
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 24 May 2021
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.668180
 Allison Ducharme-Smith1†
Allison Ducharme-Smith1† Stefan Nicolau2†
Stefan Nicolau2† C. Anwar A. Chahal1,3,4*
C. Anwar A. Chahal1,3,4* Kirstie Ducharme-Smith5Shujah Rehman1
Kirstie Ducharme-Smith5Shujah Rehman1 Keerthi Jaliparthy1
Keerthi Jaliparthy1 Nadeem Khan6Christopher G. Scott7
Nadeem Khan6Christopher G. Scott7 Erik K. St Louis2
Erik K. St Louis2 Teerin Liewluck2
Teerin Liewluck2 Virend K. Somers1Grace Lin1Peter A. Brady8
Virend K. Somers1Grace Lin1Peter A. Brady8 Margherita Milone2*
Margherita Milone2*Background: Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies and predominantly affects facial and shoulder girdle muscles. Previous case reports and cohort studies identified minor cardiac abnormalities in FSHD patients, but their nature and frequency remain incompletely characterized.
Methods: We reviewed cardiac, neurological and genetic findings of 104 patients with genetically confirmed FSHD.
Results: The most common conduction abnormality was complete (7%) or incomplete (5%) right bundle branch block (RBBB). Bifascicular block, left anterior fascicular block, complete atrioventricular block, and 2:1 atrioventricular block each occurred in 1% of patients. Atrial fibrillation or flutter were seen in 5% of patients. Eight percent of patients had heart failure with reduced ejection fraction and 25% had valvular disease. The latter included aortic stenosis in 6% (severe in 4% and moderate in 2%) and moderate aortic regurgitation in 8%. Mitral valve prolapse (MVP) was present in 9% of patients without significant mitral regurgitation. There were no significant associations between structural or conduction abnormalities and age, degree of muscle weakness, or size of the 4q deletion.
Conclusions: Both structural and conduction abnormalities can occur in FSHD. The most common abnormalities are benign (RBBB and MVP), but more significant cardiac involvement was also observed. The presence of cardiac abnormalities cannot be predicted from the severity of the neurological phenotype, nor from the genotype.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies and predominantly involves facial and shoulder girdle muscles (1), with a degree of phenotypic variability among affected individuals. There are two genetically distinct but clinically indistinguishable subtypes of FSHD, termed FSHD1 and FSHD2. The majority of patients have FSHD1, which results from a contraction of the D4Z4 microsatellite repeat containing the DUX4 pseudogene on the long arm of chromosome 4. Development of FSHD additionally requires a permissive 4qA haplotype distal to the D4Z4 microsatellite repeat (2). FSHD1 is inherited in an autosomal dominant fashion, with up to 30% of cases occurring de novo. The D4Z4 contraction leads to translational de-repression of DUX4 (3). This is generally thought to result from hypomethylation of the D4Z4 repeats, though some studies have questioned this view (4, 5). There is also evidence that additional genetic mechanisms play a role in the development of FSHD (6, 7). Approximately 5% of FSHD patients have FSHD2, which is characterized by hypomethylation of the D4Z4 locus in combination with a permissive 4qA haplotype, but without a D4Z4 contraction (8–10). Many but not all FSHD2 patients harbor mutations in SMCHD1 or, less frequently, in DNMT3B or LRIF1 (11). Mutations in SMCHD1 have also recently been associated with non-neuromuscular phenotypes (1, 12–14).
Occurrence of certain extramuscular manifestations, such as retinal vasculopathy and sensorineural hearing loss, are well-established in FSHD (15). Cardiac involvement however, which is common in many other muscular dystrophies (16), has not been fully characterized in FSHD. Cardiac screening and surveillance have not been recommended in FSHD patients without cardiac symptoms (15, 17, 18). Several case reports and cohort studies of genetically proven FSHD patients have identified cardiac abnormalities in FSHD patients. The most commonly identified cardiac abnormality has been incomplete right branch block (RBBB), detected in ~23–33% of patients (17, 19). Supraventricular tachyarrhyhmias have also been reported in approximatively 10% of FSHD patients (20, 21). A smaller number of patients exhibits ventricular arrhythmias, cardiac conduction system abnormalities such as interventricular conduction delay and atrioventricular (AV) block, focal myocardial fibrosis or fatty infiltration with preserved ejection fraction, or a hypertrophic cardiomyopathy phenotype (20–25). Limitations of the various published studies include absence of genetic confirmation in some cases, inconsistent correlation of cardiac abnormalities with the severity of the muscle disease or underlying genetic defect, association of arrhythmias with comorbidities and cardiac risk factors (26), and sometimes the small number of patients included.
