 Camille Peron1
Camille Peron1 Alessandra Maresca2
Alessandra Maresca2 Andrea Cavaliere1
Andrea Cavaliere1 Angelo Iannielli3,4
Angelo Iannielli3,4 Vania Broccoli3,4
Vania Broccoli3,4 Valerio Carelli2,5
Valerio Carelli2,5 Ivano Di Meo1
Ivano Di Meo1 Valeria Tiranti1*
Valeria Tiranti1*- 1Unit of Medical Genetics and Neurogenetics, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
- 2IRCCS Istituto delle Scienze Neurologiche di Bologna, Programma di Neurogenetica, Bologna, Italy
- 3San Raffaele Scientific Institute, Milan, Italy
- 4National Research Council (CNR), Institute of Neuroscience, Milan, Italy
- 5Department of Biomedical and Neuromotor Sciences-DIBINEM, University of Bologna, Bologna, Italy
More than 30 years after discovering Leber's hereditary optic neuropathy (LHON) as the first maternally inherited disease associated with homoplasmic mtDNA mutations, we still struggle to achieve effective therapies. LHON is characterized by selective degeneration of retinal ganglion cells (RGCs) and is the most frequent mitochondrial disease, which leads young people to blindness, in particular males. Despite that causative mutations are present in all tissues, only a specific cell type is affected. Our deep understanding of the pathogenic mechanisms in LHON is hampered by the lack of appropriate models since investigations have been traditionally performed in non-neuronal cells. Effective in-vitro models of LHON are now emerging, casting promise to speed our understanding of pathophysiology and test therapeutic strategies to accelerate translation into clinic. We here review the potentials of these new models and their impact on the future of LHON patients.
Introduction
Leber's hereditary optic neuropathy (LHON) is caused by maternally inherited missense point mutations of mitochondrial DNA (mtDNA) (1) and is estimated as the most-frequent mitochondrial disease (2). This blinding disorder is characterized by selective degeneration of retinal ganglion cells (RGCs), the retinal neurons projecting their axons, which form the optic nerve to the brain. Thus, the extended loss of RGCs and their axons leads to optic nerve atrophy, with a severe defect of central vision, in most cases leaving the patient legally blind (3, 4). Almost all LHON maternal lineages present with homoplasmic mutation (100% mtDNA copies are mutant in all tissues), having one of three frequent mtDNA mutations found in over 90% of patients worldwide (m.11778G>A/MT-ND4, m.3460G>A/MT-ND1, m.14484T>C/MT-ND6), but only some individuals develop the disease. Also, despite that the homoplasmic mtDNA mutation is present in all tissues, only a cellular type, that is, RGCs, undergoes degeneration. The pathogenic mechanism leading to cell death is thus extremely tissue and cell specific (3, 4). The phenotype of these mutations characterized by defective ATP synthesis when driven by complex I substrates (5), increased oxidative stress (6, 7), and increased propensity to undergo apoptosis (8, 9) has been thoroughly investigated in cybrids, lymphocytes, and fibroblasts but not in RGCs, the disease's target, which are not easily accessible and cannot be maintained in vitro (10).
Moreover, given the difficulties in manipulating mtDNA, very few animal models with mtDNA pathogenic mutations are available (11), preventing the possibility to study the affected tissues and organs and test therapeutic options.
To overcome these issues, we and other investigators exploited innovative approaches, based on the use of human-induced pluripotent stem cells (hiPSCs) as a faithful source of human neuronal cells and RGCs.
The use of hiPSCs to obtain terminally differentiated cells of a variety of tissues is a revolutionary approach to understanding disease mechanisms, performing drug screening, and testing gene or cell therapy (12–15).
Several studies have demonstrated the possibility to generate neurons and RGCs from plated hiPSC-derived embryoid bodies (16–19). In addition, different groups developed 3D culture systems recapitulating key steps of retinal development and allowing the generation of self-organizing retinal organoids containing RGCs (15, 20–25). These models provide a bridge between traditional 2D cell culture and mouse models, representing a paraphysiologic system with pros and cons (26), but of paramount importance for modeling mtDNA-related disorders.
Modeling LHON mutations in differentiated neurons and organoids will provide not only insights into the tissue-specific disease pathogenic mechanisms, but it will offer the unique opportunity to test in-vitro pharmacological approaches in a model system much more relevant than traditional non-neuronal cell cultures, such as fibroblasts, lymphoblasts, or cybrids. Moreover, patient-specific hiPSCs allow studying the effect of the mtDNA mutation in the context of patient-specific nuclear background, which, in LHON particularly, plays a pivotal role in the modulation of disease's presentation (27).
We here discuss our experience with the generation of hiPSCs from LHON-affected patients integrated with the data present in the literature. We particularly emphasize the translational potential for patients in exploiting LHON neuronal cells and RGCs to advance our knowledge of pathogenic mechanisms and test therapies.
Reprogramming Fibroblasts or Peripheral Blood Mononuclear Cells PBMCS From LHON Patients
Since the epochal discovery of induced pluripotent stem cells by the Yamanaka group in 2006 (28), many researchers generated hiPSC by reprogramming differentiated cells obtained from mitochondrial disease patients [MELAS syndrome (29), MERRF syndrome (30), Pearson syndrome (31); reviewed by Liang (32)]. Unexpectedly, even if LHON is the most-frequent mitochondrial disease, to date only a few groups, including ours (33), had generated LHON hiPSCs by reprogramming fibroblasts or peripheral blood mononuclear cells (PBMCs) derived from patients (34–38).
One group from Taiwan reprogrammed PBMCs from two LHON m.11778G>A patients and one LHON m.11778G>A unaffected carrier using the Sendai virus (37). The authors reported a slightly increased complex I (CI) activity, failing statistical significance, in the LHON hiPSCs, both affected and carrier, as compared to control. The authors reported a slightly increased of complex I (CI) activity in both affected and carrier LHON hiPSCs as compared to control, that failed to reach statistical significance.
