 Irene Litvan1*
Irene Litvan1* James A. Proudfoot2Eden R. Martin3
James A. Proudfoot2Eden R. Martin3 David Standaert4David Riley5
David Standaert4David Riley5 Deborah Hall6Connie Marras7
Deborah Hall6Connie Marras7 Ece Bayram1Richard M. Dubinsky8
Ece Bayram1Richard M. Dubinsky8 Yvette Bordelon9
Yvette Bordelon9 Stephen Reich10David Shprecher11,12Benzi Kluger13Christopher Cunningham14Gerard D. Schellenberg15
Stephen Reich10David Shprecher11,12Benzi Kluger13Christopher Cunningham14Gerard D. Schellenberg15 Joseph Jankovic16 on behalf of ENGENE-PSP
Joseph Jankovic16 on behalf of ENGENE-PSP- 1Department of Neurosciences, Parkinson and Other Movement Disorders Center, University of California, San Diego, La Jolla, CA, United States
- 2Clinical and Translational Research Institute, University of California, San Diego, La Jolla, CA, United States
- 3John P. Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, FL, United States
- 4Department of Neurology, University of Alabama at Birmingham, Birmingham, AL, United States
- 5InMotion, Warrensville Heights, OH, United States
- 6Department of Neurological Sciences, Rush University, Chicago, IL, United States
- 7Morto and Gloria Shulman Movement Disorders Centre and the Edmond J. Safra Program in Parkinson's Research, Toronto Western Hospital, University of Toronto, Toronto, ON, Canada
- 8Department of General Neurology, University of Kansas Medical Center, Kansas City, KS, United States
- 9Department of Neurology, University of California, Los Angeles, Los Angeles, CA, United States
- 10Department of Neurology, University of Maryland School of Medicine, Baltimore, MD, United States
- 11Banner Sun Health Research Institute, Sun City, AZ, United States
- 12Department of Neurology, University of Utah, Salt City, UT, United States
- 13Department of Neurology, University of Colorado, Denver, CO, United States
- 14Division of Movement Disorders, Department of Neurology, University of Louisville School of Medicine, Louisville, KY, United States
- 15Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, United States
- 16Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, Houston, TX, United States
Several genetic and environmental factors have been reported in progressive supranuclear palsy (PSP), although none were identified as a definitive cause. We aimed to explore potential gene-environment interactions in PSP. Two hundred and ninety two PSP cases and 292 controls matched for age, sex, and race from the ENGENE-PSP were analyzed to determine the association between PSP and minor alleles of 5 single nucleotide polymorphisms (SNPs) in 4 genes (MAPT, MOBP, EIF2AK3, and STX6), which were previously associated with PSP risk. Interactions between these SNPs and environmental factors, including previously reported occupational and agricultural risk factors for PSP, were assessed for PSP odds and age of symptom onset. Minor alleles of MAPTrs242557 and EIF2AK3rs7571971 were individually associated with increased odds; MAPTrs8070723 minor alleles were associated with lower PSP odds. There were several gene-environment interactions for PSP odds and age of symptom onset, however, they did not remain significant after FDR-correction. Larger scale studies are required to determine potential interactions.
Introduction
Progressive Supranuclear Palsy (PSP) is the second most common cause of neurodegenerative parkinsonism after Parkinson's disease (1). First described in 1964, classic PSP is characterized by early postural instability, frontal cognitive disturbances, pseudobulbar palsy, and vertical supranuclear gaze palsy, preceded by slowing of vertical saccades (2). PSP results in rapid deterioration of quality of life, with median survival time estimated at 7–8 years (3, 4). Previous estimated prevalence was 5–6 cases per 100,000 (5), but recent studies show that true PSP prevalence is higher. Recent age-adjusted prevalence estimates in Europe are 8.8–10.8 cases per 100,000 (6, 7), and in Yonogo Japan, age-adjusted PSP prevalence increased from 5.8 cases per 100,000 in 1999 (8) to 17 cases per 100,000 in 2010 (9).
Few studies have investigated the possible genetic and environmental causes of PSP. Lower levels of education (10–12), drinking well-water, prior use of firearms associated with higher blood lead levels (13), and exposure to industrial metals (14) have been reported as environmental risk factors; whereas genetic risk factors include variants of MAPT, MOBP, EIF2AK3, and STX6 (15–18). None of these risk factors have been identified as a definitive cause of PSP, and PSP is more than likely caused by a complex interaction of genetic predisposition and environmental risk factors. However, there are no prior studies investigating gene-environment interactions for PSP risk. To address this gap, we designed a large, multi-center case-control study, Environmental-Genetic PSP (ENGENE-PSP), to begin exploring the interactions of genetic and environmental factors in PSP. We also assessed whether the gene-environment interactions impact age of symptom onset in PSP. Our previous report in this cohort showed that higher PSP odds were associated with more years of drinking well-water and not having a college degree (10). As our prior analysis focused specifically on environmental and occupational risk factors in the same cohort, we did not repeat the analysis for the environmental factors, but rather focused on genetic factors and gene-environment interactions.
