 Jacqueline Dominguez1*
Jacqueline Dominguez1* Jeryl Tan Yu1
Jeryl Tan Yu1 Yi Jayne Tan2
Yi Jayne Tan2 Arlene Ng1
Arlene Ng1 Ma Fe De Guzman3Boots Natividad3
Ma Fe De Guzman3Boots Natividad3 Ma Luisa Daroy3Jemellee Cano1Justine Yu1
Ma Luisa Daroy3Jemellee Cano1Justine Yu1 Michelle M. Lian4
Michelle M. Lian4 Li Zeng5,6Weng Khong Lim7,8
Li Zeng5,6Weng Khong Lim7,8 Jia Nee Foo4,9
Jia Nee Foo4,9 Adeline S. L. Ng2,6*
Adeline S. L. Ng2,6*- 1Institute for Neurosciences, St. Luke's Medical Center, Quezon City, Philippines
- 2Department of Neurology, National Neuroscience Institute, Singapore, Singapore
- 3Research and Biotechnology Division, St Luke's Medical Centre, Quezon, Philippines
- 4Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Singapore
- 5Neural Stem Cell Research Lab, Research Department, National Neuroscience Institute, Singapore, Singapore
- 6Neuroscience and Behavioural Disorders Programme, Duke-NUS Medical School, Singapore, Singapore
- 7Singhealth Duke-NUS Institute of Precision Medicine, Singapore, Singapore
- 8Cancer & Stem Cell Biology Program, Duke-NUS Medical School, Singapore, Singapore
- 9Human Genetics, Genome Institute of Singapore, A*STAR, Singapore, Singapore
Frontotemporal Dementia (FTD) is a common cause of Young Onset Dementia and has diverse clinical manifestations involving behavior, executive function, language and motor function, including parkinsonism. Up to 50% of FTD patients report a positive family history, supporting a strong genetic basis, particularly in cases with both FTD and amyotrophic lateral sclerosis (FTD-ALS). Mutations in three genes are associated with the majority of familial FTD (fFTD) cases - microtubule associated protein tau gene (MAPT), granulin precursor (GRN), and hexanucleotide repeat expansions in chromosome 9 open reading frame 72- SMCR8complex subunit (C9orf72) while mutations in other genes such as optineurin (OPTN) have rarely been reported. Mutations in OPTN have been reported mostly in familial and sporadic cases of ALS, or in rare cases of FTD-ALS, but not in association with pure or predominant FTD and/or parkinsonian phenotype. Here, we report for the first time, a family from the Philippines with four members harboring a novel frameshift insertion at OPTN (Chr 10:13166090 G>GA) p.Lys328GluTer11, three of whom presented with FTD-related phenotypes. Additionally, one sibling heterozygous for the frameshift insertion had a predominantly parkinsonian phenotype resembling corticobasal syndrome, but it remains to be determined if this phenotype is related to the frameshift insertion. Notably, none of the affected members showed any evidence of motor neuron disease or ALS at the time of writing, both clinically and on electrophysiological testing, expanding the phenotypic spectrum of OPTN mutations. Close follow-up of mutation carriers for the development of new clinical features and wider investigation of additional family members with further genetic analyses will be conducted to investigate the possibility of other genetic modifiers in this family which could explain phenotypic heterogeneity.
Introduction
Frontotemporal Dementia (FTD) is a common cause of Young Onset Dementia defined as dementia occurring before age 65, and has diverse clinical manifestations affecting behavior, executive function or speech (1, 2). The most common clinical phenotype remains behavioral variant FTD (bvFTD) (3), with two other language-predominant phenotypes i.e., non-fluent primary progressive aphasia (nfvPPA) and semantic variant primary progressive aphasia (svPPA) (4). Between 30–50% of patients report a family history of FTD in at least one family member, supporting a strong genetic basis (1). Mutations in three genes are associated with the majority of familial FTD (fFTD) cases - microtubule associated protein tau gene (MAPT), granulin precursor (GRN), and hexanucleotide repeat expansions in chromosome 9 open reading frame 72- SMCR8complex subunit (C9orf72) (5). C9orf72 repeat expansions are the most common genetic cause of ALS-FTD spectrum disorder (6, 7), but mutations in other genes have also been linked with familial ALS-FTD (8–11).
