 Susan Harvey
Susan Harvey Mary D. King
Mary D. King Kathleen M. Gorman
Kathleen M. Gorman- 1Department of Paediatric Neurology and Clinical Neurophysiology, Children's Health Ireland at Temple Street, Dublin, Ireland
- 2School of Medicine and Medical Science, University College Dublin, Dublin, Ireland
Paroxysmal movement disorders (PxMDs) are a clinical and genetically heterogeneous group of movement disorders characterized by episodic involuntary movements (dystonia, dyskinesia, chorea and/or ataxia). Historically, PxMDs were classified clinically (triggers and characteristics of the movements) and this directed single-gene testing. With the advent of next-generation sequencing (NGS), how we classify and investigate PxMDs has been transformed. Next-generation sequencing has enabled new gene discovery (RHOBTB2, TBC1D24), expansion of phenotypes in known PxMDs genes and a better understanding of disease mechanisms. However, PxMDs exhibit phenotypic pleiotropy and genetic heterogeneity, making it challenging to predict genotype based on the clinical phenotype. For example, paroxysmal kinesigenic dyskinesia is most commonly associated with variants in PRRT2 but also variants identified in PNKD, SCN8A, and SCL2A1. There are no radiological or biochemical biomarkers to differentiate genetic causes. Even with NGS, diagnosis rates are variable, ranging from 11 to 51% depending on the cohort studied and technology employed. Thus, a large proportion of patients remain undiagnosed compared to other neurological disorders such as epilepsy, highlighting the need for further genomic research in PxMDs. Whole-genome sequencing, deep-sequencing, copy number variant analysis, detection of deep-intronic variants, mosaicism and repeat expansions, will improve diagnostic rates. Identifying the underlying genetic cause has a significant impact on patient care, modification of treatment, long-term prognostication and genetic counseling. This paper provides an update on the genetics of PxMDs, description of PxMDs classified according to causative gene rather than clinical phenotype, highlighting key clinical features and providing an algorithm for genetic testing of PxMDs.
Introduction
Paroxysmal movement disorders (PxMDs) are a clinical and genetically heterogeneous group characterized by episodic involuntary movements (dystonia, dyskinesia, chorea and/or ataxia). Historically, PxMDs were characterized clinically by the age of onset, triggers, duration and predominant movements (1, 2). Paroxysmal movement disorders are divided into paroxysmal dyskinesia or episodic ataxia (EA) depending on the main movement. However, both may co-exist with additional co-morbidities (epilepsy, headache, etc.). In both subgroups, symptoms begin in childhood or adolescence and improve or even resolve with age. Typically, between the events, neurological examination is normal. Sometimes in EA, chronic ataxia or nystagmus may co-exist. Although PxMDs can be due to secondary or acquired causes such a structural, immunological (multiple sclerosis), metabolic or neurodegenerative disorder, this review will focus on the genetic causes of PxMDs, previously referred to as primary PxMDs. Paroxysmal movement disorders are considered to be due to disruption of the network between cerebellar output, striatal dysfunction or a combination (3).
Before the era of next-generation sequencing (NGS), the clinical phenotype directed single gene testing for PxMDs. Most commonly testing of PRRT2 for paroxysmal kinesigenic dyskinesia (PKD), PNKD in paroxysmal non-kinesigenic dyskinesia (PNKD) or SLC2A1 in paroxysmal-exercised induced dyskinesia (PED). However, a large number (~50%) remained without a genetic cause, despite exhibiting the classical features of PxMDs (4). The advent of NGS has enabled the discovery of new genes associated with PxMDs (RHOBTB2, TBC1D24, PDE2A) and the expansion of the phenotypes of known PxMDs (5–8). For example, pathogenic variants in PRRT2 are now associated with PKD, PNKD, PED, hemiplegic migraine and self-limiting infantile seizures (9–11). Thus, PxMDs exhibit significant phenotypic pleiotropy and genetic heterogeneity, making it challenging to predict genotype based on the clinical phenotype.
With the identification of new phenotypes and new genes, the limits of the current classification system have become apparent. Sole identification of a trigger or the predominant movement disorder are no longer sufficient to predict the genotype. Various re-organizations have been introduced to alleviate the confusion of presentations of both adult and pediatric PxMDs (12). These systems aim to provide an accurate and criterion-based list of genetically confirmed disorders in which the naming system conveys the causative gene and maintains the connection between the phenotype and the gene. For example, Marras and colleagues suggest PxMD-PRRT2 for PKD caused by PRRT2 variants, clearly outlining its episodic nature along with the genetic cause (12). This updated system mirrors the evolution of the classification of epilepsy syndromes to now reflect the gene rather than the clinical or electroencephalogram features (13).
This paper provides an update on the genetics of PxMDs, classified according to the gene involved, highlights the key clinical features and provides an algorithm for genetic testing of PxMDs in the era of NGS.
Predominant Dyskinesia Genes
Paroxysmal dyskinesias are characterized by recurrent attacks of involuntary movement (chorea and/or dystonia often with ballismus and athetosis) without loss of consciousness. They are subdivided into three main groups depending on the key trigger for movements: sudden voluntary movements (PKD), no clear trigger (PNKD), or prolonged-exercise (PED) (1, 2). Recent reviews have provided excellent summaries on the clinical presentation, the evolution of nomenclature and terminology and treatment (2, 4). Table 1 provides a list of predominant dyskinesia genes organized according to function detailing inheritance, phenotypes and potential treatments. We have chosen to discuss eight dyskinesia genes in detail on the basis of high prevalence in clinical practice in the case of five genes (PRRT2, PNKD, SLC2A1, KCNMA1, ADCY5) and the recent discovery and newly described phenotypes (RHOBTB2, TBC1D24, PDE2A).
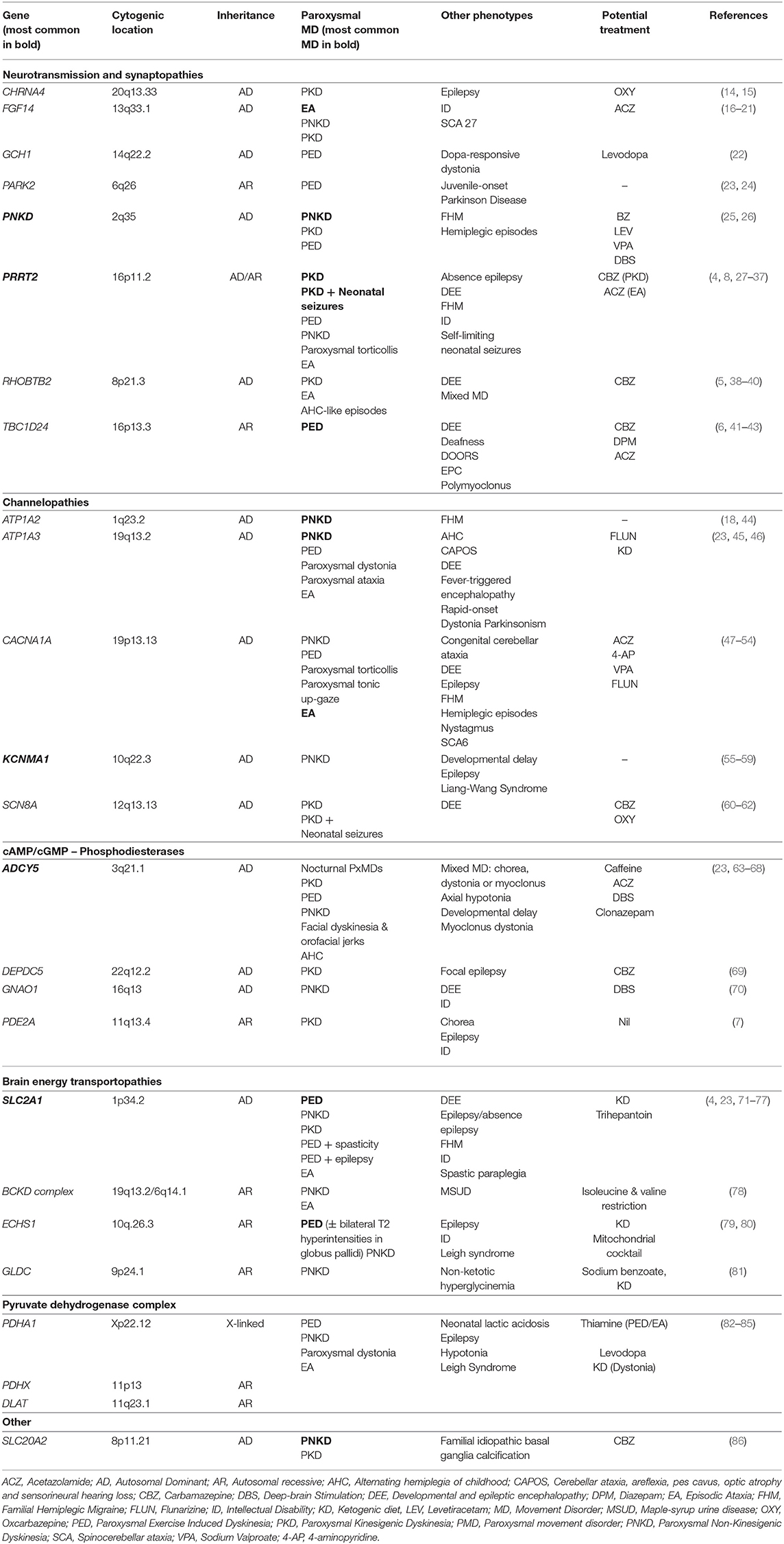
Table 1. Predominant dyskinesia genes.
PRRT2 (Chr 16:29,812,192-29,815,919, MIM: 614386)
Proline-rich transmembrane protein 2 (PRRT2) is a transmembrane protein which interacts with pre-synaptic proteins (SNAP25, VAMP2, SYT1, and SYT2) and affects neurotransmitter release. Initially reported as the first causative gene in PKD in 2011, pathogenic variants in PRRT2 are now implicated in various pediatric disorders with marked phenotypic pleiotropy (9–11).
Loss-of-function (LOF) variants in PRRT2 are the most common cause of PKD, accounting for 27–65% of all cases (25, 27). Symptoms typically begin in childhood or early adolescence [mean age of onset of 9.9 years (range: 1–40 years)]. Events are characterized by brief daily episodes of dystonia (17.6%), chorea (15.2%) or both (67.1%) which are triggered by voluntary movements that are often sudden (8). Of note, attacks can be triggered by coffee, anxiety or sleep deprivation in 40% in keeping with a PNKD phenotype. In a subset, neonatal or early-onset seizures occur (previously called infantile convulsions with choreoathetosis), with PKD emerging later, and is now viewed as part of PKD rather than a separate disorder. It occurs in 30% of PRRT2-associated PKD and is referred to as Paroxysmal Kinesigenic Dyskinesia with infantile Convulsions (PKD/IC) (8).