Another group reprogrammed fibroblasts from two LHON m.11778G>A patients as well as one LHON proband carrying two mutations m.4160T>C and m.14484T>C, using episomal vectors expressing six reprogramming factors OCT4, SOX2, KLF4, L-MYC, LIN28, and shRNA for p53 (34). They investigated hypothetical difficulties in reprogramming cell lines with OXPHOS defects since Yokota et al. reported reduced reprogramming efficiency in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome (MELAS) fibroblasts carrying more than 90% of the m.3243A>G mtDNA mutation (39). Hung and collaborators reprogrammed fibroblasts carrying the homoplasmic LHON mutations and found no significant differences in the number of hiPSC colonies between controls and LHON patients (21 colonies on average for the controls, and 13 colonies on average for the LHON patients). Differently, our own experience with LHON was more similar to what observed by Yokota, since we noticed that LHON fibroblasts or PMBCs are refractory to be reprogrammed to hiPSC.
Specifically, we attempted to reprogram different LHON cell lines: two m.3460G>A patients, four m.11778G>A patients, and two unaffected m.11778G>A carriers (Table 1). As shown in Table 1 and Figure 1A, the number of clones obtained was in general very low, even if numerous attempts were performed also in different laboratories. Conversely, using fibroblasts derived from healthy controls or disease's patients affected by mitochondrial disorders, including dominant optic atrophy (OPA1 mutation), Pearson (40), and MPAN (41), we obtained on average from 10 to 20 clones of hiPSC (Table 1 and Figure 1B) per reprogramming experiment. To overcome this issue, we tested the reprogramming efficiency of LHON cells under hypoxia laboratory conditions (5% pO2, more similar to physiological oxygen tension in vivo), a condition previously used to enhance the generation of hiPSC (42), and recently demonstrated to be specifically beneficial in several OXPHOS defects, by improving disease phenotype in mice and cells (43). In fact, under traditional culturing conditions cellular models of mitochondrial respiratory-chain disease and Friedreich's ataxia showed proliferative defects, which could be reversed by lowering oxygen tension (44). In addition, hypoxia was able to prevent and even reverse the neurological phenotype in a Leigh syndrome mouse model characterized by CI deficiency due to Ndufs4 gene ablation (45). Based on this evidence and since LHON mutations were associated with reduced CI-driven ATP synthesis and increased ROS production (46), we hypothesized that hypoxic cell culture conditions during reprogramming could increase the number of hiPSC clones generated. Thus, we recently reprogrammed PBMCs derived from one LHON m.11778G>A patient and one carrier, in parallel under normoxic (11778 4a and Carrier 2a) and hypoxic (11778 4b and Carrier 2b) conditions (5% oxygen), following published procedures (42). We found that this hypoxic condition significantly increased the number of hiPSC clones generated (Table 1 and Figure 1A). In fact, while under normoxic conditions, we obtained around nine LHON hiPSCs clones in 10 different reprogramming experiments (0.9 clones/reprogramming cycle), and this number increased, under hypoxic conditions, to 11 clones in two reprogramming experiments (5.5 clones/reprogramming cycle). Although these last results derived from only two experiments and need to be further consolidated, they indicated a statistically significant improvement of the reprogramming efficiency (Figure 1A), which remains largely below that observed for the disease control group (12.7 clones/reprogramming cycle) and for the healthy control group (19.3 clones/reprogramming cycle) (Figure 1B). This amelioration of the reprogramming efficiency is relevant not only to obtain enough biological material for further investigations but could also unravel an insight into pathogenic mechanisms, relevant for the disease, and for the development of targeted effective therapy. Remarkably, the subacute phase of LHON is hallmarked by well-known vascular changes, and ongoing discussions revolve around the issue of pseudo-hypoxic signaling that RGCs may produce as their metabolic unbalance reaches the threshold for triggering the disease, possibly underlying the microangiopathy in LHON (3, 4, 47, 48).
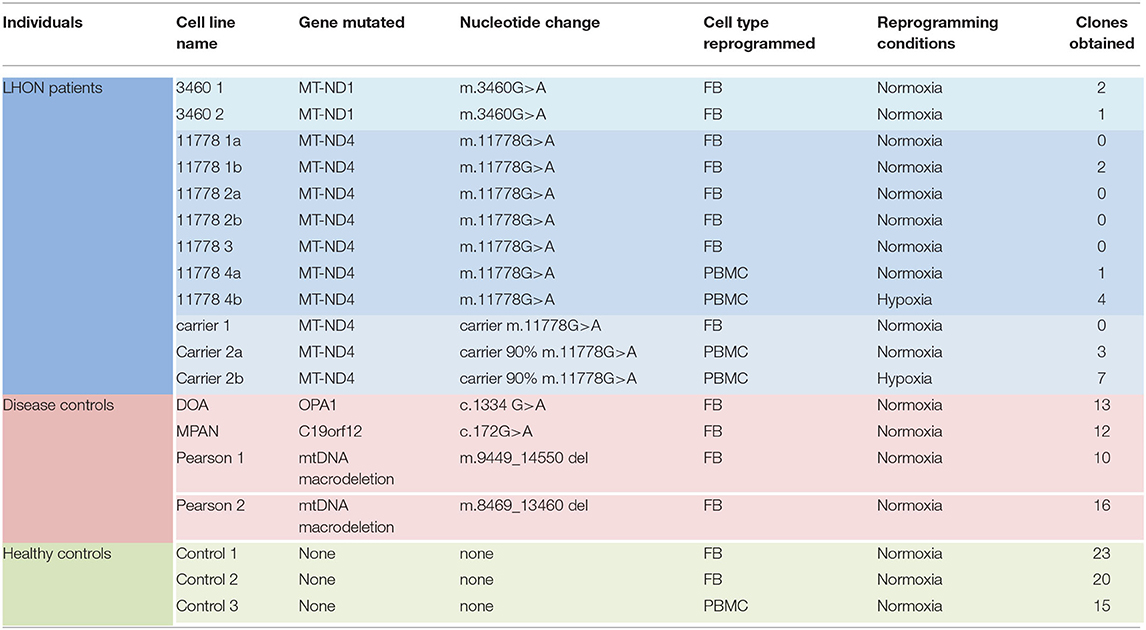
Table 1. Characteristics of cell lines subjected to reprogramming.
