 Brandon M. Fox
Brandon M. Fox Kirsten B. Dorschel
Kirsten B. Dorschel Michael T. Lawton
Michael T. Lawton John E. Wanebo
John E. Wanebo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 03 September 2021
Sec. Stroke
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.661578
This article is part of the Research TopicMoyamoya DiseaseView all 12 articles
Moyamoya disease (MMD) and moyamoya syndrome (MMS) are progressive vascular pathologies unique to the cerebrovasculature that are important causes of stroke in both children and adults. The natural history of MMD is characterized by primary progressive stenosis of the supraclinoid internal carotid artery, followed by the formation of fragile collateral vascular networks. In MMS, stenosis and collateralization occur in patients with an associated disease or condition. The pathological features of the stenosis associated with MMD include neointimal hyperplasia, disruption of the internal elastic lamina, and medial attenuation, which ultimately lead to progressive decreases in both luminal and external arterial diameter. Several molecular pathways have been implicated in the pathophysiology of stenosis in MMD with functions in cellular proliferation and migration, extracellular matrix remodeling, apoptosis, and vascular inflammation. Importantly, several of these molecular pathways overlap with those known to contribute to diseases of systemic arterial stenosis, such as atherosclerosis and fibromuscular dysplasia (FMD). Despite these possible shared mechanisms of stenosis, the contrast of MMD with other stenotic pathologies highlights the central questions underlying its pathogenesis. These questions include why the stenosis that is associated with MMD occurs in such a specific and limited anatomic location and what process initiates this stenosis. Further investigation of these questions is critical to developing an understanding of MMD that may lead to disease-modifying medical therapies. This review may be of interest to scientists, neurosurgeons, and neurologists involved in both moyamoya research and treatment and provides a review of pathophysiologic processes relevant to diseases of arterial stenosis on a broader scale.
Moyamoya disease (MMD) and moyamoya syndrome (MMS) are cerebrovascular pathologies that are characterized by progressive stenosis and eventual occlusion of the intracranial internal carotid artery (ICA) and its proximal middle cerebral artery (MCA) and anterior cerebral artery (ACA) branches. This stenosis and occlusion cause ischemia and subsequent small vessel collateralization of the brain parenchyma that is supplied by the affected arterial segments. On cerebral angiography, these collateral vascular networks are visible as regions of contrast blush in the early arterial phase, and this distinctive appearance led to the name “moyamoya,” which is Japanese for a puff of smoke (1).
When the term moyamoya is used alone, it refers to the characteristic pathologic findings of stenosis and collateralization. Moyamoya disease refers to primary moyamoya and includes this presentation in individuals who harbor the genetic risk alleles. Moyamoya syndrome refers to moyamoya that occurs in the setting of a known associated disease, condition, or exposure (2). Previously, unilateral moyamoya was classified as MMS, but this categorization has fallen out of favor due to mounting evidence that unilateral pathology frequently progresses to bilateral disease (3–8).
The most common presenting symptom of MMD and MMS is ischemic stroke, which occurs secondary to decreased perfusion downstream from moyamoya stenosis. Additionally, moyamoya frequently presents with transient ischemic attacks (TIAs), which are often recurrent, and less commonly presents with seizures or headaches (9). Intracranial hemorrhage also occurs in MMD and MMS due to the rupture of vessels in fragile collateral networks or associated aneurysms (10), and importantly, hemorrhage occurs as the presenting symptom more frequently as patient age increases (11). In 2018, Funaki et al. demonstrated that choroidal collaterals may be a bleeding source with a high risk for hemorrhagic recurrence and a predictor of recurrence in hemorrhagic MMD (12). Preliminary results obtained in 2019 by Funaki et al. (13) suggest that the presence of choroidal collaterals may affect the risk of de novo hemorrhage in nonhemorrhagic hemispheres; however, these results remain subject to verification in larger studies.
Moyamoya disease and MMS are relatively rare and occur with varying incidence in populations throughout the world. Incidence rates are highest in eastern Asia, and specifically Japan, where its incidence is reported at 0.35 per 100,000 individuals (14). By contrast, the overall incidence reported from the United States is 0.09 per 10,000 individuals, with the highest incidence occurring in Asian Americans (0.28/100,000), followed by African Americans (0.13/100,000), Caucasian Americans (0.06/100,000), and Hispanic Americans (0.03/100,000) (15). The age of onset occurs with a bimodal distribution, with an initial peak in the first decade of life and a second peak in the fifth decade, and studies suggest that the incidence in adults may be increasing (14, 16, 17).
Genetic risk factors may be involved in the development of MMD, and this idea is supported by differences in the incidence of MMD in different populations. Further support for inherited risk was provided by familial cases and a high concordance between monozygotic twins (18). However, incomplete penetrance and discordance have led some to suggest the necessity of an environmental “second hit” or possible epigenetic contribution (19, 20). Genetic linkage analysis from sequencing data identified MMD risk alleles and ultimately led to the identification of RNF213 as the first known susceptibility gene for MMD (21, 22). Despite this progress, the definitive mechanisms through which mutant RNF213 contributes to the initiation or progression of moyamoya pathology remain elusive. In 2012, Miyawaki et al. (23) performed a case-control study at a single hospital analyzing the RNF213 variant c.14576G>A in patients with non-MMD intracranial major artery stenosis/occlusion (ICASO). The authors found an association between c.14576G>A and non-MMD ICASO. They therefore advocate screening for the c.14576G>A variant in ICASO patients. In 2013, this group performed a case–control study, in which they investigated the occurrence rate of the c.14576G>A variant in 323 ICASO patients (24). They hypothesized that a subset of patients with distinct ICASO phenotypes may have a common genetic variant, RNF213 c.14576G>A, suggesting that the RNF213 c.14576G>A variant may be a high-risk allele for ICASO. In 2015, this group performed a study in 78 MMD patients and nine non-atherosclerotic MMS patients demonstrating the absence of the RNF213 c.14576G.A genetic variant in nonatherosclerotic MMS patients (25). The research group hypothesized that identification of this genetic variant could be used to help clarify the pathophysiology of vascular stenosis in the terminal ICA and associated moyamoya vessels.
Since MMD was first described in 1957, research efforts have significantly increased our understanding of its epidemiology and pathobiology. Despite this increased knowledge, medical treatments do not reverse or prevent progression of the primary disease process. In addition, definitive therapy with revascularization, although effective in reducing stroke risk and symptoms, has no effect on the progression of arterial stenosis (26, 27). Current moyamoya treatment is hindered by the lack of understanding of both the pathology process that initiates stenosis and the key signaling pathways that drive progression. In this review, we examine the unique arterial stenosis that occurs in moyamoya and explore the current state of knowledge related to its pathological characteristics, its molecular mechanisms, and the imaging methods utilized for its detection.
References were identified by use of a comprehensive, systematic computerized literature search from January 1, 1957, through January 20, 2021, performed by the authors, using the PubMed, EMBASE, BIOSIS Previews, and Medline databases and various combinations of the key words “angiopathy,” “cerebrovascular,” “moyamoya,” “stenosis,” and “stroke.” Relevant articles on MMD and supplemental basic science articles almost exclusively published in English were included. Reference lists of relevant articles were reviewed for additional references, and 165 articles were included in the final manuscript. As some aspects of MMD and MMS have been studied in greater detail compared to others, several topics receive additional attention. In spite of major progress in research in the field of MMD in recent years, the literature in great part remains descriptive. Continued collaborative clinical and basic research is fundamental to further elucidate the pathophysiology of MMD, which may lead to an increasingly differentiated diagnosis and disease-modifying treatment strategies.
The arterial stenosis of moyamoya is the primary process that sets in motion the cascade of pathological adaptations and complications that encompass MMD and MMS. Isolated stenosis without evidence of collateral vessel formation occurs first and is defined as the first of six stages in the progression of the disease (1). In most moyamoya patients, stenosis occurs in the bilateral proximal portions of the anterior cerebral circulation and involves the terminal supraclinoid ICA and the proximal MCA and ACA (1, 28). The posterior circulation is spared in most moyamoya patients, despite possessing intracranial arteries with similar caliber and proximity to affected segments of the anterior circulation. Nevertheless, involvement of the posterior circulation can occur in moyamoya (29), and studies have demonstrated posterior circulation disease in approximately one-third of pediatric and adult patients (30–33). When present, stenosis of the posterior circulation typically affects the posterior communicating artery and the P2 segment of the posterior cerebral artery (PCA), while sparing the basilar artery and P1 segment of the PCA (34). Thus, posterior involvement is likely an extension of the stenotic process from the anterior circulation and only rarely arises independently from the proximal posterior circulation, as characterized by basilar artery and P1 segment involvement. The unique properties of posterior circulation moyamoya have led investigators to suggest an embryologic basis for susceptibility of the arterial segments to develop moyamoya (34, 35). Importantly, several lines of evidence suggest that the involvement of the posterior circulation in MMD is associated with a poor prognosis (36–38). Stenosis of extracranial vessels in association with intracranial moyamoya is uncommon, but several studies have demonstrated associated stenosis of the renal artery in a minority of patients (39, 40). In addition, a few studies have demonstrated associated stenosis of external carotid artery branches, coronary arteries, and abdominal arteries (41, 42). However, other studies have specifically looked for involvement of branches of the external carotid artery or intraabdominal arteries and have failed to demonstrate stenosis in these segments (40, 43). Despite these exceptions, the restriction of moyamoya stenosis to such a limited and specific anatomic region remains one of the most fundamental questions surrounding its pathology, as other forms of medium to large artery stenosis, such as atherosclerosis and fibromuscular dysplasia (FMD), are typically far more widespread throughout the arterial tree.