Materials and Methods
Study Population
Cases and controls were recruited from 15 sites throughout North America between October 1, 2006 and February 1, 2013, as previously reported (10). Briefly, the sampling framework for the study included the catchment area surrounding each participating clinical center, and referrals from outside these areas were sent to their nearest participating site. Cases were confirmed to have PSP by the Principal Investigator of each participating site. To be included in the study, cases had to be diagnosed on-site within the past year and meet the NINDS-PSP Diagnostic Criteria for Clinically Probable or Clinically Possible PSP (2). Exclusion criteria included other central nervous system pathology, severe speech and cognitive impairment that could have interfered with recall of life events. Ninety-three percentage of cases had a Mini Mental State Examination score greater than 24. Majority of the PSP cases had PSP-Richardson's syndrome (n = 265, 90.8%) and there was a minority with PSP-parkinsonism (n = 17, 5.8%) (19).
Each subject was recruited together with two controls: an age (±10 years) and sex-matched non-blood relative (Control 1), and the subject's spouse or primary care partner (Control 2). To increase the sample size of this study we included both control 1 and control 2. Controls were age, race and sex-matched subjects, and although in-person examinations were not performed similar to the examination of cases, all controls screened negative for both parkinsonism and dementia using the Telephone Interview of Cognitive Status and Telephone Questionnaire for Parkinson's disease, respectively (20). Most cases were preferentially matched with their own recruited Control 1 (73.6%), with the remainder of cases being matched with either a different Control 1 (16.8%) or Control 2 (9.6%) (Figure 1). From an initial cohort of 350 PSP subjects (10), the analyses included 292 matched with 292 controls by race, sex, and age (±10 years, average 2.8 ± 2.5 years). Fifty-eight of the 350 cases were excluded from the analyses because of incomplete data (n = 47) or different race than the age and sex matched control (n = 11). The excluded cases were similar to the included cohort in age (68.76 ± 7.01, P = 0.125) (Table 1). However, compared to the included cases, the excluded cases had an almost-significantly larger proportion of men and were significantly different in terms of race, education level, and disease duration.
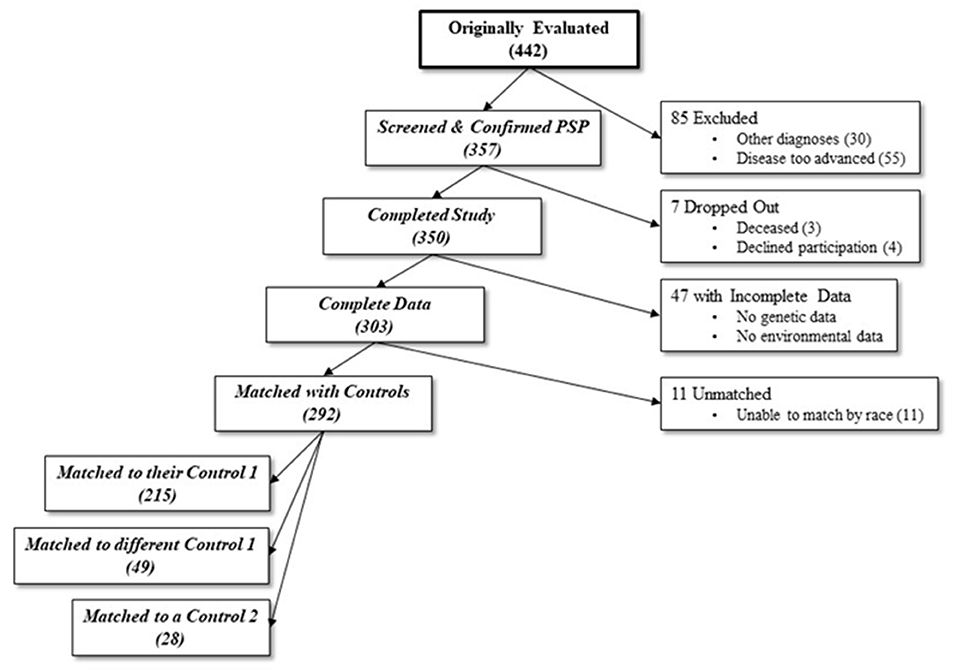
Figure 1. Number of participants at each stage of the study. From an initial sample of 442, 139 were excluded due to either incompatible diagnoses, too advanced disease, dropping out, or incomplete data. An additional 11 were excluded due to inability to match for race with a control. 292 final cases were matched with either their original control 1 (non-blood relative), a different case's control 1, or a different case's control 2 (spouse or caretaker).