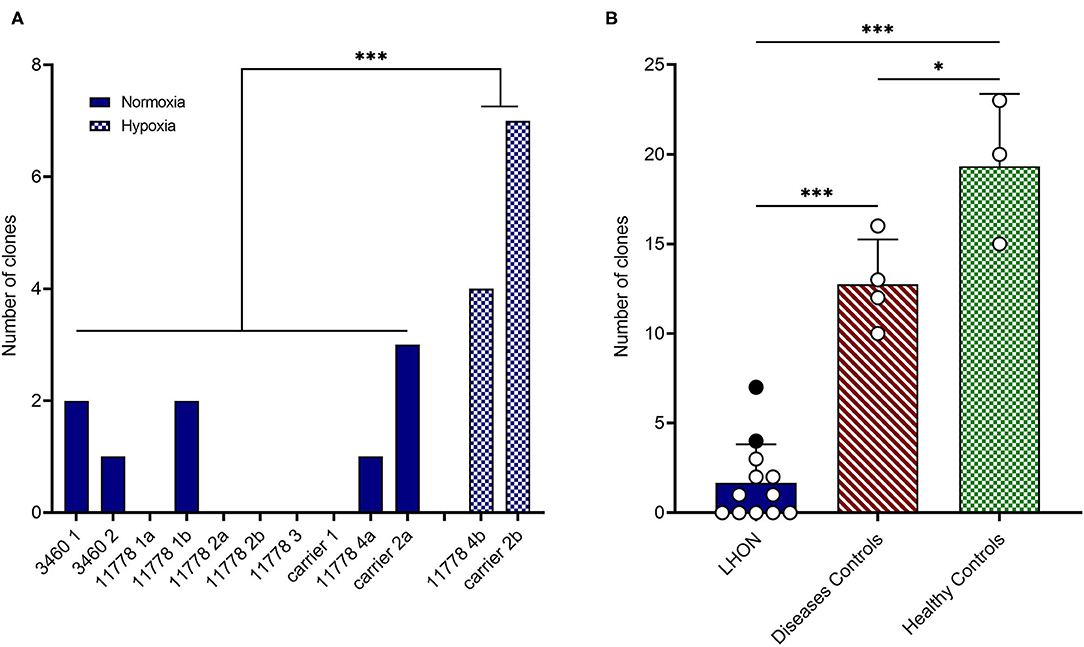
Figure 1. LHON cell lines reprogramming efficiency. (A) Number of hiPSC clones obtained by reprogramming affected or non-affected (carrier) LHON-derived fibroblasts or PBMC under normoxia (solid blue bars) or hypoxia (cross-hatched blue bars) conditions. (B) Comparison between grouped numbers of hiPSC clones obtained from LHON (solid blue bar), disease controls including non-LHON mitochondrial diseases (hatched red bar), and healthy controls (cross-hatched green bars) cell lines. The dots represent the number of clones obtained under normoxia (white dots) or hypoxia (black dots) conditions. ***p < 0.001, *p < 0.05.
Generation of RGCs From LHON Patients
In the last years, a few protocols have been developed with the purpose to differentiate RGCs directly from patients-derived hiPSCs. However, very few of these RGCs models have been produced for LHON. The first model was reported in 2017 by the Wong group, who generated RGCs from one healthy control and one patient carrying in combination the two homoplasmic mtDNA mutations m.4160T>C and m.14484T>C. Interestingly, they used cybrid technology to also generate patients' fibroblasts homoplasmic for the wild-type mtDNA, thus creating an isogenic control hiPSCs and derived RGCs (35). They found an increased level of apoptosis in LHON RGCs not observed in the healthy and isogenic corrected RGCs, demonstrating that this phenotype was a direct consequence of the LHON mutations. Another group generated hiPSCs-derived RGCs from a m.11778G>A LHON-affected and unaffected carrier, belonging both to the same family (37). They observed enhanced mitochondrial biogenesis, decreased basal respiration, and increased oxidative stress in both affected and unaffected RGCs. However, defective neurite outgrowth was only found in the affected RGCs, while carrier cells exhibited a prominently higher expression of the gene encoding γ-synuclein. Interestingly, increased CI activity was observed in RGCs derived from the asymptomatic carrier but not from the affected patient. Differences in affected and unaffected RGCs carrying homoplasmic m.11778G>A mutation were also found by Yang et al. (49). Both lines showed increased ROS production, but only the affected cells were characterized by increased apoptosis and altered mitochondrial transport pattern along the axons, with an increase in retrograde and a decrease in stationary mitochondria. Furthermore, affected RGCs displayed a significant increase of KIF5A, a member of the kinesin-1 family KIF5, involved in the transport of mitochondria along the axons. Another study carried out on hiPSC-derived RGCs by Yang et al. (50) highlighted the possible role played by AMPA receptors and excitotoxicity in m.11778G>A LHON patients. They used a modified protocol of differentiation of hiPSCs to RGCs to obtain a highly homogeneous RGCs population. They showed how the MT-ND4-mutated LHON-RGC cells exhibited significantly reduced GluR1/R2 (subunits of AMPA receptors) and their associated scaffold proteins and the resulting different pattern of response to glutamate stimulation compared to control.
Lastly, Edo et al. (51) demonstrated that hiPSC-derived RGCs can suppress the immune activity of T-cells via TGF-β, have a poor expression of HLA class I, and no expression of HLA class II (CD80 and CD86 co-stimulatory molecules), opening the possibility of using these cells in transplant without the risk of rejection.
Generation of Neurons From LHON Patients
Almost two decades ago, the Cortopassi group generated cybrids using the neuronal precursor cell line NT2, containing mitochondria from patients with m.11778G>A and m.3460G>A mutations (52). Differentiation of LHON-NT2 cells resulted in a decreased number of cells, reduction of mtDNA amount, and increased ROS production, compared to the parental line. To our knowledge, no hiPSCs-derived neuronal model different from RGCs has been generated to date. Although it is clear that RGCs represent the best model to unravel LHON pathomechanisms, hiPSCs differentiation in non-RGCs neurons could be informative as well to study the selective degeneration of RGCs in patients. To maintain the transparency of the retina to light, the retinal segment of the RGCs axon is unmyelinated, increasing the energetic demand for action potential firing along this portion and making these cells particularly susceptible to energetic deficit (53). The generation of in-vitro myelinated neurons through co-cultures of Schwann cells and hiPSCs-derived neurons (54) might be informative to establish the involvement of myelin in the pathogenesis of the disease.
