 Gabriel Torrealba-Acosta1,2*†
Gabriel Torrealba-Acosta1,2*† Eric Yu3,4†
Eric Yu3,4† Tanya Lobo-Prada2,5
Tanya Lobo-Prada2,5 Javier Ruíz-Martínez6,7,8
Javier Ruíz-Martínez6,7,8 Ana Gorostidi-Pagola7,8,9
Ana Gorostidi-Pagola7,8,9 Ziv Gan-Or3,4,10Kenneth Carazo-Céspedes11Jean-François Trempe12Ignacio F. Mata13
Ziv Gan-Or3,4,10Kenneth Carazo-Céspedes11Jean-François Trempe12Ignacio F. Mata13 Jaime Fornaguera-Trías2,5
Jaime Fornaguera-Trías2,5- 1Department of Neurology and Neurosurgery, Baylor College of Medicine, Houston, TX, United States
- 2Neurosciences Research Center, Universidad de Costa Rica, San José, Costa Rica
- 3Montreal Neurological Institute and Hospital, McGill University, Montreal, QC, Canada
- 4Department of Human Genetics, McGill University, Montreal, QC, Canada
- 5Department of Biochemistry, Medicine School, Universidad de Costa Rica, San José, Costa Rica
- 6Group of Neurodegenerative Diseases, Biodonostia Health Research Institute, San Sebastian, Spain
- 7CIBERNED, Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Madrid, Spain
- 8Movement Disorders Unit, Neurology Department, Donostialdea Integrated Health Organisation, Osakidetza Basque Health Service, San Sebastian, Spain
- 9Genomic Platform, Biodonostia Health Research Institute, San Sebastian, Spain
- 10Department of Neurology and Neurosurgery, McGill University, Montreal, QC, Canada
- 11Department of Neurology, Hospital San Juan de Dios, Caja Costarricense de Seguro Social, San José, Costa Rica
- 12Department of Pharmacology and Therapeutics and Centre de Recherche en Biologie Structurale, McGill University, Montreal, QC, Canada
- 13Cleveland Clinic Foundation, Genomic Medicine, Lerner Research Institute, Cleveland, OH, United States
Background: Most research in genomics of Parkinson's disease (PD) has been done in subjects of European ancestry, leading to sampling bias and leaving Latin American populations underrepresented. We sought to clinically characterize PD patients of Costa Rican origin and to sequence familial PD and atypical parkinsonism-associated genes in cases and controls.
Methods: We enrolled 118 PD patients with 97 unrelated controls. Collected information included demographics, exposure to risk and protective factors, and motor and cognitive assessments. We sequenced coding and untranslated regions in familial PD and atypical parkinsonism-associated genes including GBA, SNCA, VPS35, LRRK2, GCH1, PRKN, PINK1, DJ-1, VPS13C, and ATP13A2.
Results: Mean age of PD probands was 62.12 ± 13.51 years; 57.6% were male. The frequency of risk and protective factors averaged ~45%. Physical activity significantly correlated with better motor performance despite years of disease. Increased years of education were significantly associated with better cognitive function, whereas hallucinations, falls, mood disorders, and coffee consumption correlated with worse cognitive performance. We did not identify an association between tested genes and PD or any damaging homozygous or compound heterozygous variants. Rare variants in LRRK2 were nominally associated with PD; six were located between amino acids p.1620 and 1623 in the C-terminal-of-ROC (COR) domain of Lrrk2. Non-synonymous GBA variants (p.T369M, p.N370S, and p.L444P) were identified in three healthy individuals. One PD patient carried a pathogenic GCH1 variant, p.K224R.
Discussion: This is the first study that describes sociodemographics, risk factors, clinical presentation, and genetics of Costa Rican patients with PD, adding information to genomics research in a Latino population.
Introduction
Parkinson's disease (PD) is a complex and heterogeneous movement disorder caused by a progressive degeneration of dopaminergic neurons. Main clinical motor symptoms associated with PD include tremor, rigidity, bradykinesia, and postural imbalance (1). Years before motor symptoms are manifested, there can be prodromal non-motor key features that include rapid eye movement (REM) sleep disorders, anosmia, and constipation (2). Cognitive impairment involving dysexecutive dysfunction with deficits in planning (3), shifting and sharing of attention (4), and problem solving (5), together with visuospatial dysfunction (6), can be also present from early stages of the disease. PD pathophysiology involves environmental factors as well as genetic variance, which provide insight into its molecular pathogenesis. Among environmental factors that contribute to PD risk are pesticide and herbicide exposure, welding, and well water consumption. There are also protective factors such as smoking, coffee consumption, and performing physical activity that may reduce the risk of developing PD (7).
Since the description of PD-associated mutations in the SNCA (8), other genes have been linked to autosomal dominant (AD) forms of familial PD, including LRRK2 and VPS35. In addition, there are clinically and genetically diverse early-onset (EO) autosomal-recessive (AR) forms of PD with associated genes like PRKN, PINK1, and DJ-1 that exhibit phenotypes similar to idiopathic PD, while other associated genes such as VPS13C and ATP13A2 combine atypical features of parkinsonism like dystonia and early cognitive impairment, along with a poor response to levodopa (9). Large-scale genome-wide association studies (GWASs) have identified 90 variants for PD risk across 78 genomic regions, confirming SNCA and GBA as the most important ones (10). Different GBA locus present as strong risk factors for PD in both homozygous and heterozygous state, displaying a phenotype similar to idiopathic PD, yet with a faster rate of progression of cognitive and motor decline (11).
Clinical characterization of PD in Latin American and Hispanic populations has been scarce (12). Likewise, there is a lack of diversity in genomics with an overrepresentation of European-derived individuals, leading to sampling bias and leaving large populations underrepresented (13). Few genetic trials have been conducted in PD individuals from Latin American populations. Studies looking at LRRK2 mutations have shown that their frequency varies across geographic areas and ethnicity groups. For the G2019S mutation in the LRRK2 gene, frequencies range from 0.2 to 0.4% in Peruvian cohorts (14, 15), up to 4% in Uruguayans (14) and 5.45% in an Argentinian series (16–19). Likewise, the R1441G and R1441H mutations in this same gene seem to be uncommon in Latin American populations (0.3–0.8%) (14, 18). The LARGE-PD, a research consortium established among several Latin American countries, has been collecting data for what is the largest PD cohort in the region, allowing for large-scale genotyping as well as performing GWAS in these cohorts (20–22). This initiative aimed to estimate the frequency of LRRK2 mutations in the region and reported varying frequencies of the G2019S and R1441G/C mutations, which strongly correlated with the European admixture of the samples analyzed (15, 20).
GBA mutations have also been studied in few Latin American cohorts but mainly focused on most frequently reported mutations in other populations. The observed frequency of these mutations varies across regions ranging from 0.2% (p.N370S) to 0.7% (p.E326K) in Ecuadorians (23) and up to 5.5% (p.L444P) in Mexican Mestizo and Brazilian cohorts (23–27). Few studies have studied the entire GBA gene in Latin America, showing a frequency similar to those reported in individuals of European descent (4–5%), but lower than frequencies reported in Ashkenazi patients (20%) (28). Moreover, the overall frequency of GBA mutations seems to be consistently higher than LRRK2 mutations across different geographic areas, suggesting that GBA could play a more important role in PD genetics for Latin American populations. Velez-Pardo et al. found a mutation that was specific for a Colombian cohort (p.K198E) and in a much higher frequency (9.9%) highlighting the need to sequence the whole GBA gene rather than focusing only on assessing commonly reported mutations (27).
In this study, we sought to clinically characterize PD patients of Costa Rican origin and to sequence familial PD and atypical parkinsonism-associated genes in Costa Rican PD cases and controls.
Materials and Methods
Study Subjects
We enrolled 118 consecutive unrelated PD patients (68 males, 50 females) with 97 unrelated controls (28 males, 69 females), matched according to age and gender whenever possible. Thirty-five patients (16.28%) reported having a relative (≤ 2°) with any sort of movement disorder; of those, 21 (9.77%) had a formal PD diagnosis. All subjects resided and were originated from Costa Rica and were recruited at the Movement Disorders Unit of the Department of Neurology, Hospital San Juan de Dios, Caja Costarricense de Seguro Social. All patients fulfilled Gelb criteria for the clinical diagnosis of PD, while controls had no signs or personal history of any neurodegenerative disease and were mainly the spouses of the PD cases. We preferred using Gelb criteria over the United Kingdom Parkinson's Disease Society Brain Bank (UKPDSBB) as it provided different clinical diagnostic levels of certainty (possible and probable) and it has shown to have similar positive and negative predictive values, as well as sensitivity and global accuracy when compared to UKPDSBB (29). Albeit both diagnostic criteria sets have low specificity and are mainly focused on motor features, UKPDSBB criteria further err by challenging PD diagnosis in the presence of genetic risk factors (30). Our last patient was enrolled by 2011, which is 4 years earlier than when the Movement Disorder Society (MDS) task force proposed the new clinical diagnostic criteria for PD (MDS-PD criteria) (31); therefore, we were not able to use those for clinical diagnosis of patients enrolled in our study.
