 Michael Candlish
Michael Candlish Jasmin K. Hefendehl
Jasmin K. Hefendehl
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 05 May 2021
Sec. Dementia and Neurodegenerative Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.660720
This article is part of the Research TopicPrimary Glial and Immune Cell Pathology in Neurodegenerative DiseasesView all 7 articles
Microglia, the primary immune cells of the central nervous system, hold a multitude of tasks in order to ensure brain homeostasis and are one of the best predictors of biological age on a cellular level. We and others have shown that these long-lived cells undergo an aging process that impedes their ability to perform some of the most vital homeostatic functions such as immune surveillance, acute injury response, and clearance of debris. Microglia have been described as gradually transitioning from a homeostatic state to an activated state in response to various insults, as well as aging. However, microglia show diverse responses to presented stimuli in the form of acute injury or chronic disease. This complexity is potentially further compounded by the distinct alterations that globally occur in the aging process. In this review, we discuss factors that may contribute to microglial aging, as well as transcriptional microglia alterations that occur in old age. We then compare these distinct phenotypic changes with microglial phenotype in neurodegenerative disease.
Microglia originate from hematopoietic progenitor cells found in the yolk sac and, upon entering the brain, gradually adapt a homeostatic microglial phenotype (1). Homeostatic microglia feature a distinct ramified morphology and were first identified by del Rio-Hortega (2). As the primary immune cells of the brain, microglia are mostly associated with acute or chronic responses to injury. In response to these stimuli, microglia display morphological and biochemical changes that have often been grouped under the term “activation.” These changes can entail a variety of downstream effects including cytokine and chemokine production, enhanced phagocytosis, proliferation, and migration. Historically, based on in vitro experiments, this rather generalized diversion from the homeostatic cell state has led to a differentiation into two microglial groups: M1 (proinflammatory) and M2 (neuroprotective). This relatively oversimplified classification (3) of microglial reactivity is now being refined by single-cell resolution techniques that show diverse transcriptional states that can be adapted by microglia in either a gradual or acute manner. Intriguingly, we and others have shown that microglia are long-lived cells that undergo an aging process on a cellular level, altering their surveillance capacity and injury response time, and also influence neurodegenerative diseases (4–7) [reviewed in (8, 9)]. These rather slow and gradual alterations are contrasted by rapid changes brought on by acute damage. Local signals in the microglial microenvironment drive acute as well as gradual changes, leading to broad alterations in gene transcription, cell morphology, phagocytotic activity, and proliferation status (10–12). Over decades of research, a wide variety of terms have been used to describe microglial cell states (13). As research has advanced, new distinctions in microglial phenotypes have been identified based on expression of particular genes and, most recently, their transcriptome signature. In this review, we aim to uncover potential similarities in microglial phenotypes in advanced age and neurodegenerative disease (Figure 1A) (12, 15, 16).