State of the Art on Organoids Implementation
The use of 3D organoids generated in vitro from patient-derived cells may represent an important interface between in-vitro and in-vivo modeling of LHON, being more accessible and easier to obtain than mouse models and overcoming the anatomical interspecies differences between humans and rodents.
The first human brain and retinal organoids have been generated about 10 years ago from different groups (55, 56). Lancaster and colleagues successfully modeled genetic microcephaly using hiPSCs derived from patients' fibroblasts to generate brain organoids.
Only a year before, the Sasai group had generated a 3D optic structure by self-organization of cultured human embryonic stem cells (ESCs). The optic cup consisted of the retinal pigmented epithelium, and an inner neural retina correctly organized into multilayered tissue containing photoreceptors (rods and cones), interneuron precursors, and RGCs (57). Both these protocols exploited the capacity of embryoid bodies (EBs) (ESCs or hiPSCs-derived) to proceed spontaneously toward ectodermal commitment without extrinsic signaling factors, which instead are necessary for mesodermal and endodermal specifications (26, 58).
Several modifications and adjustments to the pivotal approaches of Lancaster (55, 59) and Sasai group (57, 60), have been done in the following years, essentially identifying distinct extrinsic factors to obtaining specific regions in the organoids (60, 61), or by-passing the EBs formation step (62). Moreover, improvements toward standardization are constantly evolving, such as the use of completely xeno-free culture methods (62) or the introduction of technologies allowing large-scale controlled organoids production, such as bioreactors or microfluidics chips (26). Importantly, also protocols for cryopreservation at intermediate steps of differentiation have been established, allowing the biobanking of the in-vitro-generated organoids, an additional advantage compared to animal models (26, 57, 62).
Therapeutic Approaches
Despite the numerous clinical and pre-clinical investigations carried out to date, effective therapies for LHON are still limited. Effective means that therapy should be able to tangibly modify the disease natural history either by aborting or reverting the catastrophic wave of cell death, or at least limiting the progression so that the visual function is substantially preserved based on anatomical RGCs measurable sparing. Multiple clinical trials have been conducted in recent years, essentially targeting the main pathways involved in the pathogenic mechanism (63). Several antioxidants molecules, some of which with direct effects on mitochondrial respiration, have been tested in patients: idebenone, Coenzyme Q10 (CoQ10), EPI-743, Elamipretide, curcumin (63).
To date, idebenone (Raxone®) is the only drug approved by the European Medicines Agency for LHON. It has been documented that idebenone can increase the rate of visual recovery in LHON patients after reaching a nadir of visual loss (64–66); however, its efficacy remains incomplete and variable amongst treated subjects.
The only treatment explored in LHON hiPSCs-derived RGCs was the antioxidant N-acetyl-L-cysteine, which was shown to reduce the ROS production and apoptosis, also rescuing the defective mitochondrial transport observed in the LHON cells (49).
Additional compounds targeting other pathways involved in the LHON pathogenesis (mitobiogenesis, mitophagy, mitoinflammation) have been evaluated only in patient-derived primary cells or in cybrids, such as phytoestrogens (67), rapamycin (68), papaverine, and zolpidem (69). Moreover, other potential strategies are emerging, for example, the inhibition of the miRNA181a/b, acting on both mitobiogenesis and mitophagy (70). All these pharmacological approaches should be reevaluated also in RGC to understand if they are efficacious and rapidly translatable into a therapy.
Besides pharmacological clinical trials, encouraging results are nowadays being reported by clinical trials with gene therapy for patients carrying the m.11778G>A/MT-ND4 mutation [reviewed in Amore et al. (63)], using the Adeno-Associated Virus (AAV)-mediated allotopic expression of a wild-type recoded version of the mtDNA-encoded ND4 subunit of complex I (71, 72). To better refine the efficiency of allotropic expression strategy in the context of RGCs, detailing the mitochondrial import of wild-type ND4 protein, its competition with the endogenously expressed mutant ND4, and finally the dynamics of complex I assembly of either one or the other ND4 subunits, may greatly benefit of 3D organoid modeling of LHON. This might resolve some of the criticisms previously raised by the preclinical studies (73–75). The same approach could be developed for the other LHON-related mutations, and different approaches based on gene therapy might be proposed in the future, for example, modulating the expression of modifying genes or miRNAs (70) or applying possible gene-editing strategies, as recently proposed for mtDNA (76). Similarly, the feasibility of mitochondrial import of nucleic acids, claimed by some studies (77, 78), may benefit the use of eye/brain organoids carrying LHON mutations, reproducing those experiments and possibly paving the road for further gene therapy strategies.
Discussion
In-vitro modeling of LHON through 2D cell cultures, including patient-derived hiPSCs and neurons, allowed important steps forward in the understanding of the pathogenic mechanism of this complex and fascinating disease. We here presented evidence that LHON hiPSCs are difficult to obtain as compared to other apparently more severe mitochondrial disorders, but this reduced efficiency could be improved by performing the reprogramming experiment under hypoxic conditions. This observation would deserve further investigation since obtaining a large number of hiPSCs clones is instrumental to further develop differentiated 2D cell cultures. Although 2D cell cultures show several advantages such as easy manipulation and analysis (good accessibility of nutrients and/or drugs, excellent visualization and tracking of cells at microscopy by live-cell imaging), the complex 3D architecture of in-vivo tissues is not reproduced by this method, nor are the interactions between different co-resident populations of specialized cells (79). This is particularly important for LHON, in which RGCs are the only cells affected in the retina. The application of the innovative single-cell omics on hiPSCs-derived 3D organoids can provide useful insight on the cell specificity of LHON disease. A recent study has already paved the way for this approach, performing single-cell transcriptomics on in-vitro-generated human retinal organoids and ex-vivo adult human retinas, allowing mapping of disease-associated genes to particular cell types (25). This work highlights the importance of investigating mechanisms of disease in RGCs since they could be differently regulated in the traditional cell models so far exploited. Many of the findings so far achieved in LHON should be revalidated in RGC models to assure that the right pathogenic mechanism was effectively targeted by therapies.