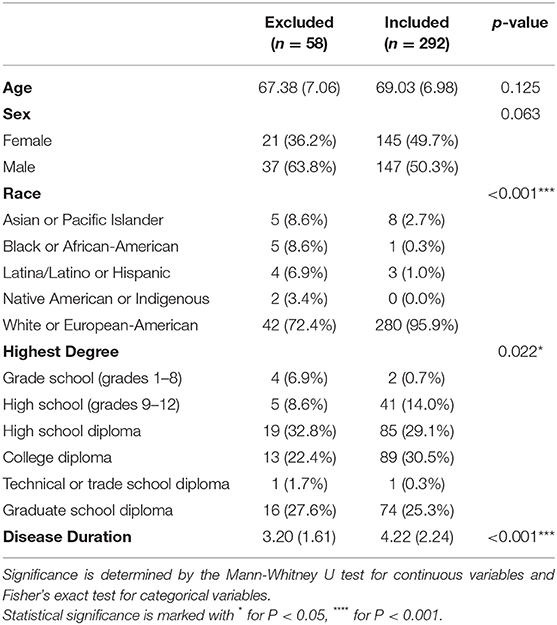
Table 1. Mean (standard deviation) and count (%) for demographic variables of included and excluded PSP cases.
Age of symptom onset was calculated from the first symptom (motor or non-motor PSP symptom) recalled by the patient and family. For controls, we used a “reference date” defined as the age of symptom onset of the matched case. PSP phenotypes of the cases were determined based on the Movement Disorders Society PSP clinical diagnostic criteria (19). This study was approved by the Institutional Review Board of each institution (IRB #111729), and each participant signed a written informed consent.
Genetic Analyses
PCR using TaqMan genotyping assays (Thermo Fisher) was used to determine expression of five single-nucleotide polymorphisms (SNPs) previously associated with PSP (17). Predesigned assays included C__1016016_1 for rs242557 (MAPT), C_29297996_10 for rs8070723 (MAPT), C__75367_10 for rs1768208 (MOBP), and C__20893_10 for rs7571971 (EIF2AK3). A custom assay (design ID AHGJ5AO, rs1411478) was used to genotype STX6, with the forward primer GGTAGGCAAAAGGTGCTATGGA, reverse primer GTCCCAGCACCCTGTCAA, reporter 1 sequence CCCAGAGAAGAAGAC, and reporter 2 sequence CCAGAGGAGAAGAC. Genotypes were recorded for each SNP and compiled into a database for further analysis. Genetic information was encoded for each genotype in terms of number of minor alleles counts per subject, with homozygous major alleles encoded as 0, heterozygous as 1, and homozygous minor alleles as 2 (Table 2). For example, for MAPT rs242557 A is the minor allele, thus, each subject A/A is encoded as 2, A/G as 1, and G/G 0.
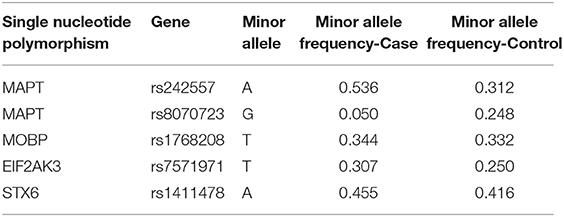
Table 2. Minor allele frequency by participant groups.
Environmental Factors
To assess occupational exposures to toxic substances, all participants were given a telephone occupational questionnaire modified from Stewart and colleagues (21), in which they listed all jobs held for over 6 months between the ages of 16 and 10 years before their PSP symptom onset (or reference date for controls). The methodology was previously reported in detail (10). Briefly, participants listed their company name, job section, position held, number of years in their position, duties, tools and equipment used, and possible chemical exposures. They also self-reported environmental exposures to various substances, including organic solvents, pesticides, herbicides, fungicides, and other chemicals. Due to usual inaccuracies with self-reported work-related toxic exposures (under or over-reporting exposures), we selected a priori a more objective assessment by an independent team consisting of a toxicologist and an industrial hygienist to review the reported lifetime occupational data and assign their own estimates of occupational exposures to chemicals in general, metals, pesticides, and organic solvents, blinded to case/control status (10). Due to the self-reported nature of the data, it wasn't possible to accurately quantify toxic exposure; exposures were instead listed as a binary yes/no values. Levels of exposure were defined as “high” for directly working with the chemical or “low” for exposure via a proximate worker working with the chemical (i.e., manager).
Other recorded environmental exposures included residential history, well-water intake, history of having lived one mile of an agricultural area, hobbies, family history of neurological disorders, military history and exposures, and specifics regarding gardening and lawn care (10).
