 Oscar B. Alcoreza
Oscar B. Alcoreza Dipan C. Patel
Dipan C. Patel Bhanu P. Tewari1
Bhanu P. Tewari1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol. , 22 March 2021
Sec. Epilepsy
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.652159
This article is part of the Research Topic Glial Dysfunction in Epileptogenesis View all 16 articles
Given the important functions that glutamate serves in excitatory neurotransmission, understanding the regulation of glutamate in physiological and pathological states is critical to devising novel therapies to treat epilepsy. Exclusive expression of pyruvate carboxylase and glutamine synthetase in astrocytes positions astrocytes as essential regulators of glutamate in the central nervous system (CNS). Additionally, astrocytes can significantly alter the volume of the extracellular space (ECS) in the CNS due to their expression of the bi-directional water channel, aquaporin-4, which are enriched at perivascular endfeet. Rapid ECS shrinkage has been observed following epileptiform activity and can inherently concentrate ions and neurotransmitters including glutamate. This review highlights our emerging knowledge on the various potential contributions of astrocytes to epilepsy, particularly supporting the notion that astrocytes may be involved in seizure initiation via failure of homeostatic responses that lead to increased ambient glutamate. We also review the mechanisms whereby ambient glutamate can influence neuronal excitability, including via generation of the glutamate receptor subunit GluN2B-mediated slow inward currents, as well as indirectly affect neuronal excitability via actions on metabotropic glutamate receptors that can potentiate GluN2B currents and influence neuronal glutamate release probabilities. Additionally, we discuss evidence for upregulation of System , a cystine/glutamate antiporter expressed on astrocytes, in epileptic tissue and changes in expression patterns of glutamate receptors.
The critical roles of astrocytes in supporting the healthy development and maintenance of a mature brain has been firmly established in the last few decades. It is well-appreciated that an imbalance between excitatory and inhibitory neurotransmission causes hyperexcitability in neuronal circuitry and underlies the processes of ictogenesis and epileptogenesis. Given the important functions that glutamate serves in excitatory neurotransmission, understanding the mechanisms regulating glutamatergic drive under physiological and pathological states provides critical insights into devising strategies to maintain glutamate homeostasis. Since 1-in-3 epilepsy patients are pharmacoresistant to currently available antiseizure drugs, that mainly target neuronal mechanisms (1, 2), elucidating the astrocytic processes involved in seizure generation and epileptogenesis in detail may help identify new targets to treat intractable forms of epilepsy.
This review serves to highlight the emerging dynamic processes that astrocytes undergo in epilepsy, in support of the notion that astrocytes play a critical role in seizure generation via homeostatic responses such as the increase in ambient glutamate through a reduction in extracellular space (ECS) following activity-dependent astrocytic potassium uptake and buffering (Figure 1-1). Importantly, we also review the mechanisms in which increased ambient glutamate can directly influence neuronal excitability, via generation of the glutamate receptor subunit GluN2B-mediated slow inward currents (SICs), as well as indirectly affect neuronal excitability via actions on metabotropic glutamate receptors (mGluRs) that can potentiate GluN2B currents, alter extracellular glutamatergic clearance, and influence neuronal glutamate release probabilities (Figure 1-2). Additionally, evidence of upregulation of System (SXC), a cystine/glutamate antiporter expressed on astrocytes, in epileptic tissue and changes in expression patterns of glutamate receptors have provided insights in how astrocytic dysregulation can contribute to seizure generation and epileptogenesis (Figure 1-3).
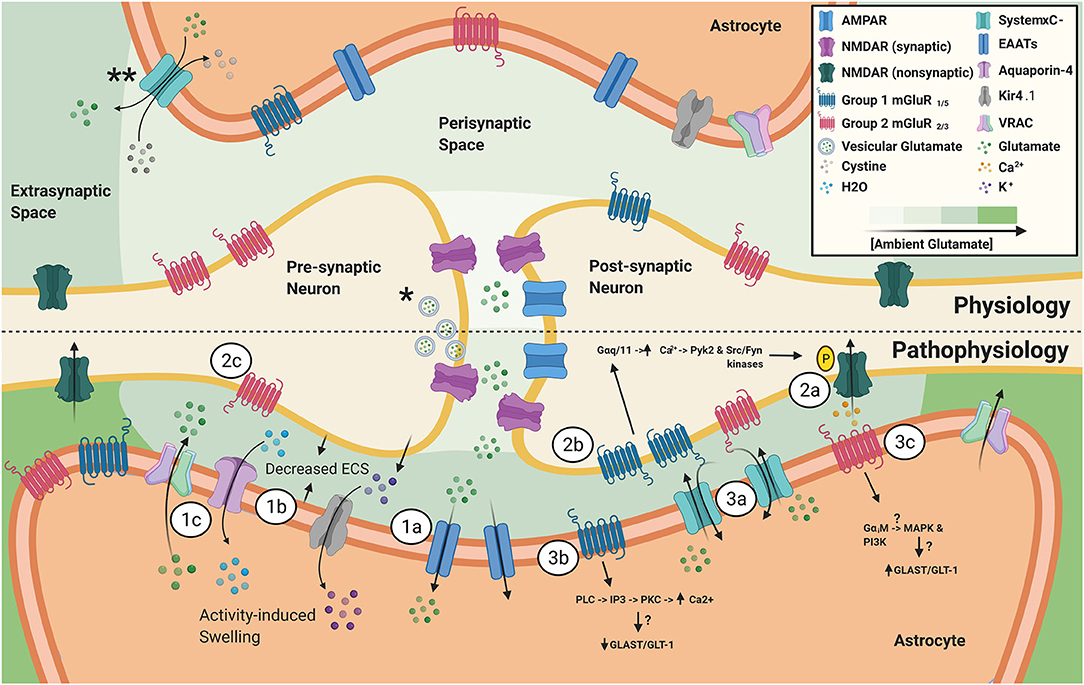
Figure 1. *Vesicular glutamate release during action potentials is the primary source of synaptic glutamate. **SXC, primarily expressed on astrocytes, is a major source of ambient, extrasynaptic glutamate. Ambient glutamate concentration around the synapse, after EAAT activity, follows a gradient with the lowest level in the synaptic cleft to the highest in the extrasynaptic compartment. (1) Astrocyte homeostatic responses to increased activity from hyperexcitable neurons. (1a) Increased vesicular glutamate release from hyperexcitable neurons leads to increased astrocytic EAAT activity. (1b) Elevated neuronal activity also causes release of K+, in attempts to maintain homeostatic neuronal resting membrane potential. Next, astrocytic buffering of extracellular K+ through elevated Kir4.1 activity, which is accompanied by increased H20 uptake through aquaporin-4, ultimately results in activity-induced astrocytic swelling and reduction in ECS. (1c) Astrocytic swelling leads to activation of VRAC and release of glutamate and other gliotransmitters into the ECS. (2) Pathophysiological effects of increased activity and changes in expression of neuronal extrasynaptic glutamate receptors. (2a) Activation of N2B-containing NMDARs leads to the generation of slow, depolarizing currents. (2b) Elevated expression and activity of group 1 mGluRs in epilepsy has been linked to increased NMDAR-mediated currents via a mechanism involving Ca2+-calmodulin dependent tyrosine phosphorylation of NMDAR subunits NR2A/B. (2c) Presynaptic group 2 mGluRs have been shown to inhibit glutamate and GABA release. Tissue from epileptic patients and animal models have revealed decreased mGluR2/3 expression, which can contribute to a pro-epileptic brain state. (3) Changes in astrocytic glutamatergic protein expression in epilepsy. (3a) SXC expression has been found to be elevated in human epileptic tissue, as well as various epilepsy animal models. SXC activity leads to the release of glutamate from astrocytes. (3b) Animal models of epilepsy have revealed that persistent upregulation of astrocytic mGluR5 was a reliable indicator of epileptogenesis. mGluR5 activation leads to altered GLAST/GLT-1 expression and induces NR2B-dependent NMDAR mediated neuronal currents. (3c) Upregulation of mGluR3 has been reported in epilepsy animal models and experimental activation of group 2 mGluRs in cultured astrocytes was shown to upregulate GLAST/GLT-1 expression, suggesting that a balance of group 1 and group 2 mGluRs on astrocytes is important in maintaining homeostatic extracellular glutamate.