Modeling mitochondrial diseases caused by mtDNA mutations in animals is still challenging due to the difficulties in manipulating the mitochondrial genome (80, 81), although a new promising method has been recently described Mok et al. (76). In 2012, the group of Doug Wallace, a pioneer in the field of mitochondrial medicine, successfully generated a mouse model carrying a mutation in the MT-ND6 gene, which developed a pathology closely resembling LHON at 2 years of age, although the mouse did not show reduced visual responses (82). This model was instrumental to reproduce some of the hallmark features observed in human post-mortem LHON retina (83, 84); however, mice, because they lack the macular region, ultimately fail to reproduce the natural history that clinically characterizes humans with the characteristic catastrophic evolution of RGC neurodegeneration (3, 4).
Thus, it will be fundamental to investigate pathogenic mechanism of LHON disease in hiPSCs-derived cell/tissue-specific models and retinal organoids might be instrumental to assess efficacy/toxicity in the pre-clinical phases. The issue of maintaining organoids in a spinning bioreactor under hypoxic conditions, with the intent of reproducing the brain endogenous developmental program, could be crucial, especially for LHON in light of our observation, but also in general for other diseases. To date, only a few brain organoids models of mitochondrial diseases have been reported, specifically for MELAS syndrome, mitochondrial neurogastrointestinal encephalomyopathy, Friedrich ataxia, and Leigh syndrome (85–88). We think that modeling LHON with retinal organoids would provide substantial progress in the understanding of the pathogenic mechanisms and in identifying the correct targets for therapy development. To this end, testing pharmacological and gene therapy approaches with human transgenes packaged in the appropriate AAV vector constructs, currently performed in animal models with obvious problematic issues (89, 90), may benefit human-patient-derived eye/brain organoids, certainly allowing to speed translation from pre-clinical science to approval for human clinical trials of regulatory agencies such as the Food and Drug Administration (FDA) and European Medicines Agency (EMA).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Fondazione IRCCS Istituto Neurologico Carlo Besta. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CP, AC, AI, and ID perform experiments, analyzed data, generated the table and figure, and analyzed the literature. AM, VB, and VC analyzed the literature. VT concept the manuscript architecture, supervised the analysis of the data, and of the literature. VT performed the final revision of the manuscript. All the authors draft the manuscript.
Funding
The financial support of Mitocon-Italy, Grant No. 2018-01 to VT and of the grant from the Italian Ministry of Health RF-2018-12366703 to VT, VB, and VC is acknowledged. CP is sustained with a fellowship of Associazione Luigi Comini ONLUS – Italy (http://www.luigicominionlus.org/).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor is currently organizing a Research Topic with one of the author VC.
Acknowledgments
This study was carried out in the Center for the Study of Mitochondrial Pediatric Diseases (http://www.mitopedia.org) funded by the Mariani Foundation. VT is member of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD).
References
1. Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. (1988) 242:1427–30. doi: 10.1126/science.3201231
2. Chinnery PF, Turnbull DM. Mitochondrial DNA mutations in the pathogenesis of human disease. Mol Med Today. (2000) 6:425–32. doi: 10.1016/S1357-4310(00)01805-0
3. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Progr Retinal Eye Res. (2004) 23:53–89. doi: 10.1016/j.preteyeres.2003.10.003
4. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
5. Baracca A, Solaini G, Sgarbi G, Lenaz G, Baruzzi A, Schapira AHV, et al. Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch Neurol. (2005) 62:730–6. doi: 10.1001/archneur.62.5.730
6. Beretta S, Mattavelli L, Sala G, Tremolizzo L, Schapira AHV, Martinuzzi A, et al. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain. (2004) 127:2183–92. doi: 10.1093/brain/awh258
7. Floreani M, Napoli E, Martinuzzi A, Pantano G, De Riva V, Trevisan R, et al. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. FEBS J. (2005) 272:1124–35. doi: 10.1111/j.1742-4658.2004.04542.x
8. Ghelli A, Zanna C, Porcelli AM, Schapira AHV, Martinuzzi A, Carelli V, et al. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem. (2003) 278:4145–50. doi: 10.1074/jbc.M210285200
9. Zanna C, Ghelli A, Porcelli AM, Martinuzzi A, Carelli V, Rugolo M. Caspase-independent death of Leber's hereditary optic neuropathy cybrids is driven by energetic failure and mediated by AIF and Endonuclease G. Apoptosis. (2005) 10:997–1007. doi: 10.1007/s10495-005-0742-5
10. Zhang X-M, Liu DT, Chiang SW-Y, Choy K-W, Pang C-P, Lam DS-C, et al. Immunopanning purification and long-term culture of human retinal ganglion cells. Mol Vis. (2010) 16:2867–72. Available online at: http://www.molvis.org/molvis/v16/a307/
11. Stewart JB. Current progress with mammalian models of mitochondrial DNA disease. J Inherited Metabolic Dis. (2021) 44:325–42. doi: 10.1002/jimd.12324
12. Llonch S, Carido M, Ader M. Organoid technology for retinal repair. Dev Biol. (2018) 433:132–43. doi: 10.1016/j.ydbio.2017.09.028