We gathered information concerning work and educational status as well as history of exposure to risk and protective factors of PD. We further obtained detailed information on PD history, comorbidities, and antiparkinsonian treatments. Additionally, motor disability of the patients was evaluated by means of the Unified Parkinson's Disease Rating Scale (UPDRS), Hoehn & Yahr (H&Y), and Schwab & England (S&E) scales. Cognitive status was assessed using the Montreal Cognitive Assessment (MoCA) test.
Genetic Analysis
Molecular inversion probes were used to sequence coding and untranslated regions in familial PD and atypical parkinsonism-associated genes including GBA, SNCA, VPS35, LRRK2, GCH1, PRKN, PINK1, DJ-1, VPS13C, and ATP13A2 at McGill University with Illumina HiSeq 4000 as previously described (32). The full protocol can be found at https://github.com/gan-orlab/MIP_protocol. All sequences were aligned using Burrows-Wheeler Aligner (BWA) using the reference genome hg19 (33). Genome Alignment Tool Kit (GATK v3.8) was used to call variants and perform quality control and ANNOVAR was used to annotate each variant (34, 35). Exons 10 and 11 of GBA were sequenced using Sanger sequencing as previously described (36), and GBA variants in other exons were also validated using Sanger sequencing. We decided to focus on genes that are involved in typical PD, as our selected cohort is of typical PD (10, 37, 38).
Quality Control
All samples and variants were filtered based on standard quality control process as previously reported (39). In brief, variants were separated into common and rare by minor allele frequency (MAF) in the cohort. Rare variants (MAF < 0.01) with a minimum depth of coverage of > 30× were included in the analysis, along with common variants (MAF ≥ 0.01) with >15× coverage. We have established that for common variants, we get reliable reads at 15×; however, to get reliable reads for rare variants, we need > 30×; otherwise, there are many false positives (40). Variant calls with a genotype frequency of <25% of the reads or genotype quality of <30 were excluded. Samples and variants with more than 10% missingness were also excluded.
In silico Structural Analysis
The atomic coordinates of the human Lrrk2 C-terminal domain structure (a.a. 1327–2527) were downloaded from the Protein Data Bank (ID 6VP6). The figure was generated using PyMol v.2.4.0.
Statistics
We used Stata® (version 14) for the statistical analysis of sociodemographic and clinical variables. Normally distributed variables are reported as mean with its standard deviation (SD), whereas continuous but non-normally distributed variables are reported as median with the 25th and 75th percentile values (interquartile range, IQR). Normally distributed variables were compared with paired or unpaired t-tests, while non-normally distributed variables were compared with Mann–Whitney U-test or Wilcoxon match-paired signed-rank test. Frequencies were compared with χ2 and Fisher's exact test. Tests were two-tailed, and significance was set at p < 0.05. We modeled through linear regression the association between demographic and clinical variables with the severity of the disease, as indexed by UPDRS and MoCA, as dependent variables in the models.
For genetic analysis, common and rare variants were analyzed separately. Association of common variants was tested using logistic regression adjusted for age and sex in PLINK v1.9. For rare variants' analysis, we examined the burden of rare variants in each gene using optimized sequence Kernel association test (SKAT-O) adjusted for age and sex (41). Rare variants were separated into different categories based on their potential pathogenicity to examine specific enrichment in different variant subgroups as described previously (40): (1) variants with Combined Annotation Dependent Depletion (CADD) score of ≥12.37 (representing the top 2% of potentially deleterious variants) (42); (2) regulatory variants predicted by ENCODE (43); (3) potentially functional variants including all non-synonymous variants, stop gain/loss variants, frameshift variants, and intronic splicing variants located within two base pairs of exon–intron junctions; (4) loss-of-function variants, which includes stop gain/loss, frameshift, and splicing variants; and (5) only non-synonymous variants. Bonferroni correction for multiple comparisons was applied as necessary.
This study was approved by the Ethics Committee of Hospital San Juan de Dios, Caja Costarricense de Seguro Social (CLOBI-HSJD #014-2015) and the University of Costa Rica (837-B5-304). Written informed consent was obtained from all participants.
Results
Sociodemographic and Clinical Variables
At enrollment, PD probands had a mean age of 62.12 ± 13.51 years (range 25–86), and the mean age at onset was 54.62 ± 13.54 (range 16–83) years. Male PD patients comprised 57.63% of the sample. Despite the fact that a significantly larger proportion of the male PD patients reported current or previous jobs involving agricultural activities (19.40% male, 2.08% female; p = 0.01), the mean number of years of education of these men was significantly higher than women (10.74 ± 3.81 vs. 8.86 ± 4.01; p = 0.03). Table 1 details subjects' baseline characteristics along with the frequency of exposure to main risk and protective factors for PD. Most of the risk and protective factors were more prevalent in men. Tables 1, 2 detail the frequency of clinical manifestations as well as the standardized scale scores reported for PD cases. The most frequent initial symptoms included resting tremor (71.30%), rigidity (24.07%), and pain (10.19%). Most of the patients had asymmetric onset (94.12%) and a good response to levodopa (89.11%). Other frequently reported motor features comprised dystonia (46.08%), falls (39.22%), and dysphagia (36.27%). Common non-motor manifestations such as hyposmia, sleep disorders and depressive/anxious mood were seen in more than 50% of the cases. Overall median score of UPDRS “ON” was 36 (22–60), most of our patients were graded in the “2.5” and “3” categories of the H&Y scale with a median for S&E score of 80% (80–90%). The median value for the MoCA test was 22 (17–25). There were no statistically significant differences between sex, regarding these scores.
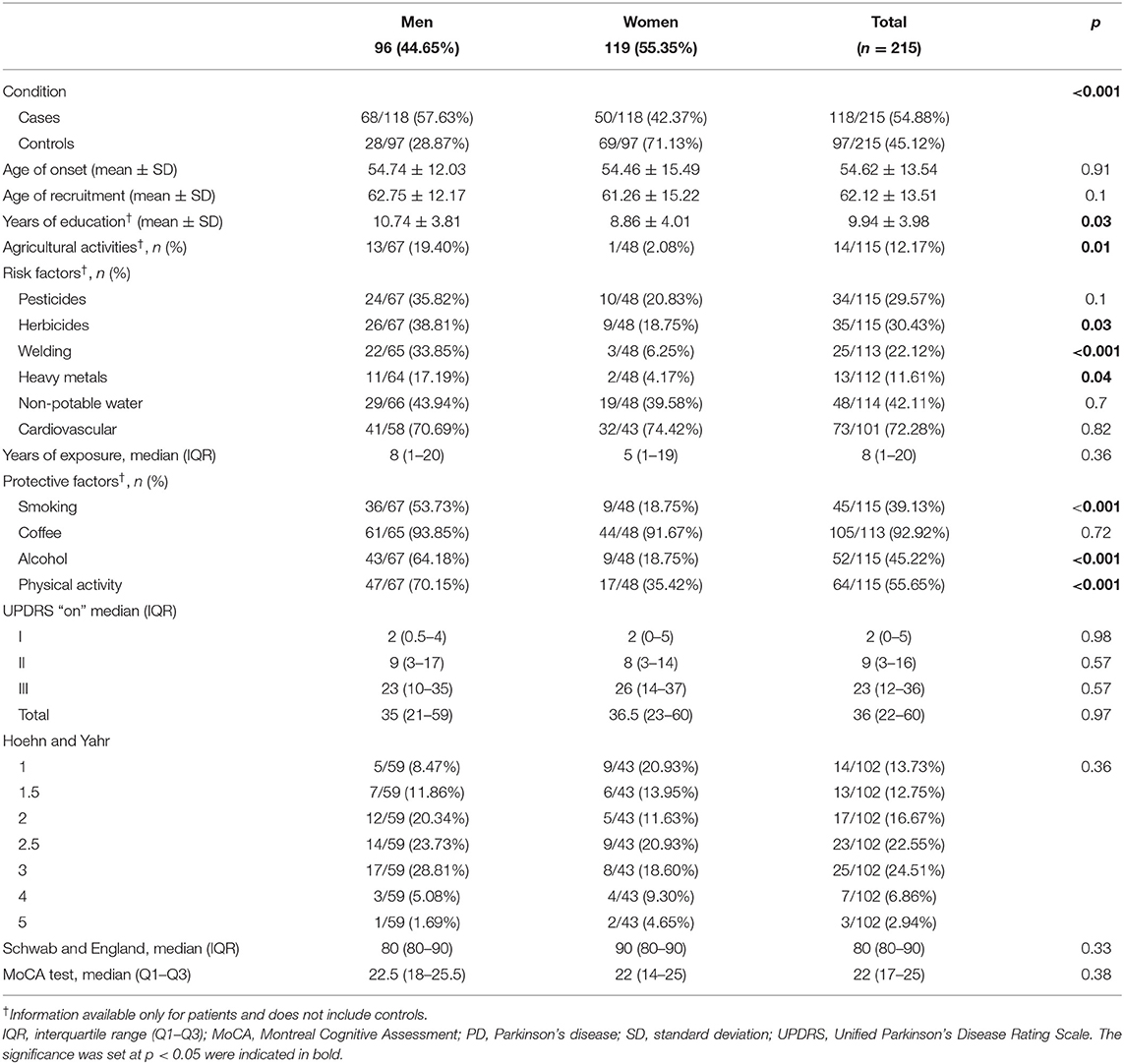
Table 1. Baseline characteristics with frequency of risk and protective factors for PD in study subjects, with sex comparison.