Before discussing the mechanisms whereby astrocytes regulate glutamatergic neurotransmission, it is important to understand the fundamental functions astrocytes play in CNS glutamate homeostasis. Glutamate or glutamic acid is a ubiquitous biological molecule serving multiple functions—mainly as an amino acid for protein synthesis, as a principal excitatory neurotransmitter in the mammalian CNS and as a source of energy (3). The use of an abundant amino acid as a neurotransmitter poses a significant challenge in the regulation of available glutamate in the CNS, since excess extracellular glutamate is highly neurotoxic. The evolution of two remarkable features of the biological system—the blood-brain barrier (BBB) and the astrocytic specializations—has solved the problem of potential “spillover” of peripheral glutamate into the CNS as a neurotransmitter. Interestingly, in both cases astrocytes are involved to the extent that glial dysfunctions can alter glutamate homeostasis and cause excitotoxicity and neuronal hyperexcitability.
The BBB is impermeable to glutamate (4), which is critical in preventing the flooding of the CNS by peripheral glutamate within the vasculature. However, this system also necessitates autonomous turnover of glutamate in the brain. The role of astrocytes in de novo synthesis of glutamate in the brain is well-established in literature (5). Astrocytes specializations, namely exclusive expression of pyruvate carboxylase and glutamine synthetase, allow them to serve critical functions in CNS glutamate homeostasis (6, 7). Since pyruvate carboxylase is essential for the synthesis of oxaloacetate that is subsequently utilized in the synthesis of α-ketoglutarate and glutamate, astrocytes are the only cell type in the brain capable of de novo synthesis of glutamate by the oxidative metabolism of glucose. Although one study recently identified another source of glutamate in the brain where glutamate is synthesized by neurons from blood urocanic acid (8), more studies are required to validate it as well as to estimate the relative contribution of neurons in total glutamate synthesis in the brain. Glutamate is subsequently amidated by glutamine synthetase into glutamine, which enters the glutamate-glutamine cycle between astrocytes and neurons.
After release into the synaptic cleft, excessive glutamate must be cleared quickly to prevent neurotoxicity. There are no extracellular enzymes that can neutralize glutamate (9), however, astrocytes can rapidly clear glutamate from the synaptic clefts via astrocytic processes that completely enclose many glutamatergic synapses. These astrocytic processes also express highly efficient excitatory amino acid transporters (10) (EAATs) that take up 80% of extracellular glutamate in the CNS (5) (Figure 1-1a). Once inside the astrocyte, it is estimated that ~85% of glutamate is converted into glutamine and returned to neurons, while the remaining ~15% is metabolized to α-ketoglutarate and further oxidized through the tricyclic acid cycle for energy production. This helps to cover the energy costs of glutamate handling as both pyruvate carboxylase and glutamine synthetase catalyze energy-dependent reactions (3). Insights into the synthesis and regulation of glutamate in the CNS serve to highlight the role that astrocytes play in health and to set up the potential consequences of disrupting astrocytic glutamate homeostasis in the pathology of epilepsy.
Several recent reviews have emphasized the relationship between cerebral edema, astrocytic swelling, and epilepsy, due to a strong link between the volume reduction of the ECS hyperexcitability (11, 12). ECS is a narrow space between CNS cells that serves as a reservoir of water, ions, and signaling molecules to maintain ionic and water homeostasis. Interestingly, ECS reduction is one of the common mechanisms of generating hyperexcitability in several forms of epilepsies and astrocytes play key role in ECS volume regulation.
Astrocytes can significantly alter the volume of the CNS ECS due to their expression of the bi-directional water channel, aquaporin-4 (AQP4), which is enriched at perivascular endfeet (13). Neuronal activity-dependent astrocytic swelling, following potassium uptake and buffering, is an important mediator in the reduction of ECS (Figure 1-1b). Indeed, measurements of the ECS before and during bath application of picrotoxin, a GABAA receptor antagonist, to induce epileptiform activity revealed that brief epileptiform discharges rapidly decreased the median ECS width, measured as the width of interstitial space separating neural structures, by over 50% (14). Rapid ECS shrinkage can inherently concentrate ions and neurotransmitters including glutamate, explaining elevated extracellular glutamate during seizures in epilepsy patients (15). ECS shrinkage due to astrocyte swelling in hypoosmolar conditions has been shown to be sufficient in evoking large excitatory slow inward currents (SICs) in neurons (16). Conversely, studies in AQP4-knockout animals have revealed that these mice have larger ECS, slower potassium kinetics and are more resistant to seizure generation using pentylenetetrazole (PTZ) (17–19), a GABAA receptor antagonist.
Another way astrocytes regulate water homeostasis and consequently ECS volume is the volume-regulated anion channel (VRAC), which are typically activated through hypotonicity-induced cell swelling (20, 21). VRAC belongs to the leucine-rich repeat-containing 8 (LRRC8) family of proteins (22, 23) and consists of a heterogenous mix of LRRC8 proteins of which LRRC8A, also known as Swell1, is an essential subunit (24). Upon astrocytic swelling, VRAC is activated and allows the release of chloride ions and other osmolytes, including glutamate (Figure 1-1c). This in turn generates an osmotic gradient that drives water out of the astrocyte. Using astrocyte specific Swell1 knockout mice and VRAC inhibitors, recent studies have identified that VRAC-mediated glutamate release can modulate NMDAR-mediated tonic currents, as well as affect neuronal excitatory vesicle release probabilities (25, 26).
