 Claudio M. de Gusmão
Claudio M. de Gusmão Lucas Garcia3
Lucas Garcia3 Mohamad A. Mikati
Mohamad A. Mikati Samantha Su
Samantha Su- 1Department of Neurology, Boston Children's Hospital and Harvard Medical School, Boston, MA, United States
- 2Department of Neurology, Universidade Estadual de Campinas (UNICAMP), São Paulo, Brazil
- 3Department of Medicine, Universidade 9 de Julho, São Paulo, Brazil
- 4Division of Pediatric Neurology and Developmental Medicine, Duke University Medical Center, Durham, NC, United States
- 5Education Unit, University College London Institute of Neurology, University College London, London, United Kingdom
Paroxysmal movement disorders include paroxysmal kinesigenic dyskinesia, paroxysmal non-kinesigenic dyskinesia, paroxysmal exercise-induced dyskinesia, and episodic ataxias. In recent years, there has been renewed interest and recognition of these disorders and their intersection with epilepsy, at the molecular and pathophysiological levels. In this review, we discuss how these distinct phenotypes were constructed from a historical perspective and discuss how they are currently coalescing into established genetic etiologies with extensive pleiotropy, emphasizing clinical phenotyping important for diagnosis and for interpreting results from genetic testing. We discuss insights on the pathophysiology of select disorders and describe shared mechanisms that overlap treatment principles in some of these disorders. In the near future, it is likely that a growing number of genes will be described associating movement disorders and epilepsy, in parallel with improved understanding of disease mechanisms leading to more effective treatments.
Introduction
The concept of neurological diseases with episodic manifestations is not foreign to clinical practice. Neurologists frequently encounter patients with “attacks” with minimal or no abnormalities on the examination in-between spells. Initially, neurological nosology relied on patient history or witnessed account from bystanders. This led to using descriptive but non-specific terms to classify disease, such as “faints,” “apoplexy,” or “convulsions.” Over time, improved understanding of the underlying physiological abnormality (or at least refinement of semiology) led to more precise nomenclature. For example, migrainous auras are characterized by transient neurologic deficits associated with cortical spreading depression while epilepsy is caused by synchronous abnormal firing of a group of cortical neurons. Altered perfusion with transient lack of oxygen supply to any central nervous system structure characterizes a transient ischemic attack. Notwithstanding nebulous pathophysiology, transient motoric phenomena attributed to abnormal basal ganglia and/or cerebellar activity are therefore labeled paroxysmal movement disorders (encompassing paroxysmal dyskinesia and episodic ataxia).
Although large efforts have been made to discriminate paroxysmal movement disorders from epileptic seizures, recent progress in neurogenetics has pointed to growing intersections between genotypes, phenotypes, and treatment in several conditions with paroxysmal neurological symptoms. In this review, we aim to revisit this relationship showing how certain gene mutations can manifest with paroxysmal dyskinesia, episodic ataxia and/or epilepsy (Table 1). Furthermore, we consider how treatment for these conditions overlaps considerably.
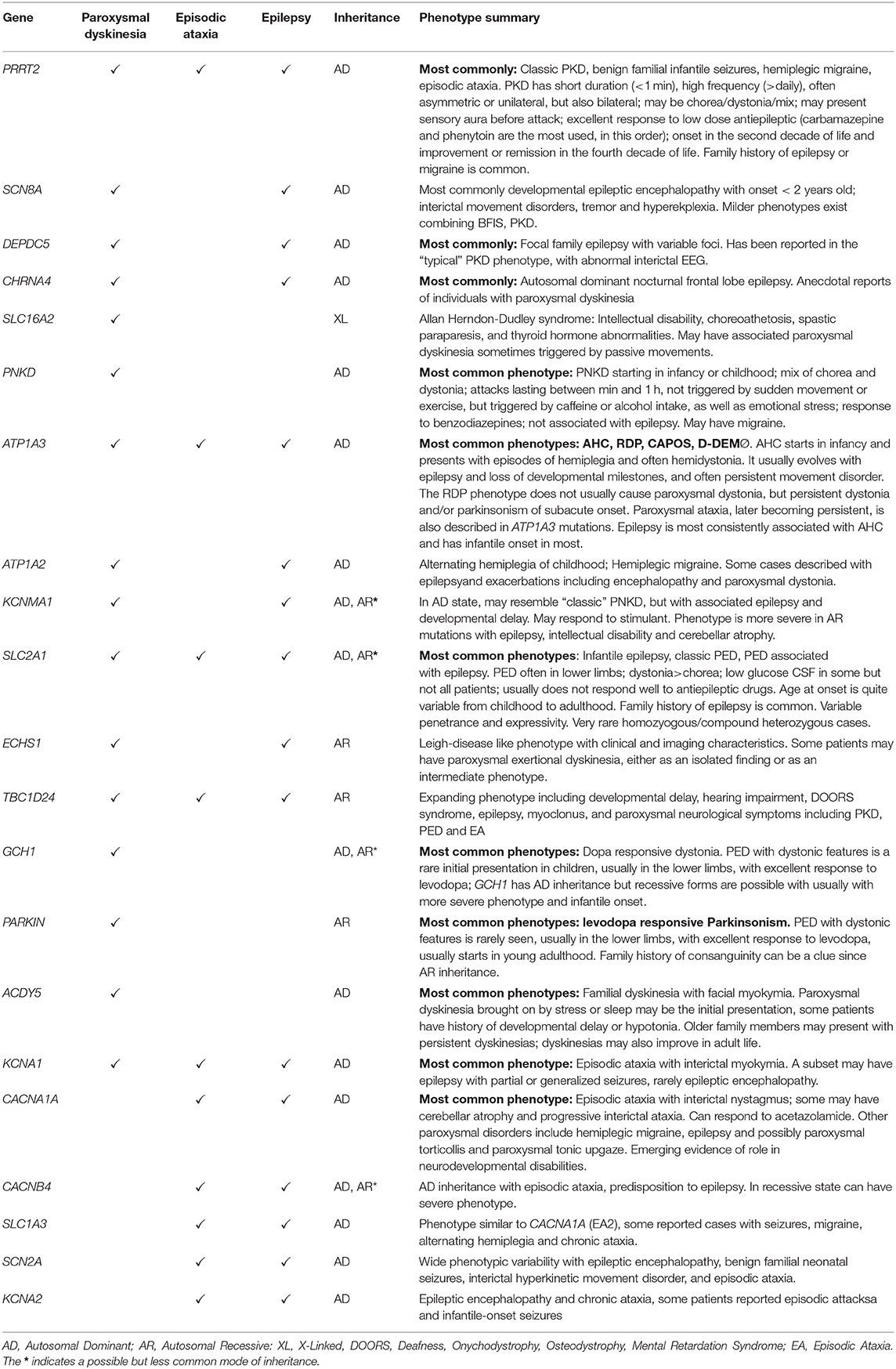
Table 1. Phenotypic clues in select neurogenetic syndromes associated with paroxysmal neurological symptoms [modified from Silveira-Moriyama et al. (1)].
Historical Overview and Underlying Genetic Conditions
The first description of paroxysmal dyskinesia is attributed to Kure, who made use of photography in the nineteenth century to document what was likely paroxysmal kinesigenic dyskinesia (PKD) (2, 3). Similar cases were described by Gowers in his seminal book on Epilepsy (4). For a period of time such spells were believed to be seizures, illustrating the reasoning why antiepileptic drugs were used for treatment of such spells (5, 6). To this day, some authors paraphrase Gowers by including these disorders on the borderland of epilepsy (7). The initial clinical classifications for paroxysmal dyskinesia attempted to classify the phenotype by motor phenomenology, using terms like “paroxysmal dystonic choreoathetosis” or “paroxysmal kinesigenic choreoathetosis” (6, 8). Subsequently, Demirkiran and Jankovic proposed a classification scheme that divided the paroxysmal dyskinesias highlighting the type of trigger as the main phenotypic characteristic, creating the kinesigenic (PKD), non-kinesigenic (PNKD), exertional (PED), and hypnogenic (PHD) subtypes (9). This classification proved useful and gained widespread acceptance; it has been the framework toward which phenotypes have been organized, leading to the unraveling of their genetic underpinnings (10). In 2004 mutations in the PNKD gene (previously called MR-1) were associated with PNKD (11). In 2008, mutations in the facilitated glucose transporter 1 (previously called GLUT1, now named SLC2A1 gene) were found in patients with PED and in 2011, mutations in PRRT2 were associated with PKD (12–14). Since its description, most cases of PHD were found to be largely attributed to nocturnal frontal lobe seizures, with rare isolated cases reported in association with PRRT2 (15–17).
The episodic ataxia phenotype was first described by Parker in 1946, who used the term “periodic” to describe a syndrome of intermittent ataxia with familial clustering and the potential for developing progressive ataxia (18, 19). Awareness of the condition increased in the ensuing decades, with descriptions of autosomal dominant inheritance and the serendipitous discovery that many patients responded favorably to acetazolamide (20–24). The clinical classification of episodic ataxia is based on presence of interictal symptoms, triggers, spell characteristics, and response to treatment. Nevertheless, they have been organized according to the suspected or known locus in the order they were described, giving rise to the terminology still used to this day (EA1, EA2, etc.) (25). Similarly to the paroxysmal dyskinesias, the clinical description has also led to the discovery of underlying genetic defects. Thus, starting in 1994, episodic ataxia type 1 (EA1) was associated with mutations in the gene KCNA1 which codes for one of the units of the potassium channel (26). Shortly afterwards, mutations in the calcium channel gene CACNA1A were associated with episodic ataxia type 2 (EA2) (26–29). In total, at least nine episodic ataxia syndromes have been described thus far. Genetic associations have been confirmed in EA1 (KCNA1), EA2 (CACNA1A), EA5 (CACNB4, another calcium channel gene), and EA 6 (SLC1A3, a glutamate transporter) (26, 29–31). More recently, EA9 was described in association with the FGF14 gene encoding for a fibroblast growth factor expressed in Purkinje cells and previously associated with spinocerebellar ataxia type 27 (32, 33). In the other episodic ataxias, cytogenetic loci are suspected but unconfirmed or unknown. EA1 and EA2 are the most frequently encountered subtypes and best characterized phenotypes. EA1 has short (seconds to minutes) attacks and interictal myokymia, while EA2 has longer attacks (hours to days) and interictal nystagmus in classic cases. Seizures have been variably reported in association with these genetic conditions, most consistently (but not exclusively) in EA2 and EA6.
While one can state that the genetic era has ushered refinement in disease classification, it is also true that it has blurred boundaries between what appeared to be well-established territories in paroxysmal movement disorders. It became widely demonstrated that a single mutated gene could appear clinically with combinations of different spells, leading to discussions about phenotypic pleiotropy and the accuracy of genotype/phenotype correlations. For example, a patient with a point mutation in CACNA1A might have a combination of different spells, including episodic ataxia, hemiplegic migraine, and/or seizures. The same applies to PRRT2 mutations. What is more, one particular phenotype (for example, episodic ataxia) could be caused by different genes, with often overlapping clinical features (a concept known as allelic heterogeneity) which justifies the ample use of gene panels in neurogenetics today (34). The combination of epilepsy and paroxysmal movement disorders is particularly interesting in this regard. Both phenomena presume transient abnormal neuronal firing leading to dysfunction of neuronal networks. These episodic disorders are generally attributed to confirmed or presumed cell membrane dysfunction, often caused by channel ion gene mutations (35).
Phenotypical Overlap
For the purposes of this review, we have organized the phenotypes according to their main characteristic features. We find it is a useful construct when applied to patient encounters. However, it is important to recognize though that these phenotypical models (kinesigenic, non-kinesigenic, and exertional dyskinesias) are not clear-cut. Some genetic conditions will exhibit more than one type of trigger in different patients. For example, in patients with PKD associated with PRRT2 mutations, non-kinesigenic episodes may be present in up to 70% of cases (36). In rare instances, PRRT2 mutations can also cause episodic ataxia (37). Patients with mutations in SLC2A1 may have multiple types of episodes often including exercise-induced and non-kinesigenic, and more rarely kinesigenic attacks (10). Kinesigenic triggers, on the other hand, may also cause episodes of ataxia in more than half of cases with EA1 associated with mutations in KCNA1 (38).