The histopathologic characteristics of stenosis in moyamoya represent a collection of pathologic processes that, in addition to the gross pathologic features described above, define moyamoya as a unique form of arterial stenosis. The primary cause of moyamoya stenosis is a concentric fibrocellular hyperplasia of the intima (44, 45). This hyperplasia is characterized by a proliferation of smooth muscle cells and extracellular matrix within the intima that leads to progressive intimal thickening (Figure 1) (46–49). In addition, the internal elastic lamina is altered such that it is frequently wavy, duplicated, and sometimes disrupted (45, 50–52). In contrast to the significant intimal thickening that occurs in moyamoya, the tunica media becomes progressively thinner as moyamoya progresses (46). This thinning occurs to the degree that, despite intimal expansion, the outer vessel diameter decreases in association with luminal stenosis (53, 54), and as moyamoya advances over time, the outer diameter progressively decreases (Figure 1) (55). In 2016, Yamamoto et al. suggested that MMS may comprise two distinct pathological subgroups, a constrictive subgroup and a nonconstrictive subgroup. Patients in the constrictive subgroup of MMS may show marked constrictive vascular changes and limited intimal hyperplasia. Patients in the non-constrictive subgroup of MMS may show profuse fibrosis (56). Luminal thrombi and microthrombi are frequently observed in autopsy specimens from moyamoya patients and are frequently associated with regions of intimal hyperplasia and stenosis (57). These thrombi contribute to arterial occlusion in moyamoya and are thought to be related to intimal pathology (2, 57).
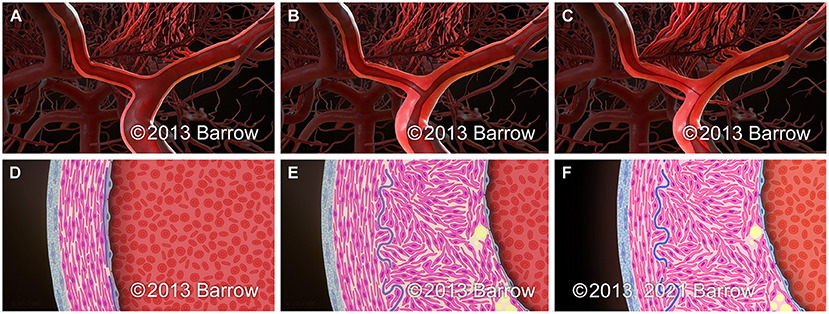
Figure 1. Schematic representation of the paradigmatic progression pattern of arterial stenosis in moyamoya on the macroscopic (A–C) and microscopic (D–F) scale. (A,D) Normal terminal ICA. (B,E) Early in the progression of moyamoya, intimal hyperplasia leads to the development of luminal stenosis without a significant change in the outer diameter of the arterial segment. (C,F) As luminal stenosis continues to progress in moyamoya, attenuation of the medial layer of the artery leads to a reduction in the outer arterial diameter. Used with permission from Barrow Neurological Institute, Phoenix, Arizona.
Like moyamoya, atherosclerosis causes stenosis with intimal expansion and intimal smooth muscle cell hyperplasia. However, atherosclerosis is marked by characteristic inflammatory infiltration and lipid accumulation that do not typically occur in moyamoya (48). Furthermore, as luminal stenosis progresses in atherosclerosis, outward vascular remodeling occurs, with increased outer vessel diameter (58, 59), in contrast to the decreased outer diameter that occurs in moyamoya. Another form of arteriopathy, the intimal fibroplasia subtype of FMD, is characterized by intimal expansion in the absence of inflammatory cell infiltration or lipid accumulation (60, 61). Like moyamoya, it causes intimal extracellular matrix expansion and abnormalities of the internal elastic lamina, but the intimal fibroplasia subtype of FMD is not characterized by hyperplasia of the intima as is seen in moyamoya (62). However, pediatric patients with FMD can also present with stroke and renal artery stenosis (62, 63). These shared pathologic features highlight the need for future research to better understand the complex relationship between these two entities.
The vascular wall pathology in MMD involves thickening of the intima without infiltration of inflammatory cells or considerable disruption of the internal elastic lamina (51, 64). Compared to typical findings in atherosclerosis, macrophage infiltration into the subintimal layer, and lipid pool accumulation are not characteristic features of moyamoya (64, 65). In contrast, a study by Masuda et al. (66) of autopsy specimens obtained from MMD patients showed proliferation of smooth muscle cells, the presence of T cells, and infiltration of macrophages into the vascular wall. In MMS associated with neurofibromatosis type I (NF-1), inflammatory cells may infiltrate the area surrounding the lesion of the arteriopathy, indicating that some types of MMS may be related to inflammation (64, 66). Kaku et al. (54) showed a reduction in both the inner and outer diameter of the ICA in the course of the moyamoya remodeling process, in contrast to the pathogenesis of atherosclerosis, which shows an outward remodeling pattern. These authors also found no reduction of the outer diameter of the circle of Willis in an MMS subgroup, and arterial shrinkage was insignificant (54, 64). Moyamoya vascular shrinkage may be detected specifically in the horizontal MCA (M1) section (64, 67). According to Yamamoto et al. (56), MMS pathology may include two distinct subgroups: a nonconstrictive subgroup as well as a constrictive subgroup. A portion of one subgroup of MMS may show excessive fibrosis. Another subgroup of MMS may show limited intimal hyperplasia and vascular constrictive changes (64). In 2015, Mikami et al. (67) showed an overall reduction in the size of the outer diameter of all vessels around C1 in MMD patients. Similar to a negative constrictive remodeling pattern, the MCA diameter was shown to be significantly smaller in patients with disease progression (67). In 2019, Choi et al. (68) used high-resolution MRI to compare plaque characteristics and the vascular remodeling pattern and hemodynamic changes related to intracranial plaques depending on the presence or absence of the RNF213 p.Arg4810Lys variant in patients without moyamoya and demonstrated that, in these patients, the RNF213 variant was associated with smaller outer vessel diameters in the distal ICA, proximal MCA, and basilar artery (68).
Moyamoya disease is idiopathic and occurs as a primary pathologic process. Conversely, MMS occurs in patients with a known associated condition, disease, or exposure that places the patient at significantly higher risk of developing moyamoya. The most frequent moyamoya-associated conditions are sickle cell disease, Down's syndrome, NF-1, and exposure to cranial radiotherapy, which are present in 10–20% of moyamoya cases (69). Less frequently associated conditions include: Alagille syndrome, Costello syndrome, microcephalic osteodysplastic primordial dwarfism type II, Noonan syndrome, Seckel syndrome, hyperthyroidism, and many others (2, 70–78). Importantly, moyamoya-associated conditions have the potential to inform the investigation into the mechanisms that may be important for the development of moyamoya. The genetic basis of many of these associated conditions is well-understood and, thus, provides important hints as to the importance of various signaling pathways in the development of moyamoya.
By identifying genes involved in MMD pathogenesis and several monogenic MMSs, investigators have associated various molecular pathways with MMD pathophysiology, including the Ras–Raf–mitogen-activated protein kinase (MEK)–extracellular-signal-related kinase (ERK) signaling pathway (NF-1, Noonan syndrome, Costello syndrome), the neurogenic locus notch homolog protein (Notch) signaling pathway (Alagille syndrome), the nitric oxide (NO)-soluble guanylyl cyclase (sGC) signaling pathway [Guanylate cyclase 1 soluble subunit alpha-3 (GUCY1A3)], and pathways involved in genomic stability [Lys-63-specific deubiquitinase BRCC36 [BRCC3], pericentrin [PCNT], and other genes involved in dwarfism] (20) (See the Supplementary Table 1 for definitions of gene symbols, proteins, and additional terminology).
The Ras-Raf-MEK-ERK signaling pathway has been shown to be associated with NF-1 (79, 80). Loss-of-function mutations in NF1 (17q11.2) lead to absent neurofibromin expression, which in turn may activate the Ras-Raf-MEK-ERK signaling pathway, leading to an increased mitogenic signaling (79, 80). In Noonan syndrome, gain-of-function mutations in PTPN11 (12q24.13), NRAS (1p13.2), SOS1 (2p22.1), RAF1 (3p25.2), BRAF (7q34), KRAS (12p12.1), and MAP2K1 (15q22.31) genes may activate the Ras-Raf-MEK-ERK signaling pathway (20, 81–84).
Costello syndrome is encoded by HRAS (11p15.5) and KRAS (12p12.1), and gain-of-function mutations influencing p.Gly13 or p.Gly12 may cause germline activation of HRAS, and thereby lead to dysregulation of the Ras-Raf-MEK-ERK signaling pathway (20, 85, 86).
Defects in the Notch signaling pathway are associated with vasculopathy. Alagille syndrome is caused by loss-of-function mutations in either JAG1 (20p12.2) or NOTCH2 (1p12-p11) (20, 87–96).
Genomic stability signaling pathways are associated with congenital dwarfing syndromes. PCNT (21q22.3) loss-of-function mutations are linked to deficient pericentrin cell cycle progression in microcephalic osteodysplastic primordial dwarfism type II. In Seckel syndrome, CEP63 (3q22.2), ATR (3q23), CENPJ (13q12.12), NIN (14q22.1), CEP152 (15q21.1), and RBBP8 (18q11.2) are associated with cycle cell progression, centrosomal function, and DNA repair (20, 71, 97–99).
BRCC3/MTCP1 is associated with MMD pathogenesis. BRCC3 (Xq28) is potentially involved in DNA repair alteration. Absent BRCC3 expression inhibits angiogenesis of trunk intersegmental vessels in the zebrafish. MTCP1 may enhance phosphorylation and activation of AKT1 and AKT2 (20, 100–102).