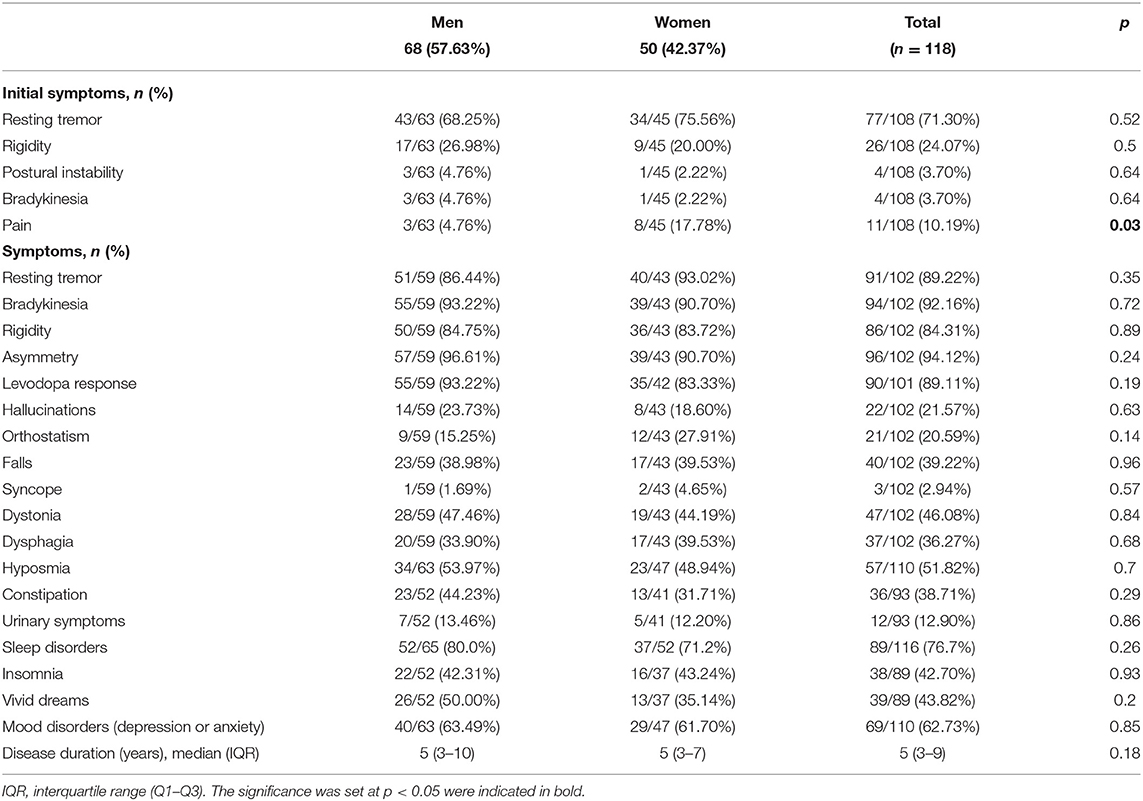
Table 2. Clinical manifestations of PD cases, with sex comparison.
We were able to establish through multivariate linear regression modeling that an increased disease duration along with the presence of orthostasis, dysphagia, and mood disorders significantly correlated with increased scores in total ON UPDRS. Furthermore, we found an interaction between performing regular physical activity and duration of disease, where despite having increased years of evolution, patients that performed regular physical activity still scored less in the total ON UPDRS (see Supplementary Figure 1). Additionally, lower scores in MoCA testing significantly correlated with increased age, coffee consumption, and the presence of hallucinations, falls, and mood disorders (depression/anxiety), whereas increased years of education correlated with better MoCA scores (see Supplementary Figure 2).
Quality of Coverage and Identified Variants
The average coverage of the 10 genes analyzed in this study was >588× for all genes. The coverage per gene and the percentage of nucleotides covered at >15× and >30× for each gene are detailed in Supplementary Table 1. There were no differences in the coverage across the samples (patients and controls). Overall, after quality control, we identified 163 rare variants (Supplementary Table 2) and 158 common variants (Supplementary Table 3) across all genes and all samples that were included in the analysis. Specific protein and DNA changes are listed in Supplementary Tables 4, 5 for rare and common exonic variants, respectively.
Rare and Common Variants in PD and Parkinsonism-Related Genes
Burden and SKAT-O analyses did not identify an association of any of the tested genes and PD (Table 3) after correction for multiple comparisons, as expected given the small sample size. We also did not identify any PD patients with potentially damaging homozygous or compound heterozygous variants in any of these genes. Rare variants in LRRK2 were nominally associated with PD, and 11 (9.2%) patients carried a rare non-synonymous variant, compared to four (4.1%) among the controls. Interestingly, six of these rare non-synonymous variants, all located between amino acids p.1620 and 1623 in the C-terminal-of-ROC (COR) domain of Lrrk2, were found in six patients and none in controls (Table 4).
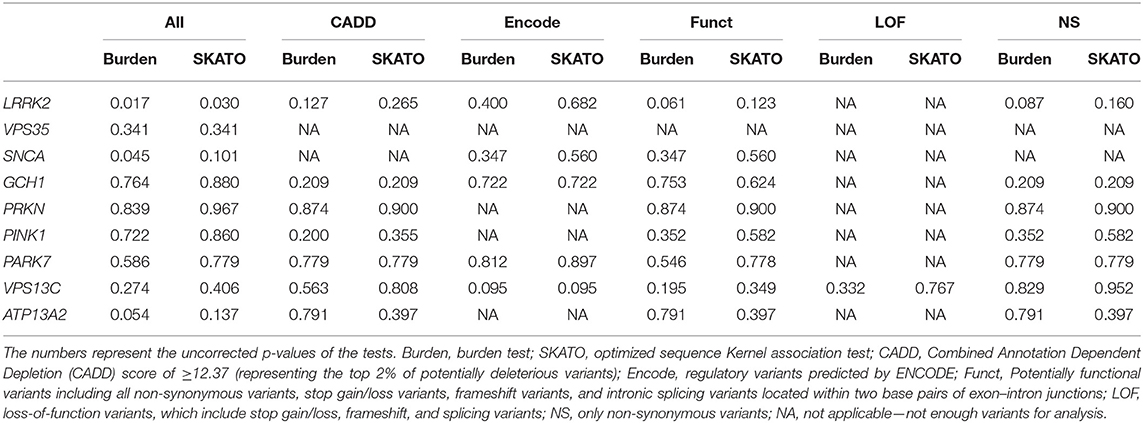
Table 3. Burden and SKAT-O analyses with no significant association found of any of the tested genes and PD, after Bonferroni correction for multiple comparisons.