13. Ahmad R, Sportelli V, Ziller M, Spengler D, Hoffmann A. Tracing early neurodevelopment in schizophrenia with induced pluripotent stem cells. Cells. (2018) 7:140. doi: 10.3390/cells7090140
14. Miltner AM, La Torre A. Retinal ganglion cell replacement: current status and challenges ahead: retinal ganglion cell replacement. Dev Dyn. (2019) 248:118–28. doi: 10.1002/dvdy.24672
15. Rabesandratana O, Chaffiol A, Mialot A, Slembrouck-Brec A, Joffrois C, Nanteau C, et al. Generation of a transplantable population of human iPSC-derived retinal ganglion cells. Front Cell Dev Biol. (2020) 8:585675. doi: 10.3389/fcell.2020.585675
16. Riazifar H, Jia Y, Chen J, Lynch G, Huang T. Chemically induced specification of retinal ganglion cells from human embryonic and induced pluripotent stem cells. Stem Cells Transl Med. (2014) 3:424–32. doi: 10.5966/sctm.2013-0147
17. Sluch VM, Davis CO, Ranganathan V, Kerr JM, Krick K, Martin R, et al. Differentiation of human ESCs to retinal ganglion cells using a CRISPR engineered reporter cell line. Sci Rep. (2015) 5:16595. doi: 10.1038/srep16595
18. Gill KP, Hung SSC, Sharov A, Lo CY, Needham K, Lidgerwood GE, et al. Enriched retinal ganglion cells derived from human embryonic stem cells. Sci Rep. (2016) 6:30552. doi: 10.1038/srep30552
19. Teotia P, Chopra DA, Dravid SM, Van Hook MJ, Qiu F, Morrison J, et al. Generation of functional human retinal ganglion cells with target specificity from pluripotent stem cells by chemically defined recapitulation of developmental mechanism. Stem Cells. (2017) 35:572–85. doi: 10.1002/stem.2513
20. Reichman S, Terray A, Slembrouck A, Nanteau C, Orieux G, Habeler W, et al. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc Natl Acad Sci USA. (2014) 111:8518–23. doi: 10.1073/pnas.1324212111
21. Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao L-H, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. (2014) 5:4047. doi: 10.1038/ncomms5047
22. Maekawa Y, Onishi A, Matsushita K, Koide N, Mandai M, Suzuma K, et al. Optimized culture system to induce neurite outgrowth from retinal ganglion cells in three-dimensional retinal aggregates differentiated from mouse and human embryonic stem cells. Curr Eye Res. (2016) 41:558–68. doi: 10.3109/02713683.2015.1038359
23. Ohlemacher SK, Sridhar A, Xiao Y, Hochstetler AE, Sarfarazi M, Cummins TR, et al. Stepwise differentiation of retinal ganglion cells from human pluripotent stem cells enables analysis of glaucomatous neurodegeneration. Stem Cells. (2016) 34:1553–62. doi: 10.1002/stem.2356
24. Fligor CM, Langer KB, Sridhar A, Ren Y, Shields PK, Edler MC, et al. Three-Dimensional retinal organoids facilitate the investigation of retinal ganglion cell development, organization and neurite outgrowth from human pluripotent stem cells. Sci Rep. (2018) 8:14520. doi: 10.1038/s41598-018-32871-8
25. Cowan CS, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, et al. Cell types of the human retina and its organoids at single-cell resolution. Cell. (2020) 182:1623–40.e34. doi: 10.1016/j.cell.2020.08.013
26. Li M, Izpisua Belmonte JC. Organoids - preclinical models of human disease. N Engl J Med. (2019) 380:569–79. doi: 10.1056/NEJMra1806175
27. Caporali L, Maresca A, Capristo M, Del Dotto V, Tagliavini F, Valentino ML, et al. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion. (2017) 36:130–37. doi: 10.1016/j.mito.2017.07.004
28. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
29. Hämäläinen RH, Manninen T, Koivumäki H, Kislin M, Otonkoski T, Suomalainen A. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc Natl Acad Sci USA. (2013) 110:E3622–30. doi: 10.1073/pnas.1311660110
30. Chou S-J, Ko Y-L, Yang Y-H, Yarmishyn AA, Wu Y-T, Chen C-T, et al. Generation of two isogenic human induced pluripotent stem cell lines from a 15 year-old female patient with MERRF syndrome and A8344G mutation of mitochondrial DNA. Stem Cell Res. (2018) 30:201–5. doi: 10.1016/j.scr.2018.05.011
31. Cherry ABC, Gagne KE, Mcloughlin EM, Baccei A, Gorman B, Hartung O, et al. Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells. (2013) 31:1287–97. doi: 10.1002/stem.1354
32. Liang X, Kristiansen CK, Vatne GH, Hong Y, Bindoff LA. Patient-specific neural progenitor cells derived from induced pluripotent stem cells offer a promise of good models for mitochondrial disease. Cell Tissue Res. (2020) 380:15–30. doi: 10.1007/s00441-019-03164-x
33. Peron C, Mauceri R, Cabassi T, Segnali A, Maresca A, Iannielli A, et al. Generation of a human iPSC line, FINCBi001-A, carrying a homoplasmic m.G3460A mutation in MT-ND1 associated with Leber's Hereditary optic Neuropathy (LHON). Stem Cell Res. (2020) 48:101939. doi: 10.1016/j.scr.2020.101939
34. Hung SSC, Van Bergen NJ, Jackson S, Liang H, Mackey DA, Hernández D, et al. Study of mitochondrial respiratory defects on reprogramming to human induced pluripotent stem cells. Aging. (2016) 8:945–57. doi: 10.18632/aging.100950
35. Wong RCB, Lim SY, Hung SSC, Jackson S, Khan S, Van Bergen NJ, et al. Mitochondrial replacement in an iPSC model of Leber's hereditary optic neuropathy. Aging. (2017) 9:1341–50. doi: 10.18632/aging.101231
36. Lu H-E, Yang Y-P, Chen Y-T, Wu Y-R, Wang C-L, Tsai F-T, et al. Generation of patient-specific induced pluripotent stem cells from Leber's hereditary optic neuropathy. Stem Cell Res. (2018) 28:56–60. doi: 10.1016/j.scr.2018.01.029
37. Wu Y-R, Wang A-G, Chen Y-T, Yarmishyn AA, Buddhakosai W, Yang T-C, et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber's hereditary optic neuropathy. Exp Cell Res. (2018) 363:299–309. doi: 10.1016/j.yexcr.2018.01.020