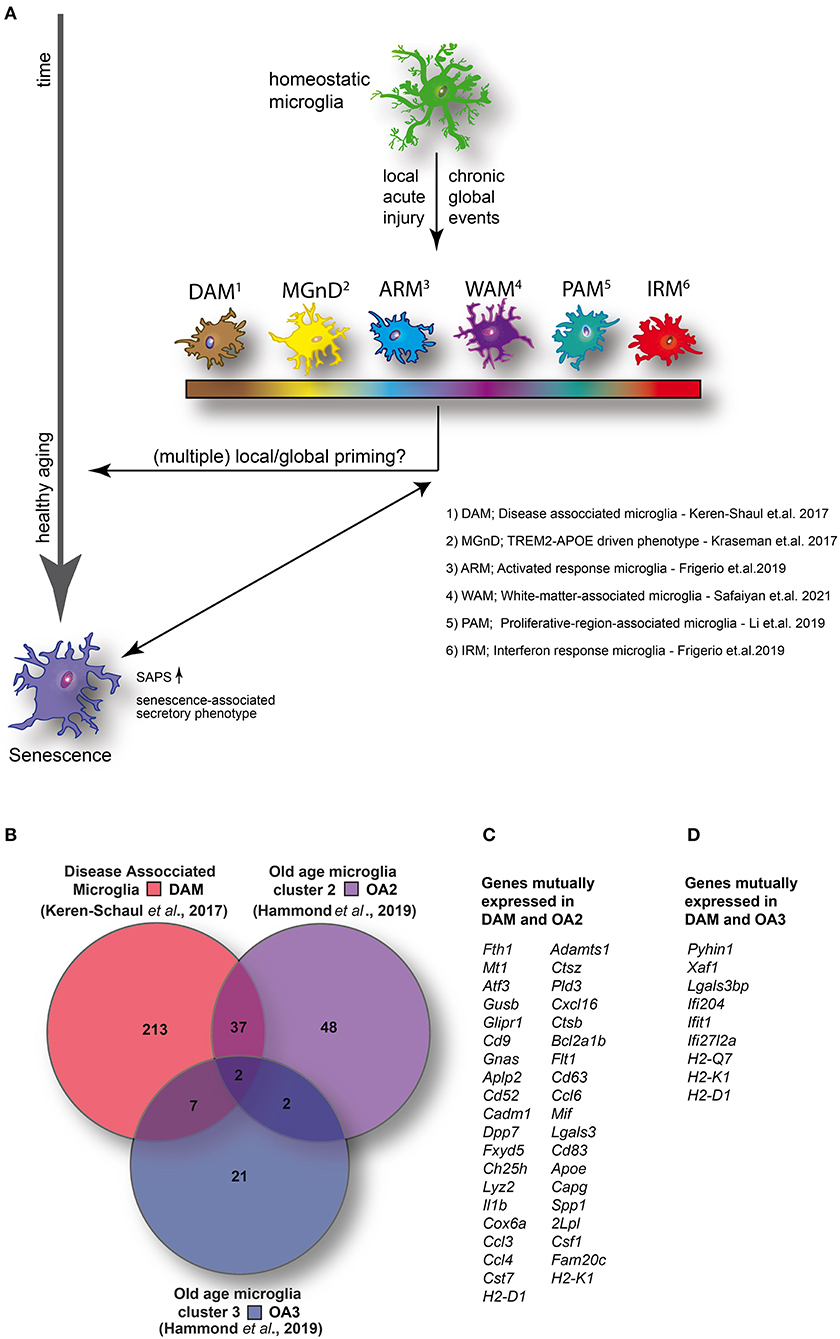
Figure 1. (A) Homeostatic microglia over the life span of the animal encounter both spatially restricted and global injuries that likely contribute to transcriptomic and functional alterations in microglia, potentially resulting in the adaption of specific disease- or injury-associated microglia phenotypes and conceivably contributing to premature cellular aging. (B) Venn diagram illustrating the number of enriched (1.5-fold or higher) mutually and exclusively expressed genes between DAM (identified in a mouse model of AD) (14) and two distinct clusters identified in aged wild-type mice [old age (OA) 2 and OA3] (7). (C) DAM and OA2 feature 39 mutually expressed genes suggesting the activation of common transcriptional programs in both neurodegenerative disease and a minority of cells in healthy aging. (D) Conversely, only nine genes were mutually expressed between DAM and OA3 suggesting that the OA3 population may be distinct from both DAM and OA2 microglia. Venn diagram generated using Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/).
It recently was shown that not only cells of the adaptive immune system but also those of the innate immune system can display memory effects (5). The priming step of the immune cells, induced by a primary insult, can result in an enhanced or dampened subsequent injury response (17–19). In vivo tracking of individual microglial cells showed that microglia can revert to a homeostatic morphology post-injury (20–22). However, even if the morphological homeostatic phenotype is reestablished, epigenetic modification may render the cells altered from their homeostatic state. As long-lived cells, microglia are very likely to be primed by signals in their microenvironment, hence memory effects might add up during the cell's life span. This poses the question if these rather individualized priming steps might be part of the cellular aging process or if they have the potential of adversely affecting healthy aging. Furthermore, as some acute injuries such as microlesions are likely to only affect surrounding microglial cells, the priming impact might be locally restricted.