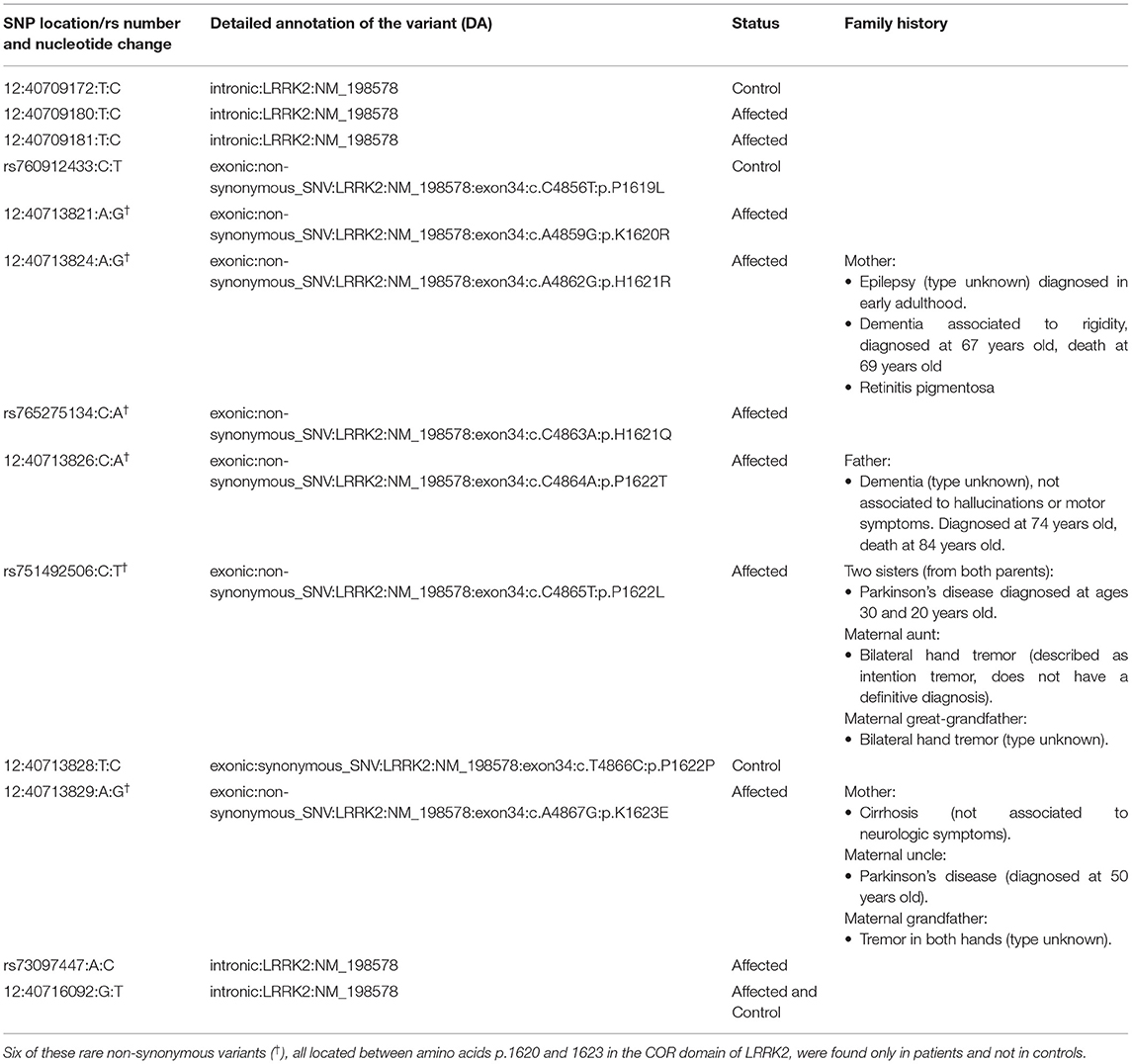
Table 4. Rare variants in LRRK2 present in patients and controls.
Non-synonymous GBA variants were identified in three individuals: p.T369M was identified in a male patient with age at onset of 48 years, p.N370S was identified in a healthy female individual recruited at the age of 78 years, and p.L444P was identified in a healthy female individual recruited at the age of 64. While we cannot rule out that these healthy individuals will develop PD in the future, it is unlikely that GBA variants have a major role in PD among Costa Rican patients. One PD patient carried a pathogenic GCH1 variant, p.K224R, further emphasizing the role of this gene in PD.
In the analysis of common variants, none of the variants was associated with PD after correction for multiple comparisons (Supplementary Table 3), which set the corrected p-value for statistical significance at p < 0.00031. One non-synonymous variant in LRRK2, p.I723V, was found with allele frequency of 0.01 in patients and 0.09 in controls (OR = 0.11, 95% CI = 0.02–0.52, p = 0.005), yet this difference was not statistically significant after correction for multiple comparisons.
Discussion
Clinical Features
PD prevalence has been increasing over time with a global age-standardized prevalence rate increase of 21.7% from the years 1990 to 2016 (44). Furthermore, PD prevalence seems to be lower in Eastern compared to Western countries (45). Few studies have explored the prevalence of PD in Latin America providing values that are similar either to other developing countries (46) or to European cohorts (47, 48). PD also becomes more common with advancing age (44, 45). Our sample average age of PD at onset and at diagnosis was lower when compared to other cohorts (49–51), although it could suggest that PD presents earlier in Costa Rica, and more epidemiological studies are needed as it could also be related to recruitment bias.
The majority of our patients fulfilled Group A Gelb criteria while up to 60% also reported at least one of Group B symptoms, the most frequent being dystonia, falls, and dysphagia. The median for years of evolution of the disease for both men and women was 5; thus, we would expect to find Group B criteria in these patients along with the evolution of the disease. Few studies have explored ethnic variations in motor symptoms of PD, suggesting increased atypical features in Black and South Asian PD patients (52, 53); however, there is not enough evidence available along with a lack of standardized methodology to determine motor subtypes across studies and to further establish ethnic patterns of motor features (12). Common non-motor manifestations such as hyposmia, sleep disorders, and depressive/anxious mood were seen in more than half of our PD cases. Regardless of ethnicity, non-motor features are commonly present in PD with subtle differences described. Gastrointestinal non-motor features along with depression seem to be high in East Asian cohorts (54, 55). Likewise, Latino populations, such as Mexican (56), Peruvian (57), and ours, also reported high frequency of mood disorders including depression and anxiety, when compared to studies from UK and USA (58, 59). We also observed in our sample a frequency of sleep disorders and hyposmia that is higher than those reported in other cohorts (12).
Overall, our patients had a low education, which has been previously associated with a higher hazard of incident parkinsonism (60). A reduced education has also been suggested as a risk factor for cognitive impairment in PD (61). A history of non-potable water consumption along with exposure to pesticides and herbicides was reported in up to 40% of our patients. This type of exposure agrees with a mostly rural origin and the fact that 12.2% of the subjects reported involvement in agricultural activities as a main income source. We did not assess the frequency of protective and risk factors in the control group; hence, we are not able to establish any comparison with PD cases. Previous exposure to pesticides and herbicides is associated with the development of PD (62); yet, the identification of a given specific agent and the exact timing and dosing of exposure are almost impossible to establish through observational studies (63, 64). Nonetheless, key work detailing specific mechanisms that render patients vulnerable to pesticide-induced injury has been elegantly shown in animal models, further establishing biologic and toxicological pathways for specific chemicals to potentially cause PD (65). A similar situation is present regarding the exposure to welding and heavy metals. Manganese (66), copper, iron (67), and mercury (68) have been proposed as possible agents associated with the development of PD. In this study, 22.1 and 11.6% of the patients reported frequent exposure to welding and other heavy metals, respectively; however, the exact timing and dosing of exposure was not possible to assess.
Other literature has underscored the presence of protective factors for PD development, among which the most notable and with the strongest evidence include tobacco (69) and coffee consumption (7, 70–73). For both protective factors, there is also a dosing effect described, where the protective effect increases along with an increasing exposure (74, 75). Paradoxically, over 90% of our PD cases had been exposed to a protective factor in the past, most of them having a regular coffee intake (two to three cups per day for over 15 years), and yet they all developed PD.
Performing regular physical activity correlated with lower ON UPDRS scores in spite of increasing age. Physical activity has been established as a possible protective factor for incident Parkinsonism (76); our data would suggest that physical activity could determine reduced severity of disease, specifically concerning motor features. Although exercise has not been proven to slow the progression of akinesia, rigidity, and gait disturbances, it promotes a feeling of physical and mental well-being, and at the same time, it can alleviate rigidity-related pain and improve patients' motor (77) and non-motor symptoms (78).
Increasing age, coffee consumption, hallucinations, falls, and mood disorders along with reduced years of education significantly correlated with worse MoCA scores. Older age and duration of PD are determinant risk factors for incidence of dementia in PD (79). Furthermore, hallucinations have been established as risk factors for cognitive impairment (79, 80) along with gait disturbances (manifested by falls) (81) and depression (82). Reduced education years also have been proposed as a risk factor for cognitive impairment in patients with PD (61). Poor global cognition has been previously associated with a higher risk of incident parkinsonism (60). Coffee consumption has been suggested to reduce risk of dementia (83) with a dosing effect (84, 85); however, there have been inconsistent findings regarding the effects of coffee consumption on specific cognitive domains. It has been suggested to be in association with improved executive performance but smaller hippocampal volume and worse memory function (86); nonetheless, this association is not sustained when cognition is analyzed longitudinally. Other literature suggested that coffee might be slightly beneficial on memory without a dose–response relationship (87). Recent large-scale genetic analysis using mendelian randomization did not find any evidence supporting any beneficial or adverse long-term effect of coffee consumption on global cognition or memory function (88) or AD incidence (89). To our knowledge, there is no literature evaluating the effect on cognition of coffee consumption, specifically for PD patients. Our findings suggest a possible deleterious effect that should be further explored in this population.
Genetic Assessment
After sequence coding familial PD and atypical parkinsonism-associated genes including GBA, SNCA, VPS35, LRRK2, GCH1, PRKN, PINK1, DJ-1, VPS13C, and ATP13A2 and correcting for multiple comparisons, burden and SKAT-O analyses did not show an association of any of the tested genes and PD. We also did not identify any homozygous or compound heterozygous pathogenic variants in any of these genes.
Non-synonymous GBA variants were identified in three individuals including one patient and two unaffected controls. While we cannot rule out that these healthy individuals will develop PD in the future, it is unlikely that GBA variants have a major role in PD among Costa Rican patients especially when compared to other European and Ashkenazi Jewish populations where we find that 8–20% of the patients harbor GBA variants (28).