38. Bahr T, Welburn K, Donnelly J, Bai Y. Emerging model systems and treatment approaches for Leber's hereditary optic neuropathy: challenges and opportunities. Biochimica et Biophysica Acta. (2020) 1866:165743. doi: 10.1016/j.bbadis.2020.165743
39. Yokota M, Hatakeyama H, Okabe S, Ono Y, Goto Y. Mitochondrial respiratory dysfunction caused by a heteroplasmic mitochondrial DNA mutation blocks cellular reprogramming. Hum Mol Genet. (2015) 24:4698–709. doi: 10.1093/hmg/ddv201
40. Peron C, Mauceri R, Iannielli A, Cavaliere A, Legati A, Rizzo A, et al. Generation of two human iPSC lines, FINCBi002-A and FINCBi003-A, carrying heteroplasmic macrodeletion of mitochondrial DNA causing Pearson's syndrome. Stem Cell Res. (2021) 50:102151. doi: 10.1016/j.scr.2020.102151
41. Panteghini C, Zorzi G, Venco P, Dusi S, Reale C, Brunetti D, et al. C19orf12 and FA2H mutations are rare in Italian patients with neurodegeneration with brain iron accumulation. Semin Pediatr Neurol. (2012) 19:75–81. doi: 10.1016/j.spen.2012.03.006
42. Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. (2009) 5:237–41. doi: 10.1016/j.stem.2009.08.001
43. Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng K-H, et al. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. PNAS. (2017) 114:E4241–50. doi: 10.1073/pnas.1621511114
44. Ast T, Meisel JD, Patra S, Wang H, Grange RMH, Kim SH, et al. Hypoxia rescues frataxin loss by restoring iron sulfur cluster biogenesis. Cell. (2019) 177:1507–21.e16. doi: 10.1016/j.cell.2019.03.045
45. Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, et al. Hypoxia as a therapy for mitochondrial disease. Science. (2016) 352:54–61. doi: 10.1126/science.aad9642
46. Kirches E. LHON: mitochondrial mutations and more. Curr Genomics. (2011) 12:44–54. doi: 10.2174/138920211794520150
47. Balducci N, Cascavilla ML, Ciardella A, Morgia CL, Triolo G, Parisi V, et al. Peripapillary vessel density changes in Leber's hereditary optic neuropathy: a new biomarker. Clin Exp Ophthalmol. (2018) 46:1055–62. doi: 10.1111/ceo.13326
48. Kousal B, Kolarova H, Meliska M, Bydzovsky J, Diblik P, Kulhanek J, et al. Peripapillary microcirculation in Leber hereditary optic neuropathy. Acta Ophthalmol. (2019) 97:e71–6. doi: 10.1111/aos.13817
49. Yang T-C, Yarmishyn AA, Yang Y-P, Lu P-C, Chou S-J, Wang M-L, et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Human Mol Genet. (2020) 29:1454–64. doi: 10.1093/hmg/ddaa063
50. Yang Y-P, Nguyen PNN, Lin T-C, Yarmishyn AA, Chen W-S, Hwang D-K, et al. Glutamate stimulation dysregulates AMPA receptors-induced signal transduction pathway in leber's inherited optic neuropathy patient-specific hiPSC-derived retinal ganglion cells. Cells. (2019) 8:625. doi: 10.3390/cells8060625
51. Edo A, Sugita S, Futatsugi Y, Sho J, Onishi A, Kiuchi Y, et al. Capacity of retinal ganglion cells derived from human induced pluripotent stem cells to suppress T-Cells. Int J Mol Sci. (2020) 21:7831. doi: 10.3390/ijms21217831
52. Wong A, Cavelier L, Collins-Schramm HE, Seldin MF, McGrogan M, Savontaus M-L, et al. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum Mol Genet. (2002) 11:431–8. doi: 10.1093/hmg/11.4.431
53. Carelli V, La Morgia C, Ross-Cisneros FN, Sadun AA. Optic neuropathies: the tip of the neurodegeneration iceberg. Hum Mol Genet. (2017) 26:R139–50. doi: 10.1093/hmg/ddx273
54. Clark AJ, Kaller MS, Galino J, Willison HJ, Rinaldi S, Bennett DLH. Co-cultures with stem cell-derived human sensory neurons reveal regulators of peripheral myelination. Brain. (2017) 140:898–913. doi: 10.1093/brain/awx012
55. Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. (2013) 501:373–9 doi: 10.1038/nature12517
56. Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, et al. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci USA. (2009) 106:16698–703. doi: 10.1073/pnas.0905245106
57. Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. (2012) 10:771–85. doi: 10.1016/j.stem.2012.05.009
58. Nam KH, Yi SA, Jang HJ, Han J-W, Lee J. In vitro modeling for inherited neurological diseases using induced pluripotent stem cells: from 2D to organoid. Arch Pharm Res. (2020) 43:877–89. doi: 10.1007/s12272-020-01260-z
59. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. (2014) 9:2329–40. doi: 10.1038/nprot.2014.158
60. Artero Castro A, Rodríguez Jimenez FJ, Jendelova P, Erceg S. Deciphering retinal diseases through the generation of three dimensional stem cell-derived organoids: concise review. Stem Cells. (2019) 37:1496–504. doi: 10.1002/stem.3089
61. Clevers H. Modeling development and disease with organoids. Cell. (2016) 165:1586–97. doi: 10.1016/j.cell.2016.05.082
62. Reichman S, Slembrouck A, Gagliardi G, Chaffiol A, Terray A, Nanteau C, et al. Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells. (2017) 35:1176–88. doi: 10.1002/stem.2586
63. Amore G, Romagnoli M, Carbonelli M, Barboni P, Carelli V, La Morgia C. Therapeutic options in hereditary optic neuropathies. Drugs. (2020) 81:57–86. doi: 10.1007/s40265-020-01428-3
64. Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, et al. Idebenone treatment in Leber's hereditary optic neuropathy. Brain. (2011) 134:e188. doi: 10.1093/brain/awr180
65. Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain. (2011) 134:2677–86. doi: 10.1093/brain/awr170
66. Catarino CB, von Livonius B, Priglinger C, Banik R, Matloob S, Tamhankar MA, et al. Real-World clinical experience with idebenone in the treatment of Leber hereditary optic neuropathy. J Neuroophthalmol. (2020) 40:558–65. doi: 10.1097/WNO.0000000000001023
