7:171. doi: 10.3389/fnagi.2015.00171
96. Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. (2010) 67:161–70. doi: 10.1001/archneurol.2009.328
97. Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. (2015) 22:745–52. doi: 10.1111/ene.12675
98. Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. (2016) 41:200 e1–5. doi: 10.1016/j.neurobiolaging.2016.02.019
99. Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. (2014) 127:845–60. doi: 10.1007/s00401-014-1262-6
100. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
101. Galimberti D, Fumagalli GG, Fenoglio C, Cioffi SMG, Arighi A, Serpente M, et al. Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. (2018) 62:245 e9–12. doi: 10.1016/j.neurobiolaging.2017.10.016
102. She A, Kurtser I, Reis SA, Hennig K, Lai J, Lang A, et al. Selectivity and Kinetic Requirements of HDAC Inhibitors as Progranulin Enhancers for Treating Frontotemporal Dementia. Cell Chem Biol. (2017) 24:892–906 e5. doi: 10.1016/j.chembiol.2017.06.010
103. Boxer A, Moebius H, Harris B, Boeve B, Borroni B, Swieten J, et al. Phase 2A randomized, double-blind, placebo-controlled trial of the histone deacetylase inhibitor (HDACI), FRM-0334, in asymptomatic carriers of, or patients with frontotemporal lobar degeneration (FTLD) due to, progranulin gene mutations. Alzheimer Dementia. (2019) 15:P1231–2. doi: 10.1016/j.jalz.2019.06.4746
104. Sha SJ, Miller ZA, Min SW, Zhou Y, Brown J, Mitic LL, et al. An 8-week, open-label, dose-finding study of nimodipine for the treatment of progranulin insufficiency from GRN gene mutations. Alzheimers Dement. (2017) 3:507–12. doi: 10.1016/j.trci.2017.08.002
105. Cenik B, Sephton CF, Dewey CM, Xian X, Wei S, Yu K, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. (2011) 286:16101–8. doi: 10.1074/jbc.M110.193433
106. Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J Neurosci. (2018) 38:2341–58. doi: 10.1523/JNEUROSCI.3081-17.2018
107. Amado DA, Rieders JM, Diatta F, Hernandez-Con P, Singer A, Mak JT, et al. AAV-mediated progranulin delivery to a mouse model of progranulin deficiency causes t cell-mediated toxicity. Mol Ther. (2019) 27:465–78. doi: 10.1016/j.ymthe.2018.11.013
108. Xu HM, Tan L, Wan Y, Tan MS, Zhang W, Zheng ZJ, et al. PGRN is associated with late-onset Alzheimer's disease: a case-control replication study and meta-analysis. Mol Neurobiol. (2017) 54:1187–95. doi: 10.1007/s12035-016-9698-4
109. Sheng J, Su L, Xu Z, Chen G. Progranulin polymorphism rs5848 is associated with increased risk of Alzheimer's disease. Gene. (2014) 542:141–5. doi: 10.1016/j.gene.2014.03.041
110. Fenoglio C, Galimberti D, Cortini F, Kauwe JS, Cruchaga C, Venturelli E, et al. Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer's disease. J Alzheimers Dis. (2009) 18:603–12. doi: 10.3233/JAD-2009-1170
111. Kamalainen A, Viswanathan J, Natunen T, Helisalmi S, Kauppinen T, Pikkarainen M, et al. GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer's disease patients. J Alzheimers Dis. (2013) 33:23–7. doi: 10.3233/JAD-2012-120946
112. Hsiung GY, Fok A, Feldman HH, Rademakers R, Mackenzie IR. rs5848 polymorphism and serum progranulin level. J Neurol Sci. (2011) 300:28–32. doi: 10.1016/j.jns.2010.10.009
113. Benussi L, Binetti G, Sina E, Gigola L, Bettecken T, Meitinger T, et al. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging. (2008) 29:427–35. doi: 10.1016/j.neurobiolaging.2006.10.028
114. Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. (2008) 71:253–9. doi: 10.1212/01.wnl.0000289191.54852.75
115. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
116. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
117. Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurol. (2018) 17:721–30. doi: 10.1016/S1474-4422(18)30232-1
118. Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodriguez-Rodriguez E, Lopez de Munain A, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. (2014) 35:444 e1–4. doi: 10.1016/j.neurobiolaging.2013.08.011
119. Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet. (2017) 49:1373–84. doi: 10.1038/ng.3916
120. Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. (2015) 10:19. doi: 10.1186/s13024-015-0016-9
121. Miyashita A, Wen Y, Kitamura N, Matsubara E, Kawarabayashi T, Shoji M, et al. Lack of genetic association between TREM2 and late-onset Alzheimer's disease in a Japanese population. J Alzheimers Dis. (2014) 41:1031–8. doi: 10.3233/JAD-140225
122. Jiao B, Liu X, Tang B, Hou L, Zhou L, Zhang F, et al. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer's disease from mainland China. Neurobiol Aging. (2014) 35:2422 e9–11. doi: 10.1016/j.neurobiolaging.2014.04.025
123. Jiang T, Tan L, Chen Q, Tan MS, Zhou JS, Zhu XC, et al. A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiol Aging. (2016) 42:217 e1–3. doi: 10.1016/j.neurobiolaging.2016.02.023
124. Bonham LW, Sirkis DW, Fan J, Aparicio RE, Tse M, Ramos EM, et al. Identification of a rare coding variant in TREM2 in a Chinese individual with Alzheimer's disease. Neurocase. (2017) 23:65–9. doi: 10.1080/13554794.2017.1294182
125. Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. (2016) 5:e20391. doi: 10.7554/eLife.20391
126. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
127. Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. (2014) 6:243ra86. doi: 10.1126/scitranslmed.3009093
128. Ma L, Allen M, Sakae N, Ertekin-Taner N, Graff-Radford NR, Dickson DW, et al. Expression and processing analyses of wild type and p.R47H TREM2 variant in Alzheimer's disease brains. Mol Neurodegener. (2016) 11:72. doi: 10.1186/s13024-016-0137-9
129. Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. (2017) 13:381–7. doi: 10.1016/j.jalz.2016.07.004
130. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81 e9. doi: 10.1016/j.immuni.2017.08.008
131. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. (2017) 170:649–63 e13. doi: 10.1016/j.cell.2017.07.023
132. Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, et al. R47H TREM2 variant increases risk of typical early-onset Alzheimer's disease but not of prion or frontotemporal dementia. Alzheimers Dement. (2014) 10:602–8 e4. doi: 10.1016/j.jalz.2014.05.1751
133. Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, Guo X, et al. R47H variant of TREM2 associated with Alzheimer disease in a large late-onset family: clinical, genetic, neuropathological study. JAMA Neurol. (2015) 72:920–7. doi: 10.1001/jamaneurol.2015.0979
134. Replogle JM, Chan G, White CC, Raj T, Winn PA, Evans DA, et al. A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Ann Neurol. (2015) 77:469–77. doi: 10.1002/ana.24337
135. Wang X, Lopez OL, Sweet RA, Becker JT, DeKosky ST, Barmada MM, et al. Genetic determinants of disease progression in Alzheimer's disease. J Alzheimers Dis. (2015) 43:649–55. doi: 10.3233/JAD-140729
136. Luis EO, Ortega-Cubero S, Lamet I, Razquin C, Cruchaga C, Benitez BA, et al. Frontobasal gray matter loss is associated with the TREM2 p.R47H variant. Neurobiol Aging. (2014) 35:2681–90. doi: 10.1016/j.neurobiolaging.2014.06.007
137. Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, Parkinson's disease. Alzheimers Dement. (2015) 11:1407–1416. doi: 10.1016/j.jalz.2014.12.009
138. Pottier C, Ravenscroft TA, Brown PH, Finch NA, Baker M, Parsons M, et al. TYROBP genetic variants in early-onset Alzheimer's disease. Neurobiol Aging. (2016) 48:222 e9–15. doi: 10.1016/j.neurobiolaging.2016.07.028
139. Ghezzi L, Carandini T, Arighi A, Fenoglio C, Arcaro M, De Riz M, et al. Evidence of CNS beta-amyloid deposition in Nasu-Hakola disease due to the TREM2 Q33X mutation. Neurology. (2017) 89:2503–5. doi: 10.1212/WNL.0000000000004747
140. Montalbetti L, Ratti MT, Greco B, Aprile C, Moglia A, Soragna D. Neuropsychological tests and functional nuclear neuroimaging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct Neurol. (2005) 20:71–5.