Finally, one PD patient carried a pathogenic variant, p.K224R, in the GCH1 gene. GCH1 encodes for GTP cyclohydrolase 1, which is a key enzyme for dopamine production in nigrostriatal neurons. Loss-of-function mutations such as p.K224R have been shown to cause Dopa-responsive dystonia (DRD); however, variants in this gene have also been implicated in PD, perhaps through regulation of GCH1 expression (90, 91). It has been suggested that late-onset DRD might present clinically with parkinsonism, or alternatively, pathogenic GCH1 mutations may predispose to both diseases and carriers will develop any or both depending on other genetic or environmental factors (92). Our patient did not present clinical features suggestive of DRD and did not have any family history of PD.
Rare variants in LRRK2 were nominally associated with PD, observed only in affected individuals; six of these rare non-synonymous variants were located between amino acids p.1620 and 1623 in the COR domain of Lrrk2. LRRK2 encodes a multiple domain protein that includes a Roc-COR tandem domain, a tyrosine kinase-like protein kinase domain, and at least four repeat domains located within the N-terminal and C-terminal regions. The Roc-COR domain classifies the Lrrk2 protein as part of the ROCO superfamily of Ras-like G proteins (93). Mutations in LRRK2 are the most common cause of late-onset hereditary PD. Most frequently reported disease-causing mutations are located in the kinase domain (i.e., G2019S), increasing kinase activity, and in the Roc-COR tandem domain (i.e., R1441C/G and Y1699C), impairing its GTPase function. Alterations of both kinase and GTPase activity may mediate neurodegeneration in these forms of PD (94). Of the six patients found to have non-synonymous variants in the COR domain, two had first-degree relatives with dementia, one had a second-degree relative with PD, and one had two sisters with PD diagnosed at a very young age (20 and 30 years old) (see Table 4).
Methodological issues, such as size and composition of the sample (i.e., number of familial and sporadic cases), might explain the variations seen in the frequency of LRRK2 mutations in case series from similar countries. However, there is a clear difference established among geographical regions, where North African Arabs (95), Ashkenazy-Jews (96) and certain Europeans cohorts (97–99) might report a higher prevalence than Latin American and Asian populations for these mutations (15, 100, 101).
Structural Analysis of LRRK2 Pathogenic Mutations
The non-synonymous missense mutations described here are all found in the COR domain of Lrrk2. To gain insight into how these mutations may affect the function of Lrrk2, we investigated their locations in the structure of Lrrk2. The high-resolution cryoelectron microscopy (cryoEM) structure of the C-terminal domains of Lrrk2 in different states have recently been reported and shed light on how allosteric interactions between different domains regulate microtubule interactions (102). The structure notably shows interactions between the ROC GTPase domain and the COR-B domain, notably involving the pathogenic mutation sites p.Arg1441 and p.Tyr1699 (Figure 1A). These interdomain interactions enable the kinase activity to be regulated by GTP binding to the ROC domain. The mutations described here, found in the segment a.a. 1619–1623, are all located in a loop of the COR-A domain. This loop, which spans a.a. 1613–1624, is disordered in the cryoEM structure, and thus, no atomic resolution model is available for that segment (Figure 1A). It is therefore not possible to gain detailed insights into the effect of each individual missense mutation.
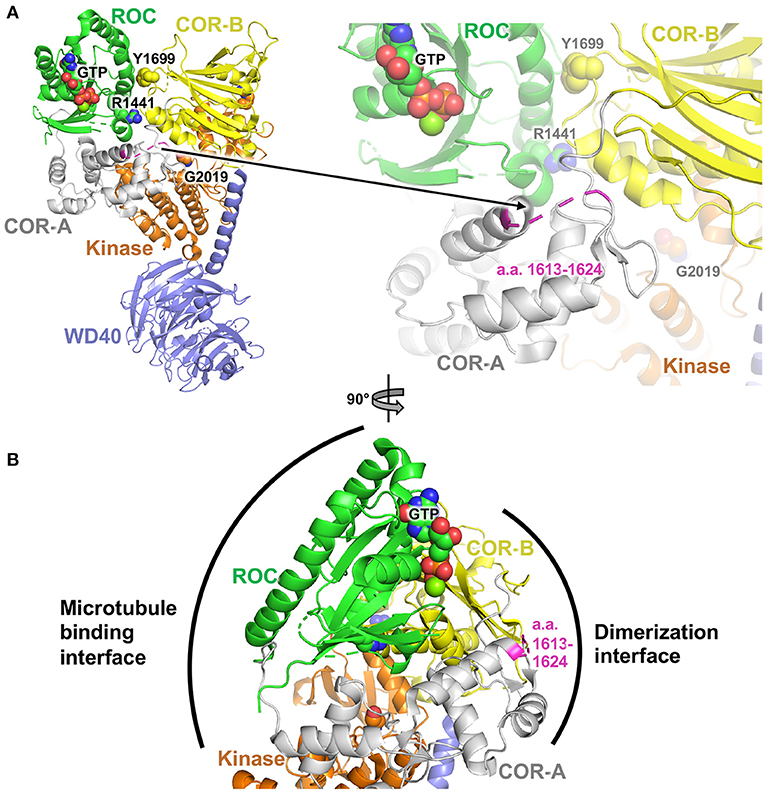
Figure 1. Structural analysis of PD variants in LRRK2. (A) Cryoelectron microscopy structure of Lrrk2 C-terminal domains, PDB code 6vp6 (102). Parkinson-linked missense mutation sites R1441, Y1699, and G2019 are shown as spheres. The loop spanning a.a. 1612–1624 in the COR-A domain is shown in magenta. (B) Rotated view (90°) of the structure in (A), showing the proposed dimerization and microtubule binding interfaces, based on integrative modeling of Lrrk2 filaments bound to microtubules (102).
However, integrative modeling, based on cryoelectron tomography (cryoET) data collected from in situ and in vitro-reconstituted Lrrk2 filaments bound to microtubules, shows how the different domains of Lrrk2 dimerize and associate with microtubules (102, 103). Dimerization is mediated via two sites through reciprocal interactions: one involving WD40–WD40 interactions and another one involving COR–COR interactions. These interactions enable Lrrk2 C-terminal domains to form extended oligomeric filaments that form a helix around the microtubule. Of particular interest here, the COR–COR dimerization interface involves both the COR-A and COR-B domains, with the loop containing a.a. 1613–1624 at the center of this interface (Figure 1B). Mutations in this loop may thus affect dimerization. Given that the kinase activity and conformation affect the ability of Lrrk2 to dimerize through the COR domain via allosteric interactions, it is possible that mutations in the COR-A loop in turn affect the kinase activity. Further experiments would be required to determine how the mutations described here affect the kinase, dimerization, and microtubule-binding activity of Lrrk2.
Limitations
Genome analysis from Mestizo populations in Latin America has previously shown in Costa Rica a European, Native American, and African admixture of 66.7, 28.7, and 4.6%, respectively (104). Therefore, we would have expected to observe a higher frequency of mutations, similar to other European series reported. However, our sample size is small and is more representative of the metropolitan area where most of the patients were recruited, thus warranting in the future a more comprehensive study involving a wider and more representative population of the whole country, particularly including more patients from the non-metropolitan and coastal zones. Moreover, the purpose of our study was to serve as an exploratory analysis in this population, which had not been studied before; likewise, we opted to cover as many genes as possible. We are aware that the sample size is limited, yet underrepresented populations with limited funding and resources that struggle to achieve large sample sizes should be studied and reported as well.
We did not gather information concerning protective and risk factors for subjects in the control group, therefore, we were not able to compare and discuss the frequency of these factors between cases and controls.
Conclusions
This is the first study that reports on sociodemographics, risk factors, clinical presentation, and genetics of Costa Rican patients with PD. We observed a high frequency of exposure to both risk factors (pesticides, herbicides, non-potable water, and low education) and protective factors (tobacco and coffee intake). Regular physical activity significantly correlated with better UPDRS scores despite years of evolution of the disease. Increased years of education were significantly associated with better MoCA test scores, whereas the presence of hallucinations, falls, and mood disorders correlated with a worse performance in the MoCA test. Interestingly, coffee consumption also correlated significantly with worse MoCA test scoring.
We did not find an association between any of the tested familial PD and atypical parkinsonism-associated genes, including GBA, SNCA, VPS35, LRRK2, GCH1, PRKN, PINK1, DJ-1, VPS13C, and ATP13A2, and PD. We also did not identify any homozygous or compound heterozygous pathogenic variants in any of these genes. Rare variants in LRRK2 were nominally associated with PD, with six of these rare non-synonymous variants all located in the COR domain of LRRK2. One PD patient carried a pathogenic GCH1 variant, p.K224R, further emphasizing the role of this gene in PD.
Data Availability Statement
The data presented in the study are deposited in the NIH-dbGAP repository, accession number phs002495.v1.p1 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002495.v1.p1).