The nitric oxide-soluble guanylyl cyclase-cyclic guanosine monophosphate (NO-sGC-cGMP) signaling pathway is a key signal transduction pathway associated with controlling vascular smooth-muscle relaxation, vascular tone, and vascular remodeling. It is hypothesized that homozygous loss-of-function mutations in GUCY1A3 (4q32.1) may be involved in an alteration of the NO pathway in vascular smooth muscle cells (SMCs), potentially leading to aberrant vascular remodeling at bifurcation and curvature regions, such as the ICA bifurcation (20, 103).
Moyamoya Syndrome in association with inflammation occurs with a frequency of 0.54–1.5% in pediatric MMD patients compared to a frequency of 1.7–4.7% in adult MMD patients (64, 104). In 1983, Suzuki et al. (105) analyzed the incidence of infections in MMD patients. They found that 61.6% of adult MMD patients and 82.6% of pediatric MMD patients suffered from infections of the face and head including both infections of unknown origin and maxillary sinusitis, otitis media, and tonsillitis (64). According to the Japanese national survey by Hayashi and colleagues (106), diseases other than atherosclerosis constitute 17.2% of all inflammatory diseases associated with MMS. For common autoimmune disorders related to MMS, meningitis constitutes 2.2%, hyperthyroidism constitutes 7.5%, and other autoimmune disorders constitute 17.2%. Less common autoimmune disorders associated with MMS may include polyarteritis nodosa, dermatomyositis, Addison's disease, Sjögren's syndrome, Kawasaki's disease, acute limbic encephalitis with anti-LGI1 antibody, antiphospholipid antibody syndrome, granulomatosis with polyangiitis, myasthenia gravis, systemic lupus erythematosus, multiple sclerosis, systemic sclerosis, primary systemic vasculitis, rheumatoid arthritis, polymyositis, and thyroiditis (64, 104, 106, 107). It may be difficult to distinguish the mechanisms associated with a distinct autoimmune disorder and from those associated with MMS because of the complexity of the differentiation between causality and correlation (64). In summary, a correlation between chronic systemic inflammation caused by an autoimmune response and MMS may be hypothesized (64).
Moyamoya Syndrome concomitant with hyperthyroidism is seen predominantly in females and may be associated with ischemia, a comparatively advanced age of disease onset, and faster disease progression compared to MMS (64, 108–111). Ischemia may worsen thyroid functioning. Ischemic symptoms caused by thyrotoxicosis were ameliorated after hyperthyroidism was controlled (64, 109). Increased thyroid hormone may cause MMS progression through an increase in vascular sensitivity to the sympathetic nervous system (64, 112). A T-cell-mediated autoimmune response may also advance MMS pathogenesis. Consequently, immunosuppressive treatment before surgery and/or treatment of a hormonal abnormality may be effective in MMS management related to autoimmune disorders (64, 113, 114). Tendler et al. (115) indicated that immunological stimulation in hyperthyroidism and vascular dysregulation and cellular proliferation in MMS may share a pathomechanism involving T-cell dysregulation. This pathomechanism mentioned above involves both vascular morphological alterations and vascular sensitivity (64, 116). In context with morphological alterations, surgical procedures including surgical revascularization may be utilized (64).
Moyamoya Syndrome caused by meningitis is rare, constituting approximately 2.2% of all disorders classified as MMS (64, 106). The associated infection may be caused by various pathogens, including Hemophilus influenzae (117, 118), Streptococcus pneumoniae (113, 119, 120), β-hemolytic group A Streptococcus (121), Mycobacterium tuberculosis (122–125), Propionibacterium acnes (126), Leptospira (127), Mycoplasma pneumoniae (128), Neisseria meningitidis with cytomegalovirus (129), neurosyphilis with human immunodeficiency virus (HIV) (130), HIV (131–133), Epstein–Barr virus (64, 129), varicella-zoster virus (134), and measles virus (135). Vascular complications related to meningitis usually become clinically manifest during the 2 weeks after disease onset (64, 136, 137). In MMS associated with meningitis, late-onset morphological alteration of the circle of Willis may be observed (113, 117–119, 123, 125). In addition, an increase in autoimmune antibodies may indicate an autoimmune trigger for the onset of MMS (64, 113). Liu et al. (138) found that the cerebrospinal fluid of MMS patients showed an immune response to leptospirosis and that MMS appears to be related to immune reactive vasculitis (64). In 2018, Takahashi et al. reported a case of MMS in familial MMD 9 years after the patient had nonherpetic acute limbic encephalitis (64, 139). The patient tested positive for anti-LGI1 antibodies, and the authors hypothesized that inflammation caused by an autoimmune disease process may have contributed to MMS progression. Consequently, chronic inflammation caused by an autoimmune disease process may lead to MMS in the course of acute inflammation. In addition, acute systemic inflammation may be related to MMD pathogenesis (64).
Pediatric MMD patients presenting with headache or fatigue as the primary symptom may be misdiagnosed as having hypochondriasis or as experiencing a developmental deficit, or they may be simply dismissed as being “lazy.” Lack of appropriate treatment in these patients may lead to severe ischemic stroke. General practitioners and physicians who do not routinely encounter MMD should be encouraged to consider a diagnosis of MMD in patients presenting with minor symptoms such as headache, fatigue, and school refusal.
Symptoms of MMD may be ascribed to changes in blood flow due to ICA stenosis. Two major etiologic categories of symptoms may be distinguished, those involving the consequences of compensatory mechanisms in response to ischemia, such as headache from dilated transdural collateral vessels and hemorrhage from fragile collateral vessels, and those caused by cerebral ischemia such as transient ischemic attacks, stroke, and seizures. Variations in the degree of arterial involvement, ischemic cortex areas, progression of stenosis, and response to a reduction in blood supply may help to clarify the broad range of symptoms in MMD (2). In Asian populations, 68% of children with MMD present with ischemic strokes or TIAs, and 2.8% present with hemorrhage (2, 140). Most children and adults with MMD in the United States present with symptoms of ischemia, with a rate of hemorrhage of 20.0% among adults compared to a rate of 2.8% among children (2, 140, 141). An increased frequency of completed strokes is evident in pediatric MMD. Presumably, children's inability to accurately report their symptoms may hinder a timely diagnosis and lead to an increased probability of completed strokes (2, 142). The symptoms of cerebral ischemia in patients with MMD are related to the areas of the brain whose blood flow is supplied by the ICAs and MCAs, specifically, the temporal, parietal, and frontal lobes. The predominant symptoms are cognitive impairment, aphasia, dysarthria, and hemiparesis (2, 140). Additional symptoms include syncope, visual deficits, seizures, and personality changes, the latter of which may be easily confused with a psychiatric disorder (2, 143). Common childhood actions, including crying with hyperventilation, may precipitate a stroke or TIA. Signs of cerebral ischemia may be temporary or permanent, and may be caused by induction of anesthesia for surgery or by exertion. A hypothesized mechanism precipitating strokes and TIAs in these patients is that the cortical vasculature, which is assumed to be fully dilated under the conditions of chronic ischemia, may constrict in response to a decrease in the partial pressure of carbon dioxide, this restriction may be caused by hyperventilation, eventually resulting in a reduction of cerebral perfusion (2, 144). Ischemic symptoms may also be precipitated by dehydration (2). Intracranial hemorrhage, which is frequently encountered in adult MMD patients, is equally evident in pediatric MMD patients (2, 140, 145). Intracranial hemorrhage occurs in the subarachnoid space, in the ventricles, and in the brain parenchyma, commonly in the basal ganglia area. In the past, intracranial hemorrhage has been ascribed to the rupture of moyamoya-associated fragile collateral vessels in the course of progressive ICA stenosis (2, 146, 147). An additional cause of hemorrhage in these patients may be alternating circulatory patterns at the base of the brain, which have been implicated in the emergence of intracranial aneurysms; these aneurysms are usually located at the apex of the basilar artery or in the PCA, which are common locations of increased shear stress in MMD (2, 148, 149). Headache is a predominant presenting symptom in MMD patients. In their 2005 review article, Seol at al. hypothesized that dilatation of leptomeningeal and meningeal collateral vessels may lead to the stimulation of dural nociceptors (2, 150). Characteristically, headache is refractory to therapy and resembles migraine in quality; despite effective surgical revascularization, headache may persist in as much as 63% of MMD patients (2, 150). Headache symptoms may abate within 1 year after surgery for MMD in some moyamoya patients, suggesting a potential regression of basal collaterals. Dilated moyamoya-related collateral vessels in the basal ganglia may be associated with the emergence of choreiform movements in pediatric MMD patients (2, 140, 151). In a study of pediatric MMD patients presenting with choreiform movements, 8 of 10 pediatric MMD patients experienced resolution of this symptom 1 year after revascularization surgery, suggesting that this surgical procedure appears to be effective in reducing the number of moyamoya-related collateral vessels in the basal ganglia (2). Another sign of MMD is the “morning glory disk,” which is an optic disk enlargement associated with retinovascular aberrations; this sign may be evaluated by vascular imaging (2, 152).
Kawabori et al. (153) studied 29 pediatric MMD patients, hypothesizing that persistent disturbance of cerebral hemodynamics may be associated with severe headache in pediatric MMD. They found that encephalo-duro-myo-arteriopericranial synangiosis and STA-MCA anastomosis may be effective surgical procedures to causally treat headache by supplying collateral blood flow to the operated hemispheres. They advocate close follow-up because of the potential for the development of postoperative headaches in these patients (153). In 2019, Riordan et al. (154) studied 59 pediatric MMD patients who had undergone surgical revascularization by pial synangiosis more than 20 years previously and found that pial synangiosis may confer long-term protection against stroke in pediatric MMD. A history of cranial irradiation was ascertained in one patient with a late stroke and in four out of five deceased patients. The authors conclude that, in the absence of significant preoperative comorbidities and neurological deficits, revascularization surgery appears to be safe and effective (154). In 2019, Kazumata et al. (155) performed a prospective, observational, single-center study in 21 pediatric MMD patients [mean age 10 (3.0) years, range 5–14 years], hypothesizing mild cognitive dysfunction caused by cerebral hypoperfusion through the association of a reduced regional cerebral blood flow (rCBF) in the left dorsolateral prefrontal cortex (DLPFC) and medial frontal cortex with a reduced processing speed index (PSI), perceptual reasoning index (PRI), and full-scale intelligence quotient (FIQ). However, they found that average intellectual ability was not reduced in pediatric MMD patients. They also found that the angiographic severity of the disease, ischemic symptoms, family history, and patient age at disease onset were not associated with inadequate cognitive performance (155). In 2019, Lee et al. (156) studied 131 surgically treated pediatric MMD patients to evaluate the risk factors and prevalence of hypertension. They demonstrated a high prevalence of hypertension in pediatric MMD patients. Consequently, they suggest that sufficient efforts should be made to achieve BP reduction in pediatric MMD patients to prevent subsequent vascular events (156).