Other paroxysmal disorders associated with PRRT2 include: PED, PNKD, paroxysmal torticollis, EA and familial hemiplegic migraine (FHM), which may co-exist with PKD or occur independently (4, 27–29). Also, pathogenic variants in PRRT2 are reported in febrile seizures and less commonly in childhood absence epilepsy in the absence of PKD (30, 87).
The majority of cases have an autosomal dominant (AD) inheritance with incomplete penetrance reported (60–90%) (8, 31). The c.649dupC is a recurrent variant reported in 53.7% of PKD cases (with or without neonatal seizures). However, biallelic variants are also reported with a more severe phenotype of PKD, self-limiting neonatal seizures, intellectual disability (ID), EA and absence epilepsy (32, 33).
PNKD (Chr 2:218,270,484-218,346,792, MIM:609023)
Metallo-beta-lactamase domain-containing protein (PNKD) has multiple alternative names: myofibrillogenesis regulator 1 (MR-1), transactivated by hepatitis C virus core protein 2 (TAHCCP2), brain protein 17 (BRP17) and KIAA1184. Located in the cell membrane, and specifically expressed in the brain, PNKD was initially identified in the stress response pathway, and recent work suggests a role in maintaining cellular redox status (88, 89).
Pathogenic variants in PNKD account for 70% of PNKD (25). There are two recurrent variants p.Ala7Val and p.Ala9Val with an AD pattern of inheritance and almost complete penetrance (95%) (4, 26, 90). Compared to PRRT2 or SLC2A1, PNKD is not associated with significant pleiotropy, although a single family with FHM and no PxMD is published (4). In PNKD, attacks consisting of chorea and/or dystonia triggered by stress, alcohol, coffee, tea or strong emotion (laughing, excitement etc.) occur. Episodes are longer than in PKD, typically between 10 min and 1 h but can last up to 12 h and are infrequent occurring only a few times per year. Onset is from childhood to early adolescence [mean age of onset 5 years (range: 6 months–35 years)] with a reduction in the frequency of attacks in adulthood (23, 55, 91). There is no associated epilepsy or neurodevelopmental disorder, in contrast to the PNKD phenotype associated with KCNMA1.
SLC2A1 (Chr 1:42,925,352-42,958,867, MIM:138140)
Solute carrier family 2 member 1 (SLC2A1) codes for glucose transporter-1 (GLUT-1), the major glucose transporter in the blood-brain barrier. Disruption of GLUT-1 causes a reduction in glucose available to the brain. Variants in SCL2A1 are responsible for a broad spectrum of neurological disorders including PxMDs (PED, PKD, EA), non-PxMDs (chorea/dystonia), chronic ataxia, classic GLUT-1 deficiency syndrome (developmental delay, hypotonia, postnatal microcephaly, epilepsy), ID, refractory absence epilepsy, FHM, and hereditary spastic paraplegia (4, 71–74).
Most variants occur de novo or are inherited from a symptomatic parent, but autosomal recessive (AR) inheritance has been published (92, 93). Genotype-phenotype correlations are observed with LOF variants (splicing, protein-truncating variants, insertions/deletions) presenting with an earlier onset and more severe presentation. Missense variants (20%) are associated with milder symptoms, as there is larger residual activity of GLUT1. However, the same variant in SLC2A1 can be implicated in different clinical phenotypes, for example, p.Arg126Cys is reported in hereditary spastic paraplegia, classic GLUT-1 deficiency and early-onset absence epilepsy. Therefore, other as yet unknown factors, such as epigenetic modifiers or environmental effects, are likely to play a role (75).
Loss-of-function variants in the SLC2A1 account for 20% of PED characterized by dystonia and/or chorea after prolonged exercise and fasting (4, 94). Attack duration (minutes-hours) is variable, as is the level of physical activity required to trigger an attack (23). The mean age of symptom onset in PED is 8.6 years (range: 1–49 years) (25). Paroxysmal-exercised induced dyskinesia may be associated with a slowly progressive paraparesis or generalized epilepsy (4, 75, 76).
KCNMA1 (Chr 10:76,869,601-77,637,968, MIM: 600150)
The alpha, pore-forming subunit of the calcium-sensitive (BK) channel (KCa1.1) is important for neuronal excitability and is encoded by potassium calcium-activated channel subfamily M Alpha 1 (KCNMA1) (91). The core features of KCNMA1-related disorders (both GOF and LOF) are epilepsy, movement disorders, neurodevelopmental disorders and ID. The majority of pathogenic variants are de novo; however, AD and AR inheritance are reported (55, 56, 91). There is intrafamilial phenotypic variability and incomplete penetrance. The movement disorder phenotype can be predicted by the functional changes in BK activity. Paroxysmal non-kinesigenic dyskinesia is associated with GOF variants and ataxia and tremor with LOF variants (91).
The initial report of pathogenic variants in KCNMA1 described the largest cohort of affected patients (15 family members) with p.Asp434Gly with a variable phenotype of PNKD and/or epilepsy. Two recurrent GOF variants (p.Asp434Gly and p.Asn1053Ser) are associated with PNKD and epilepsy (91). The mean age of onset of PxMD is 4.6 years (range: 1–15 years) (25). Overlapping with PNKD, alcohol is a trigger for some (57). Developmental delay and epilepsy frequently co-exist, in contrast to PxMD-PNKD (58, 95).
A recurrent LOF variant (p.Gly375Arg) is associated with a distinct neurodevelopmental disorder (Liang-Wang Syndrome, MIM:618729) of developmental delay, seizures, dysmorphic features, visceral and cardiac malformations but without movement disorder (59). Other LOF variants are described in neurodevelopmental disorders, cerebral atrophy, ID and ataxia (59). Homozygous variants in KCNMA1 are published in two consanguineous families with LOF variants and clinical phenotype of cerebellar atrophy, developmental delay and seizures (55, 56).
ADCY5 (Chr 3:123,282,295-123,449,089, MIM: 600293)
Adenylate cyclase 5 (ADCY5) is necessary to convert adenosine triphosphate to cyclic adenosine 3'5' monophosphate (cAMP), a secondary messenger for multiple cellular activities. The high expression of ADCY5 in the striatum has led to the hypothesis that altered dopamine signaling in response to stress is responsible for ADCY5-related dyskinesia (96). There is phenotypic variability which appears dependent on the gene domain affected. Pathogenic variants in the intracellular catalytic cyclase domains C1a and C2a are associated with moderate to severe presentations, compared to a milder phenotype and no hypotonia with variants in the second portion of the first cytoplasmic domain (C1b) (96). The p.Arg413 hotspot (p.Arg416Gln/Gly/Trp), accounts for 50% of cases (96, 97). The majority of pathogenic variants are de novo. Mosaicism is observed frequently (25–50% of cases) and associated with a milder phenotype (96, 98). Two families with biallelic variants are described (99).
Pathogenic variants in ADCY5 are associated with a childhood-onset of mixed hyperkinetic movement disorder (chorea, dystonia or myoclonus or a combination) with orofacial jerks (facial myokymia), oculomotor apraxia and sleep-related PxMD (98). The nocturnal dyskinesia, in particular, appears characteristic and distinguishes it from most other movement disorders, in which sleep abolishes dyskinesia. The abnormal nocturnal movements occur more frequently in stage two and rapid-eye-movement sleep and have a significant negative impact on the quality of sleep (100, 101). Although some experience continuous symptoms, paroxysmal exacerbations of hyperkinetic movements are common, often triggered by illness, emotions or stress. Exacerbations may last for minutes to hours, sometimes referred to as “ballistic bouts.” Caffeine may exacerbate PxMD in some, but is an effective treatment in others (63). The movement disorder is usually static or very slowly progressive and can improve with age. Hypotonia and motor delay may predate the onset of movement disorder (97, 98). Less common phenotypes include alternating hemiplegia of childhood (AHC), myoclonus-dystonia, isolated dystonia and childhood-onset chorea (96). Thus, the range of movement disorders reported with pathogenic variants in ADCY5 is expanding and other PxMDs phenotypes may emerge.
RHOBTB2 (Chr 8:22,987,250-23,020,198, MIM: 607352)
Heterozygous missense variants in Rho-related BTB domain-containing protein 2 (RHOBTB2) were reported in 2018 in 13 individuals with DEEs (102, 103). In the initial reports, 11/13 had a movement disorder (dystonia, dyskinesia, chorea and stereotypies), described as paroxysmal in 4/11 (paroxysmal dyskinesia, dystonic-athetoid attacks) (5, 38, 103). Subsequently, two case reports focusing on the PxMDs have been published, including a patient with paroxysmal dyskinesia and developmental delay without epilepsy (38, 39). Recently a cohort of 11 patients was published with a complex, polymorphic movement disorder, with both paroxysmal and non-paroxysmal features (40). Paroxysmal movements were characterized by hemiplegic episodes, focal dystonia, worsening dyskinesia, episodic ataxia, and non-epileptic myoclonus. Recurrent variants are reported (p.Arg483His, p.Arg511Gln), but numbers are too small to identify any genotype-phenotype correlations. All variants reported to date are de novo. Thus, further reports will help to elucidate the full spectrum of RHOBTB2-related movement disorder (paroxysmal and non-paroxysmal).
TBC1D24 (Chr 16:2,475,103-2,509,668, MIM: 613577)
TBC1 domain family member 24 (TBC1D24) is a member of the TBC domain-containing RAB-specific GTPase-activating proteins and is necessary for normal brain development via regulation of synaptic function and vesicle trafficking (104). There is marked phenotypic-pleiotropy associated with variants in TBC1D24 including DEE, progressive myoclonic epilepsy, non-syndromic deafness, DOORS syndrome (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures), an AHC-like phenotype, multifocal myoclonus, epilepsia partialis continua or ID (41, 105–109). Inheritance is predominantly AR or compound heterozygous. However, AD inheritance is associated with the deafness phenotype.
Paroxysmal-exercise induced movement disorder (dystonia) is described in two children with onset in infancy with paroxysmal dystonic episodes, triggered by physical exertion and co-existing with epilepsy, ataxia, dysarthria and cerebellar dysfunction (6). There was a gradual improvement over time due to trigger avoidance. Also, paroxysmal facial and limb myoclonus with onset in early infancy, triggered by fever and fatigue has been published (42).
PDE2A (Chr11:72,576,140-72,674,421, MIM 602658)
Biallelic LOF variants in phosphodiesterase 2a, cGMP-stimulated (PDE2A) is a newly reported gene associated with paroxysmal dyskinesia with developmental delay, chorea, ID and epilepsy. To date, only four patients (three families) are reported (7, 110). Thus, further expansion of this phenotype is needed.