141. Satoh JI, Kino Y, Yanaizu M, Saito Y. Alzheimer's disease pathology in Nasu-Hakola disease brains. Intractable Rare Dis Res. (2018) 7:32–6. doi: 10.5582/irdr.2017.01088
142. Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med. (2016) 8:466–76. doi: 10.15252/emmm.201506123
143. Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. (2017) 214:597–607. doi: 10.1084/jem.20160844
144. Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron. (2018) 97:1032–48 e5. doi: 10.1016/j.neuron.2018.02.002
145. Le Ber, De Septenville A, Guerreiro R, Bras J, Camuzat A, Caroppo P, et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging. (2014) 35:2419 e23–5. doi: 10.1016/j.neurobiolaging.2014.04.010
146. Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. (2013) 70:78–84. doi: 10.1001/jamaneurol.2013.579
147. Thelen M, Razquin C, Hernandez I, Gorostidi A, Sanchez-Valle R, Ortega-Cubero S, et al. Investigation of the role of rare TREM2 variants in frontotemporal dementia subtypes. Neurobiol Aging. (2014) 35:2657 e13–19. doi: 10.1016/j.neurobiolaging.2014.06.018
148. Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. (2014) 71:449–53. doi: 10.1001/jamaneurol.2013.6237
149. Benitez BA, Cruchaga C. G. United States-Spain Parkinson's disease research. TREM2 and neurodegenerative disease. N Engl J Med. (2013) 369:1567–8. doi: 10.1056/NEJMc1306509
150. Mengel D, Thelen M, Balzer-Geldsetzer M, Soling C, Bach JP, Schaeffer E, et al. TREM2 rare variant p.R47H is not associated with Parkinson's disease. Parkinsonism Relat Disord. (2016) 23:109–11. doi: 10.1016/j.parkreldis.2015.11.026
151. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. (2013) 8:19. doi: 10.1186/1750-1326-8-19
152. Chen Y, Chen X, Guo X, Song W, Cao B, Wei Q, et al. Assessment of TREM2 rs75932628 association with Parkinson's disease and multiple system atrophy in a Chinese population. Neurol Sci. (2015) 36:1903–6. doi: 10.1007/s10072-015-2279-x
153. Walton RL, Soto-Ortolaza AI, Murray ME, Lorenzo-Betancor O, Ogaki K, Heckman MG, et al. TREM2 p.R47H substitution is not associated with dementia with Lewy bodies. Neurol Genet. (2016) 2:e85. doi: 10.1212/NXG.0000000000000085
154. Jesus S, Huertas I, Bernal-Bernal I, Bonilla-Toribio M, Caceres-Redondo MT, Vargas-Gonzalez L, et al. GBA variants influence motor and non-motor features of Parkinson's disease. PLoS ONE. (2016) 11:e0167749. doi: 10.1371/journal.pone.0167749
155. Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, et al. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson's disease. J Neurol Neurosurg Psychiatry. (2020) 91:695–702. doi: 10.1136/jnnp-2020-322857
156. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. (2004) 351:1972–7. doi: 10.1056/NEJMoa033277
157. Bronner IF, Rizzu P, Seelaar H, van Mil SE, Anar B, Azmani A, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. (2007) 15:369–74. doi: 10.1038/sj.ejhg.5201772
158. Bruni AC, Momeni P, Bernardi L, Tomaino C, Frangipane F, Elder J, et al. Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology. (2007) 69:140–7. doi: 10.1212/01.wnl.0000265220.64396.b4
159. Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat. (2008) 29:1373–86. doi: 10.1002/humu.20785
160. Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. (2008) 17:3631–42. doi: 10.1093/hmg/ddn257
161. Wong TH, van der Lee SJ, van Rooij JGJ, Meeter LHH, Frick P, Melhem S, et al. EIF2AK3 variants in Dutch patients with Alzheimer's disease. Neurobiol Aging. (2019) 73:229 e11–8. doi: 10.1016/j.neurobiolaging.2018.08.016
162. Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. (2011) 43:699–705. doi: 10.1038/ng.859
163. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. (2000) 25:406–9. doi: 10.1038/78085
164. Liu QY, Yu JT, Miao D, Ma XY, Wang HF, Wang W, et al. An exploratory study on STX6, MOBP, MAPT, and EIF2AK3 and late-onset Alzheimer's disease. Neurobiol Aging. (2013) 34:1519 e13–7. doi: 10.1016/j.neurobiolaging.2012.10.004
165. Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. (2015) 130:633–42. doi: 10.1007/s00401-015-1487-z
166. Wang L, Popko B, Roos RP. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum Mol Genet. (2014) 23:2629–38. doi: 10.1093/hmg/ddt658
167. Mercado G, Castillo V, Soto P, Lopez N, Axten JM, Sardi SP, et al. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis. (2018) 112:136–48. doi: 10.1016/j.nbd.2018.01.004
Keywords: neurodegenerative, autosomal, recessive, carrier, GBA1, GRN, TREM2, EIF2AK3
Citation: Vieira SRL and Morris HR (2021) Neurodegenerative Disease Risk in Carriers of Autosomal Recessive Disease. Front. Neurol. 12:679927. doi: 10.3389/fneur.2021.679927
Received: 12 March 2021; Accepted: 14 May 2021;
Published: 04 June 2021.
Edited by:
Hassan Abolhassani, Karolinska Institute, SwedenReviewed by:
Zhang-Yu Zou, Fujian Medical University Union Hospital, ChinaCopyright © 2021 Vieira and Morris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huw R. Morris, aC5tb3JyaXNAdWNsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.