Individual local events could result in the generation of distinct spatially restricted transcriptional and phenotypic alteration as microglial phenotypes are partially regulated through membrane-bound pattern recognition receptors (PRRs), which depend on molecules released by cells in their microenvironment. In a local ischemic event, disease-associated molecular patterns are passively released from dying cells (23), which may lead to a transient activation with a priming effect, whereas the deposition of amyloid β (Aβ) in the parenchyma [a hallmark of Alzheimer disease (AD), but can also be found in aged brains on a lesser scale] may lead to a different chronic disease–associated microglial phenotype, which not only depends on PRRs, as microglia also possess a wide variety of receptors to detect other types of molecules such as hormones and neurotransmitters (23). Therefore, a multitude of factors exist that could potentially affect local and global microglial behavior both in a short- or long-term fashion. A recent study demonstrated the importance of the local milieu within the brain in this regard by transiently depleting microglia using the colony-stimulating factor 1 receptor antagonist PLX5622 (24) in aged mice (25). The authors hypothesized that withdrawal of the drug would result in the replenishment of “young” unprimed microglia (25). Conversely, they reported that the transcriptomic alterations in old age were only partially reversed, and the replenished microglia responded to lipopolysaccharide (LPS) with an exaggerated proinflammatory response, typical of primed microglia. Further in vitro experiments confirmed that media conditioned by 24-h cultivation of brain slices from aged, but not young adult, mice were sufficient to trigger an exacerbated response to LPS in neonatal microglia, elegantly demonstrating the importance of the milieu in which microglia are resident (24).
When focusing on the healthy aging process of microglia, they have been described as dystrophic or senescent [reviewed in (26)]. Historically, senescence is characterized by arrested growth caused by oxidative stress as well as elevated DNA damage. Age-related changes in the secretory profile were described to coin the term senescence-associated secretory phenotype, classifying a particular cell state in the aging brain (27, 28). The term dystrophic, on the other hand, was created by the observation of changes in microglial morphology in brain sections from elderly humans and potentially includes all visually altered microglia (20). Among other features, this phenotype includes the beading of microglial processes, which are held together by thin channels (29), and was proposed to signify microglial senescence (30). Previous studies addressing the question if one of the described phenotypes is purely age-related are controversial; some have found dystrophic microglia in aged humans without any underlying neurodegenerative disorders (30–32), whereas others gathered evidence suggesting that dystrophic microglia are associated with a variety of diseases including, e.g., AD (29, 31–34), Huntington disease (14), and multiple sclerosis (35). However, recently some light has been shed on the question whether these two terms are describing the same or two different phenotypes. To address this issue, Shahidehpour et al. (36) conducted stereological analysis of microglia in human brain tissue spanning the age of 10-90 years. The analysis revealed an increased number of dystrophic microglia with age, which, however, was much greater when neurodegenerative pathology was present as well (36). They hence conclude that aging itself is only associated with a minor increase in dystrophic microglia (36). It is thus possible that the disease event that generated an activated microglia phenotype, potentially early in life, has a priming effect on cellular aging, leading to an increase of dystrophic microglia in old age (Figure 1A).