These processes support the hypothesis that impairment in astrocyte functions may play a critical role in initial seizure generation by increasing physiologically relevant ambient glutamate through (1) a reduction of ECS, following astrocytic potassium uptake and buffering, (2) glutamate release through VRAC or (3) a dysregulated state combining both mechanisms.
Studies investigating the physiological role(s) of ambient glutamate [reviewed in (27)] have identified several diverse functions such as involvement in sleep-wakefulness cycles, synaptic plasticity, and the ability to influence neuronal resting membrane potentials via induction of NMDAR-mediated inward currents. However, the processes that maintain and regulate levels of ambient glutamate continue to be elucidated (27). Another important source of ambient, extracellular glutamate is via the sodium independent activity of the cystine/glutamate antiporter SXC. SXC is a covalently coupled heterodimeric protein complex comprised of the 4F2 heavy chain, SLC3A2, linked to the cystine/glutamate exchanger (xCT), SLC7A11. Notably, while xCT is abundantly expressed in cells throughout the body, examination of SXC knockout mice (xCT−/−) mice revealed that in the CNS, xCT is exclusively expressed in astrocytes, and absent in neurons, oligodendrocytes, and microglia (28). xCT plays a major role in the modulation of ambient extracellular glutamate as pharmacological inhibition of SXC, using (S)-4-carboxyphenylglycine (S-4-CPG), resulted in a 60% reduction in extrasynaptic glutamate in the nucleus accumbens (29). Although S-4-CPG is known to inhibit type 1 mGluRs, this study found that use of the type 1 mGluR antagonist (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) did not result in changes in ambient glutamate. This finding was confirmed recently using antisense xCT which similarly decreased extracellular glutamate in the nucleus accumbens (30). Additionally, xCT−/− mice have around 30% less hippocampal ambient glutamate (31).
In tumor-associated epilepsy, overexpression of SXC in glioblastomas and the corresponding increase in peritumoral glutamate levels have been directly associated with the development of seizures and poor patient survival (32–34). These studies also found that inhibition of SXC via sulfasalazine (SAS), an FDA approved drug used to treat inflammatory bowel disease, reduced epileptiform activity and seizure frequency both in vitro and in vivo in glioblastoma models. In addition to tumor-associated epilepsy, resected human epileptic tissue from patients with temporal lobe epilepsy (TLE) also had an elevated expression of SXC (35) (Figure 1-3a). We have also shown recently that SAS could significantly decrease the frequency and/or amplitude of evoked excitatory postsynaptic currents in multiple in vitro models of hyperexcitability (36). Furthermore, co-application of SAS with topiramate, an FDA-approved anti-seizure drug, further decreased epileptiform activity synergistically compared to topiramate alone.
The role of SXC in neuronal hyperactivity via ambient glutamate regulation is further supported by recent in vivo studies on xCT−/− mice showing delayed epileptogenesis and reduced seizures in the self-sustained status epilepticus (SSSE) and pilocarpine induced status epilepticus models (37). Additionally, this study found decreased micro- and astrogliosis in xCT−/− mice after SSSE. In contrast, wildtype mice that underwent pilocarpine-induced status epilepticus had significantly increased xCT expression during latent phase of epileptogenesis. Using another strain of SXC knockout mice, which have a spontaneous deletion in the xCT gene in subtle gray mice (xCTsut/sut), it was found that xCTsut/sut mice were significantly resistant to epileptic kindling compared to wildtype mice. Additionally, western blot analysis of plasma membrane proteins found that cortical, but not hippocampal, surface GluA1 expression was significantly decreased in xCTsut/sut mice (38).
In contrast to the above studies that show a pivotal role of SXC in hyperexcitability, a recent study using the Theiler's Murine Encephalomyelitis Virus (TMEV) model of viral-induced epilepsy found that xCT−/− mice were not protected against this form of epilepsy and had similar number and severity of behavioral seizures (39). The lack of difference in TMEV-induced seizures between xCT−/− and WT mice can be possibly explained by the fact that the proinflammatory cytokines, tumor necrosis factor-α and interleukin-6, are known as the major drivers of hyperexcitability and seizures in this epilepsy model (40, 41). Taken together, these above cited studies establish SXC as a potential astrocytic drug target. Clearly astrocytes have the capabilities to modulate ambient extracellular glutamate, yet whether astrocytic SXC exerts a direct, pro-epileptic effect by modulating ambient glutamate in acquired epilepsies requires further study.
So far, we have discussed that astrocytes not only prevent excitotoxic glutamate accumulation in the ECS, but also serve as a source of extracellular glutamate under specific conditions. However, important questions remain. How might astroglial glutamate contribute to neuronal activity, and is it sufficient to trigger neuronal hyperactivity? Additionally, how might different types of glutamate receptors at tripartite synapses and extrasynaptic spaces respond to fluctuations in ambient glutamate?
The notion of glial-neuronal crosstalk via gliotransmission has been extensively studied in the last two decades. Regardless of the mechanisms, a wealth of evidence has demonstrated that astrocytes can sense neuronal activity and respond through the release of gliotransmitters including ATP, D-serine, and glutamate (42, 43). Research into the origin and consequences of neuronal SICs revealed that these currents are generated via astrocyte-derived glutamate acting on extrasynaptic GluN2B subunit-containing N-methyl D-aspartate type of glutamate receptors (44, 45) (NMDARs) (Figure 1-2a). Extrasynaptic NMDARs are thought to have increased GluN2B-containing heterodimers, suggesting a spatially specific function. Indeed, GluN2B-containing NMDARs have a higher affinity for glutamate compared to GluN2A-containing NMDARs (46), which may facilitate sensing glutamate in the extrasynaptic space that has far lower extracellular glutamate compared to an active synapse.
SICs can be synchronized in multiple neurons over 100 microns apart in the hippocampus, raising the possibility of astrocytic synchronization of neuronal hyperactivity in epilepsy. Interestingly, blockade of glutamate uptake and exocytotic glutamate release increased the frequency and amplitude of these NMDAR-mediated SICs, suggesting that increases in ambient glutamate could play a physiologically relevant role (45). A study investigating the actions of extrasynaptic glutamate determined that SXC-mediated glutamate release preferentially activates extrasynaptic GluN2B-containing NMDARs (47). Similarly, another study determined that the glutamate responsible for generating SICs occur independently from exocytotic Ca2+-dependent glutamate release (48). Notably, a paper examining the sensitivities of glutamate receptors to glutamate predicted the percentage of glutamate receptors that would remain activated at increasing levels of ambient extracellular glutamate (49). This paper succinctly demonstrates that NMDARs and mGluRs would be preferentially activated by small, local fluctuations in astrocytic glutamate release as they are activated by glutamate concentrations around 1–30 μM, while α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid type of glutamate receptors (AMPARs) are only activated in the presence of 100–3,000 μM of glutamate. Therefore, it is evident that astrocytes possess the molecular machinery necessary to significantly alter the concentration of extrasynaptic ambient glutamate, which can act on NMDARs to induce neuronal SICs that can contribute to hyperexcitability.