Herein, we report on the frequency and nature of cardiac abnormalities observed in a large cohort of genetically proven FSHD patients evaluated at a single academic medical center. We investigated the frequency of cardiac symptoms, cardiac conduction abnormalities, and structural heart abnormalities in these patients and correlated cardiac abnormalities with the severity of muscle weakness and genetic findings.
Using both a manually maintained registry and a search of electronic medical records, we identified all cases of FSHD evaluated at Mayo Clinic from January 1st 1995 to July 31st 2018 (Figure 1). The search was performed using a combination of keywords, and ICD9 and ICD10 codes (facioscapulohumeral muscular dystrophy, FSHD, 359.1, and G71). The inclusion criterion was a genetically proven diagnosis of FSHD1 or FSHD2. We excluded patients with a clinical diagnosis of suspected FSHD without genetic confirmation. A case note abstraction form with a pre-defined data dictionary was designed and piloted in REDCap (Research Electronic Data Capture; University of Vanderbilt, Tennessee, USA). REDCap is a secure, web-based application designed to support data capture for research studies, providing: (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for importing data from external sources (27).
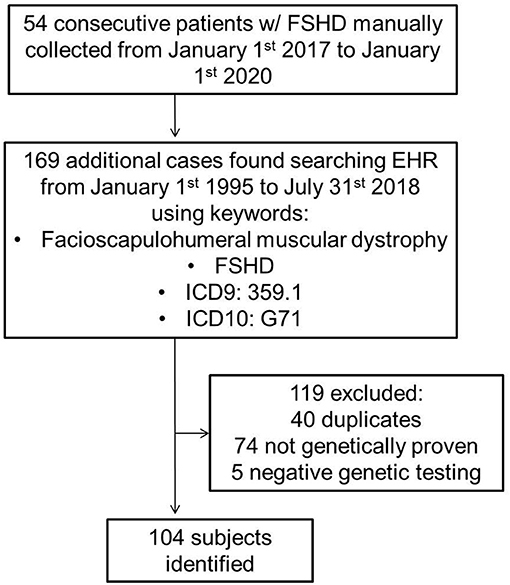
Figure 1. FSHD, facioscapulohumeral muscular dystrophy; EHR, electronic health record.
Data were manually abstracted and collected, including demographics, genetic testing results, age of onset of muscle weakness, degree of muscle weakness, age at diagnosis, cardiac symptoms and findings, and known independent cardiac risk factors (hypertension, hyperlipidemia, current or prior smoking, obesity, and diabetes mellitus). Muscle power was evaluated by manual muscle testing using the Medical Research Council (MRC) scale. A sum score was calculated using the facial muscles, weakest upper limb muscle and weakest lower limb muscle on each side (range 0–30). The following cardiac findings were collected and analyzed: echocardiograms, stress tests, and Holter monitoring data. In addition, electrocardiogram (ECG) data were electronically abstracted using MUSE (GE, Milwaukee, WI, USA). Mitral valve prolapse was defined as mitral valve displacement on echocardiogram of more than 2 mm above the mitral annulus in long-axis view (28).
Categorical variables are reported as frequencies and continuous variables with medians and interquartile ranges (IQR) (29). Continuous variables were compared using t-tests or non-parametric tests for non-Gaussian distributions. T-tests were performed between variables of interest (age, degree of muscle weakness, and size of the 4q deletion) and the cardiac outcome variables (conduction, structural abnormalities and cardiac risk). Categorical variables were compared using X2 or Fisher's exact test. A p < 0.05 was considered statistically significant for all analyses. Analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA) and R 3.4.2 (R Foundation for Statistical Computing, Vienna, Austria).
We identified 104 consecutive patients with genetically proven FSHD: 99 with FSHD1 and five with FSHD2. Table 1 summarizes demographics, symptoms, clinical and genetic findings, as well as cardiovascular risk factors of our cohort. The median age of onset was 26 years and 59% of patients were male. Seventy percent of patients were older than 18-years at symptom onset. The median age at molecular diagnosis was 45 years (IQR 29, 60). The median manual muscle testing score was 24 (IQR 24, 26). Seventy-one percent of patients had a typical distribution of weakness, involving the face and shoulder girdle muscles. Thirteen percent had shoulder girdle weakness with facial sparing. The remainder had atypical presentations, most commonly facial and predominant lower limb weakness. Among patients with FSHD1, the median EcoRI restriction fragment length was 29.5 kb (IQR 24, 33). Presence of the 4qA haplotype was confirmed in 42 patients. The remainder of patients underwent testing at a time when haplotyping was not commercially available or underwent testing in a laboratory that did not perform haplotyping. FSHD2 patients carried one of the following mutations in SMCHD1: c.724G>A (p.A242T, two patients), c.1273G>A (p.G425R), c.1787G>A (p.W596*), or c.1435C>T (p.R479*).