Statistical Analyses
All analyses were done with the R statistical programming language (version 3.6.1). We fitted conditional logistic regression models to account for the matched structure of the data using case/control status as an outcome. Two regressions were fitted; univariate models for each genetic factor, and a model which included the interactions for each genetic and environmental factor. For age at symptom onset, we used a simple linear regression and fitted two models in a similar fashion as the conditional logistic regression analysis. Model diagnostics (residual plots, qq-plots) were performed to ensure that the assumptions of the linear models were met by the data. All models were re-ran to include age, race, and sex as covariates; and the results did not change. A p-value of less than 0.05 was suggestive of significance for this study in view of evaluating the previously 5 significant SNPs. A false discovery rate (FDR) p-value correction separately for outcomes was also performed to maintain a type I error rate of 5%. Power calculation was based on detecting a significant variation in a continuous factor across genetic/case groups. With a total sample size of 584, we had an 80% power to detect an effect size (Cohen's F) of 0.02, which has previously been described as small (22).
Results
Demographics
Cases and controls were similar in age, sex, and race, reflecting the matched case-control design of this study. Overall, mean age was 69.01 years, 50.3% of the participants were male, and 95.9% were white or European-American (Table 3). Majority of the participants received a college/trade school degree or higher, and controls had higher degrees than cases (P < 0.001). Mean (standard deviation) disease duration was 4.22 (2.24) years for PSP cases.
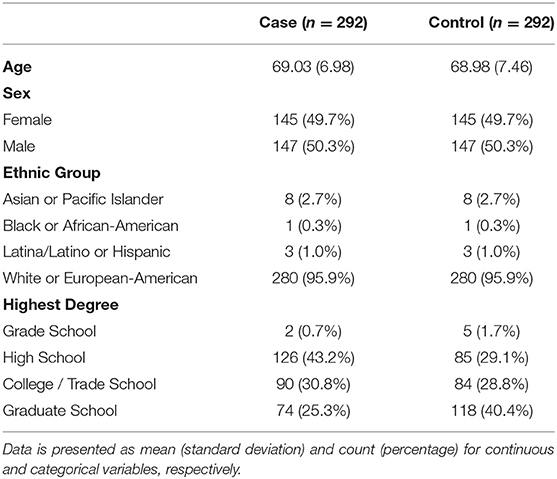
Table 3. Demographics by case status.
Genetic Associations With PSP
In univariate analysis, the minor allele of MAPTrs242557 (A) (a marker tagging the H1c sub-haplotype) and the minor allele of EIF2AK3rs7571971 (T) were significantly associated with increased odds for PSP, while the minor allele of MAPTrs8070723 (G) was associated with lower odds for PSP (Table 4).
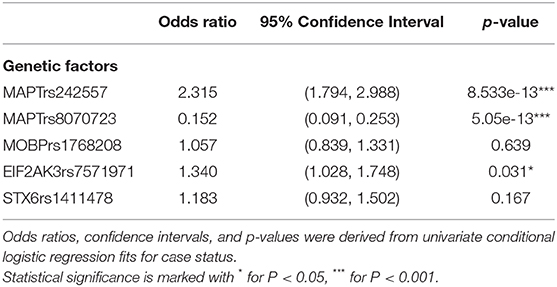
Table 4. Genetic factors associated with PSP risk.
Gene-Environment Interactions Associated With PSP
Two gene-environment interactions were significantly associated with PSP; however these interactions did not remain significant after FDR correction. Interactions between minor allele of EIF2AK3 and direct chemical exposure, as well as minor allele of EIF2AK3 and years of metal exposure were associated with increased odds for PSP. Participants who carried the minor allele of EIF2AK3 (T) and had direct chemical exposure had almost two times increased odds of PSP compared to those who did not have the minor allele and direct chemical exposure (OR = 1.91, P = 0.023, PFDR−corrected =0.71). For every year of metal exposure in individuals with the minor allele of EIF2AK3 (T), the odds of PSP increased by 8.5%, compared to those without EIF2AK3 minor allele (OR = 1.85, P = 0.049, PFDR−corrected =0.71). No significant gene-environment interactions were found for SNPs in MAPT, STX6, or MOBP.
Age of Symptom Onset
Those with the minor allele of MAPTrs8070723 had on older age of symptom onset (P = 0.029); for each additional count of minor allele (AA → AG → GG), the mean age of onset increased by 2.825 years. Other SNPs were not associated with age of symptom onset (P > 0.89 for all). Two gene-environment interactions were significantly associated with age of onset, although they did not remain significant after FDR correction. For each increased count of MAPTrs242557 minor allele (GG → AG → AA) and 1 year of living within 1 mile of agriculture, the age of onset decreased by 0.724 (P = 0.033, PFDR−corrected = 0.68) (Figure 2). For each increased count of STX6rs1411478 minor allele (GG → AG → AA) and high levels of organic solvent, the age of onset decreased by 2.889 (P = 0.042, PFDR−corrected =0.68) (Figure 3).