Predominant Episodic Ataxia Genes
Episodic ataxia is characterized by paroxysmal truncal ataxia, in-coordination, dysarthria and balance difficulties. Neurological interictal examination may be completely normal or may provide a clue to the underlying cause with myokymia (PxMD-KCNA1). Episodic ataxia was previously subdivided into nine subtypes depending on the underlying genetic cause. Episodic ataxia was common to all subtypes, but each had distinctive clinical characteristics. However, EA is now incorporated under PxMDs classification, due to the episodic nature of symptoms, overlapping clinical features and common genes with other paroxysmal disorders (12). For example, episodic ataxia type 1 is now referred to as PxMD-KCNA1. Table 2 provides a list of predominant episodic ataxia genes organized according to function detailing inheritance, phenotypes and potential treatments. We have chosen to discuss four EA genes in detail on the basis of their high prevalence in clinical practice in the case of two genes (KCNA1, CACNA1A) and the recent discovery and newly described phenotypes (SLC1A3 and FGF14).
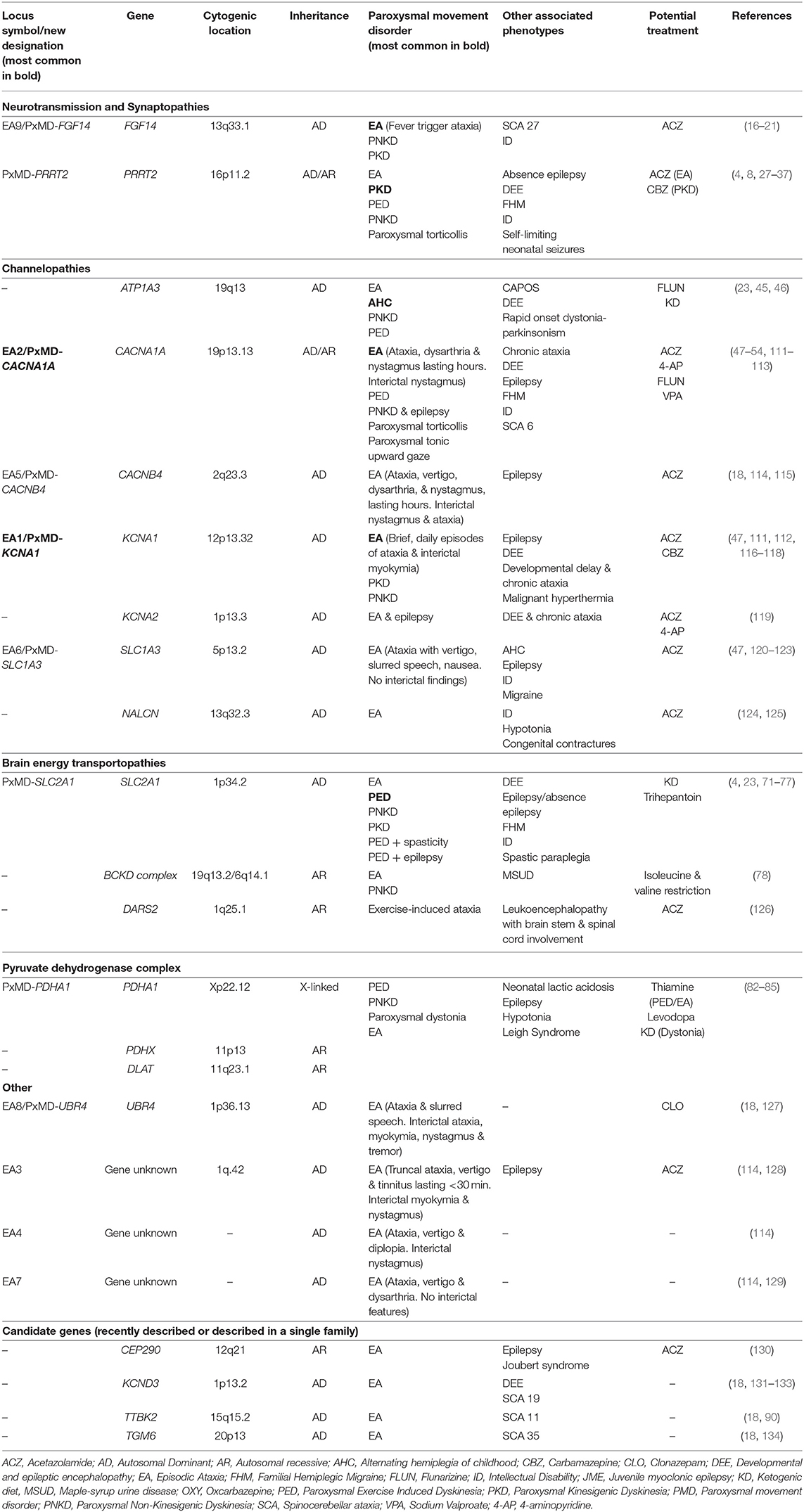
Table 2. Predominant episodic ataxia genes.
KCNA1 (Chr 12:4,909,904-4,918,255, MIM: 176260)
Potassium channel voltage-gated shaker-related subfamily member 1 (KCNA1) encodes voltage-gated potassium channel (Kv1.1), important in the repolarization of presynaptic action potentials that affect inhibitory input to Purkinje cells and cause hyperexcitability. All pathogenic variants are LOF, producing a markedly reduced inhibitory output of the cerebellum, thus causing symptoms (114). Inheritance is AD with reduced penetrance, intrafamilial and interfamilial phenotypical variability. Pathogenic variants are distributed throughout the gene in the EA phenotype, in contrast to the epilepsy phenotype where variants are limited to the pore domain. No genotype-phenotype correlations exist in the EA phenotype of KCNA1 (111, 116, 135).
Pathogenic variants in KCNA1 occur in up to 85% of EA type 1 phenotype, now referred to as PxMD-KCNA1 (116). Episodic ataxia begins in childhood (mean age of onset is 7.8 years, range: 0–20 years), attacks are frequent and last for minutes characterized by poor coordination, loss of balance, tremor and slurred speech (116). There may be multiple daily attacks and recognized triggers include exertion, emotional stress and changes in environmental temperature. Myokymia (fine twitching or intermittent cramps and stiffness) is a persistent interictal feature, which is very suggestive of KCNA1 but is recognized in other EAs including EA3 (no known causative gene, linked to cytogenic region Chr1q42) and PxMD-UBR4 (previously called EA8). Over time, persistent cerebellar dysfunction occurs in a subset of patients. Episodic ataxia may be isolated or associated with epilepsy, epileptic encephalopathy, malignant hyperthermia and neuromyotonia (117). Epilepsy, cataplexy, PKD, PNKD, myokymia and migraine are reported in the absence of EA (47, 112, 118). Non-neurological presentations include hypomagnesaemia (111).
CACNA1A (Chr19: 13,206,441-13,506,478, MIM: 601011)
Voltage-gated calcium channels are expressed throughout the central nervous system, mediating the entry of calcium into excitable cells. They are involved in multiple calcium-dependent processes, such as muscle contraction, neurotransmitter release or gene expression. Calcium channel voltage-dependent P/Q subtype, alpha 1-a subunit (CACNA1A) encodes the α1 subunit of voltage-gated P/Q calcium channel (Cav2.1). Loss-of-function variants reduce calcium entry through Cav2.1 resulting in irregular firing of the Purkinje and granule cells, where Cav2.1 is largely expressed (114).
Inheritance is AD (penetrance 80–90%) and a single family with biallelic variants is reported (136). Gain-of-function variants are associated with FHM, epilepsy and DEE (137, 138). In contrast, LOF variants occur in PxMDs, most frequently in EA but also in PKD and PED. and trinucleotide CAG repeat expansion in spinocerebellar ataxia (SCA) type 6 (139, 140). Frequently, phenotypes may overlap with migraine reported in the EA phenotype, and EA symptoms in SCA6. Intrafamilial phenotypic variability includes cases of DEE, EA, ID and asymptomatic carriers all identified within one family (141).
The most common PxMD associated with LOF variants in CACNA1A is EA, less frequently PKD and PED. CACNA1A-EA is characterized by onset, from early childhood to early adulthood, of less frequent attacks than in PxMD-KCNA1 (weekly or only a few episodes per year) with nystagmus, vertigo, nausea, vomiting and dysarthria, which last for hours. Interictal phenomena include nystagmus and migraine. Magnetic resonance imaging may demonstrate cerebellar atrophy, which may be static or progressive (139, 142). Migraine occurs in 50% of PxMD-CACNA1A cases (48, 114). Paroxysmal tonic up-gaze, nystagmus, and other eye movement disorders are all features of CACNA1A and present in up to 90% interictally (47). Eye movement disorders and paroxysmal tonic up-gaze may manifest in infancy before EA emerges years later (143). Variants in CACNA1A are associated with paroxysmal torticollis, paroxysmal benign tonic up-gaze, congenital cerebellar ataxia, autism spectrum disorder, DEE, chronic progressive ataxia, epilepsy and developmental delay (49–51, 142, 144). A severe phenotype of DEE, optic nerve atrophy and progressive cerebral and cerebellar atrophy with biallelic variants has been reported (136).
SLC1A3 (Chr 5:36,606,605-36,688,333, MIM: 600111)
The glial glutamate transporter excitatory amino acid transporter 1 (EAAT1) is present in the cerebellum and brainstem and encoded by solute carrier family 1, member 3 (SLC1A3). EAAT1 is responsible for glutamate uptake in the synapses (114). Pathogenic variants in SLC1A3 can cause both reduced and enhanced glutamate transport and thus alters channel activity. The cerebellum is sensitive to slight alterations in EAAT1 activity (145).
Heterozygous variants in SLC1A3 are responsible for PxMD-SLC1A3, previously termed episodic ataxia type 6. Less is known about PxMD-SLC1A3 than other EAs, with only 11 patients (seven families) reported to date (145). The onset of EA is variable from infancy to adulthood, and events are similar to PxMD-CACNA1A but longer in duration (hours to days) and there is no associated myokymia, nystagmus or tinnitus (146). Potential event triggers include emotional stress, physical exertion, caffeine and fever (47, 120). Additional features include seizures and migraine. Inheritance is AD. Duplications of SLC1A3 cause developmental delay and behavioral problems in the absence of cerebellar findings (147).
FGF14 (Chr: 13:101,710,803-103,402,442, MIM: 601515)
Fibroblast growth factor 14 (FGF14) has been proposed to be added to the list of distinct EA phenotypes. To date, 12 patients (seven families) are published with an identical phenotype (16–20). Ataxia episodes are triggered by fever and can be associated with dysarthria, vertigo, headache and vomiting of variable duration and frequency. Episodes have a variable age of onset from early childhood to adulthood. Inter-ictal upper limb tremor, nystagmus and ID are present. Paroxysmal kinesigenic dyskinesia and PNKD are reported in isolation or associated with EA or SCA27 (148). Inheritance is AD with intrafamilial phenotypic heterogeneity (20).