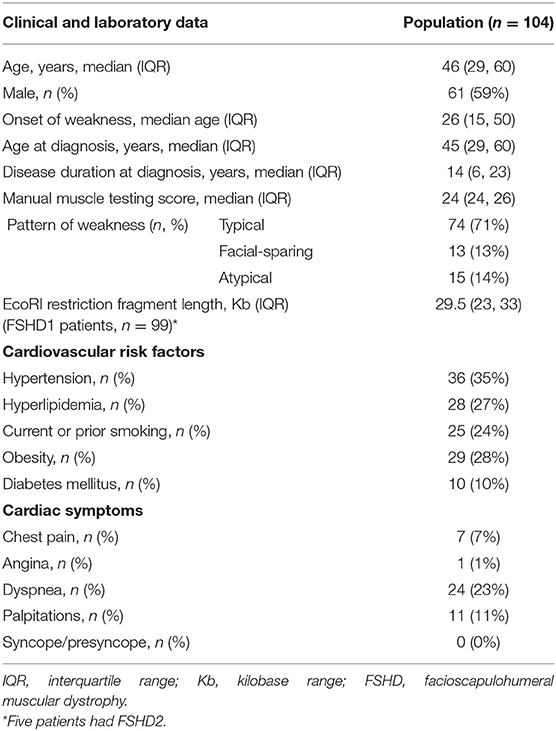
Table 1. Clinical data, genetic findings, and cardiovascular risk factors in facioscapulohumeral muscular dystrophy (FSHD) type 1 and 2 patients.
Table 2 summarizes the cardiac findings. ECGs were performed in 70% (n = 73) of patients, 12% (n = 12) of whom had also had Holter evaluation. ECGs were obtained for cardiac surveillance in the setting of FSHD or for cardiac symptoms. ECGs were abnormal in 49% of the 73 patients in whom they were obtained. The most common abnormalities were RBBB and atrial fibrillation/flutter, which were detected in 7% (n = 5) of patients and 5% (n = 4) of patients, respectively. The patients with 2:1 AV block (n = 1) and first degree AV block (n = 5) were asymptomatic. The single patient with complete AV block had a cardiac device, which was a dual chamber pacemaker for complete heart block occurring post-surgical aortic valve replacement. No patient had an implantable cardioverter defibrillator or biventricular pacemaker. Amongst those with Holter monitoring, premature ventricular complexes (PVC) were recorded in 50% (n = 6), non-sustained VT in 50% (n = 6), premature atrial complexes in 25% (n = 3), and sustained VT in 8% (n = 1). Among the patients with PVCs, one patient had a 6% burden of ventricular events, while the burden was <1% in the other five patients. Among the seven patients with non-sustained or sustained VT, five had an echocardiogram, which showed: left atrial enlargement and apical hypokinesis in one patient, severe right ventricular enlargement as well as severe aortic stenosis (trileaflet valve) and tricuspid regurgitation in one patient who had also RBBB, prior aortic valve replacement and subsequently normal ejection fraction and valvular function in one patient, and no structural abnormalities in the remaining two patients. Four patients had atrial fibrillation on either ECG or Holter.

Table 2. Cardiovascular findings in facioscapulohumeral muscular dystrophy (FSHD) type 1 and 2 patients.
Echocardiography was obtained in 51% (n = 53) of patients (of these, all 53 had ECGs and 12 also had Holter evaluation). Echocardiography was abnormal in 42% (n = 22) of these, as summarized in Table 2. Eight percent (n = 4) of these patients had heart failure with reduced ejection fraction; three of these four patients had ejection fractions of 30–40% while one had an ejection fraction of 50%. Two of the four patients had known mild bystander coronary artery disease (CAD), which was disproportionate to the degree of left ventricular dysfunction. One of these patients with heart failure had a complex medical history and the heart failure was attributed to her ischemic dilated cardiomyopathy and pulmonary hypertension. Two had sepsis-related cardiomyopathy, both of which normalized with resolution of sepsis. Valvular abnormalities were present in 24% (n = 13) and some of these consisted of more than one valvular lesion. Nine percent (n = 5) had mitral valve prolapse and 6% (n = 3) had mitral regurgitation, all of which were mild. Six percent had aortic stenosis (n = 3), two of which were severe requiring aortic valve replacement, while the third was moderate. Four percent had aortic regurgitation (n = 2) which was moderate in severity. Finally, 6% had tricuspid regurgitation (n = 3), which was mild in severity with the exception of one patient. In this case, the patient had both severe aortic stenosis and severe tricuspid regurgitation. The latter patient is the same patient described above with heart failure, reduced ejection fraction and several other structural abnormalities in the setting of scleroderma, dilated cardiomyopathy and pulmonary hypertension, as well as genetically confirmed FSHD 1.