Paroxysmal Kinesigenic Dyskinesias and Epilepsy
The clinical syndrome of PKD is the more recognizable entity among the paroxysmal movement disorders. The kinesigenic trigger was well-characterized in the 1960s (6) and clinical criteria for diagnosis have been used since the 2000s (39) (Table 2). The criteria include the presence of kinesigenic triggers, short duration of attacks, and positive response to anti-epileptic drugs. In the 1990s, 4 families were described where patients presented in the first year of life with benign familial infantile seizures (BFIS), later developing PKD in the first or second decade. The association of BFIS with PKD was coined infantile convulsion choreoathetosis syndrome (ICCA), and it was mapped to the pericentromeric region of chromosome 16p with autosomal dominant inheritance (40). Combining classic linkage with whole exome sequencing, the PKD syndrome and ICCA were mapped to mutation in the PRRT2 gene, with autosomal dominant inheritance. Mutations in PRRT2 were quickly found as the underlying cause of a large proportion of classic PKD cases, and also “pure” cases of BIFS (13, 14, 41, 42). Mutations in PRRT2 have also been associated with other paroxysmal disorders, such as episodic ataxia, and hemiplegic migraine (37).

Table 2. Criteria for Paroxysmal Kinesigenic Dyskinesia [adapted from Bruno et al. (39)].
Even in those patients with a prior a history of BFIS, the events of PKD appear to be non-epileptiform, and telemetry with EEG is negative for epileptiform discharges during the paroxysmal kinesigenic episodes. Nevertheless, the question remains whether kinesigenic activity in PKD could trigger a form of “basal ganglia seizure,” in which there is synchronous abnormal firing of the structures involved in movement disorders. This lead older authors to debate whether it was as a type of “reflex seizure” (43, 44). A study on a single patient using dural and depth electrodes during a PKD episode did indeed demonstrate focal discharges arising from the contralateral supplementary motor cortex rapidly followed by discharges in the caudate nucleus (45). However, follow-up studies using regular surface EEG electrodes did not demonstrate abnormal ictal activity, with some interictal EEG abnormalities in some patients (46, 47). It seems that the primary functional abnormality during a PKD attack is related to transient basal ganglia hypermetabolism, as demonstrated by PET (48). However, the question remains whether ongoing cortical dysfunction could somehow modulate the expression of the disease. With increased recognition of cortico-basal ganglia connectivity, it is conceivable to explore the idea that abnormalities in one node of the network could lead to symptomatic manifestations arising from other connected areas. Mutations in SCN8A exemplify this concept whereby patients with ICCA syndrome may have epileptogenic activity during BFIS and contralateral rhythmic activity in the sensory-motor region during the PKD episodes (49).
Seizures associated with mutations in PRRT2 most commonly manifest as BFIS, with mean onset at 6 months but ranging between 3 and 20 months of age (50). Seizures are focal in origin with or without secondary generalization, and are generally brief with occasional clusters up to 10 events/day (51). The seizure semiology is varied, comprising psychomotor arrest, eye and/or head deviation, unilateral tonic limb stiffening, or jerking that may generalize. Typically, BFIS responds to AED treatment and improves after age 2. Seizures associated with PRRT2 mutations can also occur in the context of febrile seizures or febrile seizures plus syndrome (GEFS+) (52, 53).
PKD has a mean age of onset at 12 years of age with attacks consistently triggered by sudden movement, often in the context of emotional, or physiologic stress (54). A sensory aura preceding the attack is commonly reported. Kinesigenic paroxysmal movements in PKD are short and frequent, with more than 90% of patients reporting duration <30 s and more than a third of patients reporting more than 20 attacks per day (39). Interestingly, the short duration and high frequency of daily events also occurs in the seizures that patients with BFIS experience. The combination of BFIS and PKD (ICCA syndrome) is associated with mutations in PRRT2 in about 80% of cases (55–57), whereas isolated PKD is associated with PRRT2 mutations in 40–90% of cases. The prevalence is increased in the setting of a positive family history (54, 58).
Mutations in PRRT2 most often occur in a hotspot where an insertion of an additional cytosine base creates a hairpin loop in the transcript causing DNA polymerase slippage (42, 52). About 80% of patients carry the c.649dupC mutation, causing a frameshift that may lead to unstable RNA or a truncated protein, subject to non-sense mediated decay. PRRT2 mutations are therefore predicted to be loss-of-function (14, 59, 60). The mechanism by which PRRT2 haploinsufficiency causes disease is not totally clear, but some translational evidence suggests that it may be related to synaptic dysfunction in glutamatergic neurons, with associated abnormalities in sodium currents (60, 61). Patients with biallelic mutations in PRRT2 have been reported to have a more severe phenotype with including different seizure types, status epilepticus, episodic ataxia, and PKD (62, 63).
Both BFIS and PKD are exquisitely responsive to antiepileptic drugs, especially low dose carbamazepine and phenytoin (54). Some authors have speculated that the sodium channel blocking properties of these AEDs may be particularly useful, and that carbamazepine also enhances the activity of certain glutamate transporters reducing its concentration on the synaptic cleft (61). Other phenotypes associated with PRRT2, such as hemiplegic migraine, respond less consistently to carbamazepine (64).
Other genes and loci have been associated with the PKD syndrome, but PRRT2 appears to be the most common. Mutations in SCN8A, encoding for the alpha subunit of the Nav1.6 voltage- gated sodium channel, are notable. Mutations in this locus were originally associated with developmental epileptic encephalopathy, but milder phenotypes exist combining BFIS, PKD, and ICCA syndrome, as well as other non-epileptic paroxysmal phenomena such as tremulousness and hyperekplexia (49, 65). In addition to SCN8A, a large study in patients with clinically defined PKD (66) identified mutations in DEPDC5 (originally described in the context of focal familial epilepsy with variable foci) as well as KCNA1, KCNMA1, SLC2A1, and PNKD (the most common phenotypes associated with these genes are discussed further in the manuscript). Finally, PKD has also been reported in association with mutations in the acetylcholine receptor gene CHRNA4 (in association with epilepsy and developmental delay), and in the thyroid hormone transporter SLC16A2 (also known as MCT8) (67–69). In the latter case, a remarkable clinical feature is that paroxysmal dyskinesias may be triggered in the context of passive manipulation of a limb.
Paroxysmal Non-Kinesigenic Dyskinesia and Epilepsy
Paroxysmal non-kinesigenic dyskinesia probably encompasses the largest group of paroxysmal disorders with overlapping epileptic features. The familial form of PNKD was originally described by Mount and Reback (5), under the term “paroxysmal familial choreoathetosis” describing a syndrome with autosomal dominant inheritance, with spells triggered by alcohol, caffeine, or fatigue. Through linkage mapping and sequencing studies, mutations in the gene MR-1 (currently called PNKD) were identified (11). Notably, mutations in PNKD appear to influence synapse formation and neurotransmitter release (61, 70), may have co-morbid migraine, but have not been implicated in epilepsy.
Paroxysmal non-kinesigenic dyskinesia may be familial or sporadic. As outlined above, familial cases are most commonly associated with mutations in PNKD and have a well-defined phenotype (Table 3) (71). However, many other genetic conditions may underlie familial cases; and acquired causes can also lead to non-kinesigenic dyskinetic episodes (72). These other causes may be called “atypical PNKD” (71). Acquired forms can be secondary to various injuries to the basal ganglia (73). Mutations in KCNMA1, a gene encoding a subunit of a potassium channel activated by calcium, resemble the classic PNKD phenotype, with associated epilepsy and developmental delay (74, 75). Although this disorder appears to be quite rare and few cases have been reported, it is worth noting that some patients may experience improvement in paroxysmal episodes with stimulants (76).

Table 3. Criteria for Paroxysmal Non-Kinesigenic Dyskinesia associated with PNKD [adapted from Bruno et al. (71)].
Perhaps the best prototypical example of non-kinesigenic dyskinesia associated with ample phenotypic variability is related to monoallelic mutations in the gene ATP1A3. The gene encodes for a subunit of the sodium-potassium (Na+/K+) ATPase responsible for establishing and maintaining the electrochemical gradient of sodium and potassium ions across neuronal plasma membranes. Mutations in this gene were originally associated with rapid-onset dystonia-parkinsonism (RDP) (77), and later were attributed to be the major cause of alternating hemiplegia of childhood (AHC), a condition which causes both motor symptoms, progressive cognitive and motor deficits, and often epilepsy (78). Although AHC is named after the episodes of hemiplegia, dyskinetic movements were noted since its original description (79). Paroxysmal dystonia is frequently reported, in addition to nystagmus, autonomic changes, and reduced awareness (80–82). In fact, paroxysmal dystonia that involves the upper extremity with flexion or extension and elevation of the limb may be a distinguishing feature (83). Although “classic” RDP does not typically present with paroxysmal dyskinesia, there is a phenotypic spectrum on the clinical expressivity of ATP1A3 mutations, with some patients exhibiting intermediary phenotypes.
The expanding spectrum of ATP1A3 disease includes patients with features of both AHC and RDP having partial features of each or fulfilling the diagnostic criteria of both; (84, 85) or patients who share features with RDP or AHC but with distinct characteristics of the D-DEMØ phenotype - Dystonia, Dysmorphism of the face, Encephalopathy with developmental delay, MRI abnormalities of the brain always including cerebellar hypoplasia, No hemiplegia (Ø), and neonatal onset (86). Finally, patients with mutations in ATP1A3 may also have CAPOS syndrome (cerebellar ataxia, areflexia, pes cavus, optic atrophy syndrome, and sensorineural hearing loss), isolated ataxia or other varied phenotypes (85, 87, 88).
The clinical characteristics of paroxysmal dystonia in AHC are of particular interest: [1] It is often precipitated by stress, hot, or cold water exposure; [2] Onset usually occurs before age 18 months (including first day of life), although later onset is possible; [3] Within the same spell, patients often have dystonia of one or more limbs/trunk with concurrent hemiplegia in other limbs, the limb can start with hemiplegia and then develop dystonia or vice versa. Dystonic and hemiplegic attacks can frequently end with an epileptic seizure or directly follow an epileptic seizure. This often represents a diagnostic challenge that can be addressed only with video EEG monitoring during an acute event to rule out ongoing electrographic seizure activity. It is important to detect such activity early on during an acute AHC event in which the distinction cannot be clearly made. This is because such events may evolve into status epilepticus, convulsive, or non-convulsive, which has the risk of developmental regression (82, 89, 90). In some patients, the hemiplegic component is more important in early childhood than later, with the opposite happening for the dystonia (89, 91, 92).
Patients with AHC may have associated motor impairments, mostly in oromotor functioning leading to dysphagia, followed by fine motor and gross motor impairments. The swallowing problems are at times, but not always, due to oral-pharyngeal dystonia (90). Associated tone abnormalities in-between AHC attacks are also seen. More often, individuals present with decreased tone, but a minority may be spastic or continuously dystonic. As the patient gets older, there is often a reduction of hypotonia and of abnormal ocular movements (93).
Paroxysmal dystonia can occur in AHC irrespective of underlying mutation in ATP1A3, including the three most common mutations (D801N, E815K, and G947R). The first two mutations are associated with more severe epileptic and developmental manifestations, dominating the clinical picture such that dystonia is often, but not always, less of a problem than the other manifestations. In individuals with the G947R mutation which has a less severe phenotype, dystonia often appears to be more noticeable and is more commonly reported in comparison to the other mutations (94, 95).
The type and severity of epilepsy in ATP1A3-related disease can vary greatly. It ranges from neonatal onset, as in the D-DEMØ and similar syndromes, to adulthood in patients with RDP (86, 96). The age of seizure onset is best studied in AHC, with an average of 1.83 ± 3.00 years (80).