From its discovery and definition, moyamoya has been a disease process that is diagnosed and classified based on vascular imaging studies. Recent advances in moyamoya imaging have furthered our understanding of the natural history of arterial stenosis in moyamoya and provide important insights into patient responses to therapeutic interventions.
In 2013, Kim et al. performed a high-resolution MRI (HR-MRI) study in patients with an MCA occlusion stroke that were angiographically confirmed to be 12 patients with MMD and 20 patients with intracranial atherosclerotic disease (ICAD). Consistent with previous pathological reports, the research group showed that MMD may be associated with smaller, concentric occlusive lesions that are rarely enhanced compared to symptomatic ICAD, suggesting that HR-MRI may assist in the evaluation of pathological changes in the vascular wall and in differentiating ICAD from MMD (157). In 2015, Kim et al. performed a case-control study in MMD patients and normal controls using magnetic resonance angiography (MRA) and computational fluid dynamics. The research group demonstrated significant morphological differences in the intracranial-extradural ICA of MMD patients. These differences may affect the hemodynamics around the ICA bifurcation. Whether these hemodynamic changes are the result or a cause of the ICA bifurcation stenosis remains to be elucidated (158).
Ladner et al. (159) proposed a scoring system for the degree of severity of moyamoya, Prior Infarcts, Reactivity, and Angiography in Moyamoya Disease (PIRAMD), which uses a combination of conventional angiography and non-invasive structural and hemodynamic 3 Tesla MRI parameters. These parameters potentially provide a measurement of the hemodynamic degree of severity of MMD, which has proved to correlate well with symptoms, and which may assist in evaluating intervention response and guiding management decisions. Limitations of this study, which was performed in 11 healthy control volunteers and 25 MMD patients with angiographically confirmed moyamoya, include its retrospective nature, and a small sample size, necessitating univariate analysis (159).
Han et al. (160) performed an HR-MRI study in 51 adult MMD patients, analyzing angiographic and HR-MRI findings as well as atherogenic risk factors. The research group suggested that HR-MRI may assist in a precise diagnosis of ICAD in MMD patients with risk factors for atherogenesis. Distinguishable symptoms observed in moyamoya patients in the presence or absence of atherosclerotic plaques may suggest a distinct pathophysiology, implying different treatment strategies (160).
Storey et al. performed a single-center study which included 204 MMD patients. Transdural collateral vessels were existent in nearly 50% of arteriograms done preoperatively on these MMD patients. Pial synangiosis was performed on these patients from 2005 to 2013. This group hypothesized that preoperative transdural collateral vessels may be utilized as radiographic biomarkers of MMD, that transdural collateral vessels may be associated with advanced moyamoya, that transdural collateral vessels may be related to stroke as a perioperative complication, and that transdural collateral vessels may be capable of inducing surgical collaterals after surgery, suggesting innovative methods to generate biomarkers that may be capable of predicting outcome and stratifying surgical candidates, and supporting the usefulness of preoperative diagnostic arteriography (161).
Song et al. performed a retrospective analysis in 90 pediatric MMD patients by MRI and digital subtraction angiography to assess the distribution of ischemic lesions and vascular abnormalities, including stenosis and occlusion, as well as diagnostic coherence between imaging modalities. Digital subtraction angiography and MRA were used to detect stenotic and/or occlusive changes of the bilateral ACA, MCA, and PCA. MRI demonstrated good diagnostic coherence regarding stenotic and/or occlusive changes in the bilateral PCA. However, it is important to note that in 6 to 11% of patients, an MRA evaluation may lead to misdiagnosis of moyamoya (162).
Zhao et al. (31) conducted a single-center study to compare the radiographic and clinical characteristics between adult and pediatric moyamoya patients, and also between moyamoya patients with and without PCA involvement. Records of 696 consecutive MMD patients from 2009 to 2015, including 574 angiograms, 434 of adult and 140 of pediatric MMD patients, were reviewed. Grading systems by Miyamoto and Suzuki were used to evaluate stenoses and/or occlusions of the anterior and posterior cerebral circulation. In both pediatric MMD and in adult MMD, occlusive and/or stenotic PCA lesions positively correlated with the ipsilateral ICA stage. In adult MMD patients, the degree of PCA involvement seemed to correlate with the frequentness of ipsilateral stroke. In comparison to adult MMD patients, pediatric MMD patients showed a tendency toward advanced PCA lesions. First, this study is limited by its retrospective nature. Second, this study may be limited by selection bias (31).
In their 2019 review article, Li et al. stated that the most acceptable and valuable imaging modality should be reliable and keep injury to the patient to a minimum. Doppler and quantitative MRI-based imaging modalities might be auspicious in establishing an accurate evaluation of moyamoya. Standardized protocols, including various imaging modalities, to evaluate the preoperative and postoperative status quo, may be advocated in future moyamoya research (163).
Hauser et al. performed a retrospective study in 20 consecutive patients with angiographically proven MMD and analyzed 160 vascular territories, indicating that CO2-triggered blood oxygen level-dependent (BOLD) MRI may be a promising, easily implementable method for the evaluation of territorial hemodynamics in MMD patients with results comparable to those attained by [15O]H2O positron emission tomography/computed tomography with acetazolamide challenge, suggesting that, after continued prospective evaluation, CO2-triggered BOLD MRI may become a routine examination method in pre- and postoperative evaluation of MMD patients (164).
In their 2019 review article, Lehman et al. discuss novel MRI imaging achievements that may further improve the analysis of moyamoya, e.g., enhanced methods of cerebral perfusion and angiography, high-resolution characterization of the vessel wall, as well as artificial intelligence. In the clinical setting, contemporary and emerging MRI imaging methods may assist in the evaluation of MMD, and help guide therapy planning and response to treatment (165).
As vascular imaging methods advance in their potential to serve as research tools, our understanding of the pathophysiology of arterial stenosis in moyamoya is very likely to grow. These imaging advances are particularly important given the lack of an animal model that reproduces the spontaneous arterial stenosis of the proximal cerebrovasculature that is seen in moyamoya.
Considerable progress has been made in moyamoya research since it was first described, leading to greater epidemiologic and pathophysiological understanding, as well as advancements in clinical practice, encompassing imaging, diagnosis, and surgical treatment. However, the proximate mechanisms that underly stenosis and progression of stenosis remain elusive. As a result, there is no clear drug target to reverse the disease process or prevent progression, and there have been no disease-modifying medical therapies developed to accomplish these goals. Such a therapy is desperately needed, as current medical therapies do not prevent progression and current surgical therapies, while effective, are associated with significant risk of morbidity and mortality. Future research should continue to aggressively pursue novel hypotheses that attempt to unify the unique aspects of moyamoya anatomic localization, pathophysiology, disease associations, and genetic risk factors to explain why stenosis develops in moyamoya. Further, as the understanding of more common stenotic arteriopathies of the distal ICA, such as atherosclerosis and FMD, is advanced, newly discovered mechanisms should continue to be investigated in moyamoya given its overlapping pathological characteristics. The initiation of arterial stenosis in moyamoya represents the biggest mystery of this disease process, and until it is mechanistically explained, patient care in moyamoya will remain insufficient.
BF and KD contributed to developing the concept of the review, developing the figures, and writing and editing the manuscript. ML oversaw the project and contributed to developing the concept of the review. JW led oversight of the project and contributed to developing the concept of the review, developing the figures, and editing the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the staff of Neuroscience Publications at Barrow Neurological Institute for assistance with manuscript preparation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.661578/full#supplementary-material
ACA, anterior cerebral artery; BOLD, blood oxygen level-dependent; HR-MRI, high-resolution magnetic resonance imaging; ICAD, intracranial atherosclerotic disease; MCA, middle cerebral artery; MMD, moyamoya disease; MMS, moyamoya syndrome; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; PCA, posterior cerebral artery. For a complete list of abbreviations and gene symbols, see Supplemental Table 1.
1. Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. (1969) 20:288–99. doi: 10.1001/archneur.1969.00480090076012
2. Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. (2009) 360:1226–37. doi: 10.1056/NEJMra0804622
3. Kelly ME, Bell-Stephens TE, Marks MP, Do HM, Steinberg GK. Progression of unilateral moyamoya disease: a clinical series. Cerebrovasc Dis. (2006) 22:109–15. doi: 10.1159/000093238
4. Kuroda S, Ishikawa T, Houkin K, Nanba R, Hokari M, Iwasaki Y. Incidence and clinical features of disease progression in adult moyamoya disease. Stroke. (2005) 36:2148–53. doi: 10.1161/01.STR.0000182256.32489.99
5. Kawano T, Fukui M, Hashimoto N, Yonekawa Y. Follow-up study of patients with “unilateral” moyamoya disease. Neurol Med Chir (Tokyo). (1994) 34:744–7. doi: 10.2176/nmc.34.744
6. Lee WJ, Jeong SK, Han KS, Lee SH Ryu YJ, Sohn CH, et al. Impact of endothelial shear stress on the bilateral progression of unilateral moyamoya disease. Stroke. (2020) 51:775–83. doi: 10.1161/STROKEAHA.119.028117
7. Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis Health Labour Sciences Research Grant for Research on Measures for Infractable Diseases. Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir (Tokyo). (2012) 52:245–66. doi: 10.2176/nmc.52.245
8. Fujimura M, Bang OY, Kim JS. Moyamoya disease. Front Neurol Neurosci. (2016) 40:204–20. doi: 10.1159/000448314
9. Smith ER, Scott RM. Moyamoya: epidemiology, presentation, and diagnosis. Neurosurg Clin N Am. (2010) 21:543–51. doi: 10.1016/j.nec.2010.03.007
10. Nah HW, Kwon SU, Kang DW, Ahn JS, Kwun BD, Kim JS. Moyamoya disease-related versus primary intracerebral hemorrhage: [corrected] location and outcomes are different. Stroke. (2012) 43:1947–50. doi: 10.1161/STROKEAHA.112.654004
11. Zipfel GJ, Fox DJ Jr, Rivet DJ. Moyamoya disease in adults: the role of cerebral revascularization. Skull Base. (2005) 15:27–41. doi: 10.1055/s-2005-868161
12. Funaki T, Takahashi JC, Houkin K, Kuroda S, Takeuchi S, Fujimura M, et al. High rebleeding risk associated with choroidal collateral vessels in hemorrhagic moyamoya disease: analysis of a nonsurgical cohort in the Japan Adult Moyamoya Trial. J Neurosurg. (2019) 130:337–673. doi: 10.3171/2017.9.JNS17576
13. Funaki T, Takahashi JC, Houkin K, Kuroda S, Fujimura M, Tomata Y, et al. Effect of choroidal collateral vessels on de novo hemorrhage in moyamoya disease: analysis of nonhemorrhagic hemispheres in the Japan Adult Moyamoya Trial. J Neurosurg. (2019) 132:408–14. doi: 10.3171/2018.10.JNS181139
14. Wakai K, Tamakoshi A, Ikezaki K, Fukui M, Kawamura T, Aoki R, et al. Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S1–5. doi: 10.1016/s0303-8467(97)00031-0
15. Uchino K, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington State and California. Neurology. (2005) 65:956–8. doi: 10.1212/01.wnl.0000176066.33797.82
16. Han DH, Kwon OK, Byun BJ, Choi BY, Choi CW, Choi JU, et al. A co-operative study: clinical characteristics of 334 Korean patients with moyamoya disease treated at neurosurgical institutes (1976–1994). The Korean Society for Cerebrovascular Disease. Acta Neurochir (Wien). (2000) 142:1263–73; discussion 73–4. doi: 10.1007/s007010070024
17. Baba T, Houkin K, Kuroda S. Novel epidemiological features of moyamoya disease. J Neurol Neurosurg Psychiatry. (2008) 79:900–4. doi: 10.1136/jnnp.2007.130666
18. Yamauchi T, Tada M, Houkin K, Tanaka T, Nakamura Y, Kuroda S, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. (2000) 31:930–5. doi: 10.1161/01.str.31.4.930
19. Koizumi A, Kobayashi H, Hitomi T, Harada KH, Habu T, Youssefian S, et al. new horizon of moyamoya disease and associated health risks explored through RNF213. Environ Health Prev Med. (2016) 21:55–70. doi: 10.1007/s12199-015-0498-7
20. Guey S, Tournier-Lasserve E, Herve D, Kossorotoff M. Moyamoya disease and syndromes: from genetics to clinical management. Appl Clin Genet. (2015) 8:49–68. doi: 10.2147/TACG.S42772
21. Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, et al. A genome-wide association study identifies RNF213 as the first moyamoya disease gene. J Hum Genet. (2011) 56:34–40. doi: 10.1038/jhg.2010.132
22. Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE. (2011) 6:e22542. doi: 10.1371/journal.pone.0022542
23. Miyawaki S, Imai H, Takayanagi S, Mukasa A, Nakatomi H, Saito N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke. (2012) 43:3371–4. doi: 10.1161/STROKEAHA.112.663864
24. Miyawaki S, Imai H, Shimizu M, Yagi S, Ono H, Mukasa A, et al. Genetic variant RNF213 c.14576G>A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke. (2013) 44:2894–7. doi: 10.1161/STROKEAHA.113.002477
25. Miyawaki S, Imai H, Shimizu M, Yagi S, Ono H, Nakatomi H, et al. Genetic analysis of RNF213 c.14576G>A variant in nonatherosclerotic quasi-moyamoya disease. J Stroke Cerebrovasc Dis. (2015) 24:1075–9. doi: 10.1016/j.jstrokecerebrovasdis.2015.01.005
26. Acker G, Fekonja L, Vajkoczy P. Surgical management of moyamoya disease. Stroke. (2018) 49:476–82. doi: 10.1161/STROKEAHA.117.018563
27. Smith JL. Understanding and treating moyamoya disease in children. Neurosurg Focus. (2009) 26:E4. doi: 10.3171/2000.01.FOCUS08306
28. Handa J, Handa H. Progressive cerebral arterial occlusive disease: analysis of 27 cases. Neuroradiology. (1972) 3:119–33. doi: 10.1007/BF00341494
29. Miyamoto S, Kikuchi H, Karasawa J, Nagata I, Ikota T, Takeuchi S. Study of the posterior circulation in moyamoya disease. Clinical and neuroradiological evaluation. J Neurosurg. (1984) 61:1032–7. doi: 10.3171/jns.1984.61.6.1032
30. Kimiwada T, Hayashi T, Shirane R, Tominaga T. Posterior cerebral artery stenosis and posterior circulation revascularization surgery in pediatric patients with moyamoya disease. J Neurosurg Pediatr. (2018) 21:632–8. doi: 10.3171/2018.1.PEDS17367
31. Zhao M, Zhang D, Wang S, Zhang Y, Wang R, Deng X, et al. Posterior circulation involvement in pediatric and adult patients with moyamoya disease: a single center experience in 574 patients. Acta Neurol Belg. (2018) 118:227–33. doi: 10.1007/s13760-017-0844-1
32. Mugikura S, Takahashi S, Higano S, Shirane R, Sakurai Y, Yamada S. Predominant involvement of ipsilateral anterior and posterior circulations in moyamoya disease. Stroke. (2002) 33:1497–500. doi: 10.1161/01.str.0000016828.62708.21
33. Yamada I, Himeno Y, Suzuki S, Matsushima Y. Posterior circulation in moyamoya disease: angiographic study. Radiology. (1995) 197:239–46. doi: 10.1148/radiology.197.1.7568830
34. Tan C, Duan R, Ye X, Zhang D, Wang R. Posterior circulation moyamoya disease versus primitive vertebral-basilar artery system moyamoya disease: new classification of moyamoya disease from the perspective of embryology. World Neurosurg. (2016) 96:222–9. doi: 10.1016/j.wneu.2016.08.099
35. Komiyama M. Moyamoya disease is a progressive occlusive arteriopathy of the primitive internal carotid artery. Interv Neuroradiol. (2003) 9:39–45. doi: 10.1177/159101990300900105
36. Mugikura S, Higano S, Shirane R, Fujimura M, Shimanuki Y, Takahashi S. Posterior circulation and high prevalence of ischemic stroke among young pediatric patients with moyamoya disease: evidence of angiography-based differences by age at diagnosis. AJNR Am J Neuroradiol. (2011) 32:192–8. doi: 10.3174/ajnr.A2216
37. Imaizumi T, Hayashi K, Saito K, Osawa M, Fukuyama Y. Long-term outcomes of pediatric moyamoya disease monitored to adulthood. Pediatr Neurol. (1998) 18:321–5. doi: 10.1016/s0887-8994(97)00209-9
38. Kim SK, Cho BK, Phi JH, Lee JY, Chae JH, Kim KJ, et al. Pediatric moyamoya disease: an analysis of 410 consecutive cases. Ann Neurol. (2010) 68:92–101. doi: 10.1002/ana.21981
39. Yamada I, Himeno Y, Matsushima Y, Shibuya H. Renal artery lesions in patients with moyamoya disease: angiographic findings. Stroke. (2000) 31:733–7. doi: 10.1161/01.str.31.3.733
40. Togao O, Mihara F, Yoshiura T, Tanaka A, Kuwabara Y, Morioka T, et al. Prevalence of stenoocclusive lesions in the renal and abdominal arteries in moyamoya disease. AJR Am J Roentgenol. (2004) 183:119–22. doi: 10.2214/ajr.183.1.1830119
41. Hoshimaru M, Kikuchi H. Involvement of the external carotid arteries in moyamoya disease: neuroradiological evaluation of 66 patients. Neurosurgery. (1992) 31:398–400. doi: 10.1227/00006123-199209000-00002
