 Pu Miao
Pu Miao Siyang Tang
Siyang Tang Jia Ye2†
Jia Ye2† Yuezhou Li
Yuezhou Li Jianhua Feng
Jianhua Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 19 May 2021
Sec. Epilepsy
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.653517
Background: Nav1.2 encoded by the SCN2A gene is a brain-expressed voltage-gated sodium channel known to be associated with neurodevelopment disorders ranging from benign familial neonatal infantile seizures (BFIS) to developmental and epileptic encephalopathy (DEE) and autism spectrum disorder. Interestingly, status epilepticus during slow sleep (ESES), which aggravates cognitive impairment, has been found in SCN2A-related epilepsy. However, the functional features and the relationship between SCN2A and ESES have not been researched.
Method: We herein investigated the functional consequences of an unpublished de novo V911A and the other two published variants in patients with SCN2A-related disorder and ESES by whole-cell patch-clamp studies in transfected HEK293T cells.
Results: The unpublished V911A and published K1933M variants detected in patients with DEE exhibited a profound gain-of-functional (GOF) change. Another published BFIS variant S863F significantly reduced current density as a loss-of-functional (LOF) change. The refractory epilepsy in the patient with V911A was controlled by using the precise treatment of oxcarbazepine (OXC) since the age of 3 months. ESES was found at 18 months during the seizure-free period. We finally chose an aggressive treatment for eliminating ESES by using methylprednisolone combined with levetiracetam and nitrazepam instead of the precise treatment of OXC.
Conclusion: Both GOF and LOF variants in the SCN2A gene can lead to ESES among the phenotypes of DEE and BFIS. We should monitor the electroencephalogram regularly in the patients with SCN2A-related epilepsy even during their seizure-free period.
The SCN2A gene can cause a wide array of phenotypes ranging from benign familial neonatal infantile seizures (BFIS) and developmental and epileptic encephalopathy (DEE) to neurodevelopmental disorders such as autism spectrum disorder (ASD) and intellectual disorder (ID) (1). In vitro functional studies in HEK293 cells revealed that pathogenic variants in the SCN2A gene could lead to both loss-of-functional (LOF) and gain-of-functional (GOF) changes in the Nav1.2 channel. SCN2A-related epilepsy (DEE and BFIS) is associated with different effects on channel function (2), and the mechanism behind this paradox is still unknown. Patients with seizure onset before the age of 3 months are often found with GOF missense variants, and they respond well to sodium channel blockers (SCBs). The specific development-dependent expression patterns of the Nav1.2 channel in myelinated and unmyelinated nerve fibers may contribute to the different ages at seizure onset (3 months) and clinical symptoms (3).
Interestingly, the SCN2A gene has also the second most pathogenic variants associated with electrical status epilepticus during slow-wave sleep (ESES) (4). ESES has a special epileptic discharge pattern that is clinically linked to different epileptic syndromes, such as the Landau–Kleffner syndrome and epileptic encephalopathies with continuous spikes and waves during slow sleep (ECSWS) (5–7). The underlying etiology is complex, including brain malformations, immune disorders, and genetic factors. Genetic factors play an important role in ESES. Multiple genes such as GRIN2A, SCN2A, KCNB1, CNKSR2, KCNQ2, KCNA2, SLC6A1, and WAC (7–14), especially genes of channelopathy, were reported (4). Case reports of SCN2A, KCNB1, KCNQ2, and KCNA2 with ESES were related to infantile epileptic encephalopathies, which underlines that ESES is not a rare electroencephalogram (EEG) pattern among DEE patients.
Seizures related to SCN2A variants are often intractable with the use of multiple antiepileptic drugs (AEDs). However, infants with seizure onset earlier than 3 months of age could be treated precisely with SCBs (15). The paradox is that oxcarbazepine (OXC) and carbamazepine (CBZ) should be avoided when patients are found with ESES, as they may worsen ESES (16). This issue has not been discussed so far even though hundreds of pathogenic variants have been discovered. In this study, we analyzed data from a patient with an unpublished de novo heterozygous variant of V911A in SCN2A gene and ESES pattern of EEG. We studied the functional changes using in vitro experiments. Meanwhile, to further understand the relationship between SCN2A variants and ESES, we then verified the functional changes of the other reported variants with ESES. Our findings outlined the phenotype–funotype of patients with ESES and the pathogenic variants of the SCN2A gene.
All procedures were approved by the Ethics Committee of the Second Affiliated Hospital of School of Medicine, Zhejiang University, China (Ethical code:2018-080). Written informed consent was obtained from the patient's parents for the publication of any potentially identifiable data included in this article.