There is evidence that more severe phenotypes are associated with mutations affecting the transmembrane and functional domains of the protein (97). Some early authors pointed out to the idea that mutations that reduced protein expressivity were more likely to cause RDP, whereas those associated with AHC did not (78). Albeit imperfect, this correlation suggested that the mechanism of disease occurred through hypomorphic effects on RDP whereas those associated with AHC did so through modulating the activity of the Na+/K+ ATPase pump.
One potential mechanism of disease in individuals with ATP1A3 mutations is that dysfunction of the Na+/K+ ATPase pump results in depolarization that leads to abnormal Na+ and K+ exchange process and disturbed proton transport (Figure 1), These abnormalities can potentially lead to subsequent opening of voltage-gated calcium channels. The increased intracellular Ca2+ concentration may then cause downstream activation of multiple signal transduction pathways (98–101). In reality, the pathophysiology is more complex with lines of evidence pointing to distinct mechanisms through which ATP1A3 mutations perturb normal physiology. Some examples include:
• Increased excitability of cortical and hippocampal neurons and impairment of firing of parvalbumin-positive fast-spiking interneurons, likely leading to epilepsy as demonstrated in the Mashlool mouse carrying the most common mutation causing AHC (D801N) (102).
• Impaired trafficking of mutant αβ complex to Golgi apparatus and plasma membrane. This was demonstrated in HEK-293 cells expressing ATP1A3 mutations associated with a severe phenotype of microcephaly, developmental delay, and dystonia, indicating competition between exogenous mutant ATP1A3 (α3) and endogenous ATP1A1 (α1) so that their sum was constant (103).
• Increased predisposition to cortical spreading depolarization (depression), a known mechanism for hemiplegic migraine, as demonstrated in the Mashlool mouse (104). Notably, spreading depolarization exerts a modulatory effect on the function of the basal ganglia (105–107). In addition, spreading depression can occur in the basal ganglia and more directly lead to dysfunction of that system (108). Thus, spreading depolarization may be a mechanism for dystonia too.
• Irregular firing of Purkinje cells and deep cerebellar nuclei neurons, resulting in dystonia as demonstrated in mice carrying the D801Y mutation (109). A few lines of evidence support the role of the cerebellum in ATP1A3- related disease. Cerebellar hypoplasia is seen in all patients with the D-DEMØ phenotype (86). Cerebellar atrophy and hypometabolism on PET scanning are noted in AHC patients (110, 111).
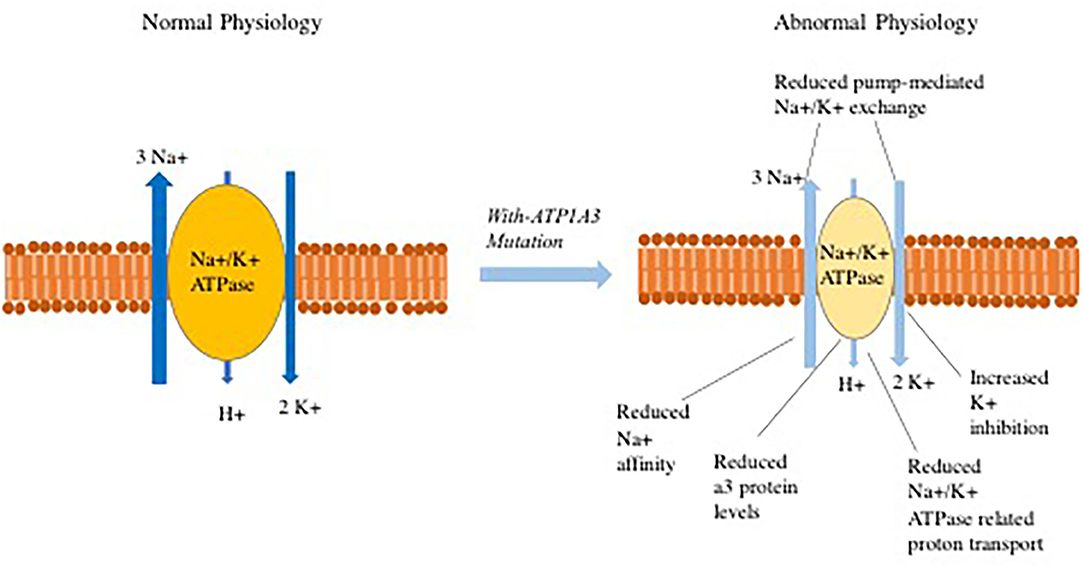
Figure 1. Abnormalities that have been observed in Na+/K+-ATPase related ion transport secondary to ATP1A3 mutations.
Paroxysmal dystonia can also occur in patients who have ATP1A2-related severe epileptic encephalopathy. For example, in a recent report of 6 such patients, 3 of them had dystonic spells in the context of exacerbation. These paroxysmal attack may include acute encephalopathy, hemiplegia, dystonia, and seizures with a duration that may span hours to days (112). There, the mechanism is presumed to be dysfunction of the glial ATPase (ATP1A2 I glial) that impairs re-uptake of glutamate which predisposes to spreading depolarization and increased excitability.
Other genetic causes of the PNKD phenotype frequently co-occur with epilepsy, such as the above cited PKD (e.g., PRRT2) or PED due to SLC2A1 mutations (10). Another disorder associated with paroxysmal non-kinesigenic dyskinesia and seizures is associated with mutations in the GNAO1 gene. The gene product encodes for a subunit of the heterotrimeric structure associated with G protein intracellular signaling, and was initially described in association with epileptic encephalopathy (particularly Ohtahara syndrome), developmental delay, and interictal dyskinetic movements (113). The baseline movement disorder may worsen in association with several physiological triggers (infection, stress, gastrointestinal issues, others) and can be life threatening. Although generalized, the dyskinetic movements may be particularly prominent in the orolingual region (114, 115). Both baseline dyskinetic movements and the paroxysmal exacerbations may respond to deep brain stimulation, although the optimal programming parameters are not yet defined (116). Additional genes in which paroxysmal dyskinetic events have been variably associated with epilepsy and/or an abnormal include SYT1 and RHOBTB2 (117–119).
Lastly, there is increasing recognition of the presence of movement disorders as a defining feature in some epileptic encephalopathies, often referred to as epilepsy-dyskinesia syndromes. In many of these disorders, the movement disorder is present without paroxysmal exacerbations, although the clinical features and/or severity may change over time. Furthermore, it may be difficult sometimes to discern whether an abrupt worsening of the underlying movements is due to their paroxysmal nature vs. worsening due to external factors (e.g., metabolic derangement, infections, etc.). Notable disorders to consider in this scenario include inborn errors of metabolism (neurotransmitter deficiencies, cerebral folate deficiency, certain lysosomal storage disorders, mitochondrial disorders, congenital disorders of glycosylation) and an extensive list of complex monoallelic disorders (e.g., mutations in SCN1A, SCN2A, KCNT1, FOXG1, ARX, GRIN1, FRSS1L, STXBP1, and many others). Reviewing all the epilepsy-dyskinesia syndromes is outside the spectrum of this review; the reader is directed to excellent reviews on this emerging topic (120, 121).
Paroxysmal Exertional Dyskinesia and Epilepsy
Paroxysmal exercise induced dyskinesias (PED) were first reported in the pre-genetic era, describing patients that would have attacks of dystonic posturing precipitated after a variable period of exercise, usually lasting 5–30 min (8). Although the lower limb seems to be most commonly involved, it may be more related to a function of the body part that has been most active in exercise. Upper extremity symptoms have also been reported (122).
An important cause of PED syndrome is related to mutations in the SCL2A1 gene, encoding for the facilitated glucose transporter type 1. The so-called “Glut 1 deficiency,” a syndrome comprising hypoglycorrhachia, seizures, acquired microcephaly, and developmental delay was first described in 1991 (123). De Vivo and colleagues first hypothesized that the fundamental defect was a genetic defect in the glucose transporter in the endothelial cells lining the blood brain barrier and erythrocytes. Treatment with the ketogenic diet, providing the brain with a means of alternative energy utilization, was remarkably successful in improving symptoms. The molecular basis for the disease, associated with monoallelic mutations in SLC2A1 was indeed confirmed a few years later (124).
Patients with mutations in SLC2A1 may have a combination of neurodevelopmental disabilities (e.g., intellectual disability, developmental delay, attentional issues), epilepsy, and movement disorders (spasticity, hypotonia, ataxia, dystonia, and chorea) (125). Most patients exhibit a combination of these features, but mildly affected individuals have also been described. In addition to seizures, several other paroxysmal events may occur in patients with SLC2A1 mutations. Besides PED, these include other paroxysmal movement disorders, paroxysmal head-eye movements in the first year of life, migraines, vomiting, hemiplegic attacks, and lethargic spells. Indeed, SCL2A1 mutations should be placed high in the differential diagnosis for most paroxysmal neurological symptoms with onset in infancy, as well as considered as a possibility in later-onset cases (126–131). Since SLC2A1 mutations cause a highly pleomorphic disease, the neurodevelopmental aspects, epilepsy, and chronic motor disturbances (spasticity, dystonia, or ataxia), may overshadow milder manifestations like isolated PED—likely to be underreported both in the literature and clinical practice. One report of 22 GLUT1 deficiency patients with confirmed SLC2A1 mutations showed that at least half presented with PED, suggesting that as a feature, it is much more common than as an isolated condition at least in SLC2A1-related disease (131). In this report, the age of onset of PED events ranged from 8 months through 14 years old, with mean onset close to 6.5 years of age.
In the majority of patients, seizures are present before age 2 and are diverse in semiology. Complex absences, focal seizures, and cyanotic spells can be seen early on, with myoclonic seizures and generalized tonic-clonic seizures observed later (126, 132). The majority of patients have more than one seizure type, and the most common ones appear to be focal and atypical absences (125, 132). In particular, suspicion for SLC2A1 mutations should be raised when there is a combination of atypical features in absence epilepsy, including early age of onset, intellectual disability, treatment refractoriness, other seizure types, or family history (133, 134).
PED is notable in the sense that it is more likely to be a feature which can compose the phenotypic spectrum of different conditions. This is in contrast to PKD and familial PNKD, in which one gene (PRRT2 for PKD and PNKD for familial cases) account for the majority of cases. In keeping with the pathophysiological construct of metabolic deficiency leading to the manifestations of SLC2A1 mutations, other genes associated with brain energy utilization have also been reported combining seizures and PED. In particular, these include mutations in ECHS1 and pyruvate dehydrogenase deficiency complex, sometimes in the context of lactic acidosis and/or developmental encephalopathy (135, 136). Nevertheless, alternative pathophysiological explanations are necessary to explain other cases of PED. Well-characterized cases of PED have been associated with dopa-responsive dystonia caused by mutations in GCH1 (137) and levodopa-responsive young onset parkinsonism caused by PARKIN mutations (138, 139). Two cases of response to levodopa in PED caused by SLC2A1 mutations (9, 140) further add to the unexpected link between these dopaminergic deficiency and PED. PED episodes associated with epilepsy can also happen in ATP1A3-related disease (in addition to chronic dystonia and parkinsonism) as well as in mutations in TBC1D24 (141–143). In clinical practice, many other cases of PED remain with undetermined etiology.
Episodic Ataxia Type 1 and Epilepsy
What is known today as Episodic Ataxia type 1 (EA1) was first identified by Van Dyke and colleagues in 1975 as hereditary myokymia and episodic ataxia (144). Almost twenty years later, autosomal dominant mutations in the KCNA1 gene were found to be responsible for the disease (26). It encodes for the α-subunit of the Kv1.1 voltage-gated potassium channel, widely expressed in the central and peripheral nervous system, including the hippocampus and cerebellum (145).
The characteristic EA1 patient will present with recurrent ataxic episodes. These episodes may be frequent, short-lasting, and often begin in the first decade of life; genetically confirmed cases often have a positive family history (146). Interictal myokymia is a hallmark of the disease, but may not always be present. Events can be triggered by kinesigenic movement, exertion, or emotional distress (38). Episodes may fade with age, but some patients may have persistent cerebellar ataxia (144, 146). A positive clinical response to acetazolamide and AEDs (mainly carbamazepine) has been reported, however no single drug appears to have widespread efficacy (147).