Forty-four percent of the cohort (n = 46) had no cardiac risk factors (which included patients who had no identified obesity, hypertension, hyperlipidemia, smoking, or diabetes). In this cohort without underlying cardiac risk factors, 13% had a conduction abnormality (n = 6) which included intermittent right bundle branch block (n = 2), frequent premature atrial contractions (n = 1), left axis deviation (n = 2). Sixteen percent (n = 7) had a structural abnormality, including MVP (n = 2), atrial enlargement (n = 3), and mitral regurgitation (n = 2).
Eight patients had early onset FSHD, defined as either facial weakness before age five or shoulder girdle weakness before age 10 (30). Six of these eight patients had both ECG and echocardiogram. One had left atrial enlargement, one had right atrial enlargement, two had biatrial enlargement, and one had right ventricular hypertrophy with right axis deviation. In addition, one of these patients had a small secundum ASD. None of these abnormalities appeared clinically relevant, as the patients were all asymptomatic and none had atrial fibrillation or flutter detected during our follow-up period.
There was no significant difference in the frequency of structural and conduction abnormalities between classical and facial-sparing or other atypical presentations. Of the five patients with FSHD2, only two had an ECG and echocardiogram. Both patients had intermittent RBBB and normal echocardiograms.
Only 11% of patients (n = 11) underwent stress testing, which was requested for reported dyspnea on exertion. Exercise stress test data revealed ischemia in two patients, aged 74 and 79 years. The first was felt to have stable and non-progressive symptoms managed well with medical therapy. The second underwent coronary artery catheterization, which demonstrated diffuse coronary artery disease requiring coronary artery bypass grafting.
There were statistically significant associations between age and presence of any cardiac risk factor (p < 0.001), presence of cardiac structural abnormalities (p = 0.01), but not presence of cardiac conduction abnormalities (p = 0.4). Individuals with cardiac risk factors and structural abnormalities tended to be older.
There were no significant associations between cardiac structural abnormalities and degree of muscle weakness (p = 0.5), or size of the 4q35 deletion in FSHD1 (p = 0.4). Similarly, there were no associations between cardiac conduction abnormalities and degree of muscle weakness (p = 0.6), or size of the 4q35 deletion in FSHD1 (p = 0.6). There were also no associations between cardiac abnormalities and degree of muscle weakness or size of the 4q35 deletion when males and females were analyzed separately, among the patients without cardiac risk factors, or among patients with early onset (p > 0.3 for all comparisons).
In our cohort, 4% (n = 4) of patients experienced a stroke, either ischemic (n = 3) or hemorrhagic (n = 1). Three of these patients had structural heart abnormalities, while one had a normal echocardiogram. As most of our patients presented to our clinic only for establishing the diagnosis of the underlying neuromuscular disease, only 43 patients were seen in follow-up one or more years after the diagnosis. Seven of these 43 patients (16%) followed in the cohort died at a median age of 68 years (IQR 56, 79). The causes of death for these seven patients were pneumonia, disseminated histoplasmosis, stroke, hypercapnic respiratory failure from muscular dystrophy, pancreatic cancer, cardiac arrest secondary to severe gastrointestinal bleed, and unknown cause (n = 1 each).
We describe the cardiac findings in a large cohort of genetically proven FSHD patients assessed at a single center. In these patients, we identified cardiac conduction and structural abnormalities, as well as a higher than expected prevalence of mitral valve prolapse. These cardiac abnormalities did not correlate with patient age or degree of muscle weakness, and, similarly to previous studies, the cardiac abnormalities did not correlate with the size of the 4q deletion in FSHD1 (17, 20, 21). We observed only an association between age and presence of cardiac risk factors, as expected in the general population. Two of 5 FSHD2 patients had incomplete RBBB but these patients were too few to assess prevalence of cardiac abnormalities in this subgroup.