The cDNA of human Nav1.2α was purchased from Addgene, and human Nav β1 and β2 subunits were generously provided by Dr. S. C. Cannon and Dr. S. G. Waxman. Wild-type (WT) Nav1.2 was subcloned into a pCMV vector. Variants were introduced into pCMV-Nav1.2 using a ClonExpress II One Step Cloning Kit (vazyme).The Nav β1 and β2 subunits were subcloned into pIRES2-EGFP and pIRES2-mCherry vectors, respectively. The ORF of all plasmids were confirmed by sequencing full length before transfection.
HEK293 cells were obtained from American Type Culture Collection (ATCC) and maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco). Expression of hNav1.2 and the accessory β1 and β2 subunits was achieved by using transient transfection with Lipofectamine 2000 (Invitrogen). Electrophysiological recordings of both green and red fluorescent cells were made 24 h after transfection.
Whole-cell voltage-clamp experiments were used to examine the voltage-gated Na+ currents. All voltage-clamp experiments were performed at room temperature. The data were collected using an Axon multiclamp 700B, Digidata1440A (Axon Instruments). Patch pipettes were pulled and fire polished to reach a pipette resistance of 1.1–2.0 MΩ. The solution in the pipette contained 10 mM NaF, 110 mM CsF, 20 mM CsCl, 2 mM EGTA, and 10 mM HEPES with a pH adjusted to 7.35 with CsOH and an osmolarity adjusted to 310 mOsmol/kg with sucrose. The bath solution contained 145 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM d-(+) glucose, and 10 mM HEPES with a pH adjusted to 7.35 with NaOH and an osmolarity adjusted to 310 mOsmol/kg with sucrose. To minimize voltage errors, we focused on data from cells expressing maximal peak Na+ current amplitudes between 1 and 8 nA. We used low-resistance pipettes and 90–95% series resistance compensation. The average series resistance in these cells was 1.8 ± 0.3 MΩ, and the estimated maximum voltage error of the recordings was 1.6 ± 0.4 mV. Cells were held at −120 mV, and sodium currents were evoked by a series of depolarizing pulses (100 ms) to potentials ranging from −80 to +90 mV in steps of 5 mV. Current densities were obtained by dividing the peak currents by the capacitance. The voltage dependence of activation was obtained by plotting the normalized conductance against test potentials with the equation G/Gmax = 1/[1 + exp(V0.5 – V)/k], where the Gmax is the maximum conductance, V0.5 is the half-maximal activation potential, and k is the slope factor. the curve is fitted to a Boltzmann function. The voltage dependence of fast inactivation was assessed by applying a double-pulse protocol: 500 ms pre-pulses were applied from −150 to 0 mV in steps of 10 mV and followed by a test pulse of −10 mV. The steady-state fast-inactivation curve was fitted using the Boltzmann equation (I/Imax = {1 + exp[(V – V0.5)/k]} – 1). Recovery from fast inactivation was assessed by a two-pulse recovery protocol with varying time intervals between a 500 ms inactivating pre-pulse and a test pulse of −10 mV. The time course of recovery from inactivation was fitted with a double-exponential function to generate τ1 and τ2. Data are presented as mean ± SEM. Statistical comparison between more than two groups was performed using one-way ANOVA followed by Bonferroni post-hoc test.
All values were expressed as means ± standard error of mean, and the one-way ANOVA by Bonferroni post-hoc test was used for statistical analysis.
The patient was a 2-year-old boy and the first child of healthy and non-consanguineous parents. He was born at full term with a normal weight of 3,250 g. The delivery was eventful. Two days after birth, he began to display gradually increasing clonic seizures, from 2–3 times per day to 15–20 times per day. Blood test, cerebrospinal fluid biochemical tests, and cranial magnetic resonance imaging (MRI) findings were normal. Tandem mass spectrometry (MS/MS) of urinary and dry blood filter paper showed no significant abnormalities. The video electroencephalography (VEEG) showed a burst-suppression pattern 10 days after birth. The patient was first treated with phenobarbital (PB) and topiramate (TPM) and then with nitrazepam (NZP) and TPM because of intractable epilepsy. When the patient was about 3 months old, the result of a next-generation sequencing panel involving epilepsy-related genes showed a variant of c.2732T>C, p.V911A(NM_001040142). We validated this result with Sanger sequencing and confirmed the de novo type by testing the parents. It was predicted to be a pathogenic variant according to the American College of Medical Genetics and Genomics (ACMG) scoring, and a functional study confirmed that it was a GOF mutation which reinforced its potential pathogenicity. The patient was treated with OXC (25 mg/kg/day) and showed a remarkably good response since then. The VEEG re-examination showed generalized spike-slow waves instead of a burst-suppression pattern. Seizures have not reoccurred to this day. At the age of 1 year and 10 months, the Griffith Mental Development Scales (GMDS) indicated profound developmental delays for all six areas as follows: the patient could not sit well and say any words. During the VEEG re-examination, the patient displayed a spike wave index of 80% during non-rapid eye movement (NREM) sleep and of <10% during wakefulness at the age of 18 months (Figure 1).