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the University of Costa Rica (VI-3668-2014 and final report 837-B5-302). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
GT-A and EY conceptualized the report and made substantial contributions to the design, drafting, and revision of the work. J-FT performed in silico structural analysis and contributed to the discussion of these results. TL-P, JR-M, AG-P, ZG-O, KC-C, IF-M, and JF-T significantly contributed to drafting and critically reviewing the paper. All authors have contributed to the work and agree with the presented findings and that the work has not been published before nor is being considered for publication in another journal. All authors approved the final version of the manuscript and assume accountabilities for all aspects of the work.
Funding
This work was supported by the Research Fund from the Vicerrectoria de Investigacion of the Universidad de Costa Rica. The genetic analysis was supported by the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), Parkinson Canada, and the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program. ZG-O is supported by the Fonds de recherche du Québec–Santé Chercheur-Boursier award and is a Parkinson Canada New Investigator awardee.
Conflict of Interest
ZG-O received consultancy fees from Lysosomal Therapeutics Inc. (LTI), Idorsia, Prevail Therapeutics, Inceptions Sciences (now Ventus), Ono Therapeutics, Denali, Handl Therapeutics, Neuron23, and Deerfield.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Jennifer Ruskey, Sandra Laurent, Lynne Krohn, Uladzislau Rudakou, D. Rochefort, H. Catoire, V. Zaharieva, and Dr. Ellen Sylvie Sánchez-Más for their assistance. This article has already been published as preprint under medRxiv server (105).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.656342/full#supplementary-material
References
1. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:17013. doi: 10.1038/nrdp.2017.13
2. Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB, et al. Update of the mDS research criteria for prodromal parkinson's disease. Mov Disord. (2019) 34:1464–70. doi: 10.1002/mds.27802
3. Taylor AE, Saint-Cyr JA, Lang AE. Frontal lobe dysfunction in parkinson's disease. The cortical focus of neostriatal outflow. Brain. (1986) 109:845–83. doi: 10.1093/brain/109.5.845
4. Cools R, Barker RA, Sahakian BJ, Robbins TW. Mechanisms of cognitive set flexibility in parkinson's disease. Brain. (2001) 124:2503–12. doi: 10.1093/brain/124.12.2503
5. Owen AM, James M, Leigh PN, Summers BA, Marsden CD, Quinn NP, et al. Fronto-striatal cognitive deficits at different stages of parkinson's disease. Brain. (1992) 115:1727–51. doi: 10.1093/brain/115.6.1727
6. Cronin-Golomb A, Braun AE. Visuospatial dysfunction and problem solving in parkinson's disease. Neuropsychology. (1997) 11:44–52. doi: 10.1037/0894-4105.11.1.44
7. Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, et al. Meta-analysis of early nonmotor features and risk factors for parkinson disease. Ann Neurol. (2012) 72:893–901. doi: 10.1002/ana.23687
8. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
9. Bonifati V. Genetics of parkinson's disease–state of the art, 2013. Parkinsonism Relat Disord. (2014) 1:S23–8. doi: 10.1016/S1353-8020(13)70009-9
10. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. (2019) 18:1091–102. doi: 10.1016/S1474-4422(19)30320-5
11. NGan-Or Z, NLiong C, NAlcalay RN. GBA-Associated parkinson's disease and other synucleinopathies. Curr Neurol Neurosci Rep. (2018) 18:44. doi: 10.1007/s11910-018-0860-4
12. Ben-Joseph A, Marshall CR, Lees AJ, Noyce AJ. Ethnic variation in the manifestation of parkinson's disease: a narrative review. J Parkinsons Dis. (2020) 10:31–45. doi: 10.3233/JPD-191763
13. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. (2016) 538:161–4. doi: 10.1038/538161a
14. Mata IF, Cosentino C, Marca V, Torres L, Mazzetti P, Ortega O, et al. LRRK2 mutations in patients with parkinson's disease from peru and uruguay. Parkinsonism Relat Disord. (2009) 15:370–3. doi: 10.1016/j.parkreldis.2008.09.002
15. Cornejo-Olivas M, Torres L, Velit-Salazar MR, Inca-Martinez M, Mazzetti P, Cosentino C, et al. Variable frequency of lRRK2 variants in the latin american research consortium on the genetics of parkinson's disease (LARGE-PD), a case of ancestry. NPJ Parkinsons Dis. (2017) 3:19. doi: 10.1038/s41531-017-0020-6
16. Perez-Pastene C, Cobb SA, Diaz-Grez F, Hulihan MM, Miranda M, Venegas P, et al. Lrrk2 mutations in south america: a study of chilean parkinson's disease. Neurosci Lett. (2007) 422:193–7. doi: 10.1016/j.neulet.2007.06.021
17. Pimentel MM, Moura KC, Abdalla CB, Pereira JS, De Rosso AL, Nicaretta DH, et al. A study of lRRK2 mutations and parkinson's disease in brazil. Neurosci Lett. (2008) 433:17–21. doi: 10.1016/j.neulet.2007.12.033
18. Yescas P, Lopez M, Monroy N, Boll MC, Rodriguez-Violante M, Rodriguez U, et al. Low frequency of common lRRK2 mutations in mexican patients with parkinson's disease. Neurosci Lett. (2010) 485:79–82. doi: 10.1016/j.neulet.2010.08.029
19. Gatto EM, Parisi V, Converso DP, Poderoso JJ, Carreras MC, Marti-Masso JF, et al. The lRRK2 g2019S mutation in a series of argentinean patients with parkinson's disease: clinical and demographic characteristics. Neurosci Lett. (2013) 537:1–5. doi: 10.1016/j.neulet.2013.01.011
20. Zabetian CP, Mata IF Latin American Research Consortium on the Genetics of PD (LARGE-PD). LARGE-PD: examining the genetics of parkinson's disease in latin america. Mov Disord. (2017) 32:1330–1. doi: 10.1002/mds.27081
21. Loesch D, Horimoto ARVR, Heilbron K, Sarihan EI, Inca-Martinez M, Mason E, et al. Characterizing the genetic architecture of parkinson's disease in latinos. medRxiv [preprint]. (2020). doi: 10.1101/2020.11.09.20227124
22. Sarihan EI, Pérez-Palma E, Niestroj LM, Loesch D, Inca-Martinez M, Horimoto ARVR, et al. Genome-wide analysis of copy number variation in latin american parkinson's disease patients. Mov. Disord. (2021) 36:434–41. doi: 10.1002/mds.28353
23. Tipton PW, Soto-Beasley AI, Walton RL, Soler-Rangel S, Romero-Osorio Ó, Díaz C, et al. Prevalence of gBA p.K198E mutation in colombian and hispanic populations. Parkinsonism Relat Disord. (2020) 73:16–18. doi: 10.1016/j.parkreldis.2020.03.008
24. Dos Santos AV, Pestana CP, Diniz K. R. D. S., Campos M, Abdalla-Carvalho CB, de Rossob ALZ, et al. Mutational analysis of gIGYF2, aTP13A2 and gBA genes in brazilian patients with early-onset parkinson's disease. Neurosci Lett. (2010) 485:121–4. doi: 10.1016/j.neulet.2010.08.083
25. González-Del Rincón MDL, Monroy Jaramillo N, Suárez Martínez A, Yescas Gómez P, Boll Woehrlen M, López López M, et al. The l444P gBA mutation is associated with early-onset parkinson's disease in mexican mestizos. Clin Genet. (2013) 84:386–7. doi: 10.1111/cge.12084
26. Abreu GM, Valença DC, Campos MJ, Da Silva CP, Pereira JS, Araujo Leite MA, et al. Autosomal dominant parkinson's disease: incidence of mutations in lRRK2, sNCA, vPS35 and gBA genes in brazil. Neurosci Lett. (2016) 635:67–70. doi: 10.1016/j.neulet.2016.10.040
27. Velez-Pardo C, Lorenzo-Betancor O, Jimenez-Del-Rio M, Moreno S, Lopera F, Cornejo-Olivas M, et al. The distribution and risk effect of gBA variants in a large cohort of pD patients from colombia and peru. Parkinsonism Relat Disord. (2019) 63:204–8. doi: 10.1016/j.parkreldis.2019.01.030
28. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
29. Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of lewy body parkinson's disease. Neurology. (2001) 57:1497–9. doi: 10.1212/WNL.57.8.1497
30. Marsili L, Rizzo G, Colosimo C. Diagnostic criteria for parkinson's disease: from james parkinson to the concept of prodromal disease. Front Neurol. (2018) 9:156. doi: 10.3389/fneur.2018.00156
31. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for parkinson's disease. Mov Disord. (2015) 30:1591–601. doi: 10.1002/mds.26424