42. Jee TK, Yeon JY, Kim SM, Bang OY, Kim JS, Hong SC. Prospective screening of extracranial systemic arteriopathy in young adults with moyamoya disease. J Am Heart Assoc. (2020) 9:e016670. doi: 10.1161/JAHA.120.016670
43. Komiyama M, Nishikawa M, Yasui T, Kitano S, Sakamoto H, Fu Y. Steno-occlusive changes in the external carotid system in moyamoya disease. Acta Neurochir (Wien). (2000) 142:421–4. doi: 10.1007/s007010050452
44. Takagi Y, Kikuta K, Nozaki K, Fujimoto M, Hayashi J, Imamura H, et al. Expression of hypoxia-inducing factor-1 alpha and endoglin in intimal hyperplasia of the middle cerebral artery of patients with moyamoya disease. Neurosurgery. (2007) 60:338–45; discussion 45. doi: 10.1227/01.NEU.0000249275.87310.FF
45. Takekawa Y, Umezawa T, Ueno Y, Sawada T, Kobayashi M. Pathological and immunohistochemical findings of an autopsy case of adult moyamoya disease. Neuropathology. (2004) 24:236–42. doi: 10.1111/j.1440-1789.2004.00550.x
46. Takagi Y, Kikuta K, Nozaki K, Hashimoto N. Histological features of middle cerebral arteries from patients treated for moyamoya disease. Neurol Med Chir (Tokyo). (2007) 47:1–4. doi: 10.2176/nmc.47.1
47. Reid AJ, Bhattacharjee MB, Regalado ES, Milewicz AL, El-Hakam LM, Dauser RC, et al. Diffuse and uncontrolled vascular smooth muscle cell proliferation in rapidly progressing pediatric moyamoya disease. J Neurosurg Pediatr. (2010) 6:244–9. doi: 10.3171/2010.5.PEDS09505
48. Fukui M, Kono S, Sueishi K, Ikezaki K. Moyamoya disease. Neuropathology. (2000) 20(Suppl):S61–4. doi: 10.1046/j.1440-1789.2000.00300.x
49. Achrol AS, Guzman R, Lee M, Steinberg GK. Pathophysiology and genetic factors in moyamoya disease. Neurosurg Focus. (2009) 26:E4. doi: 10.3171/2009.1.FOCUS08302
50. Yamashita M, Oka K, Tanaka K. Histopathology of the brain vascular network in moyamoya disease. Stroke. (1983) 14:50–8. doi: 10.1161/01.str.14.1.50
51. Hosoda Y, Ikeda E, Hirose S. Histopathological studies on spontaneous occlusion of the circle of Willis (cerebrovascular moyamoya disease). Clin Neurol Neurosurg. (1997) 99(Suppl 2):S203–8. doi: 10.1016/s0303-8467(97)00044-9
52. Lin R, Xie Z, Zhang J, Xu H, Su H, Tan X, et al. Clinical and immunopathological features of moyamoya disease. PLoS ONE. (2012) 7:e36386. doi: 10.1371/journal.pone.0036386
53. Bang OY, Fujimura M, Kim SK. The pathophysiology of moyamoya disease: an update. J Stroke. (2016) 18:12–20. doi: 10.5853/jos.2015.01760
54. Kaku Y, Morioka M, Ohmori Y, Kawano T, Kai Y, Fukuoka H, et al. Outer-diameter narrowing of the internal carotid and middle cerebral arteries in moyamoya disease detected on 3D constructive interference in steady-state MR image: is arterial constrictive remodeling a major pathogenesis? Acta Neurochir (Wien). (2012) 154:2151–7. doi: 10.1007/s00701-012-1472-4
55. Yamamoto S, Kashiwazaki D, Akioka N, Kuwayama N, Noguchi K, Kuroda S. Progressive shrinkage of involved arteries in parallel with disease progression in moyamoya disease. World Neurosurg. (2019) 122:e253–61. doi: 10.1016/j.wneu.2018.10.001
56. Yamamoto S, Koh M, Kashiwazaki D, Akioka N, Kuwayama N, Noguchi K, et al. Is quasi-moyamoya disease a uniform disease entity? A three-dimensional constructive interference in steady state imaging study. J Stroke Cerebrovasc Dis. (2016) 25:1509–16. doi: 10.1016/j.jstrokecerebrovasdis.2016.02.029
57. Yamashita M, Oka K, Tanaka K. Cervico-cephalic arterial thrombi and thromboemboli in moyamoya disease–possible correlation with progressive intimal thickening in the intracranial major arteries. Stroke. (1984) 15:264–70. doi: 10.1161/01.str.15.2.264
58. Xu WH Li ML, Gao S, Ni J, Zhou LX, Yao M, et al. In vivo high-resolution MR imaging of symptomatic and asymptomatic middle cerebral artery atherosclerotic stenosis. Atherosclerosis. (2010) 212:507–11. doi: 10.1016/j.atherosclerosis.2010.06.035
59. Shi MC, Wang SC, Zhou HW, Xing YQ, Cheng YH, Feng JC, et al. Compensatory remodeling in symptomatic middle cerebral artery atherosclerotic stenosis: a high-resolution MRI and microemboli monitoring study. Neurol Res. (2012) 34:153–8. doi: 10.1179/1743132811Y.0000000065
60. Harrison EG Jr, McCormack LJ. Pathologic classification of renal arterial disease in renovascular hypertension. Mayo Clin Proc. (1971) 46:161–7.
61. Slovut DP, Olin JW. Fibromuscular dysplasia. N Engl J Med. (2004) 350:1862–71. doi: 10.1056/NEJMra032393
62. Kirton A, Crone M, Benseler S, Mineyko A, Armstrong D, Wade A, et al. Fibromuscular dysplasia and childhood stroke. Brain. (2013) 136(Pt 6):1846–56. doi: 10.1093/brain/awt111
63. DiFazio M, Hinds SR II, Depper M, Tom B, Davis R. Intracranial fibromuscular dysplasia in a six-year-old child: a rare cause of childhood stroke. J Child Neurol. (2000) 15:559–62. doi: 10.1177/088307380001500814
64. Mikami T, Suzuki H, Komatsu K, Mikuni N. Influence of inflammatory disease on the pathophysiology of moyamoya disease and quasi-moyamoya disease. Neurol Med Chir (Tokyo). (2019) 59:361–70. doi: 10.2176/nmc.ra.2019-0059
65. Ikeda E, Hosoda Y. Distribution of thrombotic lesions in the cerebral arteries in spontaneous occlusion of the circle of Willis: cerebrovascular moyamoya disease. Clin Neuropathol. (1993) 12:44–8.
66. Masuda J, Ogata J, Yutani C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in moyamoya disease. Stroke. (1993) 24:1960–7. doi: 10.1161/01.str.24.12.1960
67. Mikami T, Noshiro S, Komatsu K, Miyata K, Akiyama Y, Wanibuchi M, et al. Vascular remodeling of the circle of Willis in moyamoya disease. Neurol Res. (2015) 37:880–5. doi: 10.1179/1743132815Y.0000000069
68. Choi EH, Lee H, Chung JW, Seo WK, Kim GM Ki CS, et al. Ring finger protein 213 variant and plaque characteristics, vascular remodeling, and hemodynamics in patients with intracranial atherosclerotic stroke: a high-resolution magnetic resonance imaging and hemodynamic study. J Am Heart Assoc. (2019) 8:e011996. doi: 10.1161/JAHA.119.011996
69. Agarwal P, Khalafallah AM, Hersh EH, Ivan ME, Mukherjee D. Impact of American association of neurological surgeons medical student interest groups on participation in organized neurosurgery, research productivity, and residency match success. World Neurosurg. (2020) 138:e437–44. doi: 10.1016/j.wneu.2020.02.153
70. Baird LC, Smith ER, Ichord R, Piccoli DA, Bernard TJ, Spinner NB, et al. Moyamoya syndrome associated with Alagille syndrome: outcome after surgical revascularization. J Pediatr. (2015) 166:470–3. doi: 10.1016/j.jpeds.2014.10.067
71. Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya. J Neurosurg Pediatr. (2009) 3:320–4. doi: 10.3171/2008.12.PEDS08205
72. Kennedy BC, McDowell MM, Yang PH, Wilson CM Li S, Hankinson TC, et al. Pial synangiosis for moyamoya syndrome in children with sickle cell anemia: a comprehensive review of reported cases. Neurosurg Focus. (2014) 36:E12. doi: 10.3171/2013.10.FOCUS13405
73. Kirkham FJ, DeBaun MR. Stroke in children with sickle cell disease. Curr Treat Options Neurol. (2004) 6:357–75. doi: 10.1007/s11940-996-0028-4
74. Koss M, Scott RM, Irons MB, Smith ER, Ullrich NJ. Moyamoya syndrome associated with neurofibromatosis type 1: perioperative and long-term outcome after surgical revascularization. J Neurosurg Pediatr. (2013) 11:417–25. doi: 10.3171/2012.12.PEDS12281
75. Moftakhar P, Smith ER, Choulakian A, Scott RM, Danielpour M. Moyamoya disease in children with congenital dwarfing conditions. Pediatr Neurosurg. (2010) 46:373–80. doi: 10.1159/000322017
76. Smith ER, McClain CD, Heeney M, Scott RM. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg Focus. (2009) 26:E10. doi: 10.3171/2009.01.FOCUS08307
77. Ullrich NJ, Robertson R, Kinnamon DD, Scott RM, Kieran MW, Turner CD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. (2007) 68:932–8. doi: 10.1212/01.wnl.0000257095.33125.48
78. Smith ER. Moyamoya biomarkers. J Korean Neurosurg Soc. (2015) 57:415–21. doi: 10.3340/jkns.2015.57.6.415
79. Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. (2000) 58:341–4. doi: 10.1034/j.1399-0004.2000.580501.x
80. Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet. (1999) 89:14–22.