Metabotropic glutamate receptors (mGluRs), which are coupled to G protein-coupled pathways and G protein-independent pathways, are another class of glutamatergic receptors whose dysregulation have been implicated in the pathology of epilepsy. Unlike ionotropic glutamate receptors, mGluR activation can trigger long-term changes in cellular signaling by regulating the expressions of various homeostatic and glutamatergic proteins in neurons and glial cells (50). Currently, eight mGluR subtypes have been described that fit into three subgroups (51, 52). Group 1 mGluRs consists of mGluR1 and mGluR5 and are coupled to Gαq/11, which stimulates the release of Ca2+ from intracellular stores upon activation. Group 1 mGluRs are thought to be distributed primarily on post-synaptic neurons, in the perisynaptic zone (53, 54). Using the kainic acid (KA) rat model of TLE, one study has reported upregulation of neuronal mGluR1 in rodent hippocampi, in addition to, mGluR1 upregulation in human TLE tissue (55). Importantly, neuronal mGluR1 activation was shown to potentiate NMDAR-mediated currents via a mechanism involving Ca2+, calmodulin and Src-dependent activation of proline-rich tyrosine kinase leading to tyrosine phosphorylation of NMDAR subunits GluN2A/B (56) (Figure 1-2b). A study investigating the efficacy of mGluR1 inhibition in epilepsy found that mGluR1 inhibition decreased PTZ-induced seizures, and this effect could be prevented by adding mGluR1 agonists (57).
mGluR5 is also expressed on cortical and hippocampal astrocytes, primarily during development (58), however, the reappearance of mGluR5 expression has been observed in astrocytes of specific epilepsy animal models, and human epileptic tissue (59–61). Intriguingly, mice with persistent astrocytic mGluR5 expression during the latent period reliably went on to develop epilepsy, whereas mice with only transient mGluR5 expression did not (59) (Figure 1-3b). Additionally, epileptic mice with astrocytic mGluR5 knocked out displayed lowered glutamate uptake kinetics during high-frequency stimulation compared to epileptic wildtype mice. Downstream effects of astrocytic mGluR5 activation include increased Ca2+-dependent, GluN2B-subunit containing NMDAR-mediated neuronal currents (61). In vivo loading of BAPTA-AM, a Ca2+chelator, selectively into astrocytes was found to be neuroprotective and significantly reduced the amount of Fluoro-Jade B labeling of dying neurons. Together, these findings suggest that reappearance of astrocytic mGluR5 may be a useful biomarker for active epileptogenesis, as well as a contributor to epileptogenesis by potentiating GluN2B-mediated inward neuronal currents.
Group 2 mGluRs consist of mGluR2 and mGluR3, while Group 3 mGluRs consist of mGluR4, mGluR6, mGluR7, and mGluR8. Both Group 2 and Group 3 inhibit adenylyl cyclase activity through Gαi activation and are thought to be largely distributed on pre-synaptic neurons, where they act to inhibit glutamate and GABA release (62, 63). Additionally, mGluR3 has been reported to be expressed on post-synaptic neurons and astrocytes (64–66). Using the pilocarpine TLE animal model, studies have shown that neuronal mGluR2/3 expression in the hippocampus (67, 68) and cortex (69) are decreased in epilepsy. Additionally, epileptic tissue from patients with TLE also have decreased mGluR2/3 expression (68) (Figure 1-2c). As pre-synaptic Group 2 mGluRs are thought to inhibit glutamate release, down-regulation of these receptors could promote glutamate release and neuronal hyperexcitability. Indeed, studies using Group 2 mGluR agonists have been shown to be neuroprotective in the models of absence epilepsy and the epilepsy models induced through amygdala kindling (57, 70). Conversely, Group 2 mGluR antagonists were shown to be pro-epileptic in an absence epilepsy model (71). Notably, these researchers also found that a mGluR1 potentiator, 9H-xanthene-9-carboxylic acid(4-trifluoromethyl-oxazol-2-yl)amide (SYN119), played a protective role against spike and wave discharges in a rat model of absence epilepsy (72).
One study investigating the localization and changes in mGluR3 and mGluR5 expression after hippocampal injury via KA-induced seizures found that mGluR3 mRNA was exclusively upregulated in astrocytes and oligodendrocytes (Figure 1-3c). Additionally, GFAP-positive astrocytes were found to be persistently upregulated from 2 days to 12 weeks post hippocampal injury (73). Although mGluR5 mRNA was not found to be upregulated in astrocytes, other studies have reported upregulated mGluR5 protein levels in animal models of epilepsy (74, 75). To understand how changes in astrocytic mGluR3 or mGluR5 activity may affect seizure generation, one study looked at the effects of adding Group I and Group II modulators to cultured astrocytes (76). This paper found that astrocytic Group 1 mGluR activation, via the use of (S)-3,5-dihydroxyphenylglycine [(S)-3,5-DHPG], led to decreased expression of glutamate transporters (GLAST and GLT-1), and that this effect could be antagonized using a selective mGluR5 antagonist, MPEP. Conversely, astrocytic Group II mGluR activation, using DCG-IV, resulted in upregulation of GLAST and GLT-1 expression, and this effect could be abolished using the Group II antagonist EGLU. As mGluR2 has not been found to be expressed by astrocytes, the authors concluded that the effects of Group II modulation in these experiments was attributable to astrocytic mGluR3 activity.
Questions remain and more work certainly needs to be done to further elucidate the role of mGluRs in epilepsy, however, it appears evident that mGluR expression and activity are significantly altered both in animal models and human epileptic tissue in ways that contribute to hyperexcitability and epileptogenesis. Although ionotropic and metabotropic glutamate receptors differ fundamentally in their structure and downstream effectors, NMDARs and mGluRs share the ability to sense changes in ambient glutamate and potentiate neuronal inward currents.
Supporting the notion that astrocytes are critically involved in the initiation of seizures through dysregulation of ambient glutamate, a recent in silico study found that increased ambient glutamate, either through increased astrocytic glutamate release or decreased uptake, was sufficient to initiate synchronous epileptiform-like discharges from neurons (77). Additionally, the use of transparent zebrafish combined with two-photon calcium imaging and local field potential recordings allowed investigators to monitor the activity and connectivity of thousands of neurons and glia revealing new insights into the role glia may play in seizure generation (78). Using PTZ, a GABAaR antagonist, to initiate seizures investigators found that pre-ictal neuronal activity did not significantly increase in synchrony, however, radial glia exhibited a significant increase in synchrony during the pre-ictal period followed by neuronal bursts. These findings support the idea that the local, pro-epileptic glial responses presented throughout this review can culminate in network-level events that can ultimately lead to neuronal hyperexcitability synchronization and seizure initiation.