Also, the opposite assumption is valid; the overall cellular aging process is likely causative for poor local injury responses, resulting in an ineffective healing process that in turn might again increase the amount of dystrophic/senescent microglia. One example is the finding that population RNAseq of murine microglia has identified a consistent age-dependent increase in genes associated with a low-grade inflammatory response (37), which might be causative for a poor local injury response by microglia to additional acute insults. We have found the injury response time to a local laser lesion of aged microglia (~2.4-year-old mice) to be reduced by ~50%. Additionally, while microglial process end-tips in young and adult mice showed an increase in local diameters after a microlaser lesion, the aged animals displayed significantly less morphological changes upon lesioning, as the process end-tips already were found to be enlarged prior to the insult (4). Further supporting a gradual overall drift into a low-grade inflammatory state during aging, Minhas et al. recently put forward evidence suggesting that a change in the metabolic state of macrophages (in the brain and periphery), signified by a reduction of the two main metabolic pathways (glycolysis and oxidative phosphorylation), is affecting brain health as acute energy demands, e.g., in order to support macrophage activation, cannot be met any longer (38). More specifically, prostaglandin E2 (PGE2), a proinflammatory signaling protein, which is known to increase not only during aging but also in AD, was investigated. The inhibition of PGE2 was shown to lead to brain rejuvenation, reducing inflammatory levels in the aged brain (38). These findings are of particular interest as they support other reports that microglia can be influenced by stimulation of peripheral immune cells (5) and that their responsiveness can be (partially) restored even in the aged brain. With the detailed mechanism leading to the reported reduction in metabolism still being unknown, a future challenge is to unravel this pathway to explore possible therapeutic interventions, aiding in a wide range of diseases, as well as aging itself.
In the effort to characterize microglia phenotype in disease, scRNAseq has become a powerful weapon and has facilitated even greater insight into the transcriptomic alterations of microglia in diverse conditions. At present, however, comparing transcriptomic data from different studies comes with inherent challenges. Variability can be introduced at many stages such as dissociation, gating for cell sorting, the scRNAseq procedure, and data analysis. Technical limitations can further result in discrepant data. For example, it was recently revealed that the technical limitations of single-nucleus (sn)RNAseq (a theoretically useful approach for postmortem human tissue as it is compatible with frozen tissue) may have resulted in data that lead inadvertently to the overstating of differences between murine and human microglial transcriptomes (16). Thrupp et al. found that many transcripts associated with microglial activation are concentrated in the cytosol as opposed to the nuclei, resulting in many transcripts remaining undetected with snRNAseq (16). Another pertinent issue is that despite recent technical advances in scRNAseq, correlating transcriptional profiles to mechanistic data remains a persistent bottleneck. Spatially resolved scRNAseq, especially at a single-cell resolution, would provide an unparalleled advantage in correlating morphological observations to distinct transcriptome signatures. At present, however, these limitations greatly hinder correlating microglial phenotypes (as evident by morphology or behavior in vivo) with specific gene-expression profiles.
Despite these difficulties, highly valuable data have been collected in several high-impact studies. Hammond et al. (12) identified two distinct microglia clusters using scRNAseq that, in the absence of overt pathology, were expanded in aged (~1.5 years old) mice. One cluster (entitled OA2) was found to up-regulate the chemokines Ccl3 and Ccl4 along with interleukin 1 beta (Il1b), indicative of a shift to a proinflammatory phenotype during aging (12). The other emerging cluster in aged animals was found to be enriched in several interferon-response genes [Ifitm3, Rtp4, and Oasl2 (entitled OA3)] (12). This shift toward expression of interferon-response genes is highly interesting, given that recent research has demonstrated that interferon signaling in a mouse model of AD triggers microglial activation, neuroinflammation, and synaptic loss in response to nucleic acid containing Aβ plaques (39).