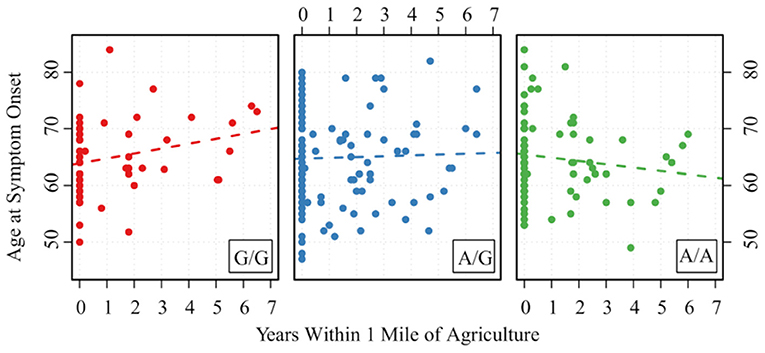
Figure 2. Association between age of PSP symptom onset and the interaction of MAPTrs242557 and years of living within 1 mile of agriculture. A is the minor allele, G is the major allele for MAPTrs242557; G/G corresponds to 0 minor allele, A/G corresponds to 1 minor allele, and A/A corresponds to 2 minor alleles.
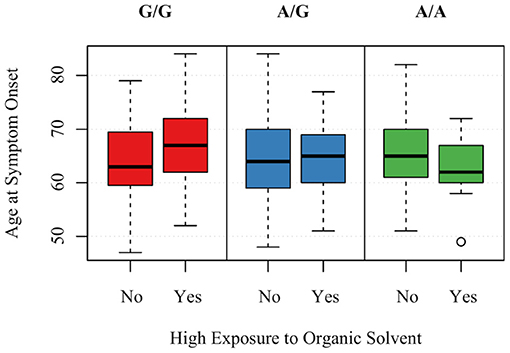
Figure 3. Association between age of PSP symptom onset and the interaction of STX6rs1411478 and exposure to organic solvents. A is the minor allele, G is the major allele for STX6rs1411478; G/G corresponds to 0 minor allele, A/G corresponds to 1 minor allele, A/A corresponds to 2 minor alleles.
Discussion
The ENGENE-PSP study used a case-control design to explore the role of gene-environment interactions in the development of PSP and provide preliminary findings for future studies. We hypothesized that sources of oxidative stress, when added to the genetic susceptibility of the MAPT H1 haplotype, may contribute to the development of PSP. Additionally, we hypothesized that sources of endoplasmic reticulum (ER) stress such as cigarette smoke and pesticides might, in conjunction with the EIF2AK3 genetic predisposition, contribute to PSP pathogenesis. We also assessed whether the associations between MOBP, STX6, and PSP would strengthen when combined with an environmental variable.
We found that minor alleles at MAPTrs242557 and EIF2AK3rs7571971 were individually associated with increased odds of developing PSP, and minor alleles of MAPTrs8070723 showed association with lower odds of developing PSP and an older age of symptom onset. The MAPT association with PSP has been long documented (15–17), but this study demonstrated that the association extends beyond the presence or absence of disease, including age of symptom onset. Additionally, prior studies have implicated EIF2AK3 in PSP, but with much larger sample sizes (17); it is interesting that this association remained strong in this study despite the smaller sample size.
We found several potential gene-environment interactions that will require further analysis, given they did not survive FDR-corrections. In unadjusted analyses, combinations of the minor allele of EIF2AK3 and direct chemical exposure, as well as minor allele of EIF2AK3 and years of metal exposure were associated with an increased risk for PSP compared to those without minor alleles of EIF2AK3 and the mentioned exposures. Additionally, the combination of MAPTrs242557 minor allele and years of living within 1 mile of agriculture, as well as STX6rs1411478 minor allele and high levels of organic solvent were associated with an earlier age of symptom onset compared to those without these genetic factors and environmental/occupational exposures.
EIF2AK3 is a gene that encodes PERK, an integral component of the ER unfolded protein response (UPR) (23). Various environmental factors can induce ER stress and activate the UPR, including formaldehyde (a substance in cigarette smoke), alcohol, and paraquat (an ingredient in herbicides) (24). Caparros-Lefebvre et al., reported a cluster of PSP patients in a city with severe environmental contamination by industrial metals in the north of France (14). Therefore, it is not unexpected to find an association between this gene, chemical and metal exposure, and the development of PSP. Finding synergy between genes and environment shown by the association between STX6 and organic solvents for age at symptom onset, despite lack of association between organic solvents and PSP in our previous study in the ENGENE-PSP sample (10) is particularly interesting. However, given the large number of associations tested, we cautiously interpret these associations, but deem them worthy of further investigation in future studies.