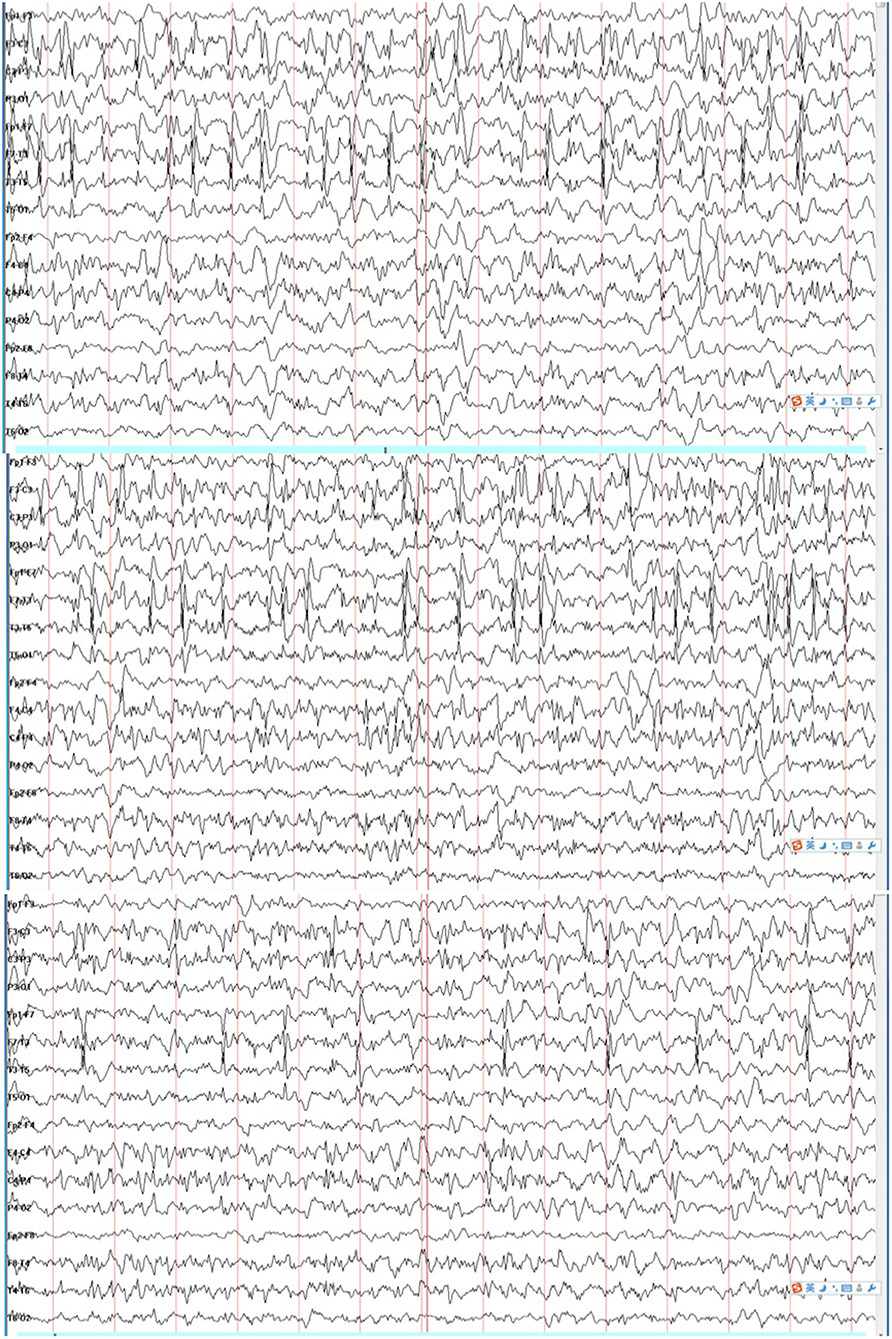
Figure 1. Electroencephalogram (EEG) showed electrical status epilepticus during sleep (ESES).