In addition to spells of episodic ataxia, patients harboring mutations in KCNA1 may also have seizures (146). Epilepsy may present with partial and/or generalized seizures. There are too few cases reported in the literature to establish a particular pattern, but the age of onset thus far reported appears to be variable, occurring in infants through childhood, and adolescence; sometimes preceding episodic ataxia (148–150). KCNA1 mutations associated with epilepsy tend to cluster in the S1/S2 helices and pore domain of the channel (145). A subset of individuals with KCNA1 mutations may have severe forms of epilepsy, where identification of episodic ataxic symptoms may prove more difficult, including a small number of children harboring KCNA1 mutations with epileptic encephalopathy (151, 152). One patient with a variant in KCNA1 had severe myoclonic epilepsy and sudden unexpected death, although it is considered whether polymorphisms in other genes could have contributed to the phenotype (153).
Mutations in KCNA1 have been associated with other phenotypes, including paroxysmal dyskinesia (56), isolated neuromyotonia (148), or myokymia (154), and hypomagnesemia (155).
Episodic Ataxia Type 2 and Epilepsy
Episodic Ataxia type 2 is probably the most prevalent amongst the episodic ataxias. The prototypical EA2 phenotype begins in infancy or early childhood with recurrent spells of unsteadiness, vertigo, and slurring of speech (34). Attacks may be triggered by exertion, fatigue or emotional arousal, and patients may exhibit interictal progressive ataxia, nystagmus, and cerebellar atrophy on imaging (34, 156, 157). EA2 is caused by autosomal dominant mutations in the CACNA1A gene, which encodes the alpha1-subunit of the Cav2.1 P/Q-type voltage gated calcium channel, widely expressed in a variety of cerebral and spinal neuronal populations (157). Mutations in this gene have been associated with different allelic disorders with autosomal dominant inheritance, albeit with phenotypic overlap: familial hemiplegic migraine, EA2, spinocerebellar ataxia type 6 (SCA6), epileptic encephalopathy, seizures, neurodevelopmental disorders, and non-progressive chronic ataxia (156–160). Because SCA6 is caused by repeat expansions of a polyglutamine tract in the CACNA1A gene, it is considered mostly as a separate, allelically related disorder from EA2. Nevertheless, up to a 1/3 of patients with SCA6 may also have episodic features (161, 162). Conversely, chronic progressive ataxia may occur in over 50% of patients with EA2 (163).
A proportion of individuals with CACNA1A mutations may have epilepsy, manifesting with febrile seizures, absence seizures, refractory tonic-clonic seizures, or multiple seizure types associated with epileptic encephalopathy (157, 158, 160, 164–166). Seizure onset in infancy and childhood is commonly reported in the literature, but it is difficult to determine if this is a publication bias vs. a notable clinical feature. The onset of CACNA1A-associated disease spectrum symptoms spans from infancy to adulthood (166). In some mutation carriers (in particular the S218L mutation), minor head trauma may lead to severe hemiplegic migraines associated with lethargy, seizures and brain edema (167). Emerging evidence suggests that mutations in CACNA1A may also be associated with neurodevelopmental disorders such as intellectual disability, learning disabilities, or autism (157, 168, 169).
Therapeutically, the episodes of ataxia in patients with CACNA1A mutations often respond well to acetazolamide (163). The pleiotropic nature of CACNA1A may give rise to other phenotypic characteristics, including migraines, and hemiplegic attacks, sometimes with overlapping features in a single individual (163, 170).
Episodic Ataxia Type 5 and Epilepsy
EA5 is caused by mutations in the voltage-gated calcium channel encoded by the gene CACNB4 (30, 171, 172). Since there are only a few kindreds described, it is difficult to draw conclusions about the phenotypic spectrum. It appears that in the heterozygous state mutations in CACNB4 lead to autosomal dominant EA5 and/or predisposition to epilepsy (particularly myoclonic and generalized types), but larger-scale human studies have not conclusively confirmed the latter (172). In the homozygous state, mutations appear cause a more severe neurodevelopmental disorder (171).
Episodic Ataxia Type 6 and Epilepsy
Episodic ataxia type 6 is caused by monoallelic mutations in SLC1A3, encoding for the excitatory amino acid transporter 1 (EAAT1) (31). This high-affinity sodium-dependent transporter is involved in regulating neurotransmitter concentrations in glutamatergic synapses and is expressed in the cerebellum, diencephalon, and brainstem. From the original description of a young boy with episodic ataxia, seizures, migraine, and alternating hemiplegia (31), the phenotype soon expanded to include individuals with EA2-like phenotypes or late-onset episodic ataxia, without epilepsy (173–175). Experimentally, distinct consequences at the functional and/or expressional level of EAAT1 have been found with different mutations (176), rendering genotype-phenotype correlations unconfirmed in this rare condition.
Other Disorders Variably Associated With Episodic Ataxias and Epilepsy
Episodic ataxia with epilepsy has also been reported in association with mutations in a few other genes, frequently encoding for ion channels. The SCN2A gene, encoding for a voltage-gated sodium channel, has a reported phenotype ranging from epileptic encephalopathy to benign familial neonatal infantile seizures, hyperkinetic movement disorders (chorea, dystonia and myoclonic jerks), neurodevelopmental disorders, and episodic ataxia, which may occasionally respond to acetazolamide (177–180). The KCNA2 gene belongs to the Kv1 family of voltage-gated potassium channels. Pathogenic mutations have been described in the context autosomal dominant epileptic encephalopathy and chronic ataxia (181–184). An episodic nature to the ataxia was described, with some patients having variable associations between attacks and infantile-onset seizures (185).
Therapeutic Overlap
Not surprisingly in light of the significant overlap amongst these conditions, drugs with anti-epileptic properties are frequently used to treat most paroxysmal movement disorders (186, 187).
Phenytoin, an anti-epileptic agent with blocking activity of voltage-gated Na+ channels, was possibly the first drug tried in paroxysmal movement disorders. It was initially reported as a failed therapy in a case of PNKD (5), but then noted to be helpful in a case of so-called reflex epilepsy (which, in retrospect, presumably reflected a case of PKD) (188). With the recognition of kinesigenic triggers, treatment with phenytoin was consolidated as the preferred treatment of the PKD syndrome in the 1960s (6).
Carbamazepine, another AED with Na+ channel blocking activity, debunked phenytoin as the treatment of choice of the PKD syndrome, probably because of its efficacy and improved side effect profile. It was first reported to be efficacious more than 50 years ago (189) and to this day remains the first-line treatment of PKD (190). Carbamazepine responsiveness is a diagnostic criterion for the disease (39). More than 98% of patients with known PRRT2 mutations may respond favorably to the drug (54), and it may also be useful in other non-PRRT2 or acquired cases of PKD (73).
Acetazolamide, a diuretic, is a carbonic anhydrase inhibitor. It blocks the breakdown of carbonic acid, thus causing acidosis. It had been known since the 1920s that starvation (causing ketoacidosis) could improve epilepsy (191). Therefore, when the drug became available it was used to treat epilepsy in the 1950s on the grounds that it could provide a similar effect (192). Initial reports of acetazolamide use were encouraging, but it soon became clear that it had limited efficacy in treating epilepsy (193). It is possible that it may be more efficient in pediatric than adult cases (194), nevertheless its use as an AED has largely been discontinued in favor of other newer and more tolerable agents. Griggs et al. (23) were the first to report successful treatment of familial episodic ataxia (then called paroxysmal ataxia) with acetazolamide. This finding was later reproduced by various other colleagues and proven to be the hallmark of episodic ataxia type 2, dubbed “acetazolamide-responsive hereditary paroxysmal ataxia” in pre-genomic era papers.
The ketogenic diet can improve both epilepsy and PED episodes in SLC2A1 mutation carriers (195), and remains a therapeutic option for refractory epilepsy of various causes (196). Triheptanoin, an odd-chain triglyceride able to produce C5-ketone bodies and provide intermediates to the Krebs cycle, was shown to improve 90% of non-epileptic paroxysmal manifestations in a study of 8 patients with SLC2A1 mutations, including paroxysmal exercise-induced dyskinesia and non-kinesigenic dyskinesia (197). Data on the response of the epileptic attacks is lacking.
Benzodiazepines (mostly clonazepam and diazepam) can be very effective as prophylactic or rescue treatment in genetically confirmed PNKD patients (71, 190), but in other causes of the PNKD syndrome their use may be limited by efficacy or intolerable side effects. Some patients with paroxysmal events associated with mutations in SLC2A1 may also benefit from benzodiazepines (190). Other AEDs, including topiramate, levetiracetam, phenobarbital, gabapentin, valproic acid, lamotrigine have all been anecdotally reported to be useful in PKD (190).
Although not in the category of anti-epileptic agents, two other drugs are worth mentioning. Levodopa has been reported to be efficacious in rare cases of PKD (198) and most consistently in PED associated with mutations in SLC2A1, GCH1, or Parkin (137, 140, 199). 4 -aminopyridine, a potassium channel blocker, was shown in a small trial to reduce the frequency of episodes and improve quality of life in patients episodic ataxia type 2 (200). It may be an option for patients who cannot tolerate acetazolamide.
Finally, despite their therapeutic usefulness, it is worth mentioning that AEDs can also be associated with drug-induced movement disorders (201–204). In most instances, the movement disorders are not paroxysmal and there is a temporal relationship to initiation of a drug, but exceptions exist. The movement disorder may occur at any time during treatment and within therapeutic serum levels. Patient and drug-specific factors may influence symptoms, including pregnancy and drug dosage changes (205–207). Many AEDs are implicated. The most frequent offenders are the older agents (205), including phenytoin (206), carbamazepine (208), and valproate (209). Newer ones such as lamotrigine, levetiracetam, and gabapentin have also been implicated (210–212). In some cases, the offending agent may be toxic and have lasting effects. For example, phenytoin may lead to cerebellar atrophy and ataxia that persists even after the drug is discontinued (213).
Tremor is a frequent abnormal movement reported in association with AEDs, accounting for 45% (89/201) of cases (214, 215). Chorea has been associated with phenytoin and valproate (216–218). Combined use of multiple AEDs may increase this risk (219). Usually seen as an anti-dyskinetic drug to treat chorea, valproate may also induce parkinsonism (209, 220), Ataxia may be induced by phenytoin, carbamazepine, valproate, and lamotrigine (207).
Conclusion
We have attempted to provide a comprehensive overview of select paroxysmal movement disorders and their associations with epilepsy. These distinct phenotypes are coalescing into established genetic etiologies with extensive pleiotropy. Several good diagnostic algorithms exist in the literature (1, 72, 221). We have refrained from designing a diagnostic algorithm for these conditions, as we believe that careful phenotyping at the bedside is just as important for diagnosis as it is for interpreting results from hypothesis-free (agnostic) genetic testing. Moreover, familiarity with paroxysmal movement disorder subtypes may allow clinicians to suspect specific genetic etiologies that, if not identified through next-generation sequencing, may be pursued through other means such as evaluating for nucleotide expansions or copy number variants (221, 222). For several disorders, although genetic causes have been identified, the molecular pathophysiology is still largely unknown. In the near future, it is likely that we will have a growing number of genes associated with movement disorders and epilepsy, in parallel with improved understanding of disease mechanisms leading to more effective treatments.
Author Contributions
CG: original idea, literature review, and first draft. LS-M: analysis and criticism of the first draft and senior input. SS: figure generation and provision of original data. MM: figure generation, provision of original data, analysis and criticism of the first draft and senior input. LG: literature review, data tabulation, and data management.
Funding
CG was supported by the Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior – Brasil (CAPES) – Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Silveira-Moriyama L, Kovac S, Kurian MA, Houlden HH, Lees AJ, Walker MC, et al. Phenotypes, genotypes, and the management of paroxysmal movement disorders. Dev Med Child Neurol. (2018) 60:559–65. doi: 10.1111/dmcn.13744
3. Kato N, Sadamatsu M, Kikuchi T, Niikawa N, Fukuyama Y. Paroxysmal kinesigenic choreoathetosis: from first discovery in 1892 to genetic linkage with benign familial infantile convulsions. Epilepsy Res. (2006) 70(Suppl.1):174–84. doi: 10.1016/j.eplepsyres.2006.02.009
4. Gowers WR. Epilepsy and other Chronic Convulsive Diseases, Their Causes, Symptoms and Treatment. London: J. & A. Churchill (1881).