Of the 73 patients (70%) who underwent an ECG, we found 7% had a complete RBBB, 5% had incomplete RBBB, and 3% had higher degree AV block. This is higher than expected in the general population, where the prevalence of incomplete and complete RBBB is 3% and 0.8%, respectively (31). Other studies have reported a much higher prevalence of incomplete RBBB (19-33%) in FSHD1 (17, 19, 20), but a slightly lower frequency of complete RBBB (4%) (17). However, none of these conduction abnormalities were clinically relevant. Meanwhile, the proportion of left bundle branch block in our cohort was similar to the general population (32). The mechanism of conduction abnormalities in FSHD is unknown. One could speculate that this may be related to abnormal vascular smooth muscle differentiation that selectively involves the His-Purkinje fibers (33). Alternatively this could be due to myocardial tissue changes, including focal fibrosis and fat infiltration described in FSHD1 via cardiovascular magnetic resonance (24), which may serve as a substrate for arrhythmias (22).
In the absence of traditional cardiac risk factors, we found a high prevalence of conduction abnormalities (13%). This is similar to Trevisan et al., who found that 12% of their cohort of 83 patients had cardiac involvement, which mostly consisted of arrhythmic disturbances (21). It is unknown whether the cardiac abnormalities detected in our patients were static or progressive, as most patients were not followed longitudinally. Limited literature is available in this regard. An 8-year follow-up study in 27 FSHD1 patients, who were mainly found to have incomplete and complete RBBB, showed no progression of the ECG abnormalities in the majority of patients (17). Longitudinal follow-up would be of interest, as the autonomic modifications reported with disease progression, such as increased sympathetic activity and decreased parasympathetic activity, could increase the risk of cardiac arrhythmias over time (34).
We also observed that four of the 73 (5%) patients who underwent ECG evaluation and six of 12 (50%) patients who underwent Holter had atrial fibrillation and non-sustained ventricular tachycardia, respectively. The non-sustained ventricular tachycardia was asymptomatic and shorter than 15 beats in all six patients, in the context of structurally normal hearts, and thus no further management was required. The prevalence of atrial fibrillation by age, in our patients, is consistent with that observed in the general population (35). Interestingly, we did not detect any supraventricular tachycardias in our cohort, which is in contrast to the literature where one of the main arrhythmogenic abnormalities observed in patients with FSHD was atrial tachycardia (21, 36). This may be due to the low frequency of Holter monitoring obtained in our cohort.
Interestingly, we found the frequency of MVP in our cohort was 9%, in comparison to 1–2% in the general population (37). This result may be skewed by the lack of echocardiographic data in all patients of this cohort. However, even assuming the remaining patients did not have MVP, its prevalence would still be higher than expected in the normal population. While MVP has been associated with Duchenne muscular dystrophy (38), to our knowledge, this is the first time an increased prevalence of MVP has been reported in FSHD. Mitral valve coaptation is complex, requiring appropriate valve leaflet length, and subvalvular apparati of chordae and papillary muscles. The mechanisms leading to prolapse include abnormalities of leaflet length, strength and shape, as well as annular disjunction. Subtle changes to cardiac muscle without overt dilatation or hypertrophy can result in papillary muscle dysfunction, which can contribute to MVP. One could speculate that minor dysfunction of the papillary muscles may have resulted in MVP. None of our patients had cardiac MRI to explore the possibility of focal fibrosis or fatty infiltration that could have led to MVP. On the other hand, the prevalence of bicuspid aortic valve (2%), aortic stenosis (6%), and tricuspid regurgitation (6%) was similar to that seen in the general population (39, 40). We observed no isolated cardiomyopathy, and the three FSHD patients with compromised ejection fraction had coexisting independent cardiovascular disease.
It is notable that four of eight patients with early-onset FSHD had atrial enlargement or ventricular hypertrophy, which had not been reported in other cohorts. Previous studies had identified conduction abnormalities in early-onset FSHD, but it remains uncertain whether these abnormalities occur at a higher frequency in early-onset FSHD compared to those with a more classical age of onset (20, 23, 41, 42).
The strength of our study is a cohort of genetically confirmed FSHD patients with comprehensively characterized neurological phenotypes. The limitations include those inherent to a retrospective design. Cardiac investigations were not obtained universally across the cohort to determine the extent of cardiac abnormalities in asymptomatic patients. As a result, the frequency of abnormalities in patients who underwent testing may be higher than in the broader FSHD population. Our study may also be underpowered to detect rare cardiac abnormalities. Follow-up data was not available for most patients, as they were evaluated for establishing the neuromuscular diagnosis, and subsequently received care locally.