The clinical examination made a conclusion of epileptic encephalopathy with continuous spikes and waves during slow sleep (CSWS). For patients with GOF mutations in SCN2A, SCBs should be considered as the first-line drug. However, for patients with CSWS, SCBs should be avoided. Ultimately, we considered withdrawing OXC and chose a more active treatment for continuous-wave square-wave (CSWS) with methylprednisolone (20 mg/kg/day for 3 days then 2 mg/kg/day orally for 4 days, three cycles above, then 1–2 mg/kg/day orally and gradually stop for 6 months), combined with levetiracetam (LEV) and NZP. The patient did not experience any seizures until the last follow-up, and the VEEG was normal after 4 months of our therapeutic schedule.
The location of the variants in the Nav1.2 channel is depicted in Figure 2A. The S863F variant is located in S4 of domain II, V911A variant is located in the pore-forming loop of domain II, and the K1933M variant is in the C-terminal domain. We performed whole-cell patch-clamp recordings to determine the functional effects of the Nav1.2 channel variants. All mutated and WT channels were functional when transiently expressed in HEK293 cells. Figure 2B illustrates the representative whole-cell currents evoked by a series of depolarizing test potentials in cells transiently expressing WT, S863F, V911A, and K1933M Nav1.2. The corresponding current density–voltage relationship showed that the current density of V911A (−793.23 ± 68.67 pA/pF) was significantly increased compared to WT (−613.85 ± 38.72 pA/pF, Figure 2C). V911A channels also produced a hyperpolarizing shift in the voltage dependence of the activation curve. Compared to WT channels (−20.59 ± 0.83 mV), the half-maximal activation potential (V0.5) of V911A (−24.8 ± 0.91 mV) shifted by 4 mV in a hyperpolarized direction (Figure 2D). Moreover, the fast-inactivation recovery time constant τ1 obtained for V911A (1.04 ± 0.08 ms) was smaller than for WT channels (1.42 ± 0.11 ms) (Figure 2F and Table 1). The current density and the voltage dependence of activation of K1933M variants were not significantly changed compared to WT (Figures 2C,D). However, the fast-inactivation curve of K1933M channels exhibited a ~4 mV depolarizing shift of V0.5 (−56.10 ± 1.58 mV) as compared to WT (−59.67 ± 1.65 mV) (Figure 2E). Furthermore, K1933M also displayed a faster recovery from fast inactivation than WT, as indicated by the smaller fast time constant τ1 (WT, 1.42 ± 0.11 ms; K1933M, 1.07 ± 0.07 ms) (Figure 2F and Table 1). These results showed that V911A and K1933M variants induced GOF changes. On the other hand, S863F shifted the fast-inactivation curve to more negative potentials (−65.08 ± 0.95 mV), slowed the recovery from fast inactivation (τ1 = 1.85 ± 0.08 ms), and significantly reduced current density (−305.53 ± 65.15 pA/pF), indicated S863F was a LOF mutant.
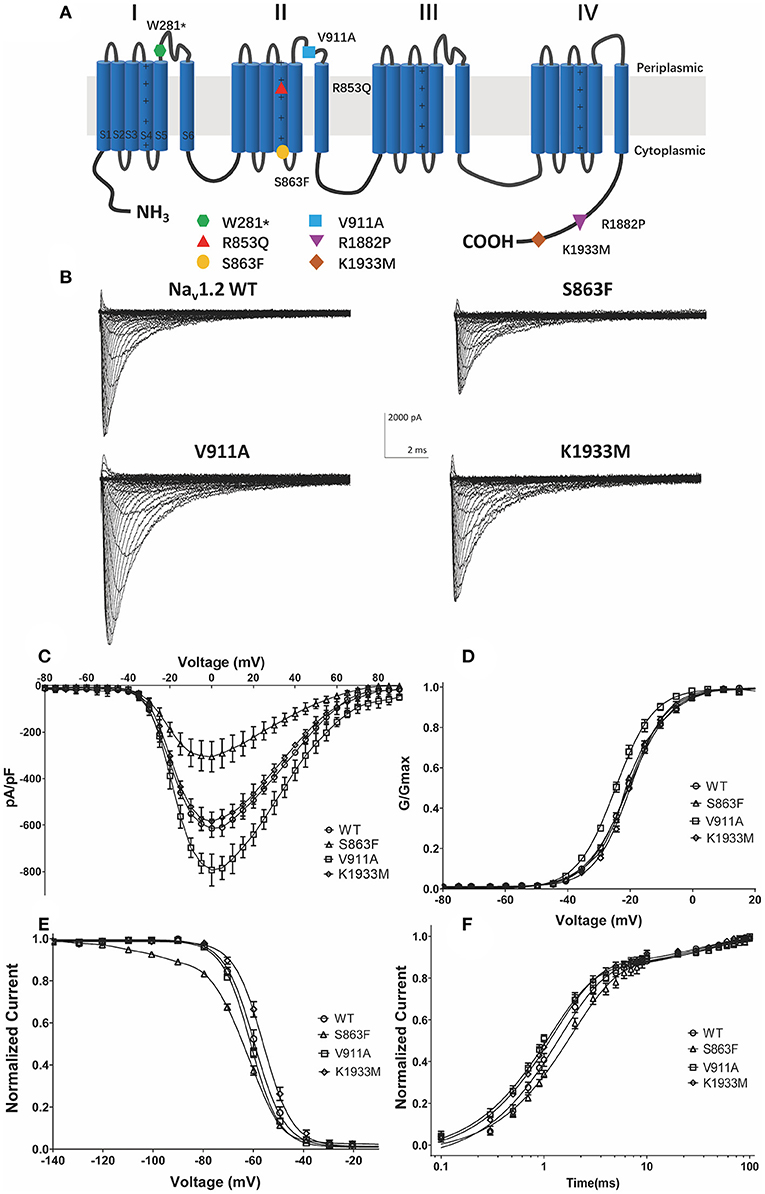
Figure 2. Functional studies reveal gain-of-function changes for p.(Val911Ala, V911A), p.(Lys1933Met, K1933M) variants, and loss-of-function changes for p.(Ser863Phe, S863F). (A) Transmembrane topology of the human Nav1.2 showing the location of the three functionally studied variants. (B) Representative whole-cell patch-clamp traces of voltage-dependent currents recorded from HEK293 cells transfected with either Nav1.2 wild-type or mutant channels (C). Normalized I–V curves of peak sodium current density (in pA/pF) vs. voltage for WT and mutations. The current density of V911A mutant was significantly increased (*p < 0.05), and S863F mutant was significantly reduced (**p < 0.01) compared to WT (D). Voltage dependence of channel activation. V911A revealed a significant hyperpolarizing shift of the activation curve compared to WT (**p < 0.01) (E). Voltage dependence of fast inactivation. K1933M revealed a depolarizing shift, and S863F revealed a polarizing shift in the voltage dependence fast inactivation compared to WT (*p < 0.05) (F). Time-dependent recovery from fast inactivation. V911A and K1933M revealed an accelerated recovery compared to WT; S863F slowed the recovery from fast inactivation (*p < 0.05). All fitting results are listed in Table 1.
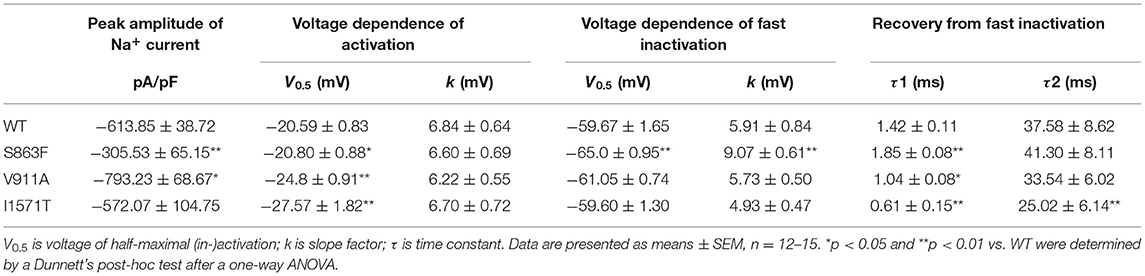
Table 1. Functional characteristics of Nav1.2 channel.
There are six patients with six variants in the SCN2A gene that have been reported to have the ESES phenotype. Combined with our patient, six patients showed DEE, and one showed BFIS. Comprehensive phenotypes of the patients, bioinformatic annotations, and functional data of their variants are presented in Table 2.