Sala Frigerio et al. (40) also identified two microglial clusters using scRNAseq that were expanded in aged mice. One cluster, entitled activated response microglia (ARM), increased from around ~3% in 3-month-old mice to ~12% of total microglia in 21-month-old mice (40). ARM microglia were found to up-regulate histocompatibility complex class II genes (Cd74, H2-Ab1, and H2-Aa) and proinflammatory genes Cst7, Clec7a, and Itgax (encoding CD11c). Notably, with the exception of Itgax and Clec7a, these genes were also found to be up-regulated in OA2 microglia (12). Another microglia cluster was also identified, dubbed interferon-response microglia (IRM) (40). This cluster was found to up-regulate Ifit3, Ifitm3, Irf7, and Oasl2, consistent with OA3 microglia (12). Furthermore, using semisupervised pseudotime analysis, the authors found that homeostatic microglia in old age can transition into either IRM or ARM. Taken together, these data suggest that in old age, microglia transition into one of two mutually exclusive states, one characterized by up-regulation of interferon-response genes and the other characterized by a shift to a proinflammatory state. However, as previously mentioned, it remains challenging to ascertain the impact that transcriptional alterations have on microglial phenotype in vivo.
Microglia play complex roles in neurodegenerative disease (41), often being beneficial in some respects, while pathogenic in others. Taking AD as an example, microglia phagocytose Aβ (42, 43) (although this becomes attenuated with aging/Aβ plaque load) and encircle Aβ plaques, prohibiting the spread of [comparably more toxic (44)] soluble amyloid species into the surrounding brain parenchyma (45). Consistent with this, ablation of microglia after amyloid deposition results in increased LAMP1 immunoreactivity surrounding Aβ plaques (46); indicative of dystrophic neurites (47). While these findings suggest that microglia play a beneficial role in AD, microglia themselves contribute to synaptic and neuronal loss in AD (48). It was also shown that abolishing microglia after Aβ plaques are well-established in the brain fails to provide any beneficial effects (49) in the APPPS1 AD mouse model. Furthermore, if microglia are abolished prior to amyloid deposition, Aβ plaques fail to develop in the 5XFAD AD mouse model (46) (although Aβ accumulates on blood vessels resulting in cerebral amyloid angiopathy, a risk factor for hemorrhagic stroke). Taken together, because of the complex matter at hand, it is hard to determine whether microglia provide a net beneficial or detrimental role in AD, and a simple binary answer is not likely.
Despite the potential for global or spatially restricted alterations in transcriptome signature in aging and pathology, recent publications seem to suggest a common—or at least highly similar—transcriptional program (50, 51) in diseased states [i.e., disease-associated microglia (DAM) (52), microglia neurogenerative phenotype (53), ARM (40), and, most recently, white matter–associated microglia (54), a recently identified microglial phenotype that expands during aging and in neurodegenerative disease]. Massively parallel single-cell analysis with chromatin profiling on immune (CD45+) cells from 5XFAD mouse brains (a well-established AD mouse model) revealed two unique microglial clusters (52). Intriguingly, these clusters expressed genes implicated in lipid metabolism and phagocytosis (52). By analyzing mice from the age of 1-8 months (as Aβ deposition is advancing), the authors uncovered an age-dependent shift from a homeostatic phenotype to a DAM phenotype with one of the two clusters identified as being a transitory stage (defined as stage 1) (52). DAM are characterized by the up-regulation of Itgax, Trem2, Axl, Cst7, Ctsl, Lpl, Cd9, Csf1, Ccl6, Clec7a, Lilrb4, and Timp2 (52). Notably, Itgax in particular was found in every cell featuring a DAM transcriptome signature (52). In addition, they down-regulate Cx3cr1, P2ry12, and Tmem119 (52), genes typically expressed in homeostatic microglia (12). The authors further identified that DAM are in close association with Aβ plaques and contain phagocytosed Aβ (52). Intriguingly, the authors identified that a transition from stage 1 DAM to stage 2 DAM was dependent on triggering receptor expressed on myeloid cells 2 (Trem2) (52); homozygous loss-of-function mutations in this gene are known to cause autosomal recessive early-onset dementia [Nasu–Hakola disease and frontotemporal dementia (FTD)–like disease] (55–57). TREM2 acts as a receptor for apolipoprotein E, Aβ, and high- and low-density lipoprotein and has been identified as crucial for triggering microglial phagocytosis, proliferation, and inflammation (58). Importantly, loss-of-function mutations in Trem2 have also been implicated in diverse neurodegenerative diseases including AD (59), amyotrophic lateral sclerosis (60), Parkinson disease (61), and FTD (62). Consistent with this, DAM transcriptomes have now been identified in diverse models of neurodegenerative disease. Based on these elegant findings, it is tempting to speculate that the DAM phenotype represents a pan-neurodegenerative disease response.