In summary, we found no association between cardiac findings and genotype or severity of skeletal myopathy in FSHD. Baseline ECG abnormalities were benign and did not require cardiac implantable electronic device and the frequency of atrial fibrillation was the same as the general population. There was a higher frequency of MVP, and RBBB in FSHD when compared to the general population. Currently, there are no specific guidelines for cardiac screening in FSHD. In light of our findings and previously reported studies, it would be prudent to screen patients with FSHD for cardiac conduction dysfunction, regardless of age, degree of muscle weakness, or cardiac symptoms. Multi-center studies with serial ECG and echocardiographic long-term follow-up would be helpful to better ascertain risk of cardiac involvement in FSHD and to assess for progression of the cardiac abnormalities.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by IRB. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
AD-S: data curation, investigation, methodology, writing—original draft, and writing—review and editing. SN: data curation, investigation, and writing—review and editing. CAAC: visualization, investigation, writing—original draft, and writing—review and editing. KD-S and CGS: software and formal analysis. SR: data curation and investigation. NK, EKSL, TL, and PAB: writing—review and editing. VKS: conceptualization, validation, writing—review and editing, supervision, and funding acquisition. GL: validation, writing—review, and editing. MM: validation, writing—review and editing, supervision, project administration, and funding. All authors contributed to the article and approved the submitted version.
This publication was made possible by support from the Research Division of the Department of CV Medicine and the Clinical and Translational Science Award Grant Number UL1 TR000135, supporting the Mayo Clinic Center for Clinical and Translational Science (CCaTS), from the National Center for Advancing Translational Sciences (NCATS), a component of NIH, and the Neurology Research departmental funds. CAAC and VKS are supported by NIH HL65176 and NIH HL134885. CAAC was supported by the American Heart Association (Award number 17POST33400211). CAAC and VKS are supported by the Paul and Ruby Tsai Foundation. MM receives research support from a Mayo Clinic benefactor and the department of Neurology. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
GL has received fees from the Pfizer Speaker's bureau. However, there was no involvement in this study in any way. VKS has served as a consultant to U-Health, GlaxoSmithKline, Price Waterhouse Coopers, Rhonda Gray, Dane Garvin, Philips, ResMed, Sorin Inc., and is working with Mayo Health Solutions and their industry partners on intellectual property related to sleep and cardiovascular disease. The Mayo Foundation has received a gift from the Philips Respironics Foundation for the study of sleep and cardiovascular disease. MM receives an honorarium to serve as associate editor of Neurology Genetics. However, none of these entities were involved in this study in any way.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Allison Schultz for assistance with identifying cases.
1. Lemmers RJ, O'Shea S, Padberg GW, Lunt PW, van der Maarel SM. Best practice guidelines on genetic diagnostics of Facioscapulohumeral muscular dystrophy: workshop 9th June 2010 LUMC, Leiden, The Netherlands. Neuromusc Disord. (2012) 22:463–70. doi: 10.1016/j.nmd.2011.09.004
2. Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. (2010) 329:1650–3. doi: 10.1126/science.1189044
3. Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy. Continuum (Minneap Minn). (2016) 22:1916–31. doi: 10.1212/CON.0000000000000399
4. Nikolic A, Jones TI, Govi M, Mele F, Maranda L, Sera F, et al. Interpretation of the epigenetic signature of facioscapulohumeral muscular dystrophy in light of genotype-phenotype studies. Int J Mol Sci. (2020) 21:2635. doi: 10.3390/ijms21072635
5. Salsi V, Magdinier F, Tupler R. Does DNA methylation matter in FSHD? Genes. (2020) 11:258. doi: 10.3390/genes11030258
6. Gaillard M-C, Roche S, Dion C, Tasmadjian A, Bouget G, Salort-Campana E, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. (2014) 83:733–42. doi: 10.1212/WNL.0000000000000708
7. Sacconi S, Briand-Suleau A, Gros M, Baudoin C, Lemmers RJLF, Rondeau S, et al. FSHD1 and FSHD2 form a disease continuum. Neurology. (2019) 92:e2273–e85. doi: 10.1212/WNL.0000000000007456
8. Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. (2012) 44:1370–4. doi: 10.1038/ng.2454
9. van den Boogaard ML, Lemmers RJLF, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, et al. Mutations in DNMT3B modify epigenetic repression of the d4z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. (2016) 98:1020–9. doi: 10.1016/j.ajhg.2016.03.013
10. Rieken A, Bossler AD, Mathews KD, Moore SA. CLIA laboratory testing for facioscapulohumeral dystrophy: a retrospective analysis. Neurology. (2021) 96:e1054–e62. doi: 10.1212/WNL.0000000000011412
11. Hamanaka K, Šikrová D, Mitsuhashi S, Masuda H, Sekiguchi Y, Sugiyama A, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. (2020) 94:e2441–e7. doi: 10.1212/WNL.0000000000009617
12. Shaw ND, Brand H, Kupchinsky ZA, Bengani H, Plummer L, Jones TI, et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat Genet. (2017) 49:238–48. doi: 10.1038/ng.3743
13. Mul K, Lemmers RJLF, Kriek M, van der Vliet PJ, van den Boogaard ML, Badrising UA, et al. FSHD type 2 and Bosma arhinia microphthalmia syndrome. Two Faces Same Mutation. (2018) 91:e562–e70. doi: 10.1212/WNL.0000000000005958
14. Gordon CT, Xue S, Yigit G, Filali H, Chen K, Rosin N, et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat Genet. (2017) 49:249–55. doi: 10.1038/ng.3765
15. Preston MK, Tawil R, Wang LH. Facioscapulohumeral muscular dystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. Seattle, WA: GeneReviews((R) (1993).