Mutations in optineurin (OPTN) have been identified mostly in familial and sporadic cases of amyotrophic lateral sclerosis (ALS) (12). OPTN mutations were first linked to ALS in the Japanese population, accounting for 3.3% of familial ALS (FALS) and 0.2% of sporadic ALS (sALS) patients (13). Rare reports of OPTN mutations linked to FTD or displaying an FTD phenotype include a Chinese patient heterozygous for a OPTN c.1546G> C (p.E516Q) mutation with a rapidly progressive ALS-FTD phenotype (14), and two male patients with onset in the 5th decade of ALS-FTD, carrying novel compound heterozygous loss-of-function mutations in OPTN (nonsense variant p.Ser262* and frameshift deletion p.Leu430Argfs*16) (15), and heterozygous for the OPTN p.E478G variant (16), respectively. A novel homozygous splice-site variant (c.1242þ1G>A) was recently reported in a patient with logopenic variant FTD (lvFTD) and whose sibling was diagnosed with non-fluent variant FTD. No ALS was reported in these siblings (17).
Here, we report for the first time, a novel frameshift insertion at OPTN (Chr 10:13166090 G>GA) p.Lys328GluTer11 in 4 members of a family from the Philippines. Affected members harboring homozygous mutation presented with FTD-related phenotypes. Notably, none of the affected members showed any evidence of motor neuron disease or ALS at the time of writing, both clinically and on electrophysiological testing.
Materials and Methods
Familial Investigation
Participants belonged to a family from Samar, region of the Visayas Islands in the Philippines, who presented to the Memory Center at St Luke's Medical Center with varying FTD phenotypes. Interviews with surviving family members were conducted. This study was approved by the St. Luke's Institutional Ethics Review Committee (Ethics Clearance IERC Ref. No. CT-14089). Written informed consent was obtained from all patients (or their caregivers) who participated in this study.
Clinical Studies
All participants underwent standardized medical history and neurologic evaluations by specialist cognitive neurologists, as well as cognitive screening at the Memory Center, including the Mini Mental State Exam (MMSE) (18), Montreal Cognitive Assessment- Philippines (MoCA-P) (19), Clinical Dementia Rating (CDR) Assessment, comprehensive psychometric evaluation and motor evaluation using the Unified Parkinson's Disease Rating Scale (UPDRS) (20).
Diagnostics and Neuroimaging Studies
Participants underwent cranial Magnetic Resonance Imaging (MRI)dementia protocol with hippocampal volumetry. Cranial PET-CT was performed using 2-F18-Fluoro-2-deoxy-D-glucose (18F-FDG). Electromyography and nerve conduction studies (EMG-NCV) and muscle ultrasound were done at the Neurophysiology Unit of the same hospital.
Molecular Genetic Studies
Genomic DNA was extracted from peripheral blood from the proband and five siblings (two symptomatic, three asymptomatic). FTD-associated genes were sequenced as part of a custom targeted exome sequencing panel of 200 neuro-degenerative disease-related genes as previously described (21) (Supplementary Table 1). Exonic sequences of these 200 genes were captured with the NimbleGenSeqCap EZ choice <7 Mb (Roche) following the manufacturer's protocol. The captured samples were pooled and submitted as one lane of 151 bp paired-end sequencing with HiSeq4000 (Illumina). Sequence reads were aligned to the human reference genome (hg19) using BWA-MEM algorithm (BWA, v0.7.15) (22) and variants were called using Genome Analysis Tool-Kit (GATK, v3.7) according to GATK best practices (23–25). Variants were annotated using ANNOVAR (26), and filtered to retain only rare (gnomADMAF <5%) non-synonymous, frameshift, stop-gain and splice site variants (27–29). Mean coverage for the OPTN gene with targeted exome sequencing was 89.8x, with an average of 98.1% of OPTN targeted exons region (5,230 bp) covered by at least 15 reads (base quality ≥10, mapping quality ≥20). OPTN frameshift mutations were further confirmed by Sanger sequencing (Sanger and C9orf72 genotyping methods and primers used are described in Supplementary Material). Four family members with OPTN frameshift mutations were then sequenced by whole-exome sequencing (WES) to elucidate any further pathogenic mutation. Details on WES can be found in Supplementary Material. In the WES data, the mean coverage for the OPTN gene was 102.4x, with an average of 100% of OPTN targeted exons region (2,817bp) covered by at least 15 reads (base quality ≥10, mapping quality ≥20).
Results
Molecular Genetic Studies
A novel frameshift insertion at OPTN (Chr 10:13166090 G>GA) p.Lys328 GluTer11 was identified in four family members (proband III-10 and two symptomatic siblings III-7 and III-8, and an asymptomatic sibling (III-6) by targeted exome sequencing (Figure 1). WES did not reveal any further pathogenic mutations. Siblings III-2 and III-9 were unaffected and did not carry the frameshift insertion. This insertion has not been seen in gnomAD (30). Genetic results, imaging findings, demographics, and clinical phenotypes are summarized in Table 1. The proband (III-10) and sibling III-8 were both homozygous for the frameshift insertion, while siblings III-7 and III-6 were heterozygous. No candidate mutations in the three most common FTD-related genes i.e., MAPT, GRN, and C9orf72 were found in any participant. In sibling III-7 with a parkinsonian phenotype, no pathogenic mutations in known PD-related genes were found.