67. Pisano A, Preziuso C, Iommarini L, Perli E, Grazioli P, Campese AF, et al. Targeting estrogen receptor β as preventive therapeutic strategy for Leber's hereditary optic neuropathy. Hum Mol Genet. (2015) 24:6921–31. doi: 10.1093/hmg/ddv396
68. Dai Y, Zheng K, Clark J, Swerdlow RH, Pulst SM, Sutton JP, et al. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum Mol Genet. (2014) 23:637–47. doi: 10.1093/hmg/ddt450
69. Datta S, Tomilov A, Cortopassi G. Identification of small molecules that improve ATP synthesis defects conferred by Leber's hereditary optic neuropathy mutations. Mitochondrion. (2016) 30:177–86. doi: 10.1016/j.mito.2016.08.002
70. Indrieri A, Carrella S, Romano A, Spaziano A, Marrocco E, Fernandez-Vizarra E, et al. miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol Med. (2019) 11:e8734. doi: 10.15252/emmm.201708734
71. Yu-Wai-Man P, Newman NJ, Carelli V, Moster ML, Biousse V, Sadun AA, et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med. (2020) 12:eaaz7423. doi: 10.1126/scitranslmed.aaz7423
72. Newman NJ, Yu-Wai-Man P, Carelli V, Moster ML, Biousse V, Vignal-Clermont C, et al. Efficacy and safety of intravitreal gene therapy for leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. (2021) 128:649–60. doi: 10.1016/j.ophtha.2020.12.012
73. Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V, et al. Rescue of a mitochondrial deficiency causing Leber hereditary optic neuropathy. Ann Neurol. (2002) 52:534–42. doi: 10.1002/ana.10354
74. Oca-Cossio J, Kenyon L, Hao H, Moraes CT. Limitations of allotopic expression of mitochondrial genes in mammalian cells. Genetics. (2003) 165:707–20. doi: 10.1093/genetics/165.2.707
75. Perales-Clemente E, Fernández-Silva P, Acín-Pérez R, Pérez-Martos A, Enríquez JA. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. (2011) 39:225–34. doi: 10.1093/nar/gkq769
76. Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. (2020) 583:631–7. doi: 10.1038/s41586-020-2477-4
77. Wang G, Chen H, Oktay Y, Zhang J, Allen E, Smith G, et al. PNPASE regulates RNA import into mitochondria. Cell. (2010) 142:456–67. doi: 10.1016/j.cell.2010.06.035
78. Yu H, Koilkonda RD, Chou T-H, Porciatti V, Ozdemir SS, Chiodo V, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber's hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA. (2012) 109:E1238–47. doi: 10.1073/pnas.1119577109
79. Mason JO, Price DJ. Building brains in a dish: Prospects for growing cerebral organoids from stem cells. Neuroscience. (2016) 334:105–18. doi: 10.1016/j.neuroscience.2016.07.048
80. Tyynismaa H, Suomalainen A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. (2009) 10:137–43. doi: 10.1038/embor.2008.242
81. Menacho C, Prigione A. Tackling mitochondrial diversity in brain function: from animal models to human brain organoids. Int J Biochem Cell Biol. (2020) 123:105760. doi: 10.1016/j.biocel.2020.105760
82. Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci USA. (2012) 109:20065–70. doi: 10.1073/pnas.1217113109
83. Sadun AA, Win PH, Ross-Cisneros FN, Walker SO, Carelli V. Leber's hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc. (2000) 98:223–32; discussion 232-235.
84. Pan BX, Ross-Cisneros FN, Carelli V, Rue KS, Salomao SR, Moraes-Filho MN, et al. Mathematically modeling the involvement of axons in Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. (2012) 53:7608–17. doi: 10.1167/iovs.12-10452
85. Winanto null, Khong ZJ, Soh B-S, Fan Y, Ng S-Y. Organoid cultures of MELAS neural cells reveal hyperactive Notch signaling that impacts neurodevelopment. Cell Death Dis. (2020) 11:182. doi: 10.1038/s41419-020-2383-6
86. Pacitti D, Bax BE. The development of an in vitro cerebral organoid model for investigating the pathomolecular mechanisms associated with the central nervous system involvement in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE). Nucleosides Nucleotides Nucleic Acids. (2018) 37:603–17. doi: 10.1080/15257770.2018.1492139
87. Mazzara PG, Muggeo S, Luoni M, Massimino L, Zaghi M, Valverde PT-T, et al. Frataxin gene editing rescues Friedreich's ataxia pathology in dorsal root ganglia organoid-derived sensory neurons. Nat Commun. (2020) 11:4178. doi: 10.1038/s41467-020-17954-3
88. Inak G, Rybak-Wolf A, Lisowski P, Jüttner R, Zink A, Mlody B, et al. SURF1 mutations causative of Leigh syndrome impair human neurogenesis. bioRxiv. (2019) 2019:551390. doi: 10.1101/551390
89. Feuer WJ, Schiffman JC, Davis JL, Porciatti V, Gonzalez P, Koilkonda RD, et al. Gene therapy for leber hereditary optic neuropathy. Ophthalmology. (2016) 123:558–70. doi: 10.1016/j.ophtha.2015.10.025
Keywords: Leber's hereditary optic neuropathy, human induced pluripotent stem cells, mitochondrial disorders, organoids, retinal ganglion cells (RGC)
Citation: Peron C, Maresca A, Cavaliere A, Iannielli A, Broccoli V, Carelli V, Di Meo I and Tiranti V (2021) Exploiting hiPSCs in Leber's Hereditary Optic Neuropathy (LHON): Present Achievements and Future Perspectives. Front. Neurol. 12:648916. doi: 10.3389/fneur.2021.648916
Received: 02 January 2021; Accepted: 26 April 2021;
Published: 08 June 2021.
Edited by:
Rustum Karanjia, University of Ottawa, CanadaReviewed by:
Manvi Goel, The Ohio State University, United StatesShlomo Dotan, Tel Aviv Sourasky Medical Center, Israel
Copyright © 2021 Peron, Maresca, Cavaliere, Iannielli, Broccoli, Carelli, Di Meo and Tiranti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Tiranti, VmFsZXJpYS5UaXJhbnRpQGlzdGl0dXRvLWJlc3RhLml0