Highly expressed in the brain, especially in granule and Purkinje cells, FGF14 regulates pre-synaptic Cav2.1 channel and vesicular recycling/synaptic transmission. FGF14 may be a risk factor for neuropsychiatric disorders (depression, addiction) and neurodegenerative diseases and thus the phenotype continues to expand (149).
Discussion
History and Examination
Although we have summarized the expanding number of genes associated with PxMDs, it is still necessary to begin with the key clinical features as a guide to requesting and interpretation of genetic testing. Thus, a detailed history of the PxMD focusing on the phenomenology, triggers (movement, exercise, alcohol or fever) alleviating factors and the duration and frequency of attacks. In addition, information on other paroxysmal (hemiplegic migraine, epilepsy), and non-paroxysmal symptoms (ID, psychiatric co-morbidities etc.) should be sought (2, 150). Frequently the PxMD, may not fit into a distinct phenotype or may have features of different paroxysmal dyskinesias (for example both kinesigenic and exercise-induced features). A family history of PxMDs or other paroxysmal phenomena (migraine, infantile seizures) may be important because of interfamilial phenotypic heterogeneity and may aid interpretation of genetic findings.
In paroxysmal dyskinesia, examination is usually normal, and the presence of focal neurological deficits or upper motor neuron signs suggest a secondary causes of PxMDs. In EA, nystagmus, myokymia, upper limb tremor or chronic ataxia maybe a clue to the underlying causes (myokymia in PxMD-KCNA1 and PxMD-UBR4).
Approach to Evaluating a Patient With Paroxysmal Movement Disorder
Before devising an investigation strategy, a thorough history and examination are essential, to determine whether or not the PxMD is primary or secondary and to define the main clinical features of the PxMD, as this will dictate how to approach investigations. Secondary or acquired causes of PxMDs, should be excluded clinically or with appropriate metabolic or radiological studies. Red flags for secondary causes of PxMDs include onset in adulthood, absence of family history, variable duration of attacks and triggering factors, progressive disease course, abnormal inter-ictal neurological examination (dystonia, upper motor neuron signs) or abnormal magnetic resonance imaging (Figure 1).
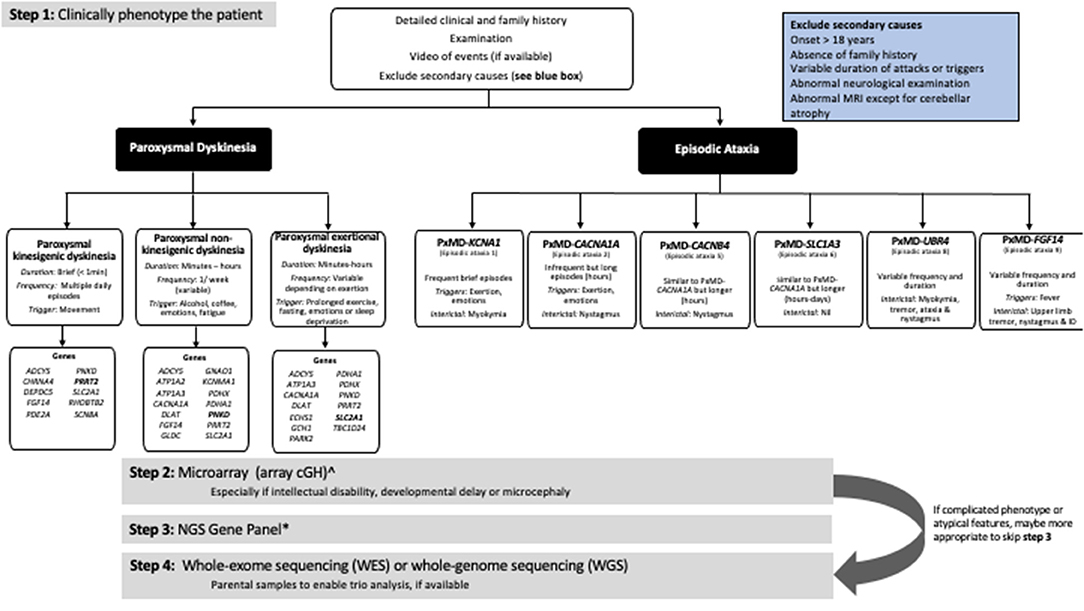
Figure 1. Approach to the genetic investigation of paroxysmal movement disorders. ∧Will not be required if using whole-genome sequencing. *The number of genes varies depending on the NGS panel used. Always check what genes are included in the panel.
Diagnostic Yield in Paroxysmal Movement Disorders
In the past 20 years, there has been a rapid advance in the understanding of the genetic basis of neurological disorders, including PxMD. However, many patients with the typical clinical features of PxMD remain without a genetic diagnosis. When NGS is utilized, the diagnostic yield is only 35–50% (Supplementary Table 1). The yield is equal or less compared to other pediatric-onset neurological disorders such as epilepsy (up to 70%), leukodystrophy (up to 90%), mixed movement disorder (51%), childhood-onset ataxia (46%) or progressive ataxia (32–53%) (87, 151–158).
Algorithm for Investigation
Traditionally in PxMDs, sequential single-gene testing was performed based on the predominant movement. For example, in PKD, single-gene testing of PRRT2 was performed, followed by SLC2A1 and PNKD. Diagnostic rates were <50% when the three genes were sequenced in a cohort with paroxysmal dyskinesias and EA (4). However, as highlighted in this article due to phenotypic pleiotropy, single-gene testing is no longer the best approach.
A microarray (arrayCGH) should be the first tier of genetic testing and is particularly relevant if epilepsy, ID or developmental delay is present (159–161). Diagnostic yield of microarray in pediatric movement disorders is reported in a single center cohort (n = 20) only. Pathogenic copy number variants (CNVs) were detected in 28% (7/20) in a heterogenous group with movement disorder and 40% (2/5) in PKD (162). The yield in children with undiagnosed neurological conditions ranges from 8.9 to 28%, depending on the cohort (163–165). Microarray should be requested even if a NGS gene panel or WES is requested as these technologies may not detect CNVs (158).
Due to genetic heterogeneity, gene-panel testing is recommended as second-tier testing, enabling parallel sequencing of multiple genes. The number of genes on NGS panels is variable, depending on the individual laboratory and when the test was performed. Gene panels for PxMDs are found under an assortment of different titles, including paroxysmal disorders, degenerative disorders, channelopathies or episodic disorders. A search of commercially available panels in European laboratories highlighted the number of genes tested varied between 6 and 186 genes, depending on the laboratory (Supplementary Table 2). Therefore, knowledge of the specific panel requested is essential as key genes and recently described genes may be missed or not included at the time of testing. For example, TBC1D24, may only be available on an epilepsy panel. An advantage of a panel approach over clinical whole-exome sequencing (WES) is that many panels include high-resolution coverage, including for exon-level deletions or duplications, which are not detected by standard sequencing techniques. Also, panel analysis rather than WES/WGS reduces the possibility of secondary reportable findings and variants of unknown significance not related to the phenotype (166).
In cases where there is a complicated clinical phenotype, WES or whole-genome sequencing (WGS) should be employed as second-tier genetic testing. If panel testing is negative, then WES or WGS may be requested. Both WES and WGS will enable identification of new genes, expansion of phenotype of known genes and recognition of new clinical phenotypes. There is a paucity of studies on the yield of WES/WGS in PxMDs (Supplementary Table 1); the majority of publications to date utilized a panel analysis of WES data (18, 69, 151, 167, 168). In the near future, WGS is likely to be the most cost and time-efficient approach to investigating patients (169).
Limitations of NGS Technology
Next-generation sequencing has revolutionized how we approach genetic-testing but the technology has several limitations. Both WES and WGS, cannot detect balanced translocations, variants are missed due to low coverage or poor sequence quality in G-C rich regions, difficulty in interpretation of short or long insertions and WES cannot detect large deletions/duplications (CNVs). Whole-genome sequencing provides advantages over WES notably continuous coverage, the ability to identify sequence variants throughout the genome (including intronic variants, intergenic and regulatory sequence variants, expanded repeats) and to detect smaller CNVs than detected on microarray (158, 170–172). However, while deep intronic, noncoding and intergenic variants are all potentially detectable by WGS they can be challenging to interpret due to the lack of functional protein to validate findings (166, 172). Triplet repeat expansions analysis is possible with WGS, but many bioinformatic pipelines are still only available on a research basis and require long-read NGS (172–174). Thus, if a trinucleotide repeat disorder is suspected, this needs to be specifically requested additional to WES analysis. High coverage of PxMDs genes is required to detect mosaic variants (174).
Mosaicism
Mosaicism is increasingly recognized within pediatric neurology as a pathogenic mechanism in monogenic disorders (~3% in neurodevelopmental disorders) (175, 176). In PxMDs, mosaic variants have been reported in ATP1A3, ADCY5, SLC2A1 and PDHA1, (98, 177–179). In ADCY5, mosaicism is reported in 25–50% of cases, associated with a milder phenotype and symptoms may not be recognized (96, 98). Parental mosaicism, if undetected, impacts on genetic counseling and risk for future pregnancy. Good read depth (>100 for WES and >30 for WGS), modification of filters/algorithms, sequencing of additional tissue or single-molecule molecular inversion probes may be required to detect somatic mosaicism (176).
Interpretation of Genetic Findings
The correct interpretation of results from microarray, gene panels and WES/WGS is essential, particularly when a novel variant is identified (180). Inheritance studies for segregation within families is helpful to support pathogenicity. However, with PxMDs reduced penetrance and variable phenotypes within families add to the complexity when evaluating findings. Functional studies and/or identification of variants within multiple individuals are needed to apply correct pathogenicity to new variants. Finally, clinical-genetic expertise is crucial for clinical phenotyping, evaluating and interpreting the genetic results, and communication with the patient and family.
Importance of Genetic Testing
The importance of identification of a genetic cause cannot be overstated. The identification of the correct genetic diagnosis enables accurate genetic counseling (including preimplantation genetic diagnosis, where applicable), prediction of disease phenotype, precision treatment strategies (the ketogenic diet in PxMD-SLC2A1), surveillance of associated co-morbidities, and engagement in family support groups (2, 181, 182). Finally, an earlier genetic diagnosis avoids invasive and expensive investigations such as muscle biopsy, multiple hospital visits and missed days from school/work (169).