16. Elliott P, Lambiase PD, Kumar D. Inherited Cardiac Disease (Oxford Specialist Handbooks in Cardiology). Oxford: Oxford University Press (2011), p. 425.
17. van Dijk GP, van der Kooi E, Behin A, Smeets J, Timmermans J, van der Maarel S, et al. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct Neurol. (2014) 29:159–65.
18. Tawil R, Kissel JT, Heatwole C, Pandya S, Gronseth G, Benatar M, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: report of the guideline development, dissemination, and implementation subcommittee of the american academy of neurology and the practice issues review panel of the american association of neuromuscular & electrodiagnostic medicine. Neurology. (2015) 85:357–64. doi: 10.1212/WNL.0000000000001783
19. Labombarda F, Maurice M, Simon JP, Legallois D, Guyant-Marechal L, Bedat-Millet AL, et al. Cardiac abnormalities in type 1 facioscapulohumeral muscular dystrophy. J Clin Neuromusc Dis. (2017) 18:199–206. doi: 10.1097/CND.0000000000000144
20. Laforet P, de Toma C, Eymard B, Becane HM, Jeanpierre M, Fardeau M, et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology. (1998) 51:1454–6. doi: 10.1212/WNL.51.5.1454
21. Trevisan CP, Pastorello E, Armani M, Angelini C, Nante G, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol. (2006) 56:1–5. doi: 10.1159/000094248
22. Galetta F, Franzoni F, Sposito R, Plantinga Y, Femia FR, Galluzzi F, et al. Subclinical cardiac involvement in patients with facioscapulohumeral muscular dystrophy. Neuromusc Disord. (2005) 15:403–8. doi: 10.1016/j.nmd.2005.02.006
23. Goselink RJM, Voermans NC, Okkersen K, Brouwer OF, Padberg GW, Nikolic A, et al. Early onset facioscapulohumeral dystrophy - a systematic review using individual patient data. Neuromusc Disord. (2017) 27:1077–83. doi: 10.1016/j.nmd.2017.09.007
24. Blaszczyk E, Grieben U, von Knobelsdorff-Brenkenhoff F, Kellman P, Schmacht L, Funk S, et al. Subclinical myocardial injury in patients with Facioscapulohumeral muscular dystrophy 1 and preserved ejection fraction - assessment by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. (2019) 21:25. doi: 10.1186/s12968-019-0541-8
25. Tsuji M, Kinoshita M, Imai Y, Kawamoto M, Kohara N. Facioscapulohumeral muscular dystrophy presenting with hypertrophic cardiomyopathy: a case study. Neuromuscul Disord. (2009) 19:140–2. doi: 10.1016/j.nmd.2008.11.011
26. Finsterer J, Stollberger C. Clinical or subclinical cardiac involvement in facioscapulohumeral muscular dystrophy. Neuromusc Disord. (2006) 16:61–2; author reply 2–3. doi: 10.1016/j.nmd.2005.10.002
27. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. (2009) 42:377–81. doi: 10.1016/j.jbi.2008.08.010
28. Hayek E, Gring CN, Griffin BP. Mitral valve prolapse. Lancet. (2005) 365:507–18. doi: 10.1016/S0140-6736(05)17869-6
29. Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. (1999) 18:695–706. doi: 10.1002/(SICI)1097-0258(19990330)18:6<695::AID-SIM60>3.0.CO;2-O
30. Brouwer OF, Padberg GW, Wijmenga C, Frants RR. Facioscapulohumeral muscular dystrophy in early childhood. Arch Neurol. (1994) 51:387–94. doi: 10.1001/archneur.1994.00540160085011
31. Bussink BE, Holst AG, Jespersen L, Deckers JW, Jensen GB, Prescott E. Right bundle branch block: prevalence, risk factors, and outcome in the general population: results from the Copenhagen City Heart Study. Eur Heart J. (2013) 34:138–46. doi: 10.1093/eurheartj/ehs291
32. Imanishi R, Seto S, Ichimaru S, Nakashima E, Yano K, Akahoshi M. Prognostic significance of incident complete left bundle branch block observed over a 40-year period. Am J Card. (2006) 98:644–8. doi: 10.1016/j.amjcard.2006.03.044
33. Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. (2007) 68:569–77. doi: 10.1212/01.wnl.0000251269.31442.d9
34. Della Marca G, Frusciante R, Scatena M, Dittoni S, Testani E, Vollono C, et al. Heart rate variability in facioscapulohumeral muscular dystrophy. Funct Neurol. (2010) 25:211–6.