Even though we and others have shown morphological and functional changes of microglial cells in aged mice, only a rather small subpopulation show a transcriptomic profile consistent with that of DAM. By comparing (Figure 1B) the top up-regulated genes (greater than 1.5-fold change) between DAM (52) and the small clusters of transcriptionally distinct cells from aged mice (OA2 and OA3) (12), a set of 39 mutually expressed genes can be identified between DAM and OA2 (Figure 1C). Conversely, only nine genes were mutually expressed between DAM and OA3 (Figure 1D). The amount of overlap in genes up-regulated in DAM and OA2 appears to support the hypothesis that a common transcriptional program is activated in both DAM and aged microglia, but only in a minority of the cell population in healthy aging. However, in various disease models, the cell population displaying transcriptomic signatures consistent with DAM is much larger, suggesting that pathological insults during the animal's life span will heavily expand the DAM population, which has a potential impact on cellular aging. Consistent with this, the ARM phenotype described by Frigerio et al. (consistent with DAM) eventually becomes the majority population of microglia at 12 months in an AD mouse model (AppNL−G−F) (40). IRM (consistent with OA3) conversely seemed to increase with age more rapidly in AppNL−G−F mice than wild-type mice but ultimately also represented only a minority of cells (< ~5%) similar to wild-type mice. Ferretin expression has been identified as a marker for dystrophic microglia (36) and senescence (26). Shahidehpour and colleagues (36) found ferretin-expressing dystrophic cells to be present, but again to a very small extent, in healthy aged humans. Conversely, the number of dystrophic microglia was significantly increased in patients suffering from neurodegenerative disorders. Consistent with the data of Shahidehpour et al. (36), ferritin is expressed in the OA2 subpopulation (12), which again is minimal in the absence of overt pathology, yet abundant in neurodegenerative disease conditions (DAM) (52). Taken together, the data suggests a disease-induced increase of cellular aging hallmarks.
Microglia with transcriptomic signatures consistent with that found in neurodegenerative diseases represent only a minority of microglia in healthy aging. It remains unclear what this subset of microglia contributes toward overall microglial dysfunction or, conversely, if they might have a beneficial impact. With regard to microglia featuring a neurodegenerative disease-associated transcriptome signature, it is possible that neurodegenerative disease causes advanced cellular aging, or conversely, advanced cellular aging may be a contributing factor to neurodegenerative disease (63). Further studies will be required to interrogate the roles of these interesting microglial populations in old age. In addition, it seems reasonable to speculate that data gathered from mice at the end of the mouse life span (~2.5 years) would be particularly valuable given the advanced ages reached by humans in contemporary society. scRNAseq studies to date seem to suggest that, despite the many factors that could potentially influence microglial phenotypes in either a global or spatially restricted manner, microglia in aged mice appear to consist of homeostatic microglia, neurodegenerative disease-like microglia, and IRM. The lack of apparent heterogeneity could conceivably be in part due to the artificial conditions that laboratory rodents reside in. This is an important caveat that should be considered when attempting to extrapolate observations from laboratory rodents to humans. With highly individualized lifestyles, disease backgrounds, and environmental factors, microglia in humans are likely to be primed more diversely and extensively given human longevity in comparison to laboratory mice. Hence, much remains to be discovered that could potentially bring valuable mechanistic insights into both aging and neurodegenerative disease.