5. Mount LA, Reback S. Familial paroxysmal choreoathetosis: preliminary report on a hitherto undescribed clinical syndrome. Arch Neurol Psychiatry. (1940) 44:841–7. doi: 10.1001/archneurpsyc.1940.02280100143011
6. Kertesz A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology. (1967) 17:680–90. doi: 10.1212/WNL.17.7.680
7. Crompton DE, Berkovic SF. The borderland of epilepsy: clinical and molecular features of phenomena that mimic epileptic seizures. Lancet Neurol. (2009) 8:370–81. doi: 10.1016/S1474-4422(09)70059-6
8. Lance JW. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol. (1977) 2:285–93. doi: 10.1002/ana.410020405
9. Demirkiran M, Jankovic J. Paroxysmal dyskinesias : clinical features and classlfication. Ann Neurol. (1995) 38:571–9. doi: 10.1002/ana.410380405
10. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain. (2015) 138:3567–80. doi: 10.1093/brain/awv310
11. Lee HY, Xu Y, Huang Y, Ahn AH, Auburger GWJ, Pandolfo M, et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. (2004) 13:3161–70. doi: 10.1093/hmg/ddh330
12. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. (2008) 131:1831–44. doi: 10.1093/brain/awn113
13. Wang JL, Cao L, Li XH, Hu ZM, Li Da J, Zhang JG, et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain. (2011) 134(Pt 12):3493–501. doi: 10.1093/brain/awr289
14. Chen W-J, Lin Y, Xiong Z-Q, Wei W, Ni W, Tan G-H, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. (2011) 43:1252–5. doi: 10.1038/ng.1008
15. Provini F, Plazzi G, Lugaresi E. From nocturnal paroxysmal dystonia to nocturnal frontal lobe epilepsy. Clin Neurophysiol. (2000) 111(Suppl 2):S2–8. doi: 10.1016/S1388-2457(00)00396-5
16. Tinuper P, Bisulli F, Provini F, Lugaresi E. Familial frontal lobe epilepsy and its relationship with other nocturnal paroxysmal events. Epilepsia. (2010) 51:51–3. doi: 10.1111/j.1528-1167.2009.02446.x
17. Liu X-R, Huang D, Wang J, Wang Y-F, Sun H, Tang B, et al. Paroxysmal hypnogenic dyskinesia is associated with mutations in the PRRT2 gene. Neurol Genet. (2016) 2:e66. doi: 10.1212/NXG.0000000000000066
19. Klaas JP, Burkholder DB, Singer W, Boes CJ. Harry lee parker and paroxysmal dysarthria and ataxia. Neurology. (2013) 80:311–4. doi: 10.1212/WNL.0b013e31827dec0f
20. Farmer TW, Mustian VM. Vestibulocerebellar Ataxia: a newly defined hereditary syndrome with periodic manifestations. Arch Neurol. (1963) 8:471–80. doi: 10.1001/archneur.1963.00460050021002
21. White JC. Familial periodic nystagmus, vertigo, and Ataxia. Arch Neurol. (1969) 20:276–80. doi: 10.1001/archneur.1969.00480090064010
22. Hanson PA, Martinez LB, Cassidy R. Contractures, continuous muscle discharges, and titubation. Ann Neurol. (1977):120–4. doi: 10.1002/ana.410010203
23. Griggs RC, Moxley RT, Lafrance RA, McQuillen J. Hereditary paroxysmal ataxia: response to acetazolamide. Neurology. (1978) 28:1259–64. doi: 10.1212/WNL.28.12.1259
24. Donat JR, Auger R. Familial periodic Ataxia. Arch Neurol. (1979) 36:568–9. doi: 10.1001/archneur.1979.00500450062011
25. Gancher ST, Nutt JG. Autosomal dominant episodic ataxia: a heterogeneous syndrome. Mov Disord. (1986) 1:239–53. doi: 10.1002/mds.870010404
26. Browne DL, Gancher ST, Nutt JG, Brunt ERP, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. (1994) 8:136–40. doi: 10.1038/ng1094-136
27. Brederlow Von B, Hahn AF, Koopman WJ, Ebers GC, Bulman DE. Mapping the gene for acetazolamide responsive hereditary paryoxysmal cerebellar ataxia to chromosome 19p. Hum Mol Genet. (1995) 4:279–84. doi: 10.1093/hmg/4.2.279
28. Vahedi K, Joutel A, van Bogaert P, Ducros A, Maciazeck J, Bach JFO, et al. A gene for hereditary paroxysmal cerebellar ataxia maps to chromosome 19p. Ann Neurol. (1995) 37:289–93. doi: 10.1002/ana.410370304
29. Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. (1996) 87:543–52. doi: 10.1016/S0092-8674(00)81373-2
30. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel β4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic Ataxia. Am J Hum Genet. (2000) 66:1531–9. doi: 10.1086/302909
31. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. (2005) 65:529–34. doi: 10.1212/01.WNL.0000172638.58172.5a
32. Choquet K, La Piana R, Brais B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics. (2015) 16:233–6. doi: 10.1007/s10048-014-0436-7
33. Piarroux J, Riant F, Humbertclaude V, Remerand G, Hadjadj J, Rejou F, et al. FGF14-related episodic ataxia: delineating the phenotype of Episodic Ataxia type 9. Ann Clin Transl Neurol. (2020) 7:565–72. doi: 10.1002/acn3.51005
34. Jen JC, Wan J. Episodic ataxias. In: Handbook of Clinical Neurology. Elsevier (2018). p. 205–15. Available online at: https://www-sciencedirect-com.ezp-prod1.hul.harvard.edu/science/article/pii/B9780444641892000135?via%3Dihub doi: 10.1016/B978-0-444-64189-2.00013-5 (accessed May 2, 2019).
35. Russell JF, Fu YH, Ptáček LJ. Episodic neurologic disorders: Syndromes, genes, and mechanisms. Annu Rev Neurosci. (2013) 36:25–50. doi: 10.1146/annurev-neuro-062012-170300
36. Silveira-Moriyama L, Gardiner AR, Meyer E, King MD, Smith M, Rakshi K, et al. Clinical features of childhood-onset paroxysmal kinesigenic dyskinesia with PRRT2 gene mutations. Dev Med Child Neurol. (2013) 55:327–34. doi: 10.1111/dmcn.12056
37. Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, et al. PRRT2 gene mutations from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology. (2012) 8:657. doi: 10.1212/WNL.0b013e3182752c5a
38. de Gusmao CM, Garcia LR, Jesuthasan A, Muir M, Paciorkowski A, Mink JW, et al. Kinesigenic triggers in episodic ataxia type 1. Movement Disorders Clinical Practice, Vol. 7. Wiley-Blackwell (2020). p. 723–4. Available online at: https://onlinelibrary.wiley.com/doi/abs/10.1002/mdc3.13008 (accessed Sep 15, 2020).
39. Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. (2004) 63:2280–7. doi: 10.1212/01.WNL.0000147298.05983.50
40. Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet. (1997) 61:889–98. doi: 10.1086/514877
41. Ono S, Yoshiura K, Kinoshita A, Kikuchi T, Nakane Y, Kato N, et al. Mutations in PRRT2 responsible for paroxysmal kinesigenic dyskinesias also cause benign familial infantile convulsions. J Hum Genet. (2012) 57:338–41. doi: 10.1038/jhg.2012.23
42. Heron SE, Grinton BE, Kivity S, Afawi Z, Zuberi SM, Hughes JN, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. (2012) 90:152–60. doi: 10.1016/j.ajhg.2011.12.003
43. Whitty CWM, Lishman WA, Fitzgibbon JP. Seizures induced by movement: a form of reflex epilepsy. Lancet. (1964) 283:1403–6. doi: 10.1016/S0140-6736(64)91980-4
44. Perez-Borja C, Tassinari AC, Swanson AG. Paroxysmal choreoathetosis and seizures induced by movement (Reflex Epilepsy). Epilepsia. (1967) 8:260–70. doi: 10.1111/j.1528-1157.1967.tb04442.x
45. Lombroso CT. Paroxysmal choreoathetosis: an epileptic or non-epileptic disorder? Ital J Neurol Sci. (1995) 16:271–7. doi: 10.1007/BF02249102
46. Ohmori I, Ohtsuka Y, Ogino T, Yoshinaga H, Kobayashi K, Oka E. The relationship between paroxysmal kinesigenic choreoathetosis and epilepsy. Neuropediatrics. (2002) 33:15–20. doi: 10.1055/s-2002-23594
47. van Strien TW, van Rootselaar AF, Hilgevoord AAJ, Linssen WHJP, Groffen AJA, Tijssen MAJ. Paroxysmal kinesigenic dyskinesia: cortical or non-cortical origin. Park Relat Disord. (2012) 18:645–8. doi: 10.1016/j.parkreldis.2012.03.006
48. Ko CH, Kong CK, Ngai WT, Ma KM. Ictal 99mTc ECD SPECT in paroxysmal kinesigenic choreoathetosis. Pediatr Neurol. (2001) 24:225–7. doi: 10.1016/S0887-8994(00)00257-5
49. Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LHG, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. (2016) 79:428–36. doi: 10.1002/ana.24580
50. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138:3476–95. doi: 10.1093/brain/awv317
51. Chahine LM, Mikati MA. Benign pediatric localization-related epilepsies: part II. Syndromes in childhood. Epileptic Disord. (2006) 8:243–58.