81. Ganesan V, Kirkham FJ. Noonan syndrome and moyamoya. Pediatr Neurol. (1997) 16:256–8. doi: 10.1016/s0887-8994(97)89980-8
82. Hung PC, Wang HS, Wong AM. Moyamoya syndrome in a child with Noonan syndrome. Pediatr Neurol. (2011) 45:129–31. doi: 10.1016/j.pediatrneurol.2011.03.007
83. Schuster JM, Roberts TS. Symptomatic moyamoya disease and aortic coarctation in a patient with Noonan's syndrome: strategies for management. Pediatr Neurosurg. (1999) 30:206–10. doi: 10.1159/000028797
84. Yamashita Y, Kusaga A, Koga Y, Nagamitsu S, Matsuishi T. Noonan syndrome, moyamoya-like vascular changes, and antiphospholipid syndrome. Pediatr Neurol. (2004) 31:364–6. doi: 10.1016/j.pediatrneurol.2004.05.015
85. Shiihara T, Kato M, Mitsuhashi Y, Hayasaka K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr Neurol. (2005) 32:361–3. doi: 10.1016/j.pediatrneurol.2004.12.010
86. Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M, et al. mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest. (2008) 118:2169–79. doi: 10.1172/JCI34385
87. Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. (2012) 49:138–44. doi: 10.1136/jmedgenet-2011-100544
88. Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. (1997) 16:235–42. doi: 10.1038/ng0797-235
89. Connor SE, Hewes D, Ball C, Jarosz JM. Alagille syndrome associated with angiographic moyamoya. Childs Nerv Syst. (2002) 18:186–90. doi: 10.1007/s00381-001-0518-3
90. Emerick KM, Krantz ID, Kamath BM, Darling C, Burrowes DM, Spinner NB, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. (2005) 41:99–107. doi: 10.1097/01.mpg.0000162776.67758.2f
91. Gaba RC, Shah RP, Muskovitz AA, Guzman G, Michals EA. Synchronous moyamoya syndrome and ruptured cerebral aneurysm in Alagille syndrome. J Clin Neurosci. (2008) 15:1395–8. doi: 10.1016/j.jocn.2007.05.033
92. Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. (2004) 109:1354–8. doi: 10.1161/01.CIR.0000121361.01862.A4
93. Rocha R, Soro I, Leitao A, Silva ML, Leao M. Moyamoya vascular pattern in Alagille syndrome. Pediatr Neurol. (2012) 47:125–8. doi: 10.1016/j.pediatrneurol.2012.04.014
94. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. (2012) 20:251–7. doi: 10.1038/ejhg.2011.181
95. Woolfenden AR, Albers GW, Steinberg GK, Hahn JS, Johnston DC, Farrell K. Moyamoya syndrome in children with Alagille syndrome: additional evidence of a vasculopathy. Pediatrics. (1999) 103:505–8. doi: 10.1542/peds.103.2.505
96. Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. (1997) 16:243–51. doi: 10.1038/ng0797-243
97. Munot P, Crow YJ, Ganesan V. Paediatric stroke: genetic insights into disease mechanisms and treatment targets. Lancet Neurol. (2011) 10:264–74. doi: 10.1016/S1474-4422(10)70327-6
98. Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. (2008) 319:816–9. doi: 10.1126/science.1151174
99. Rauch A. The shortest of the short: pericentrin mutations and beyond. Best Pract Res Clin Endocrinol Metab. (2011) 25:125–30. doi: 10.1016/j.beem.2010.10.015
100. Herve D, Touraine P, Verloes A, Miskinyte S, Krivosic V, Logeart D, et al. A hereditary moyamoya syndrome with multisystemic manifestations. Neurology. (2010) 75:259–64. doi: 10.1212/WNL.0b013e3181e8ee3f
101. Laine J, Kunstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. (2000) 6:395–407. doi: 10.1016/s1097-2765(00)00039-3
102. Miskinyte S, Butler MG, Herve D, Sarret C, Nicolino M, Petralia JD, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. (2011) 88:718–28. doi: 10.1016/j.ajhg.2011.04.017
103. Herve D, Philippi A, Belbouab R, Zerah M, Chabrier S, Collardeau-Frachon S, et al. Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet. (2014) 94:385–94. doi: 10.1016/j.ajhg.2014.01.018
104. Mejia-Munne JC, Ellis JA, Feldstein NA, Meyers PM, Connolly ES. Moyamoya and inflammation. World Neurosurg. (2017) 100:575–8. doi: 10.1016/j.wneu.2017.01.012
105. Suzuki J, Kodama N. Moyamoya disease–a review. Stroke. (1983) 14:104–9. doi: 10.1161/01.str.14.1.104
106. Hayashi K, Horie N, Izumo T, Nagata I. Nationwide survey on quasi-moyamoya disease in Japan. Acta Neurochir (Wien). (2014) 156:935–40. doi: 10.1007/s00701-014-2013-0
107. Houkin K, Ito M, Sugiyama T, Shichinohe H, Nakayama N, Kazumata K, et al. Review of past research and current concepts on the etiology of moyamoya disease. Neurol Med Chir (Tokyo). (2012) 52:267–77. doi: 10.2176/nmc.52.267
108. Malik S, Russman AN, Katramados AM, Silver B, Mitsias PD. Moyamoya syndrome associated with Graves' disease: a case report and review of the literature. J Stroke Cerebrovasc Dis. (2011) 20:528–36. doi: 10.1016/j.jstrokecerebrovasdis.2010.03.006
109. Ohba S, Nakagawa T, Murakami H. Concurrent Graves' disease and intracranial arterial stenosis/occlusion: special considerations regarding the state of thyroid function, etiology, and treatment. Neurosurg Rev. (2011) 34:297–304; discussion. doi: 10.1007/s10143-011-0311-z
110. Chen JB, Lei D, He M, Sun H, Liu Y, Zhang H, et al. Clinical features and disease progression in moyamoya disease patients with Graves disease. J Neurosurg. (2015) 123:848–55. doi: 10.3171/2014.10.JNS141140
111. Lee R, Sung K, Park YM Yu JJ, Koh YC, Chung S. A case of moyamoya disease in a girl with thyrotoxicosis. Yonsei Med J. (2009) 50:594–8. doi: 10.3349/ymj.2009.50.4.594
112. Liu JS, Juo SH, Chen WH, Chang YY, Chen SS. A case of Graves' diseases associated with intracranial moyamoya vessels and tubular stenosis of extracranial internal carotid arteries. J Formos Med Assoc. (1994) 93:806–9.
113. Czartoski T, Hallam D, Lacy JM, Chun MR, Becker K. Postinfectious vasculopathy with evolution to moyamoya syndrome. J Neurol Neurosurg Psychiatry. (2005) 76:256–9. doi: 10.1136/jnnp.2004.041046
114. Im SH, Oh CW, Kwon OK, Kim JE, Han DH. Moyamoya disease associated with Graves disease: special considerations regarding clinical significance and management. J Neurosurg. (2005) 102:1013–7. doi: 10.3171/jns.2005.102.6.1013
115. Tendler BE, Shoukri K, Malchoff C, MacGillivray D, Duckrow R, Talmadge T, et al. Concurrence of Graves' disease and dysplastic cerebral blood vessels of the moyamoya variety. Thyroid. (1997) 7:625–9. doi: 10.1089/thy.1997.7.625
116. Ni J, Gao S, Cui LY Li SW. Intracranial arterial occlusive lesion in patients with Graves' disease. Chin Med Sci J. (2006) 21:140–4.
117. Palacio S, Hart RG, Vollmer DG, Kagan-Hallet K. Late-developing cerebral arteropathy after pyogenic meningitis. Arch Neurol. (2003) 60:431–3. doi: 10.1001/archneur.60.3.431
118. Kerr L, Filloux FM. Cerebral infarction as a remote complication of childhood Haemophilus influenzae meningitis. West J Med. (1992) 157:179–82.
119. Pinardi F, Stracciari A, Spinardi L, Guarino M. Postpneumococcal moyamoya syndrome case report and review of the postinfective cases. BMJ Case Rep. (2013) 2013:bcr2012006726. doi: 10.1136/bcr-2012-006726
120. Pugin D, Copin JC, Goodyear MC, Landis T, Gasche Y. Persisting vasculitis after pneumococcal meningitis. Neurocrit Care. (2006) 4:237–40. doi: 10.1385/NCC:4:3:237
121. Cardenas JF, Chapman K. Sydenham's chorea as a presentation of moyamoya disease. Semin Pediatr Neurol. (2010) 17:30–4. doi: 10.1016/j.spen.2010.01.004
122. Stoeter P, Voigt K. [Moyamoya syndrome of tuberculous cerebral arteritis]. Rofo. (1976) 124:516–9. doi: 10.1055/s-0029-1230382
123. Kitahara M, Kaneko U, Fujiwara S, Suzuki J. [A case showing angiographically moyamoya vessels combined with intracranial calcification]. No Shinkei Geka. (1986) 14(3 Suppl):367–72.
124. Kashiwagi S, Abiko S, Harada K, Yamashita K, Ito H. [Ischemic cerebrovascular complication in tuberculous meningitis; a case of Frohlich syndrome and hemiparesis]. No Shinkei Geka. (1990) 18:1141–5.
125. Nakayama Y, Tanaka A, Nagasaka S, Ikui H. [Intracerebral hemorrhage in a patient with moyamoya phenomenon caused by tuberculous arteritis: a case report]. No Shinkei Geka. (1999) 27:751–5.
126. Yamada H, Deguchi K, Tanigawara T, Takenaka K, Nishimura Y, Shinoda J, et al. The relationship between moyamoya disease and bacterial infection. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S221–4. doi: 10.1016/s0303-8467(97)00048-6
127. Matsushima Y, Qian L, Aoyagi M. Comparison of moyamoya disease in Japan and moyamoya disease (or syndrome) in the People's Republic of China. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S19–22. doi: 10.1016/s0303-8467(97)00034-6
128. Greco F, Castellano Chiodo D, Sorge A, Perrini S, Sorge G. [Multiple arterial ischemic strokes in a child with moyamoya disease and Mycoplasma pneumoniae infection]. Minerva Pediatr. (2006) 58:63–8.