32. Ross JP, Dupre N, Dauvilliers Y, Strong S, Ambalavanan A, Spiegelman D, et al. Analysis of dNAJC13 mutations in french-Canadian/French cohort of parkinson's disease. Neurobiol Aging. (2016) 45:213–217. doi: 10.1016/j.neurobiolaging.2016.04.023
33. Li H, Durbin R. Fast and accurate short read alignment with burrows–Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
34. Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapReduce framework for analyzing next-generation dNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
35. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
36. Ruskey JA, Greenbaum L, Roncière L, Alam A, Spiegelman D, Liong C, et al. Increased yield of full gBA sequencing in ashkenazi jews with parkinson's disease. Eur J Med Genet. (2019) 62:65–9. doi: 10.1016/j.ejmg.2018.05.005
37. Yang X, Xu Y. Mutations in the ATP13A2 gene and parkinsonism: a Preliminary review. BioMed Res Int. (2014) 2014:371256. doi: 10.1155/2014/371256
38. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, Van Der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new parkinson's disease risk loci. Nat Genet. (2017) 49:1511–6. doi: 10.1038/ng.3955
39. Mufti K, Rudakou U, Yu E, Krohn L, Ruskey JA, Asayesh F, et al. Comprehensive analysis of familial parkinsonism genes in rapid-eye-movement sleep behavior disorder. Mov Disord. (2021) 36:235–40. doi: 10.1002/mds.28318
40. Rudakou U, Yu E, Krohn L, Ruskey JA, Asayesh F, Dauvilliers Y, et al. Targeted sequencing of parkinson's disease loci genes highlights sYT11, fGF20 and other associations. Brain. (2021) 144:462–72. doi: 10.1093/brain/awaa401
41. Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. (2012) 91:224–37. doi: 10.1016/j.ajhg.2012.06.007
42. Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. (2015) 25:305–15. doi: 10.1101/gr.183483.114
43. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. (2011) 473:43–9. doi: 10.1038/nature09906
44. GBD 2016 Parkinson's Disease Collaborators. Global, regional, and national burden of Parkinson's disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018) 17:939–53. doi: 10.1016/S1474-4422(18)30295-3
45. Abbas MM, Xu Z, Tan LCS. Epidemiology of parkinson's disease-East versus west. Mov Disord Clin Pract. (2017) 5:14–28. doi: 10.1002/mdc3.12568
46. Nicoletti A, Sofia V, Bartoloni A, Bartalesi F, Gamboa Barahon H, Giuffrida S, et al. Prevalence of parkinson's disease: a door-to-door survey in rural bolivia. Parkinsonism Relat Disord. (2003) 10:19–21. doi: 10.1016/S1353-8020(03)00066-X
47. Melcon MO, Anderson DW, Vergara RH, Rocca WA. Prevalence of parkinson's disease in junin, buenos aires province, argentina. Mov Disord. (1997) 12:197–205. doi: 10.1002/mds.870120210
48. Barbosa MT, Caramelli P, Maia DP, Cunningham MC, Guerra HL, Lima-Costa MF, et al. Parkinsonism and parkinson's disease in the elderly: a community-based survey in brazil (the bambui study). Mov Disord. (2006) 21:800–8. doi: 10.1002/mds.20806
49. Viñes JJ, Larumbe R, Gaminde I, Artázcoz MT. [Incidence of idiopathic and secondary parkinson disease in navarre. Population-based case registry]. Neurologia. (1999) 14:16–22.
50. Mizuno Y. [Progress in the basic and clinical aspects of parkinson's disease]. Rinsho Shinkeigaku. (2004) 44:741–50.
51. Gan-Or Z, Rao T, Leveille E, Degroot C, Chouinard S, Cicchetti F. The quebec parkinson network: a Researcher-Patient matching platform and multimodal biorepository. J Parkinsons Dis. (2020) 10:301–13. doi: 10.3233/JPD-191775
52. Chaudhuri KR, Hu MT, Brooks DJ. Atypical parkinsonism in afro-Caribbean and indian origin immigrants to the uK. Mov Disord. (2000) 15:18–23. doi: 10.1002/1531-8257(200001)15:1<18::AID-MDS1005>3.0.CO;2-Z
53. Yu RL, Wu RM, Chan AY, Mok V, Wu YR, Tilley BC, et al. Cross-Cultural differences of the non-Motor symptoms studied by the traditional chinese version of the international parkinson and movement disorder society- unified parkinson's disease rating scale. Mov Disord Clin Pract. (2017) 4:68–77. doi: 10.1002/mdc3.12349
54. Cheon SM, Ha MS, Park MJ, Kim JW. Nonmotor symptoms of parkinson's disease: prevalence and awareness of patients and families. Parkinsonism Relat Disord. (2008) 14:286–90. doi: 10.1016/j.parkreldis.2007.09.002
55. Li HJ, Zhang MF, Chen MX, Hu AL, Li JB, Zhang B, et al. Validation of the nonmotor symptoms questionnaire for parkinson's disease: results from a chinese pilot study. Int J Neurosci. (2015) 125:929–35. doi: 10.3109/00207454.2014.986573
56. Rodríguez-Violante M, Cervantes-Arriaga A, Villar-Velarde A, Corona T. Relationship between the type and side of motor symptoms with the prevalence of non-motor symptoms in parkinson's disease. Neurología (English Edition). (2011) 26:319–24. doi: 10.1016/S2173-5808(11)70076-1
57. Cosentino C, Nuñez Y, Torres L. Frequency of non-motor symptoms in peruvian patients with parkinson's disease. Arq Neuropsiquiatr. (2013) 71:216–9. doi: 10.1590/0004-282X20130005
58. Romenets SR, Wolfson C, Galatas C, Pelletier A, Altman R, Wadup L, et al. Validation of the non-motor symptoms questionnaire (NMS-Quest). Parkinsonism Relat Disord. (2012) 18:54–8. doi: 10.1016/j.parkreldis.2011.08.013
59. Duncan GW, Khoo TK, Yarnall AJ, O'brien JT, Coleman SY, Brooks DJ, et al. Health-related quality of life in early parkinson's disease: the impact of nonmotor symptoms. Mov Disord. (2014) 29:195–202. doi: 10.1002/mds.25664
60. Darweesh SKL, Wolters FJ, Postuma RB, Stricker BH, Hofman A, Koudstaal PJ, et al. Association between poor cognitive functioning and risk of incident parkinsonism: the rotterdam study. JAMA Neurol. (2017) 74:1431–8. doi: 10.1001/jamaneurol.2017.2248
61. Kierzynka A, Kazmierski R, Kozubski W. Educational level and cognitive impairment in patients with parkinson disease. Neurol Neurochir Pol. (2011) 45:24–31. doi: 10.1016/S0028-3843(14)60056-6
62. Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and parkinson's disease: a metaanalysis. Environ Res. (2001) 86:122–7. doi: 10.1006/enrs.2001.4264
63. Firestone JA, Smith-Weller T, Franklin G, Swanson P, Longstreth WT, Jr, Checkoway H, et al. Pesticides and risk of parkinson disease: a population-based case-control study. Arch Neurol. (2005) 62:91–5. doi: 10.1001/archneur.62.1.91
64. Kasten M, Chade A, Tanner CM. Epidemiology of parkinson's disease. Handb Clin Neurol. (2007) 83:129–51. doi: 10.1016/S0072-9752(07)83006-5
65. Ritz BR, Paul KC, Bronstein JM. Of pesticides and men: a california story of genes and environment in parkinson's disease. Curr Environ Health Rep. (2016) 3:40–52. doi: 10.1007/s40572-016-0083-2
66. Powers KM, Smith-Weller T, Franklin GM, Longstreth WT Jr, Swanson PD, Checkoway H, et al. Parkinson's disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology. (2003) 60:1761–6. doi: 10.1212/01.WNL.0000068021.13945.7F
67. Kaur D, Andersen J. Does cellular iron dysregulation play a causative role in parkinson's disease? Ageing Res Rev. (2004) 3:327–43. doi: 10.1016/j.arr.2004.01.003
68. Ngim CH, Devathasan G. Epidemiologic study on the association between body burden mercury level and idiopathic parkinson's disease. Neuroepidemiology. (1989) 8:128–41. doi: 10.1159/000110175
69. Grandinetti A, Morens DM, Reed D, Maceachern D. Prospective study of cigarette smoking and the risk of developing idiopathic parkinson's disease. Am J Epidemiol. (1994) 139:1129–38. doi: 10.1093/oxfordjournals.aje.a116960
70. Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of parkinson's disease. Ann Neurol. (2002) 52:276–84. doi: 10.1002/ana.10277
71. Ascherio A, Chen H. Caffeinated clues from epidemiology of parkinson's disease. Neurology. (2003) 61:S51–4. doi: 10.1212/01.WNL.0000095213.86899.21
72. Liu R, Guo X, Park Y, Huang X, Sinha R, Freedman ND, et al. Caffeine intake, smoking, and risk of parkinson disease in men and women. Am J Epidemiol. (2012) 175:1200–7. doi: 10.1093/aje/kwr451