The unfavorable effect of combining a MAPT allele with years living within 1 mile of agriculture fits well into the larger narrative of PSP research on etiopathogenesis. MAPT has been well-documented as a significant predisposing gene in PSP, and oxidative stress has been shown to cause tau accumulation (25). Research has implicated various aspects of agriculture as environmental risk factors for PSP development, and though this study did not directly find an association between MAPT and exposure to any specific agents, this combined analysis of MAPT and living within a mile of agriculture does lend further support to a possible interaction (10).
Despite this study being the first to suggest possible gene-environment interactions in PSP, most of these associations did not achieve significance using multivariate analysis. This likely reflects the limited sample size of the study population; although this is the largest case-control study of PSP conducted to date, larger cohorts will be needed to fully examine these interactions. We included five SNPs previously reported to increase the PSP risk in our analysis, and other SNPs reported in PSP (18, 26) should be explored in future studies. Additionally, true gene-environment interactions are likely due to more specific exposures, which we were unable to delineate in this study (i.e., a particular herbicide rather than herbicides in general). Amount of exposure to risk factors also needs to be taken into account as PSP risk can be dose-dependent. Future studies should consider quantification of exposure in association with disease risk. Race is an important factor in genetic studies, as different allele frequencies are observed across different ethnicities (27) and the same SNP may not have the same impact in different races (28). Although we attempted to control for the confounding impact of race in our sample and found the results unchanged, the majority of our sample consisted of White or European-Americans (95.9%) and future studies with more diverse samples are required to determine the race impact for risk factors in PSP. Given that majority of PSP cases had PSP-Richardson's syndrome and a very low number of cases had PSP-parkinsonism, we were unable to assess the gene-environment associations with specific phenotypes in PSP. This is an important point to assess in the future as a GWAS study has suggested TRIM11 locus as a genetic modifier of PSP phenotype (29). Although there is still a need to determine the clinical utility of biomarkers for PSP, future studies would also benefit from incorporating neuroimaging, biofluid and electrophysiological biomarkers [e.g., PET with tau-specific ligands, cerebrospinal fluid tau RT-QuIC (30, 31)] to provide more insight to gene-environment interactions and to strengthen the findings in clinically-diagnosed patients.
In terms of data collection, excluded cases had a significantly shorter disease duration than the included cohort, which may have affected disease severity. For our analysis focusing on gene-environment interactions, we matched cases and controls for race to account for the differences in environmental exposures across different racial groups in the U.S (32). The excluded cases also differed from those included in this study in terms of education and race. The significantly increased proportion of minorities in the excluded cases reflected the challenges of matching such patients with controls for race. These differences in education and race between the included and excluded cases may affect the overall generalizability of the study. Since nearly 10% of the controls in this study were drawn from the spouses or care partners of other participants, this could have artificially masked environmental differences that may have been seen if controls were truly chosen at random. Age of symptom onset was determined based on patient and family report, which may have led to an unreliable estimation of onset as subtle or non-specific symptoms associated with PSP may have been unnoticed before impacting daily life activities significantly. However, the strengths of this study lie in its overall large sample size, careful matching of controls by age, sex, and race, and thorough assessment of several complex genes and environmental exposures that could contribute to PSP pathogenesis.
This case-control, multicenter study is the largest to date to assess gene-environment interactions in PSP and will pave the way for future research into these complex associations. Our explorative study indicates potential interactions between EIF2AK3rs7571971 minor allele and direct chemical exposure, and EIF2AK3rs7571971 minor allele and years of metal exposure for PSP odds, and between MAPTrs242557 minor allele and years of living within 1 mile of agriculture, and STX6rs1411478 minor allele and high levels of organic solvent for age of symptom onset. Studies of larger cohorts are needed to confirm these results and to identify additional interactions. Understanding the specific gene-environment interactions that lead to the development of PSP is integral for our overall understanding of this disease, and for future therapeutic and preventive interventions.
Data Availability Statement
The raw data supporting the conclusions of this article are available from the corresponding author upon reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Boards of each participating institution (IRB #111729). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
IL: conceptualization, obtaining the funding, data collection, and drafting the manuscript. EM, DSt, DR, DH, CM, RD, YB, SR, DSh, BK, CC, GS, and JJ: collection of data. JP and EB: data analysis, and drafting the manuscript. All authors contributed to the interpretation of data and critical review of the manuscript.
Funding
The ENGENE-PSP study was funded by US National Institutes of Health/ National Institute on Aging (NIA) with the grant number 5R01AG024040. The NIA had no role in the study design, data collection, data analysis and interpretation, or writing of this report.