Knowing that current anti-seizure therapeutics are ineffective for 1-in-3 people living with epilepsy (1, 2), elucidating the astrocytic processes involved in epileptogenesis may help identify new therapeutic targets that offer relief to patients with intractable epilepsy. Table 1 summarizes current FDA-approved and investigational pharmacotherapies targeting glutamate signaling for epilepsy. In summary, astrocytes exclusively possess the enzymatic activity to generate de novo glutamate in the CNS, as well as the molecular machinery to determine ambient glutamate through (1) a reduction of ECS, (2) glutamate release through SXC or VRAC or (3) a dysregulated state combining these mechanisms. Importantly, ambient glutamate can directly influence neuronal excitability, via generation of GluN2B-mediated SICs, as well as indirectly affect neuronal excitability via actions on mGluRs that can potentiate GluN2B-mediated currents, alter extracellular glutamatergic clearance, and influence neuronal glutamate release probabilities. Although inherent challenges exist in ubiquitously targeting glutamatergic mechanisms in the CNS, it appears clear that astrocytes are uniquely positioned to act as a master regulator of glutamate in health, and when dysregulated, can mediate pro-epileptic changes that warrant further investigation in hopes of unveiling novel therapeutic targets.
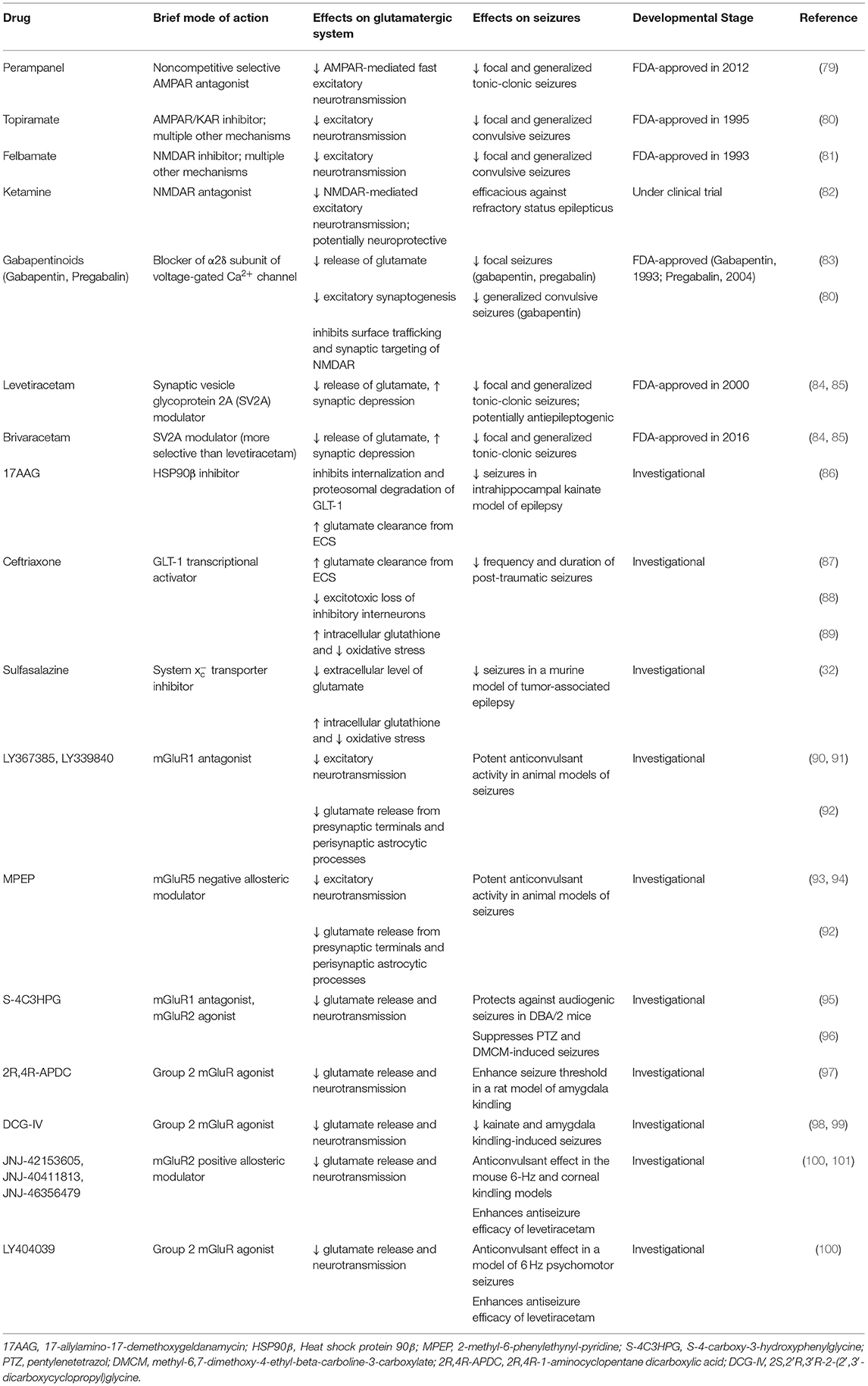
Table 1. Pharmacotherapies targeting glutamate signaling for epilepsy.
OA did an exhaustive literature search, generated a complete draft of the review, and prepared the figure. DP contributed written material and prepared Table 1. DP, BT, and HS also reviewed the literature, provided detailed comments and edits to the review, figure, and table. All authors contributed to the article and approved the submitted version.