73. Palacios N, Gao X, Mccullough ML, Schwarzschild MA, Shah R, Gapstur S, et al. Caffeine and risk of parkinson's disease in a large cohort of men and women. Mov Disord. (2012) 27:1276–82. doi: 10.1002/mds.25076
74. Quik M. Smoking, nicotine and parkinson's disease. Trends Neurosci. (2004) 27:561–8. doi: 10.1016/j.tins.2004.06.008
75. Costa J, Lunet N, Santos C, Santos J, Vaz-Carneiro A. Caffeine exposure and the risk of parkinson's disease: a systematic review and meta-analysis of observational studies. J Alzheimers Dis. (2010) 1:S221–38. doi: 10.3233/JAD-2010-091525
76. Fang X, Han D, Cheng Q, Zhang P, Zhao C, Min J, et al. Association of levels of physical activity with risk of parkinson disease: a Systematic review and meta-analysis. JAMA Netw Open. (2018) 1:e182421. doi: 10.1001/jamanetworkopen.2018.2421
77. Shulman LM, Katzel LI, Ivey FM, Sorkin JD, Favors K, Anderson KE, et al. Randomized clinical trial of 3 types of physical exercise for patients with parkinson disease. JAMA Neurol. (2013) 70:183–90. doi: 10.1001/jamaneurol.2013.646
78. Amara AW, Memon AA. Effects of exercise on non-motor symptoms in parkinson's disease. Clin Ther. (2018) 40:8–15. doi: 10.1016/j.clinthera.2017.11.004
79. Hobson P, Meara J. Risk and incidence of dementia in a cohort of older subjects with parkinson's disease in the united kingdom. Mov Disord. (2004) 19:1043–9. doi: 10.1002/mds.20216
80. Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P. Prevalence and characteristics of dementia in parkinson disease: an 8-year prospective study. Arch Neurol. (2003) 60:387–92. doi: 10.1001/archneur.60.3.387
81. Reid WG, Hely MA, Morris JG, Broe GA, Adena M, Sullivan DJ, et al. A longitudinal of parkinson's disease: clinical and neuropsychological correlates of dementia. J Clin Neurosci. (1996) 3:327–33. doi: 10.1016/S0967-5868(96)90028-4
82. Stern Y, Marder K, Tang MX, Mayeux R. Antecedent clinical features associated with dementia in parkinson's disease. Neurology. (1993) 43:1690–2. doi: 10.1212/WNL.43.9.1690
83. Santos C, Costa J, Santos J, Vaz-Carneiro A, Lunet N. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. (2010) 1:S187–204. doi: 10.3233/JAD-2010-091387
84. Liu QP, Wu YF, Cheng HY, Xia T, Ding H, Wang H, et al. Habitual coffee consumption and risk of cognitive decline/dementia: a systematic review and meta-analysis of prospective cohort studies. Nutrition. (2016) 32:628–36. doi: 10.1016/j.nut.2015.11.015
85. Wu L, Sun D, He Y. Coffee intake and the incident risk of cognitive disorders: a dose-response meta-analysis of nine prospective cohort studies. Clin Nutr. (2017) 36:730–6. doi: 10.1016/j.clnu.2016.05.015
86. Araujo LF, Mirza SS, Bos D, Niessen WJ, Barreto SM, Van Der Lugt A, et al. Association of coffee consumption with mRI markers and cognitive function: a Population-Based study. J Alzheimers Dis. (2016) 53:451–61. doi: 10.3233/JAD-160116
87. Araujo LF, Giatti L, Reis RC, Goulart AC, Schmidt MI, Duncan BB, et al. Inconsistency of association between coffee consumption and cognitive function in adults and elderly in a cross-Sectional study (ELSA-Brasil). Nutrients. (2015) 7:9590–601. doi: 10.3390/nu7115487
88. Zhou A, Taylor AE, Karhunen V, Zhan Y, Rovio SP, Lahti J, et al. Habitual coffee consumption and cognitive function: a mendelian randomization meta-analysis in up to 415,530 participants. Sci Rep. (2018) 8:7526. doi: 10.1038/s41598-018-25919-2
89. Kwok MK, Leung GM, Schooling CM. Habitual coffee consumption and risk of type 2 diabetes, ischemic heart disease, depression and alzheimer's disease: a mendelian randomization study. Sci Rep. (2016) 6:36500. doi: 10.1038/srep36500
90. Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson's disease in gTP cyclohydrolase 1 mutation carriers. Brain. (2014) 137:2480–92. doi: 10.1093/brain/awu179
91. Guella I, Sherman HE, Appel-Cresswell S, Rajput A, Rajput AH, Farrer MJ. Parkinsonism in gTP cyclohydrolase 1 mutation carriers. Brain. (2015) 138:e349. doi: 10.1093/brain/awu341
92. Rudakou U, Ouled Amar Bencheikh B, Ruskey JA, Krohn L, Laurent SB, Spiegelman D, et al. Common and rare gCH1 variants are associated with parkinson's disease. Neurobiol Aging. (2019) 73 231–236. doi: 10.1016/j.neurobiolaging.2018.09.008
93. Bosgraaf L, Van Haastert PJ. Roc, a ras/GTPase domain in complex proteins. Biochim Biophys Acta. (2003) 1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008
94. Tsika E, Moore DJ. Contribution of gTPase activity to lRRK2-associated parkinson disease. Small GTPases. (2013) 4:164–70. doi: 10.4161/sgtp.25130
95. Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, et al. G2019S lRRK2 mutation in french and north african families with parkinson's disease. Ann Neurol. (2005) 58:784–7. doi: 10.1002/ana.20636
96. Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 g2019S as a cause of Parkinson's disease in Ashkenazi jews. N Engl J Med. (2006) 354:424–5. doi: 10.1056/NEJMc055509
97. Infante J, Rodriguez E, Combarros O, Mateo I, Fontalba A, Pascual J, et al. LRRK2 g2019S is a common mutation in spanish patients with late-onset parkinson's disease. Neurosci Lett. (2006) 395:224–6. doi: 10.1016/j.neulet.2005.10.083
98. Gonzalez-Fernandez MC, Lezcano E, Ross OA, Gomez-Esteban JC, Gomez-Busto F, Velasco F, et al. Lrrk2-associated parkinsonism is a major cause of disease in northern spain. Parkinsonism Relat Disord. (2007) 13:509–15. doi: 10.1016/j.parkreldis.2007.04.003
99. Gorostidi A, Ruiz-Martinez J, Lopez De Munain A, Alzualde A, Marti Masso JF. LRRK2 g2019S and r1441G mutations associated with parkinson's disease are common in the basque country, but relative prevalence is determined by ethnicity. Neurogenetics. (2009) 10:157–9. doi: 10.1007/s10048-008-0162-0
100. Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E, et al. The g2019S lRRK2 mutation is uncommon in an asian cohort of parkinson's disease patients. Neurosci Lett. (2005) 384:327–9. doi: 10.1016/j.neulet.2005.04.103
101. Cho JW, Kim SY, Park SS, Kim HJ, Ahn TB, Kim JM, et al. The g2019S lRRK2 mutation is rare in korean patients with parkinson's disease. Can J Neurol Sci. (2007) 34:53–5. doi: 10.1017/S0317167100005783
102. Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, et al. Structure of lRRK2 in parkinson's disease and model for microtubule interaction. Nature. (2020) 588:344–9. doi: 10.1038/s41586-020-2673-2
103. Watanabe R, Buschauer R, Bohning J, Audagnotto M, Lasker K, Lu TW, et al. The in situ structure of parkinson's disease-Linked lRRK2. Cell. (2020) 182:1508–18. doi: 10.1016/j.bpj.2019.11.2690
104. Wang S, Ray N, Rojas W, Parra MV, Bedoya G, Gallo C, et al. Geographic patterns of genome admixture in latin american mestizos. PLoS Genet. (2008) 4:e1000037. doi: 10.1371/journal.pgen.1000037
Keywords: Parkinson's disease, genotype, phenotype, Costa Rica, Latin America
Citation: Torrealba-Acosta G, Yu E, Lobo-Prada T, Ruíz-Martínez J, Gorostidi-Pagola A, Gan-Or Z, Carazo-Céspedes K, Trempe J-F, Mata IF and Fornaguera-Trías J (2021) Clinical and Genetic Analysis of Costa Rican Patients With Parkinson's Disease. Front. Neurol. 12:656342. doi: 10.3389/fneur.2021.656342
Received: 20 January 2021; Accepted: 18 June 2021;
Published: 04 August 2021.
Edited by:
Andrew Anthony Hicks, Eurac Research, ItalyReviewed by:
Mario Reynaldo Cornejo-Olivas, National Institute of Neurological Sciences (INCN), PeruKenya Nishioka, Juntendo University, Japan
Lucía Fernández Cardo, Cardiff University, United Kingdom
Dayne A. Beccano-Kelly, Cardiff University, United Kingdom
Kinya Ishikawa, Tokyo Medical and Dental University, Japan
Copyright © 2021 Torrealba-Acosta, Yu, Lobo-Prada, Ruíz-Martínez, Gorostidi-Pagola, Gan-Or, Carazo-Céspedes, Trempe, Mata and Fornaguera-Trías. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel Torrealba-Acosta, Z2FicmllbC50b3JyZWFsYmFhY29zdGFAYmNtLmVkdQ==
†These authors have contributed equally to this work and share first authorship