35. Chamberlain AM, Gersh BJ, Alonso A, Chen LY, Berardi C, Manemann SM, et al. Decade-long trends in atrial fibrillation incidence and survival: a community study. Am J Med. (2015) 128:260–7.e1. doi: 10.1016/j.amjmed.2014.10.030
36. Stevenson WG, Perloff JK, Weiss JN, Anderson TL. Facioscapulohumeral muscular dystrophy: evidence for selective, genetic electrophysiologic cardiac involvement. J Am Coll Card. (1990) 15:292–9. doi: 10.1016/S0735-1097(10)80052-X
37. Freed LA, Levy D, Levine RA, Larson MG, Evans JC, Fuller DL, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. (1999) 341:1–7. doi: 10.1056/NEJM199907013410101
38. Sanyal SK, Johnson WW, Dische MR, Pitner SE, Beard C. Dystrophic degeneration of papillary muscle and ventricular myocardium. A basis for mitral valve prolapse in Duchenne's muscular dystrophy. Circulation. (1980) 62:430–8. doi: 10.1161/01.CIR.62.2.430
39. Osnabrugge RL, Mylotte D, Head SJ, Van Mieghem NM, Nkomo VT, LeReun CM, et al. Aortic stenosis in the elderly: disease prevalence and number of candidates for transcatheter aortic valve replacement: a meta-analysis and modeling study. J Am Coll Cardiol. (2013) 62:1002–12. doi: 10.1016/j.jacc.2013.05.015
40. Topilsky Y, Maltais S, Medina Inojosa J, Oguz D, Michelena H, Maalouf J, et al. Burden of tricuspid regurgitation in patients diagnosed in the community setting. JACC Cardiovasc Imaging. (2019) 12:433–42. doi: 10.1016/j.jcmg.2018.06.014
41. Chen T-H, Lai Y-H, Lee P-L, Hsu J-H, Goto K, Hayashi YK, et al. Infantile facioscapulohumeral muscular dystrophy revisited: Expansion of clinical phenotypes in patients with a very short EcoRI fragment. Neuromusc Disord. (2013) 23:298–305. doi: 10.1016/j.nmd.2013.01.005
Keywords: arrhythima, facioscapulohumeral dystrophy, FSHD, mitral valve prolapse, conduction
Citation: Ducharme-Smith A, Nicolau S, Chahal CAA, Ducharme-Smith K, Rehman S, Jaliparthy K, Khan N, Scott CG, St Louis EK, Liewluck T, Somers VK, Lin G, Brady PA and Milone M (2021) Cardiac Involvement in Facioscapulohumeral Muscular Dystrophy (FSHD). Front. Neurol. 12:668180. doi: 10.3389/fneur.2021.668180
Received: 15 February 2021; Accepted: 26 April 2021;
Published: 24 May 2021.
Edited by:
Edoardo Malfatti, INSERM U1179 Handicap neuromusculaire: Physiopathologie, Biothérapie et Pharmacologie appliquées (END-ICAP), FranceReviewed by:
Lucia Ruggiero, Università di Napoli Federico II, ItalyCopyright © 2021 Ducharme-Smith, Nicolau, Chahal, Ducharme-Smith, Rehman, Jaliparthy, Khan, Scott, St Louis, Liewluck, Somers, Lin, Brady and Milone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita Milone, bWlsb25lLm1hcmdoZXJpdGFAbWF5by5lZHU=; C. Anwar A. Chahal, Y2hhaGFsLmFud2FyQG1heW8uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.