Future Directions
Though new genes associated with PxMDs have been discovered, only 50% of individuals with PxMDs have an underlying genetic diagnosis. Therefore, further work is needed to elucidate a cause for the remainder. Episodic ataxia types 3 and 7 are only linked to chromosomal regions (EA3 linked to 1q42, EA7 to 19q13) and EA4 is not linked to a known locus. However, a single gene has not been identified in these regions to explain the phenotype. Adults with symptom onset in childhood should have previous genetic testing re-evaluated in light of the evolution of NGS and the discovery of new genes in the last decade. Re-interrogation of WES and WGS data for “negative patients,” with time is necessary. Several groups have demonstrated the increased yield of 5.8% in DEE and 7% in ataxia (157, 183, 184) after re-analysis.
As discussed in the limitations paragraph, WGS provides advantages over WES/gene panel analysis with better coverage, additional analysis including repeat expansions, CNV analysis and identification of deep intronic variants (166). The application of transcriptome analysis/transcriptome data to WGS findings improves the diagnosis of WGS findings by aiding the interpretation of variants. This has already been employed in neuromuscular disorders with an increased diagnostic yield (166, 185).
Additional features must contribute to the clinical presentation of PxMDs given the infrafamilial heterogeneity and reduced penetrance. Epigenetics refers to cellular processes that influence gene expression outside of direct changes to the DNA and includes methylation, histone modification, microRNAs, transcriptome and the microbiome (158, 186–189). Further work is needed to fully understand the role of epigenetics in PxMDs (187).
Impact of Treatment
Discussion regarding specific treatment of PxMDs is beyond the scope of this review and is discussed in detail elsewhere (2, 190). Identification of a genetic cause is the first step toward the development of precision therapy targeting specific pathophysiological mechanisms and potential gene therapy for PxMDs (181). Currently, treatment is often based on the clinical phenotype rather than genotype as in the use of carbamazepine in PKD. Despite small numbers reported with certain pathogenic variants, patterns are emerging, showing better efficacy of some medications in certain genotypes. For example, in SCN2A GOF variants respond to sodium channel blockers which are ineffective in patients with LOF variants (191). Thus, knowledge of the causative variants in successfully treated patients will aid the treatment approach in the future.
Conclusion
Paroxysmal movement disorders are a genetically heterogeneous group of movement disorders with phenotypic pleiotropy. Next-generation sequencing has revolutionized how we classify and investigate PxMDs. However, a large proportion of patients remain undiagnosed compared to other neurological diseases. Thus, further research is needed in PxMDs to identify new genes, understand disease mechanisms and develop precision treatments.
Author Contributions
SH, MK, and KG contributed to the design, writing, and review of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.659064/full#supplementary-material
Abbreviations
AD, Autosomal dominant; AHC, Alternating hemiplegia of childhood; AR, Autosomal recessive; DEE, Developmental and epileptic encephalopathy; GLUT-1, Glucose-transport-1; EA, Episodic Ataxia; FHM, Familial hemiplegic migraine; GOF, Gain-of-function; ID, Intellectual disability; LOF, Loss-of-function; NGS, Next-generation sequencing; PED, Paroxysmal exercise induced dyskinesia; PKD, Paroxysmal kinesigenic dyskinesia; PxMD, Paroxysmal movement disorders; PNKD, Paroxysmal non-kinesigenic dyskinesia; SCA, Spinocerebellar ataxia; WES, Whole-exome sequencing; WGS, Whole-genome sequencing.
References
1. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. (1995) 38:571–9. doi: 10.1002/ana.410380405
2. Silveira-Moriyama L, Kovac S, Kurian MA, Houlden H, Lees AJ, Walker MC, et al. Phenotypes, genotypes, and the management of paroxysmal movement disorders. Dev Med Child Neurol. (2018) 60:559–65. doi: 10.1111/dmcn.13744
3. Delorme C, Giron C, Bendetowicz D, Méneret A, Mariani LL, Roze E. Current challenges in the pathophysiology, diagnosis and treatment of paroxysmal movement disorders. Expert Rev Neurother. (2020) 21:81–97. doi: 10.1080/14737175.2021.1840978
4. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain. (2015) 138 (Pt 12):3567–80. doi: 10.1093/brain/awv310
5. Straub J, Konrad EDH, Grüner J, Toutain A, Bok LA, Cho MT, et al. Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in drosophila. Am J Hum Genet. (2018) 102:44–57. doi: 10.1016/j.ajhg.2017.11.008
6. Steel D, Heim J, Kruer MC, Sanchis-Juan A, Raymond LF, Eunson P, et al. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov Disord. (2020) 35:372–3. doi: 10.1002/mds.27981
7. Doummar D, Dentel C, Lyautey R, Metreau J, Keren B, Drouot N, et al. Biallelic PDE2A variants: a new cause of syndromic paroxysmal dyskinesia. Eur J Hum Genet. (2020) 28:1403–13. doi: 10.1038/s41431-020-0641-9
8. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138 (Pt 12):3476–95. doi: 10.1093/brain/awv317
9. Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. (2011) 43:1252–5. doi: 10.1038/ng.1008
10. Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang JG, et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain. (2011) 134(Pt 12):3493–501. doi: 10.1093/brain/awr289
11. Doring JH, Saffari A, Bast T, Brockmann K, Ehrhardt L, Fazeli W, et al. The phenotypic spectrum of PRRT2-associated paroxysmal neurologic disorders in childhood. Biomedicines. (2020) 8:456. doi: 10.3390/biomedicines8110456
12. Marras C, Lang A, van de Warrenburg BP, Sue CM, Tabrizi SJ, Bertram L, et al. Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov Disord. (2016) 31:436–57. doi: 10.1002/mds.26527
13. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
14. Jiang YL, Yuan F, Yang Y, Sun XL, Song L, Jiang W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure. (2018) 56:88–91. doi: 10.1016/j.seizure.2018.02.005
15. Conti V, Aracri P, Chiti L, Brusco S, Mari F, Marini C, et al. Nocturnal frontal lobe epilepsy with paroxysmal arousals due to CHRNA2 loss of function. Neurology. (2015) 84:1520–8. doi: 10.1212/WNL.0000000000001471
16. Choquet K, La Piana R, Brais B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics. (2015) 16:233–6. doi: 10.1007/s10048-014-0436-7
17. Coebergh JA, Fransen van de Putte DE, Snoeck IN, Ruivenkamp C, van Haeringen A, Smit LM. A new variable phenotype in spinocerebellar ataxia 27 (SCA 27) caused by a deletion in the FGF14 gene. Eur J Paediatr Neurol. (2014) 18:413–5. doi: 10.1016/j.ejpn.2013.10.006
18. Choi KD, Kim JS, Kim HJ, Jung I, Jeong SH, Lee SH, et al. Genetic variants associated with episodic ataxia in Korea. Sci Rep. (2017) 7:13855. doi: 10.1038/s41598-017-14254-7
19. Schesny M, Joncourt F, Tarnutzer AA. Acetazolamide-Responsive episodic ataxia linked to novel splice site variant in FGF14 gene. Cerebellum. (2019) 18:649–53. doi: 10.1007/s12311-018-0997-3
20. Piarroux J, Riant F, Humbertclaude V, Remerand G, Hadjadj J, Rejou F, et al. FGF14-related episodic ataxia: delineating the phenotype of episodic ataxia type 9. Ann Clin Transl Neurol. (2020) 7:565–72. doi: 10.1002/acn3.51005
21. Amado A, Blanco MO, Repáraz-Andrade A. Spinocerebellar ataxia 27: clinical phenotype of twin sisters with FGF14 deletion. Neuropediatrics. (2017) 48:131. doi: 10.1055/s-0037-1598110
22. Dale RC, Melchers A, Fung VS, Grattan-Smith P, Houlden H, Earl J. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. (2010) 52:583–6. doi: 10.1111/j.1469-8749.2010.03619.x
23. Meneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol. (2016) 172:433–45. doi: 10.1016/j.neurol.2016.07.005
24. Yoshimura K, Kanki R. Child-onset paroxysmal exercise-induced dystonia as the initial manifestation of hereditary Parkinson's disease. Parkinsonism Relat Disord. (2018) 49:108–9. doi: 10.1016/j.parkreldis.2018.01.004
25. Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord. (2014) 29:1108–16. doi: 10.1002/mds.25933
26. Ghezzi D, Viscomi C, Ferlini A, Gualandi F, Mereghetti P, DeGrandis D, et al. Paroxysmal non-kinesigenic dyskinesia is caused by mutations of the MR-1 mitochondrial targeting sequence. Hum Mol Genet. (2009) 18:1058–64. doi: 10.1093/hmg/ddn441
27. Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology. (2012) 79:2115–21. doi: 10.1212/WNL.0b013e3182752c5a
28. Liu Q, Qi Z, Wan XH, Li JY, Shi L, Lu Q, et al. Mutations in PRRT2 result in paroxysmal dyskinesias with marked variability in clinical expression. J Med Genet. (2012) 49:79–82. doi: 10.1136/jmedgenet-2011-100653
29. Riant F, Roze E, Barbance C, Méneret A, Guyant-Maréchal L, Lucas C, et al. PRRT2 mutations cause hemiplegic migraine. Neurology. (2012) 79:2122–4. doi: 10.1212/WNL.0b013e3182752cb8
30. Méneret A, Gaudebout C, Riant F, Vidailhet M, Depienne C, Roze E. PRRT2 mutations and paroxysmal disorders. Eur J Neurol. (2013) 20:872–8. doi: 10.1111/ene.12104
31. Liu XR, Wu M, He N, Meng H, Wen L, Wang JL, et al. Novel PRRT2 mutations in paroxysmal dyskinesia patients with variant inheritance and phenotypes. Genes Brain Behav. (2013) 12:234–40. doi: 10.1111/gbb.12008
32. Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. (2012) 53:e196–9. doi: 10.1111/epi.12009
33. Delcourt M, Riant F, Mancini J, Milh M, Navarro V, Roze E, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry. (2015) 86:782–5. doi: 10.1136/jnnp-2014-309025
34. Heron SE, Dibbens LM. Role of PRRT2 in common paroxysmal neurological disorders: a gene with remarkable pleiotropy. J Med Genet. (2013) 50:133–9. doi: 10.1136/jmedgenet-2012-101406
35. Nobile C, Striano P. PRRT2: a major cause of infantile epilepsy and other paroxysmal disorders of childhood. Prog Brain Res. (2014) 213:141–58. doi: 10.1016/B978-0-444-63326-2.00008-9