MC and JKH wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-403813475, 419157387, the Alzheimer's Association (AARF-17-529810), and the Alzheimer Forschung Initiative e.V. (20041).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797-805. doi: 10.1038/ni.3423
2. del Rio-Hortega P. Cytology and cellular pathology of the nervous system. Arch Intern Med (Chic). (1932) 50:508–9. doi: 10.1001/archinte.1932.00150160159019
3. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987-91. doi: 10.1038/nn.4338
4. Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. (2014) 13:60-9. doi: 10.1111/acel.12149
5. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332-8. doi: 10.1038/s41586-018-0023-4
6. Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermuller U, et al. Microglia turnover with aging and in an Alzheimer's model via long-term in vivo single-cell imaging. Nat Neurosci. (2017) 20:1371-6. doi: 10.1038/nn.4631
7. Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci. (2017) 20:793-803. doi: 10.1038/nn.4547
8. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. (2014) 88:594-604. doi: 10.1016/j.bcp.2014.01.008
9. Spittau B. Aging microglia-phenotypes, functions and implications for age-related neurodegenerative diseases. Front Aging Neurosci. (2017) 9:194. doi: 10.3389/fnagi.2017.00194
10. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. (2014) 159:1327-40. doi: 10.1016/j.cell.2014.11.023
11. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131-43. doi: 10.1038/nn.3599
12. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253-71.e6. doi: 10.1016/j.immuni.2018.11.004
13. Stratoulias V, Venero JL, Tremblay ME, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. (2019) 38:e101997. doi: 10.15252/embj.2019101997
14. Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. (2007) 55:1074-84. doi: 10.1002/glia.20526
15. Rexach JE, Polioudakis D, Yin A, Swarup V, Chang TS, Nguyen T, et al. Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. (2020) 33:108398. doi: 10.1016/j.celrep.2020.108398
16. Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, et al. Single-nucleus RNA-Seq is not suitable for detection of microglial activation genes in humans. Cell Rep. (2020) 32:108189. doi: 10.1016/j.celrep.2020.108189
17. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
18. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
19. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475-87. doi: 10.1016/j.it.2009.07.009
20. Miller EB, Zhang P, Ching K, Pugh EN, Jr., Burns ME. In vivo imaging reveals transient microglia recruitment and functional recovery of photoreceptor signaling after injury. Proc Natl Acad Sci U S A. (2019) 116:16603-12. doi: 10.1073/pnas.1903336116
21. Hierro-Bujalance C, Bacskai BJ, Garcia-Alloza M. In vivo imaging of microglia with multiphoton microscopy. Front Aging Neurosci. (2018) 10:218. doi: 10.3389/fnagi.2018.00218
22. Onuska KM. The dual role of microglia in the progression of Alzheimer's disease. J Neurosci. (2020) 40:1608-10. doi: 10.1523/JNEUROSCI.2594-19.2020
23. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. (2011) 91:461-553. doi: 10.1152/physrev.00011.2010
24. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. (2014) 82:380-97. doi: 10.1016/j.neuron.2014.02.040
25. O'Neil SM, Witcher KG, McKim DB, Godbout JP. Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun. (2018) 6:129. doi: 10.1186/s40478-018-0636-8
26. Angelova DM, Brown DR. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J Neurochem. (2019) 151:676-88. doi: 10.1111/jnc.14860
27. Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK. Cellular senescence and the aging brain. Exp Gerontol. (2015) 68:3-7. doi: 10.1016/j.exger.2014.09.018
28. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853-68. doi: 10.1371/journal.pbio.0060301
29. Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, et al. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer's disease. Glia. (2016) 64:1562-72. doi: 10.1002/glia.23024
30. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. (2004) 45:208-12. doi: 10.1002/glia.10319
31. Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun. (2015) 3:32. doi: 10.1186/s40478-015-0209-z
32. Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer's disease brain is associated with ferritin immunoreactivity. Glia. (2008) 56:1048-60. doi: 10.1002/glia.20678
33. Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, et al. Soluble phospho-tau from Alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathol. (2016) 132:897-916. doi: 10.1007/s00401-016-1630-5
34. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. (2009) 118:475-85. doi: 10.1007/s00401-009-0556-6
35. Hametner S, Wimmer I, Haider L, Pfeifenbring S, Bruck W, Lassmann H. Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol. (2013) 74:848-61. doi: 10.1002/ana.23974
36. Shahidehpour RK, Higdon RE, Crawford NG, Neltner JH, Ighodaro ET, Patel E, et al. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol Aging. (2021) 99:19-27. doi: 10.1016/j.neurobiolaging.2020.12.003
37. Pan J, Ma N, Yu B, Zhang W, Wan J. Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation. (2020) 17:97. doi: 10.1186/s12974-020-01774-9
38. Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. (2021) 590:122-8. doi: 10.1038/s41586-020-03160-0
39. Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. (2020) 130:1912-30. doi: 10.1172/JCI133737
40. Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. (2019) 27:1293-306.e6. doi: 10.1016/j.celrep.2019.03.099
41. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. (2010) 6:193-201. doi: 10.1038/nrneurol.2010.17
42. Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer's amyloid beta-protein by microglia in culture. J Neurosci Res. (1996) 43:190-202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B
43. Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. (1996) 17:553-65. doi: 10.1016/S0896-6273(00)80187-7
44. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. (2008) 14:837-42. doi: 10.1038/nm1782
45. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. (2015) 6:6176. doi: 10.1038/ncomms7176
46. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
47. Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc Nat Acad Sci U S A. (2015) 112:E3699-708. doi: 10.1073/pnas.1510329112
48. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712-6. doi: 10.1126/science.aad8373
49. Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. (2009) 12:1361-3. doi: 10.1038/nn.2432
50. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. (2015) 3:31. doi: 10.1186/s40478-015-0203-5
51. Benmamar-Badel A, Owens T, Wlodarczyk A. Protective microglial subset in development, aging, and disease: lessons from transcriptomic studies. Front Immunol. (2020) 11:430. doi: 10.3389/fimmu.2020.00430
52. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276-90.e17. doi: 10.1016/j.cell.2017.05.018
53. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566-81.e9. doi: 10.1016/j.immuni.2017.08.008
54. Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, et al. White matter aging drives microglial diversity. Neuron. (2021) 109:1100-17.e10. doi: 10.1016/j.neuron.2021.01.027
55. Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Megarbane A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat. (2008) 29:E194-204. doi: 10.1002/humu.20836
56. Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J, et al. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol Aging. (2013) 34:2077.e11-8. doi: 10.1016/j.neurobiolaging.2013.02.016
57. Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. (2013) 70:78-84. doi: 10.1001/jamaneurol.2013.579
58. Wolfe CM, Fitz NF, Nam KN, Lefterov I, Koldamova R. The role of APOE and TREM2 in Alzheimer's disease-current understanding and perspectives. Int J Mol Sci. (2018) 20:81. doi: 10.3390/ijms20010081
59. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107-16. doi: 10.1056/NEJMoa1211103
60. Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. (2014) 71:449-53. doi: 10.1001/jamaneurol.2013.6237
61. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. (2013) 8:19. doi: 10.1186/1750-1326-8-19
62. Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging. (2014) 35:934.e7-10. doi: 10.1016/j.neurobiolaging.2013.09.017
Keywords: microglia, aging, neurodegeneration, alzheimer's disease, senescence
Citation: Candlish M and Hefendehl JK (2021) Microglia Phenotypes Converge in Aging and Neurodegenerative Disease. Front. Neurol. 12:660720. doi: 10.3389/fneur.2021.660720
Received: 29 January 2021; Accepted: 19 March 2021;
Published: 05 May 2021.
Edited by:
Gabor Petzold, Helmholtz Association of German Research Centers (HZ), GermanyReviewed by:
Karen Gertz, Charité – Universitätsmedizin Berlin, GermanyCopyright © 2021 Candlish and Hefendehl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jasmin K. Hefendehl, aGVmZW5kZWhsQGJpby51bmktZnJhbmtmdXJ0LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.