Conflict of Interest
IL supported by the National Institutes of Health grants: 2R01AG038791-06A, U01NS090259, U01NS100610, U01NS80818, R25NS098999, P20GM109025; U19 AG063911-1; 1R21NS114764-01A1; Michael J Fox Foundation, Lewy Body Association, Abbvie, Biogen, Centogene, Roche, EIP-Pharma and Biohaven Pharmaceuticals. She was member of a Lundbeck Advisory Board. She receives her salary from the University of California San Diego and as Chief Editor of Frontiers in Neurology. DSt supported by the Abbvie, Inc., the American Parkinson Disease Association, the Michael J. Fox Foundation for Parkinson Research, Alabama Department of Commerce, the Department of Defense, and NIH grants P50NS108675, R25NS079188, and T32NS095775. He has a clinical practice and is compensated for these activities through the University of Alabama Health Services Foundation. He has served as a consultant for or received honoraria from Abbvie Inc., Sutter Health, the International Parkinson Disease and Movement Disorder Society, Theravance, McGraw Hill, and Sanofi-Aventis. He is a member of the faculty of the University of Alabama at Birmingham and is supported by endowment and University funds. CM received research funding from The Michael J Fox Foundation, Canadian Institutes of Health Research, Parkinson's Foundation (US), International Parkinson and Movement Disorders Society. She is employed by University Health Network, contracted by Grey Matter Technologies, and receives financial compensation as a steering committee member from the Michael J Fox Foundation. DSh employed by Banner Health, received research support from the Arizona Alzheimer's Consortium, Abbvie, Acadia, Aptinyx, Axovant, Biogen, Eisai, Eli Lilly, Enterin, Neurocrine, Michael J Fox Foundation, NIH, Nuvelution, Theravance and Teva; consultant fees from Amneal, Forensis and Neurocrine; speaker honoraria from Acorda, Neurocrine, Sunovion, Teva and US World Meds.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the participants and their caregivers who kindly gave their time to participate this study. They would also like to thank Dr. Peter Lees for his significant help in modifying the Stewart questionnaire; Susan Slifer and Evadnie Rampersaud for assisting in early statistical analysis; Dr. Laura B. Robertson for her contribution to the earlier drafts of the manuscript. ENGENE-PSP Consortium Site Investigators and coordinators who recruited 9 patients or more, listed according to number of cases and controls recruited: Irene Litvan, MD and Cassandra Shepherd, MPH from the University of Louisville; Jorge Juncos, MD from Emory University; David Riley, MD from Case Western Reserve University; Yvette Bordelon, MD, PhD and Arik Johnson, PsyD from UC Los Angeles; Stephen Reich, MD and Katherine Holmes, MPH from the University of Maryland; Deborah Hall, MD, PhD from Rush University and University of Colorado; David Standaert, MD, PhD and Rebecca McMurray, RN from the University of Alabama in Birmingham; David Shprecher, DO, MSci and Paola Wall from the University of Utah; Connie Marras, MD, PhD and Brandon Rothberg from the University of Toronto; Benzi Kluger, MD and Carol Hennessy, NP, MSN from the University of Colorado and Joseph Jankovic, MD and Christine Hunter, RN, BSN from Baylor College of Medicine. We also thank the referral of fewer than 9 patients from the ENGENE Co-Investigators: Richard Dubinsky, MD, MPH, Kansas University; Claire Henchcliffe MD, DPhil, Cornell University; Ryan Uitti, MD, Mayo Clinic Jacksonville and James Leverenz, MD, University of Washington.
References
1. Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. (1999) 354:1771–5. doi: 10.1016/S0140-6736(99)04137-9
2. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. (1996) 47:1–9. doi: 10.1212/WNL.47.1.1
3. Chiu WZ, Kaat LD, Seelaar H, Rosso SM, Boon AJW, Kamphorst W, et al. Survival in progressive supranuclear palsy and frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2010) 81:441–5. doi: 10.1136/jnnp.2009.195719
4. Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain. (2007) 130:1552–65. doi: 10.1093/brain/awm032
5. Nath U, Ben-Shlomo Y, Thomson RG, Morris HR, Wood NW, Lees AJ, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. (2001) 124:1438–49. doi: 10.1093/brain/124.7.1438
6. Coyle-Gilchrist ITS, Dick KM, Patterson K, Rodríquez PV, Wehmann E, Wilcox A, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. (2016) 86:1736–43. doi: 10.1212/WNL.0000000000002638
7. Fleury V, Brindel P, Nicastro N, Burkhard PR. Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Park Relat Disord. (2018) 54:30–39. doi: 10.1016/j.parkreldis.2018.03.030
8. Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord. (2004) 19:1239–40. doi: 10.1002/mds.20149