This work was supported by NIH grants 1R01CA227149-01A1 and 1R01NS036692-01A1, and DOD grant W81XWH-18-1-0521.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. (2012) 78:1548–54. doi: 10.1212/WNL.0b013e3182563b19
2. Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. (2011) 52:657–78. doi: 10.1111/j.1528-1167.2011.03024.x
3. McKenna MC. Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne). (2013) 4:191. doi: 10.3389/fendo.2013.00191
4. Hawkins RA. The blood-brain barrier and glutamate. Am J Clin Nutr. (2009). 90:867S−74S. doi: 10.3945/ajcn.2009.27462BB
5. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. (2018) 98:239–389. doi: 10.1152/physrev.00042.2016
6. Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. (1999) 57:417–28. doi: 10.1002/(SICI)1097-4547(19990815)57:4<417::AID-JNR1>3.0.CO;2-N
7. Anlauf E, Derouiche A. Glutamine synthetase as an astrocytic marker: its cell type and vesicle localization. Front Endocrinol (Lausanne). (2013) 4:144. doi: 10.3389/fendo.2013.00144
8. Zhu H, et al. Moderate UV exposure enhances learning and memory by promoting a novel glutamate biosynthetic pathway in the brain. Cell. (2018) 173:1716–27.e17. doi: 10.1016/j.cell.2018.04.014
9. Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC. Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol. (2014) 11:13–30. doi: 10.1007/978-3-319-08894-5_2
10. Chung WS, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. (2015) 7:a020370. doi: 10.1101/cshperspect.a020370
11. Murphy TR, Binder DK, Fiacco TA. Turning down the volume: astrocyte volume change in the generation and termination of epileptic seizures. Neurobiol Dis. (2017) 104:24–32. doi: 10.1016/j.nbd.2017.04.016
12. Colbourn R, Naik A, Hrabetova S. ECS dynamism and its influence on neuronal excitability and seizures. Neurochem Res. (2019) 44:1020–36. doi: 10.1007/s11064-019-02773-w
13. Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. (1997) 17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997
14. Tønnesen J, Inavalli VVGK, Nägerl UV. Super-resolution imaging of the extracellular space in living brain tissue. Cell. (2018) 172:1108–21.e15. doi: 10.1016/j.cell.2018.02.007
15. During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. (1993) 341:1607–10. doi: 10.1016/0140-6736(93)90754-5
16. Lauderdale K, Murphy T, Tung T, Davila D, Binder DK, Fiacco TA. Osmotic edema rapidly increases neuronal excitability through activation of NMDA receptor-dependent slow inward currents in juvenile and adult hippocampus. ASN Neuro. (2015) 7:1759091415605115. doi: 10.1177/1759091415605115
17. Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport. (2004) 15:259–62. doi: 10.1097/00001756-200402090-00009
18. Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci. (2004) 24:8049–56. doi: 10.1523/JNEUROSCI.2294-04.2004
19. Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. (2006) 53:631–6. doi: 10.1002/glia.20318
20. Parkerson KA, Sontheimer H. Contribution of chloride channels to volume regulation of cortical astrocytes. Am J Physiol Cell Physiol. (2003) 284:C1460–7. doi: 10.1152/ajpcell.00603.2002
21. Parkerson KA, Sontheimer H. Biophysical and pharmacological characterization of hypotonically activated chloride currents in cortical astrocytes. Glia. (2004) 46:419–36. doi: 10.1002/glia.10361
22. Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. (2014) 344:634–8. doi: 10.1126/science.1252826
23. Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. (2014) 157:447–58. doi: 10.1016/j.cell.2014.03.024
24. Formaggio F, Saracino E, Mola MG, Rao SB, Amiry-Moghaddam M, Muccini M, et al. LRRC8A is essential for swelling-activated chloride current and for regulatory volume decrease in astrocytes. FASEB J. (2019) 33:101–13. doi: 10.1096/fj.201701397RR
25. Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z. Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron. (2019) 102:813–27.e6. doi: 10.1016/j.neuron.2019.03.029
26. Zhou JJ, Luo Y, Chen SR, Shao JY, Sah R, Pan HL. LRRC8A-dependent volume-regulated anion channels contribute to ischemia-induced brain injury and glutamatergic input to hippocampal neurons. Exp Neurol. (2020) 332:113391. doi: 10.1016/j.expneurol.2020.113391
27. Pál B. Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell Mol Life Sci. (2018) 75:2917–49. doi: 10.1007/s00018-018-2837-5
28. Ottestad-Hansen S, Hu QX, Follin-Arbelet VV, Bentea E, Sato H, Massie A, et al. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia. (2018) 66:951–70. doi: 10.1002/glia.23294
29. Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. (2002) 22:9134–41. doi: 10.1523/JNEUROSCI.22-20-09134.2002
30. LaCrosse AL, O'Donovan SM, Sepulveda-Orengo MT, McCullumsmith RE, Reissner KJ, Schwendt M, et al. Contrasting the role of xCT and GLT-1 upregulation in the ability of ceftriaxone to attenuate the cue-induced reinstatement of cocaine seeking and normalize AMPA receptor subunit expression. J Neurosci. (2017) 37:5809–21. doi: 10.1523/JNEUROSCI.3717-16.2017
31. De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. (2011) 31:5792–803. doi: 10.1523/JNEUROSCI.5465-10.2011
32. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. (2011) 17:1269–74. doi: 10.1038/nm.2453
33. Campbell SL, Buckingham SC, Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia. (2012) 53:1360–70. doi: 10.1111/j.1528-1167.2012.03557.x
34. Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med. (2015) 7:289ra86. doi: 10.1126/scitranslmed.aaa8103
35. Lewerenz J, Baxter P, Kassubek R, Albrecht P, Van Liefferinge J, Westhoff MA, et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2α and activating transcription factor 4 - A pathway active in glioblastomas and epilepsy. Antioxid Redox Signal. (2014) 20:2907–22. doi: 10.1089/ars.2013.5455
36. Alcoreza O, Tewari BP, Bouslog A, Savoia A, Sontheimer H, Campbell SL. Sulfasalazine decreases mouse cortical hyperexcitability. Epilepsia. (2019) 60:1365–77. doi: 10.1111/epi.16073
37. Leclercq K, Liefferinge JV, Albertini G, Neveux M, Dardenne S, Mairet-Coello G, et al. Anticonvulsant and antiepileptogenic effects of system. Epilepsia. (2019) 60:1412–23. doi: 10.1111/epi.16055
38. Sears SMS, Hewett JA, Hewett SJ. Decreased epileptogenesis in mice lacking the System x. Epilepsia Open. (2019) 4:133–43. doi: 10.1002/epi4.12307
39. Loewen JL, Albertini G, Dahle EJ, Sato H, Smolders IJ, Massie A, et al. Genetic and pharmacological manipulation of glial glutamate transporters does not alter infection-induced seizure activity. Exp Neurol. (2019) 318:50–60. doi: 10.1016/j.expneurol.2019.04.010