36. Becker F, Schubert J, Striano P, Anttonen AK, Liukkonen E, Gaily E, et al. PRRT2-related disorders: further PKD and ICCA cases and review of the literature. J Neurol. (2013) 260:1234–44. doi: 10.1007/s00415-012-6777-y
37. Lee HY, Huang Y, Bruneau N, Roll P, Roberson ED, Hermann M, et al. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. (2012) 1:2–12. doi: 10.1016/j.celrep.2011.11.001
38. Spagnoli C, Soliani L, Caraffi SG, Baga M, Rizzi S, Salerno GG, et al. Paroxysmal movement disorder with response to carbamazepine in a patient with RHOBTB2 developmental and epileptic encephalopathy. Parkinsonism Relat Disord. (2020) 76:54–5. doi: 10.1016/j.parkreldis.2020.05.031
39. Necpál J, Zech M, Valachová A, Sedláček Z, Bendová Š, Hančárová M, et al. Severe paroxysmal dyskinesias without epilepsy in a RHOBTB2 mutation carrier. Parkinsonism Relat Disord. (2020) 77:87–8. doi: 10.1016/j.parkreldis.2020.06.028
40. Zagaglia S, Steel D, Krithika S, Hernandez-Hernandez L, Custodio HM, Gorman KM, et al. Mutations expand the phenotypic spectrum of alternating hemiplegia of childhood. Neurology. (2021) 96:e1539–50. doi: 10.1212/WNL.0000000000011543
41. Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology. (2016) 87:77–85. doi: 10.1212/WNL.0000000000002807
42. Zimmern V, Riant F, Roze E, Ranza E, Lehmann-Horn F, de Bellescize J, et al. Infantile-onset paroxysmal movement disorder and episodic ataxia associated with a TBC1D24 mutation. Neuropediatrics. (2019) 50:308–12. doi: 10.1055/s-0039-1688410
43. Zhou Q, Lin Y, Ye J, Li L, Hu N, Wang D, et al. Homozygous. Front Neurol. (2017) 8:750. doi: 10.3389/fneur.2017.00750
44. Pelzer N, Haan J, Stam AH, Vijfhuizen LS, Koelewijn SC, Smagge A, et al. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology. (2018) 90:e575–82. doi: 10.1212/WNL.0000000000004966
45. Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatr Neurol. (2015) 52:56–64. doi: 10.1016/j.pediatrneurol.2014.09.015
46. Yano ST, Silver K, Young R, DeBrosse SD, Ebel RS, Swoboda KJ, et al. Fever-induced paroxysmal weakness and encephalopathy, a new phenotype of ATP1A3 mutation. Pediatr Neurol. (2017) 73:101–5. doi: 10.1016/j.pediatrneurol.2017.04.022
47. Kipfer S, Strupp M. The clinical spectrum of autosomal-dominant episodic ataxias. Mov Disord Clin Pract. (2014) 1:285–90. doi: 10.1002/mdc3.12075
48. Duque KR, Marsili L, Sturchio A, Mahajan A, Merola A, Espay AJ, et al. Progressive ataxia with hemiplegic migraines: a phenotype of CACNA1A missense mutations, Not CAG repeat expansions. Cerebellum. (2020) 20:134–9. doi: 10.1007/s12311-020-01185-9
49. Indelicato E, Nachbauer W, Karner E, Eigentler A, Wagner M, Unterberger I, et al. The neuropsychiatric phenotype in CACNA1A mutations: a retrospective single center study and review of the literature. Eur J Neurol. (2019) 26:66–e7. doi: 10.1111/ene.13765
50. Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. (2015) 23:1505–12. doi: 10.1038/ejhg.2015.21
51. Roubertie A, Echenne B, Leydet J, Soete S, Krams B, Rivier F, et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol. (2008) 255:1600–2. doi: 10.1007/s00415-008-0982-8
52. Scoggan KA, Friedman JH, Bulman DE. CACNA1A mutation in a EA-2 patient responsive to acetazolamide and valproic acid. Can J Neurol Sci. (2006) 33:68–72. doi: 10.1017/S0317167100004728
53. Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. (2004) 62:1623–5. doi: 10.1212/01.WNL.0000125691.74109.53
54. Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, et al. Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol. (2014) 261:983–91. doi: 10.1007/s00415-014-7310-2
55. Yeşil G, Aralaşmak A, Akyüz E, Içagasioglu D, Uygur Sahin T, Bayram Y. Expanding the phenotype of homozygous. Balkan Med J. (2018) 35:336–9. doi: 10.4274/balkanmedj.2017.0986
56. Tabarki B, AlMajhad N, AlHashem A, Shaheen R, Alkuraya FS. Homozygous KCNMA1 mutation as a cause of cerebellar atrophy, developmental delay and seizures. Hum Genet. (2016) 135:1295–8. doi: 10.1007/s00439-016-1726-y
57. Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. (2005) 37:733–8. doi: 10.1038/ng1585
58. Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord. (2015) 30:1290–2. doi: 10.1002/mds.26216
59. Liang L, Li X, Moutton S, Schrier Vergano SA, Cogne B, Saint-Martin A, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. (2019) 28:2937–51. doi: 10.1093/hmg/ddz117
60. Gardella E, Møller RS. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia. (2019) 60(Suppl. 3):S77–85. doi: 10.1111/epi.16319
61. Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LH, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. (2016) 79:428–36. doi: 10.1002/ana.24580
62. Papandreou A, Danti FR, Spaull R, Leuzzi V, Mctague A, Kurian MA. The expanding spectrum of movement disorders in genetic epilepsies. Dev Med Child Neurol. (2020) 62:178–91. doi: 10.1111/dmcn.14407
63. Méneret A, Gras D, McGovern E, Roze E. Caffeine and the dyskinesia related to Mutations in the ADCY5 gene. Ann Intern Med. (2019) 171:439. doi: 10.7326/L19-0038
64. Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol. (2012) 69:630–5. doi: 10.1001/archneurol.2012.54
65. Chen YZ, Friedman JR, Chen DH, Chan GC, Bloss CS, Hisama FM, et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol. (2014) 75:542–9. doi: 10.1002/ana.24119
66. Friedman JR, Méneret A, Chen DH, Trouillard O, Vidailhet M, Raskind WH, et al. ADCY5 mutation carriers display pleiotropic paroxysmal day and nighttime dyskinesias. Mov Disord. (2016) 31:147–8. doi: 10.1002/mds.26494
67. Dy ME, Chang FC, Jesus SD, Anselm I, Mahant N, Zeilman P, et al. Treatment of ADCY5-associated dystonia, chorea, and hyperkinetic disorders with deep brain stimulation: a multicenter case series. J Child Neurol. (2016) 31:1027–35. doi: 10.1177/0883073816635749
68. de Almeida Marcelino AL, Mainka T, Krause P, Poewe W, Ganos C, Kühn AA. Deep brain stimulation reduces (nocturnal) dyskinetic exacerbations in patients with ADCY5 mutation: a case series. J Neurol. (2020) 267:3624–31. doi: 10.1007/s00415-020-09871-8
69. Tian WT, Huang XJ, Mao X, Liu Q, Liu XL, Zeng S, et al. Proline-rich transmembrane protein 2-negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov Disord. (2018) 33:459–67. doi: 10.1002/mds.27274
70. Danti FR, Galosi S, Romani M, Montomoli M, Carss KJ, Raymond FL, et al. GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol Genet. (2017) 3:e143. doi: 10.1212/NXG.0000000000000143
71. Vermeer S, Koolen DA, Visser G, Brackel HJ, van der Burgt I, de Leeuw N, et al. A novel microdeletion in 1(p34.2p34.3), involving the SLC2A1 (GLUT1) gene, and severe delayed development. Dev Med Child Neurol. (2007) 49:380–4. doi: 10.1111/j.1469-8749.2007.00380.x
72. Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. (2010) 25:275–81. doi: 10.1002/mds.22808
73. Nicita F, Schirinzi T, Stregapede F, Vasco G, Bertini E, Travaglini L. SLC2A1 mutations are a rare cause of pediatric-onset hereditary spastic paraplegia. Eur J Paediatr Neurol. (2019) 23:329–32. doi: 10.1016/j.ejpn.2018.12.004
74. Castellotti B, Ragona F, Freri E, Solazzi R, Ciardullo S, Tricomi G, et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: identification of novel variants and associated phenotypes. J Neurol. (2019) 266:1439–48. doi: 10.1007/s00415-019-09280-6
75. Weber YG, Kamm C, Suls A, Kempfle J, Kotschet K, Schüle R, et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology. (2011) 77:959–64. doi: 10.1212/WNL.0b013e31822e0479
76. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. (2008) 131 (Pt 7):1831–44. doi: 10.1093/brain/awn113
78. Pode-Shakked N, Korman SH, Pode-Shakked B, Landau Y, Kneller K, Abraham S, et al. Clues and challenges in the diagnosis of intermittent maple syrup urine disease. Eur J Med Genet. (2020) 63:103901. doi: 10.1016/j.ejmg.2020.103901
79. Olgiati S, Skorvanek M, Quadri M, Minneboo M, Graafland J, Breedveld GJ, et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. (2016) 31:1041–8. doi: 10.1002/mds.26610
80. Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. (2017) 264:185–7. doi: 10.1007/s00415-016-8381-z
81. Zima L, Ceulemans S, Reiner G, Galosi S, Chen D, Sahagian M, et al. Paroxysmal motor disorders: expanding phenotypes lead to coalescing genotypes. Ann Clin Transl Neurol. (2018) 5:996–1010. doi: 10.1002/acn3.597
82. Castiglioni C, Verrigni D, Okuma C, Diaz A, Alvarez K, Rizza T, et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur J Paediatr Neurol. (2015) 19:497–503. doi: 10.1016/j.ejpn.2015.04.008
83. Head RA, de Goede CG, Newton RW, Walter JH, McShane MA, Brown RM, et al. Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev Med Child Neurol. (2004) 46:710–2. doi: 10.1017/S0012162204001197
84. Brown GK, Otero LJ, LeGris M, Brown RM. Pyruvate dehydrogenase deficiency. J Med Genet. (1994) 31:875–9. doi: 10.1136/jmg.31.11.875
85. Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. (2010) 52:e1–9. doi: 10.1111/j.1469-8749.2009.03541.x
86. Zhu M, Zhu X, Wan H, Hong D. Familial IBGC caused by SLC20A2 mutation presenting as paroxysmal kinesigenic dyskinesia. Parkinsonism Relat Disord. (2014) 20:353–4. doi: 10.1016/j.parkreldis.2013.12.006
87. Symonds JD, Zuberi SM, Stewart K, McLellan A, O'Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. (2019) 142:2303–18. doi: 10.1093/brain/awz195
88. Lee HY, Xu Y, Huang Y, Ahn AH, Auburger GW, Pandolfo M, et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. (2004) 13:3161–70. doi: 10.1093/hmg/ddh330
89. Shen Y, Lee HY, Rawson J, Ojha S, Babbitt P, Fu YH, et al. Mutations in PNKD causing paroxysmal dyskinesia alters protein cleavage and stability. Hum Mol Genet. (2011) 20:2322–32. doi: 10.1093/hmg/ddr125
90. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. GeneReviews, Seattle, WA: University of Washington (1993).