129. Tanigawara T, Yamada H, Sakai N, Andoh T, Deguchi K, Iwamura M. Studies on cytomegalovirus and Epstein-Barr virus infection in moyamoya disease. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S225–8. doi: 10.1016/s0303-8467(97)00049-8
130. Morgello S, Laufer H. Quaternary neurosyphilis in a Haitian man with human immunodeficiency virus infection. Hum Pathol. (1989) 20:808–11. doi: 10.1016/0046-8177(89)90078-6
131. Hsiung GY, Sotero de. Menezes M. Moyamoya syndrome in a patient with congenital human immunodeficiency virus infection. J Child Neurol. (1999) 14:268–70. doi: 10.1177/088307389901400412
132. Galati V, Grilli E, Sallustio F, Petrosillo N. An adult HIV patient with unilateral moyamoya syndrome. Clin Neurol Neurosurg. (2010) 112:76–8. doi: 10.1016/j.clineuro.2009.09.002
133. Sharfstein SR, Ahmed S, Islam MQ, Najjar MI, Ratushny V. Case of moyamoya disease in a patient with advanced acquired immunodeficiency syndrome. J Stroke Cerebrovasc Dis. (2007) 16:268–72. doi: 10.1016/j.jstrokecerebrovasdis.2007.07.001
134. Ueno M, Oka A, Koeda T, Okamoto R, Takeshita K. Unilateral occlusion of the middle cerebral artery after varicella-zoster virus infection. Brain Dev. (2002) 24:106–8. doi: 10.1016/s0387-7604(02)00005-0
135. Takasugi H, Maemoto T, Kitazawa K, Honda A. [A case of Down syndrome with moyamoya syndrome presenting extensive multiple cerebral infarction during measles infection]. No To Hattatsu. (2000) 32:39–43. doi: 10.11251/ojjscn1969.32.39
136. Weststrate W, Hijdra A, de Gans J. Brain infarcts in adults with bacterial meningitis. Lancet. (1996) 347:399. doi: 10.1016/s0140-6736(96)90577-2
137. Igarashi M, Gilmartin RC, Gerald B, Wilburn F, Jabbour JT. Cerebral arteritis and bacterial meningitis. Arch Neurol. (1984) 41:531–5. doi: 10.1001/archneur.1984.04050170077022
138. Liu XM, Ruan XZ, Cai Z, Yu BR, He SP, Gong YH. Moyamoya disease caused by leptospiral cerebral arteritis. Chin Med J (Engl). (1980) 93:599–604.
139. Takahashi Y, Mikami T, Suzuki H, Komatsu K, Yamamoto D, Shimohama S, et al. Development of moyamoya disease after non-herpetic acute limbic encephalitis: a case report. J Clin Neurosci. (2018) 53:250–3. doi: 10.1016/j.jocn.2018.04.042
140. Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. (2004) 100(2 Suppl Pediatrics):142–9. doi: 10.3171/ped.2004.100.2.0142
141. Hallemeier CL, Rich KM, Grubb RL Jr, Chicoine MR, Moran CJ, Cross DT III, et al. Clinical features and outcome in North American adults with moyamoya phenomenon. Stroke. (2006) 37:1490–6. doi: 10.1161/01.STR.0000221787.70503.ca
142. Jea A, Smith ER, Robertson R, Scott RM. Moyamoya syndrome associated with Down syndrome: outcome after surgical revascularization. Pediatrics. (2005) 116:e694–701. doi: 10.1542/peds.2005-0568
143. Lubman DI, Pantelis C, Desmond P, Proffitt TM, Velakoulis D. Moyamoya disease in a patient with schizophrenia. J Int Neuropsychol Soc. (2003) 9:806–10. doi: 10.1017/S1355617703950132
144. Tagawa T, Naritomi H, Mimaki T, Yabuuchi H, Sawada T. Regional cerebral blood flow, clinical manifestations, and age in children with moyamoya disease. Stroke. (1987) 18:906–10. doi: 10.1161/01.str.18.5.906
145. Han DH, Nam DH, Oh CW. Moyamoya disease in adults: characteristics of clinical presentation and outcome after encephalo-duro-arterio-synangiosis. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S151–5. doi: 10.1016/s0303-8467(97)00058-9
146. Iwama T, Morimoto M, Hashimoto N, Goto Y, Todaka T, Sawada M. Mechanism of intracranial rebleeding in moyamoya disease. Clin Neurol Neurosurg. (1997) 99(Suppl 2):S187–90.
147. Irikura K, Miyasaka Y, Kurata A, Tanaka R, Fujii K, Yada K, et al. A source of haemorrhage in adult patients with moyamoya disease: the significance of tributaries from the choroidal artery. Acta Neurochir (Wien). (1996) 138:1282–6. doi: 10.1007/BF01411056
148. Kawaguchi S, Sakaki T, Morimoto T, Kakizaki T, Kamada K. Characteristics of intracranial aneurysms associated with moyamoya disease. A review of 111 cases. Acta Neurochir (Wien). (1996) 138:1287–94. doi: 10.1007/BF01411057
149. Kuroda S, Houkin K, Kamiyama H, Abe H. Effects of surgical revascularization on peripheral artery aneurysms in moyamoya disease: report of three cases. Neurosurgery. (2001) 49:463–7; discussion 7–8. doi: 10.1097/00006123-200108000-00039
150. Seol HJ, Wang KC, Kim SK, Hwang YS, Kim KJ, Cho BK. Headache in pediatric moyamoya disease: review of 204 consecutive cases. J Neurosurg. (2005) 103(5 Suppl):439–42. doi: 10.3171/ped.2005.103.5.0439
151. Parmar RC, Bavdekar SB, Muranjan MN, Limaye U. Chorea: an unusual presenting feature in pediatric moyamoya disease. Indian Pediatr. (2000) 37:1005–9.
152. Massaro M, Thorarensen O, Liu GT, Maguire AM, Zimmerman RA, Brodsky MC. Morning glory disc anomaly and moyamoya vessels. Arch Ophthalmol. (1998) 116:253–4.
153. Kawabori M, Kuroda S, Nakayama N, Hirata K, Shiga T, Houkin K, et al. Effective surgical revascularization improves cerebral hemodynamics and resolves headache in pediatric moyamoya disease. World Neurosurg. (2013) 80:612–9. doi: 10.1016/j.wneu.2012.08.005
154. Riordan CP, Storey A, Cote DJ, Smith ER, Scott RM. Results of more than 20 years of follow-up in pediatric patients with moyamoya disease undergoing pial synangiosis. J Neurosurg Pediatr. (2019) 2019:1–7. doi: 10.3171/2019.1.PEDS18457
155. Kazumata K, Tokairin K, Sugiyama T, Ito M, Uchino H, Osanai T, et al. Association of cognitive function with cerebral blood flow in children with moyamoya disease. J Neurosurg Pediatr. (2019) 2019:1–7. doi: 10.3171/2019.7.PEDS19312
156. Lee J Kim SK Kang HG Ha IS Wang KC Lee JY . High prevalence of systemic hypertension in pediatric patients with moyamoya disease years after surgical treatment. J Neurosurg Pediatr. (2019) 2019:1–7. doi: 10.3171/2019.9.PEDS1986
157. Kim YJ, Lee DH, Kwon JY, Kang DW, Suh DC, Kim JS, et al. High resolution MRI difference between moyamoya disease and intracranial atherosclerosis. Eur J Neurol. (2013) 20:1311–8. doi: 10.1111/ene.12202
158. Kim T, Bang JS, Kwon OK, Hwang G, Kim JE, Kang HS, et al. Morphology and related hemodynamics of the internal carotid arteries of moyamoya patients. Acta Neurochir (Wien). (2015) 157:755–61. doi: 10.1007/s00701-015-2367-y
159. Ladner TR, Donahue MJ, Arteaga DF, Faraco CC, Roach BA, Davis LT, et al. Prior Infarcts, Reactivity, and Angiography in Moyamoya Disease (PIRAMD): a scoring system for moyamoya severity based on multimodal hemodynamic imaging. J Neurosurg. (2017) 126:495–503. doi: 10.3171/2015.11.JNS15562
160. Han C, Li ML, Xu YY, Ye T, Xie CF, Gao S, et al. Adult moyamoya-atherosclerosis syndrome: clinical and vessel wall imaging features. J Neurol Sci. (2016) 369:181–4. doi: 10.1016/j.jns.2016.08.020
161. Storey A, Michael Scott R, Robertson R, Smith E. Preoperative transdural collateral vessels in moyamoya as radiographic biomarkers of disease. J Neurosurg Pediatr. (2017) 19:289–95. doi: 10.3171/2016.9.PEDS16161
162. Song P, Qin J, Lun H, Qiao P, Xie A, Li G. Magnetic resonance imaging (MRI) and digital subtraction angiography investigation of childhood moyamoya disease. J Child Neurol. (2017) 32:1027–34. doi: 10.1177/0883073817736161
163. Li J, Jin M, Sun X, Li J, Liu Y, Xi Y, et al. Imaging of moyamoya disease and moyamoya syndrome: current status. J Comput Assist Tomogr. (2019) 43:257–63. doi: 10.1097/RCT.0000000000000834
164. Hauser TK, Seeger A, Bender B, Klose U, Thurow J, Ernemann U, et al. Hypercapnic BOLD MRI compared to H2(15)O PET/CT for the hemodynamic evaluation of patients with moyamoya disease. Neuroimage Clin. (2019) 22:101713. doi: 10.1016/j.nicl.2019.101713
Keywords: angiopathy, cerebrovascular, moyamoya, stenosis, stroke
Citation: Fox BM, Dorschel KB, Lawton MT and Wanebo JE (2021) Pathophysiology of Vascular Stenosis and Remodeling in Moyamoya Disease. Front. Neurol. 12:661578. doi: 10.3389/fneur.2021.661578
Received: 31 January 2021; Accepted: 09 August 2021;
Published: 03 September 2021.
Edited by:
Yuxiang Gu, Fudan University, ChinaReviewed by:
Masahito Kawabori, Hokkaido University, JapanCopyright © 2021 Fox, Dorschel, Lawton and Wanebo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John E. Wanebo, TmV1cm9wdWJAYmFycm93bmV1cm8ub3Jn
†ORCID: Kirsten B. Dorschel orcid.org/0000-0002-8133-3880
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.