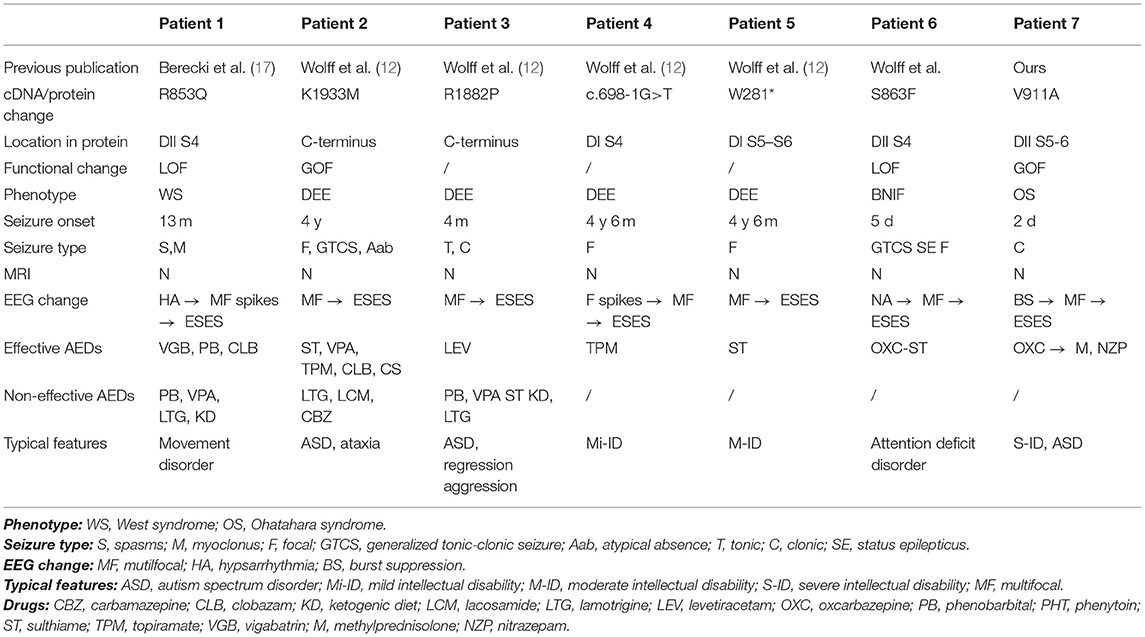
Table 2. Patients' phenotype, molecular features, and functional results.
ESES is a special type of EEG pattern with a prevalence of 0.5%, and structural or genetic abnormalities are the most common etiologies of ESES. GRIN2A, SCN2A, KCNQ2, KCNB1, and KCNA2, which encode significant channels in the brain, contributed to the etiology of ESES solely (4). However, the relationship between phenotype, genotype, and funotype among patients diagnosed as the SCN2A-related disorder and ESES has not been studied.
In addition to the six reported cases, we describe the seventh patient harboring a new de novo missense pathogenic variant in the SCN2A gene with ESES (12). We selected our patient's V911A variant and the published K1933M variant with DEE, as well as the S863F variant with BFIS for the further study of genotype–phenotype–funotype correlation. All these three variants from patients with SCN2A-related epilepsy and an ESES pattern here showed distinct functional changes. A recent study found GOF variants R230 and R227 in KCNQ3 that showed multifocal status epilepticus during sleep (18). However, in our study, both DEE and BFIS phenotypes and both GOF and LOF variants in the SCN2A gene can combine with ESES.
Most functional studies revealed that variants detected in neonatal and early infantile epilepsies, including some DEE cases and almost all BFIE cases, lead to hyperexcitability of sodium channel activity (19). In our study, S863F was found in a patient with BFIS who had seizure onset at 5 days after birth, while the electrophysiological result of S863F exhibited LOF effects. S863F owns its particularity, and it is the only reported variant in SCN2A BFIS with ESES (12). R853Q, also in dominant II(S4), was detected in a patient with DEE and ESES, and a published study recognized it as another LOF variant (17). Protein truncating variant W281* and splice variant C.698-1G>T, the other published variants in SCN2A with ESES, always lead to decreased neuronal excitability. These four LOF variants in SCN2A combine with the ESES phenotype. The detailed knowledge of basic phenotype mechanisms behind variant characteristics is obscure. Recent studies reinforced the understanding of epilepsy as a network disease (20, 21). Researchers have discovered that an impaired cortico-striatal excitatory transmission can trigger epilepsy among SCN2A haplodeficient mice. This shows that the networks are also crucial to the pathophysiology and genetic etiology of epilepsy (22). The overlap of the cortico-striatal-thalamus network between ESES (23) and SCN2A might explain the reason why LOF variants in the SCN2A gene showed ESES.