91. Bailey CS, Moldenhauer HJ, Park SM, Keros S, Meredith AL. -linked channelopathy. J Gen Physiol. (2019) 151:1173–89. doi: 10.1085/jgp.201912457
92. Klepper J, Scheffer H, Elsaid MF, Kamsteeg EJ, Leferink M, Ben-Omran T. Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics. (2009) 40:207–10. doi: 10.1055/s-0030-1248264
93. Gordon N, Newton RW. Glucose transporter type1 (GLUT-1) deficiency. Brain Dev. (2003) 25:477–80. doi: 10.1016/S0387-7604(03)00058-5
94. Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, Quinn NP, Weber YG, Lerche H, et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord. (2009) 24:1684–8. doi: 10.1002/mds.22507
95. Buckley C, Williams J, Munteanu T, King M, Park SM, Meredith AL, et al. Status dystonicus, oculogyric crisis and paroxysmal dyskinesia in a 25 year-old woman with a novel. Tremor Other Hyperkinet Mov. (2020) 10:49. doi: 10.5334/tohm.549
96. Vijiaratnam N, Bhatia KP, Lang AE, Raskind WH, Espay AJ. ADCY5-related dyskinesia: improving clinical detection of an evolving disorder. Mov Disord Clin Pract. (2019) 6:512–20. doi: 10.1002/mdc3.12816
97. Carecchio M, Mencacci NE, Iodice A, Pons R, Panteghini C, Zorzi G, et al. ADCY5-related movement disorders: frequency, disease course and phenotypic variability in a cohort of paediatric patients. Parkinsonism Relat Disord. (2017) 41:37–43. doi: 10.1016/j.parkreldis.2017.05.004
98. Chen DH, Méneret A, Friedman JR, Korvatska O, Gad A, Bonkowski ES, et al. ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology. (2015) 85:2026–35. doi: 10.1212/WNL.0000000000002058
99. Bohlega SA, Abou-Al-Shaar H, AlDakheel A, Alajlan H, Bohlega BS, Meyer BF, et al. Autosomal recessive ADCY5-Related dystonia and myoclonus: expanding the genetic spectrum of ADCY5-Related movement disorders. Parkinsonism Relat Disord. (2019) 64:145–9. doi: 10.1016/j.parkreldis.2019.02.039
100. Balint B, Antelmi E, Mencacci NE, Batla A, Eriksson SH, Walker MC, et al. Oculomotor apraxia and disrupted sleep with nocturnal ballistic bouts in ADCY5-related disease. Parkinsonism Relat Disord. (2018) 54:103–6. doi: 10.1016/j.parkreldis.2018.04.011
101. Méneret A, Roze E, Maranci JB, Dodet P, Doummar D, Riant F, et al. Sleep in. J Clin Sleep Med. (2019) 15:1021–9. doi: 10.5664/jcsm.7886
102. Berthold J, Schenková K, Ramos S, Miura Y, Furukawa M, Aspenström P, et al. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes–evidence for an autoregulatory mechanism. Exp Cell Res. (2008) 314:3453–65. doi: 10.1016/j.yexcr.2008.09.005
103. Belal H, Nakashima M, Matsumoto H, Yokochi K, Taniguchi-Ikeda M, Aoto K, et al. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat. (2018) 39:1070–5. doi: 10.1002/humu.23550
104. Finelli MJ, Aprile D, Castroflorio E, Jeans A, Moschetta M, Chessum L, et al. The epilepsy-associated protein TBC1D24 is required for normal development, survival and vesicle trafficking in mammalian neurons. Hum Mol Genet. (2019) 28:584–97. doi: 10.1093/hmg/ddy370
105. Campeau PM, Kasperaviciute D, Lu JT, Burrage LC, Kim C, Hori M, et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol. (2014) 13:44–58. doi: 10.1016/S1474-4422(13)70265-5
106. Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, et al. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum Mutat. (2014) 35:819–23. doi: 10.1002/humu.22557
107. Ragona F, Castellotti B, Salis B, Magri S, DiFrancesco JC, Nardocci N, et al. Alternating hemiplegia and epilepsia partialis continua: a new phenotype for a novel compound TBC1D24 mutation. Seizure. (2017) 47:71–3. doi: 10.1016/j.seizure.2017.03.003
108. Li J, Liu R, Feng H, Zhang J, Wang D, Wang Y, et al. Novel TBC1D24 mutations in a case of nonconvulsive status epilepticus. Front Neurol. (2018) 9:623. doi: 10.3389/fneur.2018.00623
109. Ngoh A, Bras J, Guerreiro R, McTague A, Ng J, Meyer E, et al. Mutations in a sibship with multifocal polymyoclonus. Tremor Other Hyperkinet Mov. (2017) 7:452. doi: 10.5334/tohm.357
110. Salpietro V, Perez-Duenas B, Nakashima K, San Antonio-Arce V, Manole A, Efthymiou S, et al. A homozygous loss-of-function mutation in PDE2A associated to early-onset hereditary chorea. Mov Disord. (2018) 33:482–8. doi: 10.1002/mds.27286
111. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy Comorbidity. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21082802
112. Verdura E, Fons C, Schlüter A, Ruiz M, Fourcade S, Casasnovas C, et al. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J Med Genet. (2020) 57:132–7. doi: 10.1136/jmedgenet-2019-106373
113. Marti S, Baloh RW, Jen JC, Straumann D, Jung HH. Progressive cerebellar ataxia with variable episodic symptoms–phenotypic diversity of R1668W CACNA1A mutation. Eur Neurol. (2008) 60:16–20. doi: 10.1159/000127974
114. Choi KD, Choi JH. Episodic ataxias: clinical and genetic features. J Mov Disord. (2016) 9:129–35. doi: 10.14802/jmd.16028
115. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. (2000) 66:1531–9. doi: 10.1086/302909
116. Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. (2014) 137 (Pt 4):1009–18. doi: 10.1093/brain/awu012
117. Mestre TA, Manole A, MacDonald H, Riazi S, Kraeva N, Hanna MG, et al. A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia. Neurogenetics. (2016) 17:245–9. doi: 10.1007/s10048-016-0486-0
118. Brownstein CA, Beggs AH, Rodan L, Shi J, Towne MC, Pelletier R, et al. Clinical heterogeneity associated with KCNA1 mutations include cataplexy and nonataxic presentations. Neurogenetics. (2016) 17:11–6. doi: 10.1007/s10048-015-0460-2
119. Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology. (2016) 87:1975–84. doi: 10.1212/WNL.0000000000003309
120. Choi KD, Jen JC, Choi SY, Shin JH, Kim HS, Kim HJ, et al. Late-onset episodic ataxia associated with SLC1A3 mutation. J Hum Genet. (2017) 62:443–6. doi: 10.1038/jhg.2016.137
121. Iwama K, Iwata A, Shiina M, Mitsuhashi S, Miyatake S, Takata A, et al. A novel mutation in SLC1A3 causes episodic ataxia. J Hum Genet. (2018) 63:207–11. doi: 10.1038/s10038-017-0365-z
122. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. (2005) 65:529–34. doi: 10.1212/01.WNL.0000172638.58172.5a
123. de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. (2009) 66:97–101. doi: 10.1001/archneurol.2008.535
124. Aoyagi K, Rossignol E, Hamdan FF, Mulcahy B, Xie L, Nagamatsu S, et al. A gain-of-function mutation in NALCN in a child with intellectual disability, ataxia, and arthrogryposis. Hum Mutat. (2015) 36:753–7. doi: 10.1002/humu.22797
125. Chong JX, McMillin MJ, Shively KM, Beck AE, Marvin CT, Armenteros JR, et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet. (2015) 96:462–73. doi: 10.1016/j.ajhg.2015.01.003
126. Synofzik M, Schicks J, Lindig T, Biskup S, Schmidt T, Hansel J, et al. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J Med Genet. (2011) 48:713–5. doi: 10.1136/jmg.2011.090282
127. Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia:UBR4 the likely candidate. Eur J Hum Genet. (2014) 22:505–10. doi: 10.1038/ejhg.2013.173
128. Kotagal V. Acetazolamide-responsive ataxia. Semin Neurol. (2012) 32:533–7. doi: 10.1055/s-0033-1334475
129. Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. (2007) 64:749–52. doi: 10.1001/archneur.64.5.749
130. Hamed M, Shetty A, Dzwiniel T, Buller M, Koskinen L, Suchowersky O. Episodic ataxia secondary to CEP290 compound heterozygous mutations: a case report. Mov Disord Clin Pract. (2020) 7:104–6. doi: 10.1002/mdc3.12872
131. Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. (2012) 72:870–80. doi: 10.1002/ana.23700
132. Wang J, Wen Y, Zhang Q, Yu S, Chen Y, Wu X, et al. Gene mutational analysis in a cohort of Chinese children with unexplained epilepsy: identification of a new KCND3 phenotype and novel genes causing Dravet syndrome. Seizure. (2019) 66:26–30. doi: 10.1016/j.seizure.2019.01.025
133. Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. (2018) 75:591–9. doi: 10.1001/jamaneurol.2017.5121
134. Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. (2010) 133 (Pt 12):3510–8. doi: 10.1093/brain/awq323
135. D'Adamo MC, Gallenmüller C, Servettini I, Hartl E, Tucker SJ, Arning L, et al. Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol. (2014) 5:525. doi: 10.3389/fphys.2014.00525
136. Reinson K, Õiglane-Shlik E, Talvik I, Vaher U, Õunapuu A, Ennok M, et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. (2016) 170:2173–6. doi: 10.1002/ajmg.a.37678
137. Labrum RW, Rajakulendran S, Graves TD, Eunson LH, Bevan R, Sweeney MG, et al. Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J Med Genet. (2009) 46:786–91. doi: 10.1136/jmg.2009.067967
138. Jiang X, Raju PK, D'Avanzo N, Lachance M, Pepin J, Dubeau F, et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. (2019) 60:1881–94. doi: 10.1111/epi.16316
139. Sintas C, Carreño O, Fernàndez-Castillo N, Corominas R, Vila-Pueyo M, Toma C, et al. Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci Rep. (2017) 7:2514. doi: 10.1038/s41598-017-02554-x
140. Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. (1997) 6:1973–8. doi: 10.1093/hmg/6.11.1973
141. Angelini C, Van Gils J, Bigourdan A, Jouk PS, Lacombe D, Menegon P, et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur J Med Genet. (2019) 62:103530. doi: 10.1016/j.ejmg.2018.08.011
142. Humbertclaude V, Riant F, Krams B, Zimmermann V, Nagot N, Annequin D, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol. (2020) 62:330–7. doi: 10.1111/dmcn.14261
143. Tantsis EM, Gill D, Griffiths L, Gupta S, Lawson J, Maksemous N, et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev Med Child Neurol. (2016) 58:639–44. doi: 10.1111/dmcn.13033