9. Takigawa H, Kitayama M, Wada-Isoe K, Kowa H, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago: change throughout a decade. Brain Behav. (2016) 6:e00557. doi: 10.1002/brb3.557
10. Litvan I, Lees PSJ, Cunningham CR, Rai SN, Cambon AC, Standaert DG, et al. Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord. (2016) 31:644–52. doi: 10.1002/mds.26512
11. Vidal J-S, Vidailhet M, Derkinderen P, de Gaillarbois TD, Tzourio C, Alpérovitch A. Risk factors for progressive supranuclear palsy: a case-control study in France. J Neurol Neurosurg Psychiatry. (2009) 80:1271–4. doi: 10.1136/jnnp.2008.149849
12. Golbe LI, Rubin RS, Cody RP, Belsh JM, Duvoisin RC, Grosmann C, et al. Follow-up study of risk factors in progressive supranuclear palsy. Neurology. (1996) 47:148–54. doi: 10.1212/WNL.47.1.148
13. Kelley KD, Checkoway H, Hall DA, Reich SG, Cunningham C, Litvan I. Traumatic brain injury and firearm use and risk of progressive supranuclear palsy among veterans. Front Neurol. (2018) 9:474. doi: 10.3389/fneur.2018.00474
14. Caparros-Lefebvre D, Golbe LI, Deramecourt V, Maurage CA, Huin V, Buée-Scherrer V, et al. A geographical cluster of progressive supranuclear palsy in northern France. Neurology. (2015) 85:1293–300. doi: 10.1212/WNL.0000000000001997
15. Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. (1999) 8:711–5. doi: 10.1093/hmg/8.4.711
16. Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. (2015) 6:7247. doi: 10.1038/ncomms8247
17. Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. (2011) 43:699–705. doi: 10.1038/ng.859
18. Sanchez-Contreras MY, Kouri N, Cook CN, Serie DJ, Heckman MG, Finch NA, et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener. (2018) 13:37. doi: 10.1186/s13024-018-0267-3
19. Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. (2017) 32:853–64. doi: 10.1002/mds.26987
20. Rocca WA, Maraganore DM, McDonnell SK, Schaid DJ. Validation of a telephone questionnaire for Parkinson's disease. J Clin Epidemiol. (1998) 51:517–23. doi: 10.1016/S0895-4356(98)00017-1
21. Stewart PA, Stewart WF, Heineman EF, Dosemeci M, Linet M, Inskip PD. A novel approach to data collection in a case-control study of cancer and occupational exposures. Int J Epidemiol. (1996) 25:744–52. doi: 10.1093/ije/25.4.744
22. Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale, NJ: Lawrence Erlbaum Associates (1988).
23. Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. (2006) 65:348–57. doi: 10.1097/01.jnen.0000218445.30535.6f
24. Kitamura M. The unfolded protein response triggered by environmental factors. Semin Immunopathol. (2013) 35:259–75. doi: 10.1007/s00281-013-0371-y
25. Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear Tau, a key player in neuronal DNA protection. J Biol Chem. (2011) 286:4566–75. doi: 10.1074/jbc.M110.199976
26. Chen JA, Chen Z, Won H, Huang AY, Lowe JK, Wojta K, et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener. (2018) 13:41. doi: 10.1186/s13024-018-0270-8
27. Cross DS, Ivacic LC, Stefanski EL, McCarty CA. Population based allele frequencies of disease associated polymorphisms in the Personalized Medicine Research Project. BMC Genet. (2010) 11:51. doi: 10.1186/1471-2156-11-51
28. Camuzat A, Romana M, Dürr A, Feingold J, Brice A, Ruberg M, et al. The PSP-associated MAPT H1 subhaplotype in Guadeloupean atypical parkinsonism. Mov Disord. (2008) 23:2384–91. doi: 10.1002/mds.22297
29. Jabbari E, Woodside J, Tan MMX, Shoai M, Pittman A, Ferrari R, et al. Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann Neurol. (2018) 84:485–96. doi: 10.1002/ana.25308
30. Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. (2017) 16:552–63. doi: 10.1016/S1474-4422(17)30157-6
31. Saijo E, Metrick MA, Koga S, Parchi P, Litvan I, Spina S, et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. (2020) 139:63–77. doi: 10.1007/s00401-019-02080-2
Keywords: progressive supranuclear palsy, gene, environment, epidemiology, risk factors
Citation: Litvan I, Proudfoot JA, Martin ER, Standaert D, Riley D, Hall D, Marras C, Bayram E, Dubinsky RM, Bordelon Y, Reich S, Shprecher D, Kluger B, Cunningham C, Schellenberg GD and Jankovic J (2021) Gene-Environment Interactions in Progressive Supranuclear Palsy. Front. Neurol. 12:664796. doi: 10.3389/fneur.2021.664796
Received: 06 February 2021; Accepted: 15 March 2021;
Published: 09 April 2021.
Edited by:
Steven Frucht, Mount Sinai Hospital, United StatesReviewed by:
Daniele Belvisi, Istituto Neurologico Mediterraneo Neuromed (IRCCS), ItalyPratap Chand, Saint Louis University, United States
Copyright © 2021 Litvan, Proudfoot, Martin, Standaert, Riley, Hall, Marras, Bayram, Dubinsky, Bordelon, Reich, Shprecher, Kluger, Cunningham, Schellenberg and Jankovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Litvan, aWxpdHZhbkB1Y3NkLmVkdQ==