The major goal of ESES syndrome therapy should be to prevent or reduce associated cognitive and neurodevelopmental deficits. Age at onset, duration of ESES, etiology, and response to treatment are the possible predictors of outcomes (24, 25). The patient with the unpublished GOF V911A de novo variant displayed a typical phenotype–genotype–funotype, which at first led to no confusion about the use of SCBs as a valid treatment option. A patient's motor and cognitive abilities may worsen with the use of SCBs during an ESES pattern. Finding an effective treatment for these patients is an important and complex issue. OXC and CBZ should be avoided as they may worsen ESES (26). The contradiction was highlighted when the patients treated with OXC experienced aggravation of ESES symptoms at their final control. According to this, OXC therapy is not the best option for patients with ESES as it can cause persistent damage and worsen symptoms and foreseeable improvement of cognitive functions after remission. The treatment of ESES in SCN2A DEE patients may be more challenging for some patients as an early stop of the onset of ESES was a significant predictor of treatment success in all analyses (24, 27, 28). Steroids and surgery are the most effective treatments for ESES. We chose steroids initially, as our patient did not have a surgical indication. Two months after treatment with steroids, the patient's EEG did not show any improvements. We added levetiracetam and benzodiazepines actively, and 3 months after stopping OXC, the patient had remission of ESES. Therefore, for DEE patients with ESES, the treatment should be more active and intensive to shorten the duration of ESES.
ESES is found in patients expressing the GOF or LOF variants of SCN2A. This indicates that we should monitor EEG, especially among seizure-free patients. For patients with GOF variants, we should reinforce a treatment combining methylprednisolone and benzodiazepines to decrease ESES symptoms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: NCBI BioProject, PRJNA716238.
The studies involving human participants were reviewed and approved by the Second Affiliated Hospital of Zhejiang University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
PM and JF designed the experiments. JT supplied and did the EEG test and CZ followed up the patient. ST and JY carried out experiments. YL and ST analyzed the experimental results. PM and JW analyzed the experimental data. PM, ST, and JY wrote the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by grants from the National Natural Science Foundation of China (31871060 and 81527901).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Cannon (University of California Los Angeles) and Dr. S. G. Waxman (Yale University School of Medicine) for the gift of NaV-β1 and β2 vector. We are thankful to core facilities of Institute of Neuroscience, core facilities of Zhejiang University School of Medicine, for the technique support.
1. Claire R, Mary DK, Gorman KM. The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol. (2020) 24:117–22. doi: 10.1016/j.ejpn.2019.12.016
2. Ulrike B SH, Stephan L, Lerche H. SCN2A channelopathies: mechanisms and models. Epilepsia. (2019) 12:S68–S76. doi: 10.1111/epi.14731
3. Brunklaus A, Lal D. Sodium channel epilepsies and neurodevelopmental disorders: from disease mechanisms to clinical application. Dev Med Child Neurol. (2020) 62:784–92. doi: 10.1111/dmcn.14519
4. Kessi M, Peng J, Yang L, Xiong J, Duan HL, Pang N, et al. Genetic etiologies of the electrical status epilepticus during slow wave sleep: systematic review. BMC Genetics. (2018) 19:40. doi: 10.1186/s12863-018-0628-5
5. Rubboli G, Tassinari CA. Linking epilepsy, sleep disruption and cognitive impairment in Encephalopathy related to Status Epilepticus during slow Sleep (ESES). Epileptic Disord. (2019) 21:1–2. doi: 10.1684/epd.2019.1049
6. Gaetan L, Gabrielle R, Audrey L, Edouard H, Alexis A, Pierre G, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. (2012) 53:1526–38. doi: 10.1111/j.1528-1167.2012.03559.x
7. Gemma LC, Brigid MR, Simone CY, Brian JO, Natalia L, Nadine B, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. (2013) 45:1073–6. doi: 10.10382/ng.2727
8. Carolien GF, Steffen S, Eva HB, Nienke V, Bronwyn K, Holly D, et al. Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. (2017) 74:1228–36. doi: 10.1001/jamaneurol.2017.1714
9. Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, et al. Early-onset epileptic encephalopathy caused by gain-offunction mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. (2015) 35:3782–93. doi: 10.1523/JNEUROSCI.4423-14.2015
10. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotypephenotype associations of KCNA2-related encephalopathies. Brain. (2017) 140:2337–54. doi: 10.1093/brain/awx184
11. Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki AE, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. (2018) 59:389–402. doi: 10.1111/epi.13986
12. Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. (2017) 140:1316–36. doi: 10.1093/brain/awx054
13. Bonardi CM, Mignot C, Serratosa JM, Giraldez BG, Moretti R, Rudolf G, et al. Expanding the clinical and EEG spectrum of CNKSR2-related encephalopathy with status epilepticus during slow sleep (ESES). Clin Neurophysiol. (2020) 131:1030–9. doi: 10.1016/j.clinph.2020.01.020
14. Leonardi E, Bellini M, Aspromonte MC, Giraldez BG, Moretti R, Rudolf G, et al. A novel WAC loss of function mutation in an individual presenting with encephalopathy related to status epilepticus during sleep (ESES). Genes (Basel). (2020) 11:344. doi: 10.3390/genes11030344
15. Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. (2018) 41:442–56. doi: 10.1016/j.tins.2018.03.011
16. van den Munckhof B, van Dee V, Sagi L, Caraballo RH, Veggiotti P, Liukkonen E, et al. Treatment of electrical status epilepticus in sleep: a pooled analysis of 575 cases. Epilepsia. (2015) 56:1738–46. doi: 10.1111/epi.13128
17. Berecki G, Howell K, Deerasooriya Y, Cilio MR, Oliva MK, Kaplan D, et al. Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc Natl Acad Sci U S A. (2018) 115:E5516–E25. doi: 10.1073/pnas.1800077115
18. Sands TT, Miceli F, Lesca G, Beck AE, Sadleir LG, Arrington DK, et al. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann Neurol. (2019) 86:181–92. doi: 10.1002/ana.25522
19. Abela L, Plecko B, Kroell-Seger J, Baumer A, Yamakawa K, Inoue Y, et al. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol Med. (2019) 25:6. doi: 10.1186/s10020-019-0073-6
20. Lagarde S, Roehri N, Lambert I, Trebuchon A, McGonigal A, Carron R, et al. Interictal stereotactic-EEG functional connectivity in refractory focal epilepsies. Brain. (2018) 141:2966–80. doi: 10.1093/brain/awy214
21. Shih JJ. It's all about the networks. Epilepsy Curr. (2019) 19:165–7. doi: 10.1177/1535759719843301
22. Miyamoto H, Tatsukawa T, Shimohata A, Yamagata T, Suzuki T, Amano K, et al. Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat Commun. (2019) 10:1917. doi: 10.1038/s41467-019-09954-9
23. Tassinari CA, Rubboli G. Encephalopathy related to Status Epilepticus during slow Sleep-current concepts and future directions. Epileptic Disord. (2019) 21:S82–S7. doi: 10.1684/epd.2019.1062
24. Kramer U, Sagi L, Goldberg-Stern H, Zelnik N, Nissenkorn A, Ben-Zeev B. Clinical spectrum and medical treatment of children with electrical status epilepticus in sleep (ESES). Epilepsia. (2009) 50:1517–24. doi: 10.1111/j.1528-1167.2008.01891.x
25. Seegmüller C, Deonna T, Dubois CM, Valenti-Hirsch MP, Hirsch E, Metz-Lutz MN, et al. Long-term outcome after cognitive and behavioral regression in nonlesional epilepsy with continuous spike-waves during slow-wave sleep. Epilepsia. (2012) 53:1067–76. doi: 10.1111/j.1528-1167.2012.03465.x
26. Prakash K Current status of treatments for children with electrical status in slow-wave sleep (ESES/CSWS). Epilepsy Curr. (2017) 17:214–6. doi: 10.5698/1535-7597.17.4.214
27. Elina L, Elisa KS, Ritva P, Eija G, Maria P, Granström ML. Long-term outcome of 32 children with encephalopathy with status epilepticus during sleep,or ESES syndrome. Epilepsia. (2010) 51:2023–32. doi: 10.1111/j.1528-1167.2010.02578.x
Keywords: SCN2A, developmental epileptic encephalopathy, gain-of-function, treatment, status epilepticus during sleep
Citation: Miao P, Tang S, Ye J, Tang J, Wang J, Zheng C, Li Y and Feng J (2021) Differential Functional Changes of Nav1.2 Channel Causing SCN2A-Related Epilepsy and Status Epilepticus During Slow Sleep. Front. Neurol. 12:653517. doi: 10.3389/fneur.2021.653517
Received: 14 January 2021; Accepted: 24 March 2021;
Published: 19 May 2021.
Edited by:
Eliane Kobayashi, McGill University, CanadaReviewed by:
Gaetan Lesca, Université Claude Bernard Lyon 1, FranceCopyright © 2021 Miao, Tang, Ye, Tang, Wang, Zheng, Li and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuezhou Li, eXVlemhvdS1saUB6anUuZWR1LmNu; Jianhua Feng, aHpoejg3MDgzODg2QHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.