52. Scheffer IE, Grinton BE, Heron SE, Kivity S, Afawi Z, Iona X, et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile seizures. Neurology. (2012) 79:2104–8. doi: 10.1212/WNL.0b013e3182752c6c
53. He Z-W, Qu J, Zhang Y, Mao C-X, Wang Z-B, Mao X-Y, et al. PRRT2 mutations are related to febrile seizures in epileptic patients. Int J Mol Sci. (2014) 15:23408–17. doi: 10.3390/ijms151223408
54. Huang X-J, Wang T, Wang JL, Liu XL, Che XQ, Li J, et al. Paroxysmal Kinesigenic Dyskinesia. Clinical and genetic analysis of 110 patients. Neurology. (2015) 85:1546–53. doi: 10.1212/WNL.0000000000002079
55. Van Vliet R, Breedveld G, De Rijk-Van Andel J, Brilstra E, Verbeek N, et al. PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology. (2012) 79:777–84. doi: 10.1212/WNL.0b013e3182661fe3
56. Yin XM, Lin JH, Cao L, Zhang TM, Zeng S, Zhang KL, et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum Mol Genet. (2018) 27:625–37. doi: 10.1093/hmg/ddx430
57. Erro R, Bhatia KP. Unravelling of the paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry. (2019) 90:227–34. doi: 10.1136/jnnp-2018-318932
58. Méneret A, Grabli D, Depienne C, Gaudebout C, Picard F, Dürr A, et al. PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia in the European population. Neurology. (2012) 79:170–4. doi: 10.1212/WNL.0b013e31825f06c3
59. Lee YC, Lee MJ, Yu HY, Chen C, Hsu CH, Lin KP, et al. PRRT2 mutations in paroxysmal kinesigenic dyskinesia with infantile convulsions in a Taiwanese cohort. PLoS ONE. (2012) 7:e38543. doi: 10.1371/journal.pone.0038543
60. Fruscione F, Valente P, Sterlini B, Romei A, Baldassari S, Fadda M, et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. (2018) 141:1000–16. doi: 10.1093/brain/awy051
61. Erro R, Bhatia KP, Espay AJ, Striano P. The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: channelopathies, synaptopathies, and transportopathies. Mov Disord. (2017) 32:310–8. doi: 10.1002/mds.26901
62. El Achkar CM, Rosen Sheidley B, O'Rourke D, Takeoka M, Poduri A. Compound heterozygosity with PRRT2: pushing the phenotypic envelope in genetic epilepsies. Epilepsy Behav Case Rep. (2017) 11:125–8. doi: 10.1016/j.ebcr.2016.12.001
63. Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. (2012) 53:e196–9. doi: 10.1111/epi.12009
64. Dale RC, Gardiner A, Branson JA, Houlden H. Benefit of carbamazepine in a patient with hemiplegic migraine associated with PRRT2 mutation. Dev Med Child Neurol. (2014) 56:910. doi: 10.1111/dmcn.12396
65. Pons L, Lesca G, Sanlaville D, Chatron N, Labalme A, Manel V, et al. Neonatal tremor episodes and hyperekplexia-like presentation at onset in a child with SCN8A developmental and epileptic encephalopathy. Epileptic Disord. (2018) 20:289–94. doi: 10.1684/epd.2018.0988
66. Tian WT, Huang XJ, Mao X, Liu Q, Liu XL, Zeng S, et al. Proline-rich transmembrane protein 2–negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov Disord. (2018) 33:459–67. doi: 10.1002/mds.27274
67. Jiang li Y, Yuan F, Yang Y, Sun long X, Song L, Jiang W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure. (2018) 56:88–91. doi: 10.1016/j.seizure.2018.02.005
68. Fuchs O, Pearr N, Pohlensz J, Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. (2009) 51:240–4. doi: 10.1111/j.1469-8749.2008.03125.x
69. Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. (2005) 252:663–6. doi: 10.1007/s00415-005-0713-3
70. Shen Y, Ge W-P, Li Y, Hirano A, Lee H-Y, Rohlmann A, et al. Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc Natl Acad Sci USA. (2015) 112:2935–41. doi: 10.1073/pnas.1501364112
71. Bruno MK, Lee HY, Auburger GWJ, Friedman A, Nielsen JE, Lang AE, et al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology. (2007) 68:1782–9. doi: 10.1212/01.wnl.0000262029.91552.e0
72. Meneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol. (2016) 172:433–45. doi: 10.1016/j.neurol.2016.07.005
73. Blakeley J, Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord. (2002) 17:726–34. doi: 10.1002/mds.10178
74. Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord. (2015) 30:1290–2. doi: 10.1002/mds.26216
75. Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. (2005) 37:733–8. doi: 10.1038/ng1585
76. Zhang G, Gibson RA, McDonald M, Liang P, Kang PW, Shi J, et al. A Gain-of-function mutation in KCNMA1 causes dystonia spells controlled with stimulant therapy. Mov Disord. (2020) 35:1868–73. doi: 10.1002/mds.28138
77. de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, et al. Mutations in the Na+/K+-ATPase α3 Gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. (2004) 43:169–75. doi: 10.1016/j.neuron.2004.06.028
78. Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, De Vries B, Tiziano FD, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. (2012) 44:1030–4. doi: 10.1038/ng.2358
79. Verret S, Steele JC. Alternating hemiplegia in childhood: a report of eight patients with complicated migraine beggining in infancy. Pediatrics. (1971) 47:675–80.
80. Uchitel J, Helseth A, Prange L, McLean M, Ghusayni R, Sachdev M, et al. The epileptology of alternating hemiplegia of childhood. Neurology. (2019) 93:e1248–59. doi: 10.1212/WNL.0000000000008159
81. Marzin P, Mignot C, Dorison N, Dufour L, Ville D, Kaminska A, et al. Early-onset encephalopathy with paroxysmal movement disorders and epileptic seizures without hemiplegic attacks: about three children with novel ATP1A3 mutations. Brain Dev. (2018) 40:768–74. doi: 10.1016/j.braindev.2018.05.008
82. Stagnaro M, Pisciotta L, Gherzi M, Di Rocco M, Gurrieri F, Parrini E, et al. ATP1A3 spectrum disorders: a video-documented history of 7 genetically confirmed early onset cases. Eur J Paediatr Neurol. (2018) 22:264–71. doi: 10.1016/j.ejpn.2018.01.010
83. Balint B, Stephen CD, Udani V, Sankhla CS, Barad NH, Lang AE, et al. Paroxysmal asymmetric dystonic arm posturing—a less recognized but characteristic manifestation of ATP1A3-related disease. Mov Disord Clin Pract. (2019) 6:212–5. doi: 10.1002/mdc3.12747
84. Tran L, Richards J, McDonald M, McConkie-Rosell A, Stong N, Jasien J, et al. Epileptic encephalopathy with features of rapid-onset dystonia Parkinsonism and alternating hemiplegia of childhood: a novel combination phenotype associated with ATP1A3 mutation. Epileptic Disord. (2020) 22:103–9. doi: 10.1684/epd.2020.1127
85. Fernandes C, Mikati MA. The expanding spectrum of ATP1A3 related disease. Eur J Paediatr Neurol. (2019) 23:345–6. doi: 10.1016/j.ejpn.2019.05.007
86. Prange L, Pratt M, Herman K, Schiffmann R, Mueller DM, McLean M, et al. D-DEMØ, a distinct phenotype caused by ATP1A3 mutations. Neurol Genet. (2020) 6:e466. doi: 10.1212/NXG.0000000000000466
87. Demos MK, van Karnebeek CD, Ross CJ, Adam S, Shen Y, Zhan SH, et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J Rare Dis. (2014) 9:15. doi: 10.1186/1750-1172-9-15
88. de Gusmao CM, Dy M, Sharma N. Beyond dystonia-parkinsonism: chorea and Ataxia with ATP1A3 mutations. Mov Disord Clin Pract. (2016) 3:402–4. doi: 10.1002/mdc3.12317
89. Mikati MA, Kramer U, Zupanc ML, Shanahan RJ. Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr Neurol. (2000) 23:134–41. doi: 10.1016/S0887-8994(00)00157-0
90. Masoud M, Gordon K, Hall A, Jasien J, Lardinois K, Uchitel J, et al. Motor function domains in alternating hemiplegia of childhood. Dev Med Child Neurol. (2017) 59:822–8. doi: 10.1111/dmcn.13443
91. Mikati MA, Maguire H, Barlow CF, Ozelius L, Breakefield XO, Klauck SM, et al. A syndrome of autosomal dominant alternating hemiplegia: clinical presentation mimicking intractable epilepsy; chromosomal studies; and physiologic investigations. Neurology. (1992) 42:2251–7. doi: 10.1212/WNL.42.12.2251
92. Masoud M, Prange L, Wuchich J, Hunanyan A, Mikati MA. Diagnosis and treatment of alternating hemiplegia of childhood. Curr Treat Options Neurol. (2017) 19:8. doi: 10.1007/s11940-017-0444-7
93. Kansagra S, Mikati MA, Vigevano F. Alternating hemiplegia of childhood. Handb Clin Neurol. (2013) 112:821–6. doi: 10.1016/B978-0-444-52910-7.00001-5
94. Panagiotakaki E, Doummar D, Nogue E, Nagot N, Lesca G, Riant F, et al. Movement disorders in patients with alternating hemiplegia: “soft” and “stiff” at the same time. Neurology. (2020) 94:e1378–85. doi: 10.1212/WNL.0000000000009175
95. Panagiotakaki E, de Grandis E, Stagnaro M, Heinzen EL, Fons C, Sisodiya S, et al. Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood - a study of 155 patients. Orphanet J Rare Dis. (2015) 10:123. doi: 10.1186/s13023-015-0335-5
96. Haq IU, Snively BM, Sweadner KJ, Suerken CK, Cook JF, Ozelius LJ, et al. Revising rapid-onset dystonia–parkinsonism: broadening indications for ATP1A3 testing. Mov Disord. (2019) 34:1528–36. doi: 10.1002/mds.27801
97. Rosewich H, Ohlenbusch A, Huppke P, Schlotawa L, Baethmann M, Carrilho I, et al. The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology. (2014) 82:945–55. doi: 10.1212/WNL.0000000000000212
98. Helseth AR, Hunanyan AS, Adil S, Linabarger M, Sachdev M, Abdelnour E, et al. Novel E815K knock-in mouse model of alternating hemiplegia of childhood. Neurobiol Dis. (2018) 119:100–12. doi: 10.1016/j.nbd.2018.07.028
99. Heinzen EL, Arzimanoglou A, Brashear A, Clapcote SJ, Gurrieri F, Goldstein DB, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. (2014) 13:503–14. doi: 10.1016/S1474-4422(14)70011-0
100. Holm TH, Lykke-Hartmann K. Insights into the pathology of the α3 Na+/K+-ATPase ion pump in neurological disorders; lessons from animal models. Front Physiol. (2016) 7:209. doi: 10.3389/fphys.2016.00209
101. Simmons CQ, Thompson CH, Cawthon BE, Westlake G, Swoboda KJ, Kiskinis E, et al. Direct evidence of impaired neuronal Na/K-ATPase pump function in alternating hemiplegia of childhood. Neurobiol Dis. (2018) 115:29–38. doi: 10.1016/j.nbd.2018.03.009
102. Hunanyan AS, Helseth AR, Abdelnour E, Kherallah B, Sachdev M, Chung L, et al. Mechanisms of increased hippocampal excitability in the Mashl +/− mouse model of Na + /K + -ATPase dysfunction. Epilepsia. (2018) 59:1455–68. doi: 10.1111/epi.14441
103. Arystarkhova E, Haq IU, Luebbert T, Mochel F, Saunders-Pullman R, Bressman SB, et al. Factors in the disease severity of ATP1A3 mutations: impairment, misfolding, and allele competition. Neurobiol Dis. (2019) 132:104577. doi: 10.1016/j.nbd.2019.104577
104. Hunanyan AS, Fainberg NA, Linabarger M, Arehart E, Leonard AS, Adil SM, et al. Knock-in mouse model of alternating hemiplegia of childhood: behavioral and electrophysiologic characterization. Epilepsia. (2015) 56:82–93. doi: 10.1111/epi.12878
105. de Iure A, Napolitano F, Beck G, Quiroga Varela A, Durante V, Sciaccaluga M, et al. Striatal spreading depolarization: possible implication in levodopa-induced dyskinetic-like behavior. Mov Disord. (2019) 34:832–44. doi: 10.1002/mds.27632
106. Seghatoleslam M, Ghadiri MK, Ghaffarian N, Speckmann E-J, Gorji A. Cortical spreading depression modulates the caudate nucleus activity. Neuroscience. (2014) 267:83–90. doi: 10.1016/j.neuroscience.2014.02.029
107. Arabia A-M, Shen P-J, Gundlach AL. Increased striatal proenkephalin mRNA subsequent to production of spreading depression in rat cerebral cortex: activation of corticostriatal pathways? Mol Brain Res. (1998) 61:195–202. doi: 10.1016/S0169-328X(98)00189-2
108. Albe-Fessard D, Sanderson P, Mavoungou R. The influence of striatum on the substantia nigra: a study using the spreading depression technique. Brain Res Bull. (1990) 24:213–9. doi: 10.1016/0361-9230(90)90208-H
109. Isaksen TJ, Kros L, Vedovato N, Holm TH, Vitenzon A, Gadsby DC, et al. Hypothermia-induced dystonia and abnormal cerebellar activity in a mouse model with a single disease-mutation in the sodium-potassium pump. Petrou S, editor. PLoS Genet. (2017) 13:e1006763. doi: 10.1371/journal.pgen.1006763
110. Ghusayni R, Richardson JP, Uchitel J, Abdelnour E, McLean M, Prange L, et al. Magnetic resonance imaging volumetric analysis in patients with Alternating hemiplegia of childhood: a pilot study. Eur J Paediatr Neurol. (2020) 26:15–9. doi: 10.1016/j.ejpn.2020.02.001
111. Mikati M. Alternating hemiplegia of childhood. Vol. 21, Pediatric Neurology. Elsevier Inc (1999). p. 764. Available online at: https://linkinghub.elsevier.com/retrieve/pii/S0887899499000909
112. Moya-Mendez M, Mueller D, Pratt M, Bonner M, Elliott C, Hunanyan A, et al. ATP1A2 encephalopathy: case series to expand the clinical spectrum. Epilepsy Behav. (2021).
113. Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, et al. De novo mutations in GNAO1, encoding a gαo subunit of heterotrimeric g proteins, cause epileptic encephalopathy. Am J Hum Genet. (2013) 93:496–505. doi: 10.1016/j.ajhg.2013.07.014
114. Ananth AL, Robichaux-Viehoever A, Kim Y-M, Hanson-Kahn A, Cox R, Enns GM, et al. Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr Neurol. (2016) 59:81–4. doi: 10.1016/j.pediatrneurol.2016.02.018
115. Waak M, Mohammad SS, Coman D, Sinclair K, Copeland L, Silburn P, et al. GNAO1-related movement disorder with life-threatening exacerbations: movement phenomenology and response to DBS. J Neurol Neurosurg Psychiatr. (2017) jnnp-2017–315653. doi: 10.1136/jnnp-2017-315653
116. Koy A, Cirak S, Gonzalez V, Becker K, Roujeau T, Milesi C, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci. (2018) 391:31–9. doi: 10.1016/j.jns.2018.05.018
117. Baker K, Gordon SL, Grozeva D, Van Kogelenberg M, Roberts NY, Pike M, et al. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J Clin Invest. (2015) 125:1670–8. doi: 10.1172/JCI79765
118. Straub J, Konrad EDH, Grüner J, Toutain A, Bok LA, Cho MT, et al. Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in Drosophila. Am J Hum Genet. (2018) 102:44–57. doi: 10.1016/j.ajhg.2017.11.008
119. Necpál J, Zech M, Valachová A, Sedláček Z, Bendová Š, Hančárová M, et al. Severe paroxysmal dyskinesias without epilepsy in a RHOBTB2 mutation carrier. Park Relat Disord. (2020) 77:87–8. doi: 10.1016/j.parkreldis.2020.06.028
120. Papandreou A, Danti FR, Spaull R, Leuzzi V, Mctague A, Kurian MA. The expanding spectrum of movement disorders in genetic epilepsies. Dev Med Child Neurol. (2020) 62:178–91. doi: 10.1111/dmcn.14407
121. Carecchio M, Mencacci NE. Emerging monogenic complex hyperkinetic disorders. Vol. 17. Current Neurology and Neuroscience Reports. Current Medicine Group LLC 1. (2017). doi: 10.1007/s11910-017-0806-2
122. Clark CN, Weber YW, Lerche H, Warner TT. Paroxysmal exercise-induced dyskinesia of the hands. Mov Disord. (2012) 27:1579–80. doi: 10.1002/mds.25200
123. De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. (1991) 325:703–9. doi: 10.1056/NEJM199109053251006
124. Seidner G, Alvarez MG, Yeh JI, O'Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood- brain barrier hexose carrier. Nat Genet. (1998) 18:188–91. doi: 10.1038/ng0298-188
125. Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. (2013) 13:342. doi: 10.1007/s11910-013-0342-7
126. Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, Van Engelen BG, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. (2010) 133:655–70. doi: 10.1093/brain/awp336
127. Pearson TS, Pons R, Engelstad K, Kane SA, Goldberg ME, De Vivo DC. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology. (2017) 88:1666–73. doi: 10.1212/WNL.0000000000003867
128. Urbizu A, Cuenca-León E, Raspall-Chaure M, Gratacòs M, Conill J, Redecillas S, et al. Paroxysmal exercise-induced dyskinesia, writer's cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J Neurol Sci. (2010) 295:110–3. doi: 10.1016/j.jns.2010.05.017
129. Weller CM, Leen WG, Neville BGR, Duncan JS, De Vries B, Geilenkirchen MA, et al. A novel SLC2A1 mutation linking hemiplegic migraine with alternating hemiplegia of childhood. Cephalalgia. (2015) 35:10–5. doi: 10.1177/0333102414532379
130. Rotstein M, Doran J, Yang H, Ullner PM, Engelstad K, De Vivo DC. Glut1 deficiency and alternating hemiplegia of childhood. Neurology. (2009) 73:2042–4. doi: 10.1212/WNL.0b013e3181c55ebf
131. De Giorgis V, Teutonico F, Cereda C, Balottin U, Bianchi M, Giordano L, et al. Sporadic and familial glut1ds Italian patients: a wide clinical variability. Seizure. (2015) 24:28–32. doi: 10.1016/j.seizure.2014.11.009
132. Hully M, Vuillaumier-Barrot S, Le Bizec C, Boddaert N, Kaminska A, Lascelles K, et al. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet. (2015) 58:443–54. doi: 10.1016/j.ejmg.2015.06.007
133. Soto-Insuga V, López RG, Losada-Del Pozo R, Rodrigo-Moreno M, Cayuelas EM, Giráldez BG, et al. Glut1 deficiency is a rare but treatable cause of childhood absence epilepsy with atypical features. Epilepsy Res. (2019) 154:39–41. doi: 10.1016/j.eplepsyres.2019.04.003
134. Arsov T, Mullen SA, Damiano JA, Lawrence KM, Huh LL, Nolan M, et al. Early onset absence epilepsy: 1 in 10 cases is caused by GLUT1 deficiency. Epilepsia. (2012) 53:e204–7. doi: 10.1111/epi.12007
135. Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. (2017) 264:185–7. doi: 10.1007/s00415-016-8381-z
136. Friedman J, Feigenbaum A, Chuang N, Silhavy J, Gleeson JG. Pyruvate Dehydrogenase Complex-e2 Deficiency Causes Paroxysmal Exerciseinduced Dyskinesia. Vol. 89. Neurology. Lippincott Williams and Wilkins (2017). p. 2297–8. Available online at: https://pubmed.ncbi.nlm.nih.gov/29093066/ doi: 10.1212/WNL.0000000000004689
137. Dale RC, Melchers A, Fung VS, Grattan-Smith P, Houlden H, Earl J. Familial paroxysmal exercise-induced dystonia: Atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. (2010) 52:583–6. doi: 10.1111/j.1469-8749.2010.03619.x
138. Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain. (2003) 126:1279–92. doi: 10.1093/brain/awg142
139. Bozi M, Bhatia KP. Paroxysmal exercise-induced dystonia as a presenting feature of young-onset Parkinson's disease. Mov Disord. (2003) 18:1545–7. doi: 10.1002/mds.10597
140. Baschieri F, Batla A, Erro R, Ganos C, Cordivari C, Bhatia KP. Paroxysmal exercise-induced dystonia due to GLUT1 mutation can be responsive to levodopa: a case report. J Neurol. (2014) 261:615–6. doi: 10.1007/s00415-014-7250-x
141. Roubergue A, Roze E, Vuillaumier-Barrot S, Fontenille M-J, Méneret A, Vidailhet M, et al. The multiple faces of the ATP1A3 -related dystonic movement disorder. Mov Disord. (2013) 28:1457–9. doi: 10.1002/mds.25396
142. Steel D, Heim J, Kruer MC, Sanchis-Juan A, Raymond LF, Eunson P, et al. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov Disord. (2020) 35:372–3. doi: 10.1002/mds.27981
143. Lüthy K, Mei D, Fischer B, De Fusco M, Swerts J, Paesmans J, et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: Rescue by antioxidants in a disease model. Brain. (2019) 142:2319–35. doi: 10.1093/brain/awz175
144. Van Dyke DH, Griggs RC, Murphy MJ, Goldstein MN. Hereditary myokymia and periodic ataxia. J Neurol Sci. (1975) 25:109–18. doi: 10.1016/0022-510X(75)90191-4
145. Paulhus K, Ammerman L, Glasscock E. Clinical Spectrum of KCNA1 Mutations: New Insights Into Episodic Ataxia and Epilepsy Comorbidity. Vol. 21 International Journal of Molecular Sciences. MDPI AG (2020). Available online at: https://pubmed.ncbi.nlm.nih.gov/32316562/ doi: 10.3390/ijms21082802 (accessed Dec 6, 2020).
146. Graves TD, Cha Y-H, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. (2014) 137:1009–18. doi: 10.1093/brain/awu012
147. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. (2007) 130:2484–93. doi: 10.1093/brain/awm126
148. Eunson L, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. (2000) 48:647–56. doi: 10.1002/1531-8249(200010)48:4<647::AID-ANA12>3.0.CO;2-Q
149. Zuberi SM, Eunson LH, Spauschus A, Silva De R, Tolmie J, Wood NW, et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1. 1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. (1999) 122:817–25. doi: 10.1093/brain/122.5.817
150. Tristán-Clavijo E, Scholl FG, Macaya A, Iglesias G, Rojas AM, Lucas M, et al. Dominant-negative mutation p.Arg324Thr in KCNA1 impairs Kv1.1 channel function in episodic ataxia. Mov Disord. (2016) 31:1743–8. doi: 10.1002/mds.26737
151. Rogers A, Golumbek P, Cellini E, Doccini V, Guerrini R, Wallgren-Pettersson C, et al. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am J Med Genet Part A. (2018) 176:1748–52. doi: 10.1002/ajmg.a.38840
152. Verdura E, Fons C, Schlüter A, Ruiz M, Fourcade S, Casasnovas C, et al. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J Med Genet. (2020) 57:132–7. doi: 10.1136/jmedgenet-2019-106373
153. Klassen TL, Bomben VC, Patel A, Drabek J, Chen TT, Gu W, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. (2014) 55:e6–12. doi: 10.1111/epi.12489
154. Chen H, Von Hehn C, Kaczmarek LK, Ment LR, Pober BR, Hisama FM. Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia. Neurogenetics. (2007) 8:131–5. doi: 10.1007/s10048-006-0071-z
155. Van Der Wijst J, Konrad M, Verkaart SAJ, Tkaczyk M, Latta F, Altmüller J, et al. A de novo KCNA1 mutation in a patient with tetany and hypomagnesemia. Nephron. (2018) 139:359–66. doi: 10.1159/000488954
156. Travaglini L, Nardella M, Bellacchio E, D'Amico A, Capuano A, Frusciante R, et al. Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur J Paediatr Neurol. (2017) 21:450–6. doi: 10.1016/j.ejpn.2016.11.005
157. Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. (2015) 23:1505–12. doi: 10.1038/ejhg.2015.21
158. Myers CT, McMahon JM, Schneider AL, Petrovski S, Allen AS, Carvill GL, et al. De Novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. (2016) 99:287–98. doi: 10.1016/j.ajhg.2016.06.003
159. Tantsis EM, Gill D, Griffiths L, Gupta S, Lawson J, Maksemous N, et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev Med Child Neurol. (2016) 58:639–44. doi: 10.1111/dmcn.13033
160. Epperson MV, Haws ME, Standridge SM, Gilbert DL. An atypical rett syndrome phenotype due to a novel missense mutation in CACNA1A. J Child Neurol. (2018) 33:286–9. doi: 10.1177/0883073818754987
161. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansion in the α1A-voltage-dependent calcium channel. Nat Genet. (1997) 15:62–9. doi: 10.1038/ng0197-62
162. Geschwind DH, Perlman S, Figueroa KP, Karrim J, Baloh RW, Pulst SM. Spinocerebellar ataxia type 6 frequency of the mutation and genotype-phenotype correlations. Neurology. (1997) 49:1247–51. doi: 10.1212/WNL.49.5.1247
163. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Vol. 62, Neurology. Lippincott Williams & Wilkins (2004). p. 17–22. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/14718690 (accessed Feb 27, 2017).
164. Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. (2010) 588:1905–13. doi: 10.1113/jphysiol.2009.186437
165. Hayashida T, Saito Y, Ishii A, Yamada H, Itakura A, Minato T, et al. CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. (2018) 40:130–3. doi: 10.1016/j.braindev.2017.08.006
166. Gur-Hartman T, Berkowitz O, Yosovich K, Roubertie A, Zanni G, Macaya A, et al. Clinical phenotypes of infantile onset CACNA1A-related disorder. Eur J Paediatr Neurol. (2020) 30:1–8. doi: 10.1016/j.ejpn.2020.10.004
167. Stam AH, Luijckx GJ, Poll-Thé BT, Ginjaar IB, Frants RR, Haan J, et al. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatr. (2009) 80:1125–9. doi: 10.1136/jnnp.2009.177279
168. Li J, You Y, Yue W, Jia M, Yu H, Lu T, et al. Genetic evidence for possible involvement of the calcium channel gene CACNA1A in Autism pathogenesis in chinese han population. Hu VW, editor. PLoS ONE. (2015) 10:e0142887. doi: 10.1371/journal.pone.0142887
169. Humbertclaude V, Riant F, Krams B, Zimmermann V, Nagot N, Annequin D, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol. (2020) 62:330–7. doi: 10.1111/dmcn.14261
170. Imbrici P, Eunson LH, Graves TD, Bhatia KP, Wadia NH, Kullmann DM, et al. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology. (2005) 65:944–6. doi: 10.1212/01.wnl.0000176069.64200.28
171. Coste de Bagneaux P, von Elsner L, Bierhals T, Campiglio M, Johannsen J, Obermair GJ, et al. A homozygous missense variant in CACNB4 encoding the auxiliary calcium channel beta4 subunit causes a severe neurodevelopmental disorder and impairs channel and non-channel functions. Helbig I, editor. PLoS Genet. (2020) 16:e1008625. doi: 10.1371/journal.pgen.1008625
172. Heyne HO, Artomov M, Battke F, Bianchini C, Smith DR, Liebmann N, et al. Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genet Med. (2019) 21:2496–503. doi: 10.1038/s41436-019-0531-0
173. de Vries B, Mamsa H, Stam AH, Wan J, Bakker SLM, Vanmolkot KRJ, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. (2009) 66:97–101. doi: 10.1001/archneurol.2009.68
174. Choi K-D, Jen JC, Choi SY, Shin J-H, Kim H-S, Kim J-S, et al. Late-onset episodic ataxia associated with SLC1A3 mutation. J Hum Genet. (2017) 62:443–6. doi: 10.1038/jhg.2016.137
175. Iwama K, Iwata A, Shiina M, Mitsuhashi S, Miyatake S, Takata A, et al. A novel mutation in SLC1A3 causes episodic ataxia. J Hum Genet. (2018) 63:207–11. doi: 10.1038/s10038-017-0365-z
176. Chivukula AS, Suslova M, Kortzak D, Kovermann P, Fahlke C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum Mutat. (2020) 41:1892–905. doi: 10.1002/humu.24089
177. Liao Y, Anttonen A-K, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. (2010) 75:1454–8. doi: 10.1212/WNL.0b013e3181f8812e
178. Hackenberg A, Baumer A, Sticht H, Schmitt B, Kroell-Seger J, Wille D, et al. Infantile epileptic encephalopathy, transient choreoathetotic movements, and hypersomnia due to a de novo missense mutation in the scn2a gene. Neuropediatrics. (2014) 45:261–4. doi: 10.1055/s-0034-1372302
179. Reynolds C, King MD, Gorman KM. The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol. (2020) 24:117–22. doi: 10.1016/j.ejpn.2019.12.016
180. Schwarz N, Bast T, Gaily E, Golla G, Gorman KM, Griffiths LR, et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur J Paediatr Neurol. (2019) 23:438–47. doi: 10.1016/j.ejpn.2019.03.001
181. Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, et al. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. (2010) 285:32160–73. doi: 10.1074/jbc.M110.153676
182. Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, et al. De novo loss-or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. (2015) 47:393–9. doi: 10.1038/ng.3239
183. Pena SDJ, Coimbra RLM. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin Genet. (2015) 87:1–3. doi: 10.1111/cge.12542
184. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain. (2017) 140:2337–54. doi: 10.1093/brain/awx184
185. Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology. (2016) 87:1975–84. doi: 10.1212/WNL.0000000000003309
186. Siniscalchi A, Gallelli L, De Sarro G. Use of antiepileptic drugs for hyperkinetic movement disorders. Curr Neuropharmacol. (2010) 8:359–66. doi: 10.2174/157015910793358187
187. Zaremba PD, Bialek M, Blaszczyk B, Cioczek P, Czuczwar SJ. Non-epilepsy uses of antiepileptic drugs. Pharmacol Rep. (2006) 58:1–12.
188. Michaux L, Granier M. Epilepsie bravais-jacksonienne réflexe. Ann Med Psychol. (1945) 103:172–7.
189. Kato M, Araki S. Paroxysmal kinesigenic choreoathetosis. report of a case relieved by Carbamazepine. Arch Neurol. (1969) 20:513. doi: 10.1001/archneur.1969.00480110072006
190. Latorre A, Bhatia KP. Treatment of paroxysmal dyskinesia. Neurol Clin. (2020) 38, 433–47. doi: 10.1016/j.ncl.2020.01.007
192. Ansell B, Clarke E. Acetazolamide in treatment of epilepsy. BMJ. (1956) 1:650–4. doi: 10.1136/bmj.1.4968.650
193. Livingston S, Peterson D, Boks L. Ineffectiveness of diamox in the treatment of childhood epilepsy. Pediatrics. (1956) 17:541.
195. Friedman JRL, Thiele EA, Wang D, Levine KB, Cloherty EK, Pfeifer HH, et al. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov Disord. (2006) 21:241–4. doi: 10.1002/mds.20660
196. Schoeler NE, Cross JH. Ketogenic dietary therapies in adults with epilepsy: a practical guide. Pract Neurol. (2016) 16:208–14. doi: 10.1136/practneurol-2015-001288
197. Mochel F, Hainque E, Gras D, Adanyeguh IM, Caillet S, Héron B, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatr. (2016) 87:550–3. doi: 10.1136/jnnp-2015-311475
198. Loong SC, Ong YY. Paroxysmal kinesigenic choreoathetosis. Report of a case relieved by L dopa. J Neurol Neurosurg Psychiatr. (1973) 36:921–4. doi: 10.1136/jnnp.36.6.921
199. Yoshimura K, Kanki R. Child-onset paroxysmal exercise-induced dystonia as the initial manifestation of hereditary Parkinson's disease. Park Relat Disord. (2018) 49:108–9. doi: 10.1016/j.parkreldis.2018.01.004
200. Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology. (2011) 77:269–75. doi: 10.1212/WNL.0b013e318225ab07
201. Lazaro RP. Involuntary movements induced by anticonvulsants drugs. Mt Sinai J Med. (1982) 49:274–81.
202. Chadwick D, Reynolds EH, Marsden CD. Anticonvulsant-induced dyskinesias: a comparison with dyskinesias induced by neuroleptics. J Neurol Neurosurg Psychiatr. (1976) 39:1210–8. doi: 10.1136/jnnp.39.12.1210
203. Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. (2015) 33:153–74. doi: 10.1016/j.ncl.2014.09.011
204. Zádori D, Veres G, Szalárdy L, Klivényi P, Vécsei L. Drug-induced movement disorders. Exp Opin Drug Safety. (2015) 14:877–90. doi: 10.1517/14740338.2015.1032244
205. Zaccara G, Cincotta M, Borgheresi A, Balestrieri F. Adverse motor effects induced by antiepileptic drugs. Epileptic Disord. (2004) 6:153–68.
206. Montenegro MA, Scotoni AE, Cendes F. Dyskinesia induced by phenytoin. Arq Neuropsiquiatr. (1999) 57:356–60. doi: 10.1590/S0004-282X1999000300002
207. van Gaalen J, Kerstens FG, Maas RPPWM, Härmark L, van de Warrenburg BPC. Drug-induced cerebellar ataxia: a systematic review. CNS Drugs. (2014) 28:1139–53. doi: 10.1007/s40263-014-0200-4
208. Rissardo JP, Caprara ALF. Carbamazepine-, oxcarbazepine-, eslicarbazepine-associated movement disorder: a literature review. Clinical Neuropharmacology Vol. 43. Lippincott Williams and Wilkins (2020). p. 66–80. Available online at: http://journals.lww.com/10.1097/WNF.0000000000000387 (accessed Dec 26, 2020).
209. Mahmoud F, Tampi RR. Valproic acid Induced parkinsonism in the elderly: a comprehensive review of the literature. J Geriatr Pharmacother. (2011) 9:405–12. doi: 10.1016/j.amjopharm.2011.09.002
210. Reeves AL, So EL, Sharbrough FW, Krahn LE. Movement disorders associated with the use of gabapentin. Epilepsia. (1996) 37:988–90. doi: 10.1111/j.1528-1157.1996.tb00537.x
211. Yim SH, Choi YH, Heo K, Cho ho K. A case of dyskinesia after levetiracetam administration. BMC Neurol. (2019) 19:292. doi: 10.1186/s12883-019-1519-8
212. Peters J, Vijiaratnam N, Angus-Leppan H. Tics induced by antiepileptic drugs: a pragmatic review. J Neurol. (2020). 268:321–33. doi: 10.1007/s00415-020-10153-6
213. Del Negro A, De Rosalmeida Dantas C, Zanardi V, Montenegro MA, Cendes F. Dose-related cerebellar atrophy in patients with epilepsy using phenytoin. Arq Neuropsiquiatr. (2000) 58:276–81. doi: 10.1590/s0004-282x2000000200012
214. Zadikoff C, Munhoz RP, Asante AN, Politzer N, Wennberg R, Carlen P, et al. Movement disorders in patients taking anticonvulsants. J Neurol Neurosurg Psychiatr. (2007) 78:147–51. doi: 10.1136/jnnp.2006.100222
215. Morgan JC, Sethi KD. Drug-induced tremors. Lancet Neurol. (2005) 4:866–76. doi: 10.1016/S1474-4422(05)70250-7
216. Gill D, Lyons M, Allam F. Phenytoin induced chorea. Am J Ther. (2018) 25:e390. doi: 10.1097/MJT.0000000000000562
217. Srinivasan S, Lok AW. Valproate-induced reversible hemichorea. Mov Disord. (2010) 25:1511–2. doi: 10.1002/mds.23119
218. Gunal DI, Guleryuz M, Bingol CA. Reversible valproate-induced choreiform movements. Seizure. (2002) 11:205–6. doi: 10.1053/seiz.2001.0576
219. Zaatreh M, Tennison M, D'Cruz O, Beach RL. Anticonvulsants-induced chorea: a role for pharmacodynamic drug interaction? Seizure. (2001) 10:596–9. doi: 10.1053/seiz.2001.0555
220. Munhoz RP, Werneck LC, Teive HAG. The differential diagnoses of parkinsonism: findings from a cohort of 1528 patients and a 10 years comparison in tertiary movement disorders clinics. Clin Neurol Neurosurg. (2010) 112:431–5. doi: 10.1016/j.clineuro.2010.03.003
221. Zima L, Ceulemans S, Reiner G, Galosi S, Chen D, Sahagian M, et al. Paroxysmal motor disorders: expanding phenotypes lead to coalescing genotypes. Ann Clin Transl Neurol. (2018) 5:996–1010. doi: 10.1002/acn3.597
Keywords: paroxysmal kinesigenic dyskinesia, paroxysmal non-kinesigenic dyskinesia, paroxysmal exercise induced dyskinesia, episodic ataxia, infantile convulsions and choreoathetosis syndrome, generalized epilepsy and paroxysmal dyskinesia
Citation: de Gusmão CM, Garcia L, Mikati MA, Su S and Silveira-Moriyama L (2021) Paroxysmal Genetic Movement Disorders and Epilepsy. Front. Neurol. 12:648031. doi: 10.3389/fneur.2021.648031
Received: 31 December 2020; Accepted: 22 February 2021;
Published: 23 March 2021.
Edited by:
Emmanuel Roze, Assistance Publique Hopitaux de Paris, FranceReviewed by:
Giacomo Garone, Bambino Gesù Children Hospital (IRCCS), ItalyAgathe Roubertie, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Li Cao, Shanghai Jiao Tong University, China
Copyright © 2021 de Gusmão, Garcia, Mikati, Su and Silveira-Moriyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio M. de Gusmão, Y2xhdWRpby5kZWd1c21hb0BjaGlsZHJlbnMuaGFydmFyZC5lZHU=