144. Hayashida T, Saito Y, Ishii A, Yamada H, Itakura A, Minato T, et al. CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. (2018) 40:130–3. doi: 10.1016/j.braindev.2017.08.006
145. Chivukula AS, Suslova M, Kortzak D, Kovermann P, Fahlke C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum Mutat. (2020) 41:1892–905. doi: 10.1002/humu.24089
146. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. (2007) 130 (Pt 10):2484–93. doi: 10.1093/brain/awm126
147. van Amen-Hellebrekers CJ, Jansen S, Pfundt R, Schuurs-Hoeijmakers JH, Koolen DA, Marcelis CL, et al. Duplications of SLC1A3: associated with ADHD and autism. Eur J Med Genet. (2016) 59:373–6. doi: 10.1016/j.ejmg.2016.06.003
148. Shimojima K, Okumura A, Natsume J, Aiba K, Kurahashi H, Kubota T, et al. Spinocerebellar ataxias type 27 derived from a disruption of the fibroblast growth factor 14 gene with mimicking phenotype of paroxysmal non-kinesigenic dyskinesia. Brain Dev. (2012) 34:230–3. doi: 10.1016/j.braindev.2011.04.014
149. Di Re J, Wadsworth PA, Laezza F. Intracellular fibroblast growth factor 14: emerging risk factor for brain disorders. Front Cell Neurosci. (2017) 11:103. doi: 10.3389/fncel.2017.00103
150. Manso-Calderón R. Clinical features and treatment in the spectrum of paroxysmal dyskinesias: an observational study in South-West Castilla y Leon, Spain. Neurol Res Int. (2019) 2019:4191796. doi: 10.1155/2019/4191796
151. Montaut S, Tranchant C, Drouot N, Rudolf G, Guissart C, Tarabeux J, et al. Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurol. (2018) 75:1234–45. doi: 10.1001/jamaneurol.2018.1478
152. Ashrafi MR, Amanat M, Garshasbi M, Kameli R, Nilipour Y, Heidari M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev Neurother. (2020) 20:65–84. doi: 10.1080/14737175.2020.1699060
153. Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J, et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat. (2014) 35:45–9. doi: 10.1002/humu.22451
154. Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol. (2020) 24:15–23. doi: 10.1016/j.ejpn.2019.12.008
155. Fogel BL. Genetic and genomic testing for neurologic disease in clinical practice. Handb Clin Neurol. (2018) 147:11–22. doi: 10.1016/B978-0-444-63233-3.00002-6
156. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kobayashi J, Cohn RD, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4:e265. doi: 10.1212/NXG.0000000000000265
157. Ngo KJ, Rexach JE, Lee H, Petty LE, Perlman S, Valera JM, et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum Mutat. (2020) 41:487–501. doi: 10.1002/humu.23946
158. Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. (2018) 19:253–68. doi: 10.1038/nrg.2017.116
159. Ueda K, Shinawi M, Pearson TS. Paroxysmal kinesigenic dyskinesia in twins with chromosome 16p11.2 duplication syndrome. Neurol Genet. (2021) 7:e549. doi: 10.1212/NXG.0000000000000549
160. Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. (2013) 17:589–99. doi: 10.1016/j.ejpn.2013.04.010
161. Ezugha H, Anderson CE, Marks HG, Khurana D, Legido A, Valencia I. Microarray analysis in children with developmental disorder or epilepsy. Pediatr Neurol. (2010) 43:391–4. doi: 10.1016/j.pediatrneurol.2010.06.019
162. Dale RC, Grattan-Smith P, Nicholson M, Peters GB. Microdeletions detected using chromosome microarray in children with suspected genetic movement disorders: a single-centre study. Dev Med Child Neurol. (2012) 54:618–23. doi: 10.1111/j.1469-8749.2012.04287.x
163. Howell KB, Kornberg AJ, Harvey AS, Ryan MM, Mackay MT, Freeman JL, et al. High resolution chromosomal microarray in undiagnosed neurological disorders. J Paediatr Child Health. (2013) 49:716–24. doi: 10.1111/jpc.12256
164. Mullen SA, Carvill GL, Bellows S, Bayly MA, Trucks H, Lal D, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology. (2013) 81:1507–14. doi: 10.1212/WNL.0b013e3182a95829
165. Misra S, Peters G, Barnes E, Ardern-Holmes S, Webster R, Troedson C, et al. Yield of comparative genomic hybridization microarray in pediatric neurology practice. Neurol Genet. (2019) 5:e367. doi: 10.1212/NXG.0000000000000367
166. Rexach J, Lee H, Martinez-Agosto JA, Németh AH, Fogel BL. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. (2019) 18:492–503. doi: 10.1016/S1474-4422(19)30033-X
167. Maksemous N, Sutherland HG, Smith RA, Haupt LM, Griffiths LR. Comprehensive exonic sequencing of known ataxia genes in episodic ataxia. Biomedicines. (2020) 8:134. doi: 10.3390/biomedicines8050134
168. Graziola F, Garone G, Stregapede F, Bosco L, Vigevano F, Curatolo P, et al. Diagnostic yield of a targeted next-generation sequencing gene panel for pediatric-onset movement disorders: a 3-year cohort study. Front Genet. (2019) 10:1026. doi: 10.3389/fgene.2019.01026
169. van Egmond ME, Lugtenberg CHA, Brouwer OF, Contarino MF, Fung VSC, Heiner-Fokkema MR. A post hoc study on gene panel analysis for the diagnosis of dystonia. Mov Disord. (2017) 32:569–75. doi: 10.1002/mds.26937
170. Thiffault I, Farrow E, Zellmer L, Berrios C, Miller N, Gibson M, et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet Med. (2018) 21:303–10. doi: 10.1038/s41436-018-0075-8
171. Smith HS, Swint JM, Lalani SR, Yamal JM, de Oliveira Otto MC, Castellanos S, et al. Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: a scoping review of the literature. Genet Med. (2018) 21:3–16. doi: 10.1038/s41436-018-0024-6
172. Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: from raw data to diagnosis with lifetime value. Clinical Genetics. (2018) 93:508–19. doi: 10.1111/cge.13190
173. Olgiati S, Quadri M, Bonifati V. Genetics of movement disorders in the next-generation sequencing era. Mov Disord. (2016) 31:458–70. doi: 10.1002/mds.26521
174. Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, et al. Somatic mutations in cerebral cortical malformations. New Engl J Med. (2014) 371:733–43. doi: 10.1056/NEJMoa1314432
175. Wright CF, Prigmore E, Rajan D, Handsaker J, McRae J, Kaplanis J, et al. Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat Commun. (2019) 10:2985. doi: 10.1038/s41467-019-11059-2
176. Myers CT, Hollingsworth G, Muir AM, Schneider AL, Thuesmunn Z, Knupp A, et al. Parental mosaicism in “de novo” epileptic encephalopathies. N Engl J Med. (2018) 378:1646–8. doi: 10.1056/NEJMc1714579
177. Hully M, Ropars J, Hubert L, Boddaert N, Rio M, Bernardelli M, et al. Mosaicism in ATP1A3-related disorders: not just a theoretical risk. Neurogenetics. (2017) 18:23–8. doi: 10.1007/s10048-016-0498-9
178. Deeb KK, Bedoyan JK, Wang R, Sremba L, Schroeder MC, Grahame GJ, et al. Somatic mosaicism for a novel. Mol Genet Metab Rep. (2014) 1:362–7. doi: 10.1016/j.ymgmr.2014.08.001
179. Stosser MB, Lindy AS, Butler E, Retterer K, Piccirillo-Stosser CM, Richard G, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med. (2018) 20:403–10. doi: 10.1038/gim.2017.114
180. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
181. Erro R, Bhatia KP, Espay AJ, Striano P. The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: Channelopathies, synaptopathies, and transportopathies. Mov Disord. (2017) 32:310–8. doi: 10.1002/mds.26901
182. Rosell AM, Pena LD, Schoch K, Spillmann R, Sullivan J, Hooper SR, et al. Not the end of the odyssey: parental perceptions of whole exome sequencing (WES) in pediatric undiagnosed disorders. J Genet Couns. (2016) 25:1019–31. doi: 10.1007/s10897-016-9933-1
183. Epilepsy Genetics I. The epilepsy genetics initiative: systematic reanalysis of diagnostic exomes increases yield. Epilepsia. (2019) 60:797–806. doi: 10.1111/epi.14698
184. Rochtus A, Olson HE, Smith L, Keith LG, El Achkar C, Taylor A, et al. Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia. (2020) 61:249–58. doi: 10.1111/epi.16427
185. Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. (2017) 9:eaal5209. doi: 10.1126/scitranslmed.aal5209
186. Ye J, Wu W, Li Y, Li L. Influences of the gut microbiota on DNA methylation and histone modification. Digest Dis Sci. (2017) 62:1155–64. doi: 10.1007/s10620-017-4538-6
187. Hauser RM, Henshall DC, Lubin FD. The epigenetics of epilepsy and its progression. Neuroscientist. (2018) 24:186–200. doi: 10.1177/1073858417705840
188. Bernardo BC, Charchar FJ, Lin RC, McMullen JR. A microRNA guide for clinicians and basic scientists: background and experimental techniques. Heart Lung Circ. (2012) 21:131–42. doi: 10.1016/j.hlc.2011.11.002
189. Enright N, Simonato M, Henshall DC. Discovery and validation of blood microRNAs as molecular biomarkers of epilepsy: ways to close current knowledge gaps. Epilepsia Open. (2018) 3:427–36. doi: 10.1002/epi4.12275
190. Xu Z, Lim CK, Tan LCS, Tan EK. Paroxysmal movement disorders: recent advances. Curr Neurol Neurosci Rep. (2019) 19:48. doi: 10.1007/s11910-019-0958-3
Keywords: paroxysmal movement disorders, genetics, next-generation sequencing, paroxysmal dyskinesia, episodic ataxia
Citation: Harvey S, King MD and Gorman KM (2021) Paroxysmal Movement Disorders. Front. Neurol. 12:659064. doi: 10.3389/fneur.2021.659064
Received: 26 January 2021; Accepted: 20 April 2021;
Published: 11 June 2021.
Edited by:
Anna De Rosa, Università di Napoli Federico II, ItalyReviewed by:
Victor Fung, Westmead Hospital, AustraliaRoberto Erro, University of Salerno, Italy
Beisha Tang, Central South University, China
Copyright © 2021 Harvey, King and Gorman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen M. Gorman, a2F0aGxlZW4uZ29ybWFuQGN1aC5pZQ==