40. Cusick MF, Libbey JE, Patel DC, Doty DJ, Fujinami RS. Infiltrating macrophages are key to the development of seizures following virus infection. J Virol. (2013) 87:1849–60. doi: 10.1128/JVI.02747-12
41. Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, et al. Hippocampal TNFα signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. eNeuro. (2017) 4:ENEURO.0105-17.2017. doi: 10.1523/ENEURO.0105-17.2017
42. Papouin T, Dunphy J, Tolman M, Foley JC, Haydon PG. Astrocytic control of synaptic function. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160154. doi: 10.1098/rstb.2016.0154
43. Patel DC, Tewari BP, Chaunsali L, Sontheimer H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. (2019) 20:282–97. doi: 10.1038/s41583-019-0126-4
44. Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. (2004) 43:729–43. doi: 10.1016/j.neuron.2004.08.011
45. Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. (2004) 24:6920–7. doi: 10.1523/JNEUROSCI.0473-04.2004
46. Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. (2011) 33:1351–65. doi: 10.1111/j.1460-9568.2011.07628.x
47. Soria FN, Pérez-Samartín A, Martin A, Gona KB, Llop J, Szczupak B, et al. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J Clin Invest. (2014) 124:3645–55. doi: 10.1172/JCI71886
48. Gómez-Gonzalo M, Zehnder T, Requie LM, Bezzi P, Carmignoto G. Insights into the release mechanism of astrocytic glutamate evoking in neurons NMDA receptor-mediated slow depolarizing inward currents. Glia. (2018) 66:2188–99. doi: 10.1002/glia.23473
49. Featherstone DE, Shippy SA. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist. (2008) 14:171–81. doi: 10.1177/1073858407308518
50. Reiner A, Levitz J. Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron. (2018) 98:1080–98. doi: 10.1016/j.neuron.2018.05.018
51. Ribeiro FM, Vieira LB, Pires RG, Olmo RP, Ferguson SS. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res. (2017) 115:179–91. doi: 10.1016/j.phrs.2016.11.013
52. Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. (2010) 50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533
53. Nusser Z, Mulvihill E, Streit P, Somogyi P. Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localization. Neuroscience. (1994) 61:421–7. doi: 10.1016/0306-4522(94)90421-9
54. Luján R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. (1997) 13:219–41. doi: 10.1016/S0891-0618(97)00051-3
55. Blümcke I, Becker AJ, Klein C, Scheiwe C, Lie AA, Beck H, et al. Temporal lobe epilepsy associated up-regulation of metabotropic glutamate receptors: correlated changes in mGluR1 mRNA and protein expression in experimental animals and human patients. J Neuropathol Exp Neurol. (2000) 59:1–10. doi: 10.1093/jnen/59.1.1
56. Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, et al. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci. (2002) 22:5452–61. doi: 10.1523/JNEUROSCI.22-13-05452.2002
57. Watanabe Y, Kaida Y, Fukuhara S, Takechi K, Uehara T, Kamei C. Participation of metabotropic glutamate receptors in pentetrazol-induced kindled seizure. Epilepsia. (2011) 52:140–50. doi: 10.1111/j.1528-1167.2010.02764.x
58. Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. (2013). 339:197–200. doi: 10.1126/science.1226740
59. Umpierre AD, West PJ, White JA, Wilcox KS. Conditional knock-out of mGluR5 from astrocytes during epilepsy development impairs high-frequency glutamate uptake. J Neurosci. (2019) 39:727–42. doi: 10.1523/JNEUROSCI.1148-18.2018
60. Kelly E, Schaeffer SM, Dhamne SC, Lipton JO, Lindemann L, Honer M, et al. mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology. (2018) 43:1457–65. doi: 10.1038/npp.2017.295
61. Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, et al. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. (2007) 27:10674–84. doi: 10.1523/JNEUROSCI.2001-07.2007
62. Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. (1997) 37:205–37. doi: 10.1146/annurev.pharmtox.37.1.205
63. Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. (2008) 9:423–36. doi: 10.1038/nrn2379
64. Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M, et al. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. (2007) 27:8297–308. doi: 10.1523/JNEUROSCI.1889-07.2007
65. Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. (2011) 60:1017–41. doi: 10.1016/j.neuropharm.2010.10.022
66. Di Menna L, Joffe ME, Iacovelli L, Orlando R, Lindsley CW, Mairesse J, et al. Functional partnership between mGlu3 and mGlu5 metabotropic glutamate receptors in the central nervous system. Neuropharmacology. (2018) 128:301–13. doi: 10.1016/j.neuropharm.2017.10.026
67. Pacheco Otalora LF, Couoh J, Shigamoto R, Zarei MM, Garrido Sanabria ER. Abnormal mGluR2/3 expression in the perforant path termination zones and mossy fibers of chronically epileptic rats. Brain Res. (2006) 1098:170–85. doi: 10.1016/j.brainres.2006.04.124
68. Tang FR, Lee WL, Gao H, Chen Y, Loh YT, Chia SC. Expression of different isoforms of protein kinase C in the rat hippocampus after pilocarpine-induced status epilepticus with special reference to CA1 area and the dentate gyrus. Hippocampus. (2004) 14:87–98. doi: 10.1002/hipo.10156
69. Garrido-Sanabria ER, Otalora LF, Arshadmansab MF, Herrera B, Francisco S, Ermolinsky BS. Impaired expression and function of group II metabotropic glutamate receptors in pilocarpine-treated chronically epileptic rats. Brain Res. (2008) 1240:165–76. doi: 10.1016/j.brainres.2008.08.084
70. Moldrich RX, Jeffrey M, Talebi A, Beart PM, Chapman AG, Meldrum BS. Anti-epileptic activity of group II metabotropic glutamate receptor agonists (–)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate (LY379268) and (–)-2-thia-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate (LY389795). Neuropharmacology. (2001) 41:8–18. doi: 10.1016/S0028-3908(01)00044-2
71. Ngomba RT, Biagioni F, Casciato S, Willems-van Bree E, Battaglia G, Bruno V, et al. The preferential mGlu2/3 receptor antagonist, LY341495, reduces the frequency of spike-wave discharges in the WAG/Rij rat model of absence epilepsy. Neuropharmacology. (2005) 49(Suppl 1):89–103. doi: 10.1016/j.neuropharm.2005.05.019
72. Ngomba RT, Santolini I, Biagioni F, Molinaro G, Simonyi A, van Rijn CM, et al. Protective role for type-1 metabotropic glutamate receptors against spike and wave discharges in the WAG/Rij rat model of absence epilepsy. Neuropharmacology. (2011) 60:1281–91. doi: 10.1016/j.neuropharm.2011.01.007
73. Mudo G, Trovato-Salinaro A, Caniglia G, Cheng Q, Condorelli DF. Cellular localization of mGluR3 and mGluR5 mRNAs in normal and injured rat brain. Brain Res. (2007) 1149:1–13. doi: 10.1016/j.brainres.2007.02.041
74. Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. (2000) 12:2333–44. doi: 10.1046/j.1460-9568.2000.00131.x
75. Ferraguti F, Corti C, Valerio E, Mion S, Xuereb J. Activated astrocytes in areas of kainate-induced neuronal injury upregulate the expression of the metabotropic glutamate receptors 2/3 and 5. Exp Brain Res. (2001) 137:1–11. doi: 10.1007/s002210000633
76. Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. (2003) 17:2106–18. doi: 10.1046/j.1460-9568.2003.02657.x
77. Li J, Tang J, Ma J, Du M, Wang R, Wu Y. Dynamic transition of neuronal firing induced by abnormal astrocytic glutamate oscillation. Sci Rep. (2016) 6:32343. doi: 10.1038/srep32343
78. Diaz Verdugo C, Myren-Svelstad S, Aydin E, Van Hoeymissen E, Deneubourg C, Vanderhaeghe S, et al. Glia-neuron interactions underlie state transitions to generalized seizures. Nat Commun. (2019) 10:3830. doi: 10.1038/s41467-019-11739-z
79. Frampton JE. Perampanel: a review in drug-resistant epilepsy. Drugs. (2015) 75:1657–68. doi: 10.1007/s40265-015-0465-z
80. Klein P, Friedman A, Hameed MQ, Kaminski RM, Bar-Klein G, Klitgaard H, et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia. (2020) 61:359–86. doi: 10.1111/epi.16450
81. Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. (2016) 6:a022780. doi: 10.1101/cshperspect.a022780
82. Rosati A, De Masi S, Guerrini R. Ketamine for refractory status epilepticus: a systematic review. CNS Drugs. (2018) 32:997–1009. doi: 10.1007/s40263-018-0569-6
83. Dooley DJ, Taylor CP, Donevan S, Feltner D. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci. (2007) 28:75–82. doi: 10.1016/j.tips.2006.12.006
84. Löscher W, Gillard M, Sands ZA, Kaminski RM, Klitgaard H. Synaptic vesicle glycoprotein 2A ligands in the treatment of epilepsy and beyond. CNS Drugs. (2016) 30:1055–77. doi: 10.1007/s40263-016-0384-x
85. Yang X, Bognar J, He T, Mohammed M, Niespodziany I, Wolff C, et al. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia. (2015) 56:1899–909. doi: 10.1111/epi.13223
86. Sha L, Wang X, Li J, Shi X, Wu L, Shen Y, et al. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J Exp Med. (2017) 214:547–63. doi: 10.1084/jem.20160667
87. Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma. (2013) 30:1434–41. doi: 10.1089/neu.2012.2712
88. Hameed MQ, Hsieh TH, Morales-Quezada L, Lee HHC, Damar U, MacMullin PC, et al. Ceftriaxone treatment preserves cortical inhibitory interneuron function via transient salvage of GLT-1 in a rat traumatic brain injury model. Cereb Cortex. (2019) 29:4506–18. doi: 10.1093/cercor/bhy328
89. Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. (2009) 111:332–43. doi: 10.1111/j.1471-4159.2009.06347.x
90. Kingston AE, Griffey K, Johnson MP, Chamberlain MJ, Kelly G, Tomlinson R, et al. Inhibition of group I metabotropic glutamate receptor responses in vivo in rats by a new generation of carboxyphenylglycine-like amino acid antagonists. Neurosci Lett. (2002) 330:127–30. doi: 10.1016/S0304-3940(02)00751-6
91. Celli R, Santolini I, Van Luijtelaar G, Ngomba RT, Bruno V, Nicoletti F. Targeting metabotropic glutamate receptors in the treatment of epilepsy: rationale and current status. Expert Opin Ther Targets. (2019) 23:341–51. doi: 10.1080/14728222.2019.1586885
92. Álvarez-Ferradas C, Morales JC, Wellmann M, Nualart F, Roncagliolo M, Fuenzalida M, et al. Enhanced astroglial Ca2+ signaling increases excitatory synaptic strength in the epileptic brain. Glia. (2015) 63:1507–21. doi: 10.1002/glia.22817
93. Smolders I, Lindekens H, Clinckers R, Meurs A, O'Neill MJ, Lodge D, et al. In vivo modulation of extracellular hippocampal glutamate and GABA levels and limbic seizures by group I and II metabotropic glutamate receptor ligands. J Neurochem. (2004) 88:1068–77. doi: 10.1046/j.1471-4159.2003.02251.x
94. Qian F, Tang FR. Metabotropic glutamate receptors and interacting proteins in epileptogenesis. Curr Neuropharmacol. (2016) 14:551–62. doi: 10.2174/1570159X14666160331142228
95. Thomsen C, Klitgaard H, Sheardown M, Jackson HC, Eskesen K, Jacobsen P, et al. (S)-4-carboxy-3-hydroxyphenylglycine, an antagonist of metabotropic glutamate receptor (mGluR) 1a and an agonist of mGluR2, protects against audiogenic seizures in DBA/2 mice. J Neurochem. (1994) 62:2492–5. doi: 10.1046/j.1471-4159.1994.62062492.x
96. Dalby NO, Thomsen C. Modulation of seizure activity in mice by metabotropic glutamate receptor ligands. J Pharmacol Exp Ther. (1996) 276:516–22.
97. Attwell PJ, Koumentaki A, Abdul-Ghani AS, Croucher MJ, Bradford HF. Specific group II metabotropic glutamate receptor activation inhibits the development of kindled epilepsy in rats. Brain Res. (1998) 787:286–91. doi: 10.1016/S0006-8993(97)01500-X
98. Miyamoto M, Ishida M, Shinozaki H. Anticonvulsive and neuroprotective actions of a potent agonist (DCG-IV) for group II metabotropic glutamate receptors against intraventricular kainate in the rat. Neuroscience. (1997) 77:131–40. doi: 10.1016/S0306-4522(96)00442-3
99. Attwell PJ, Singh Kent N, Jane DE, Croucher MJ, Bradford HF. Anticonvulsant and glutamate release-inhibiting properties of the highly potent metabotropic glutamate receptor agonist (2S,2′R, 3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV). Brain Res. (1998) 805:138–43. doi: 10.1016/S0006-8993(98)00698-2
100. Metcalf CS, Klein BD, Smith MD, Pruess T, Ceusters M, Lavreysen H, et al. Efficacy of mGlu. Epilepsia. (2017) 58:484–93. doi: 10.1111/epi.13659
101. Metcalf CS, Klein BD, Smith MD, Ceusters M, Lavreysen H, Pype S, et al. Potent and selective pharmacodynamic synergy between the metabotropic glutamate receptor subtype 2-positive allosteric modulator JNJ-46356479 and levetiracetam in the mouse 6-Hz (44-mA) model. Epilepsia. (2018) 59:724–35. doi: 10.1111/epi.14005
Keywords: glutamate homeostasis, System , epilepsy, astroglia, metabotrophic glutamate receptor, NMDAR
Citation: Alcoreza OB, Patel DC, Tewari BP and Sontheimer H (2021) Dysregulation of Ambient Glutamate and Glutamate Receptors in Epilepsy: An Astrocytic Perspective. Front. Neurol. 12:652159. doi: 10.3389/fneur.2021.652159
Received: 11 January 2021; Accepted: 24 February 2021;
Published: 22 March 2021.
Edited by:
Christian Steinhaeuser, Universität Bonn, GermanyReviewed by:
Rheinallt Parri, Aston University, United KingdomCopyright © 2021 Alcoreza, Patel, Tewari and Sontheimer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Sontheimer, c29udGhlaW1AdnQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.