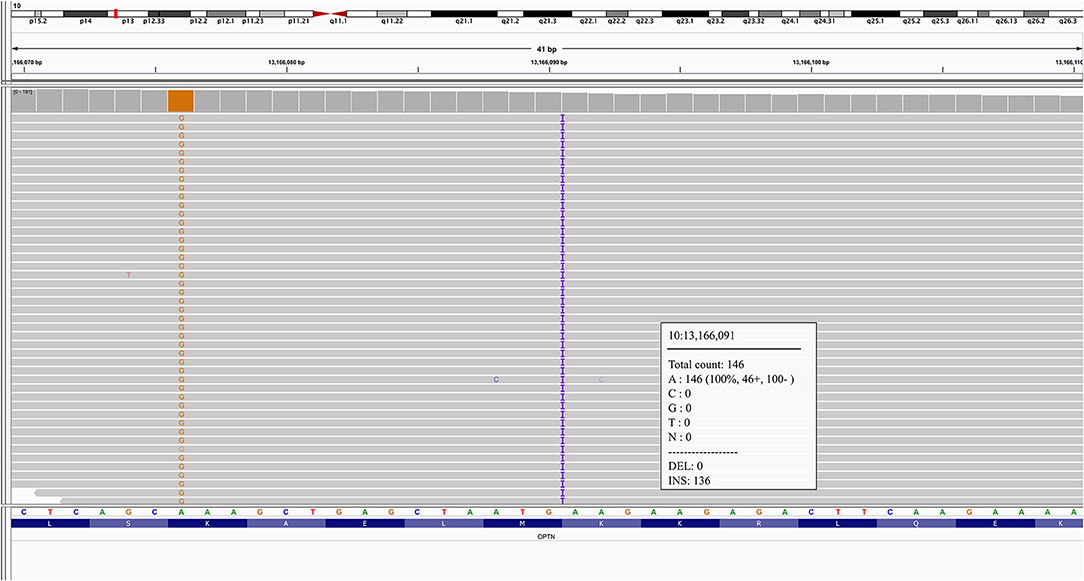
Figure 1. Integrative Genomics Viewer (IGV) inspection of proband III-10 at chr10:13166090, G>GA.
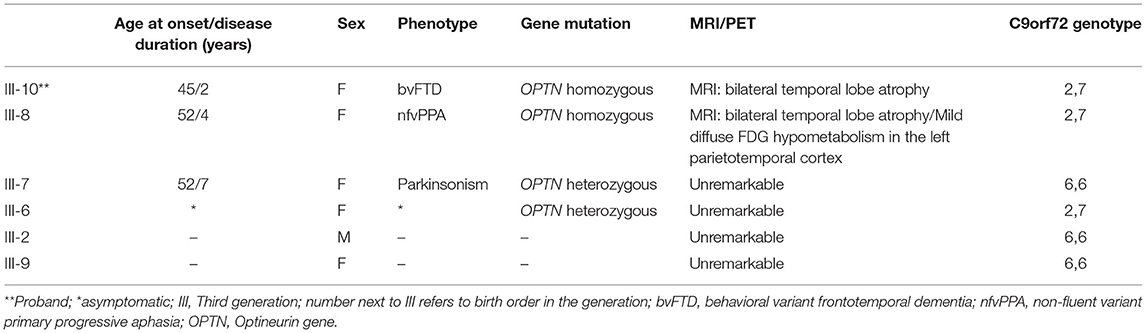
Table 1. Summary of demographic, clinical and genetic profile of family members.
Clinical Information
The family consists of 62 members originally from Basey, Samar, Philippines, spanning four generations (Figure 2). Four individuals carried the OPTN frameshift mutation, of which three were symptomatic. This family belongs to category 2 based on the Goldman criteria for familial frontotemporal dementia. The proband, III-10 (Figure 2), presented with changes in behavior at 45 years of age, which included facing backwards while sitting as a pillion rider on the motorcycle, plucking her hair in bunches, and wandering around her village. Executive tasks became challenging and she was eventually home bound. She developed poor personal hygiene and lost interest in social interaction, eventually becoming no longer functional. In the clinic, she was garrulous and laughed inappropriately. At initial presentation, she had an MMSE score of 13/30 and a MoCA-P score of 4/30. Her comprehension was impaired, and she had difficulty following complex sequential commands. Cranial MRI showed mild atrophy of both temporal lobes (Figure 3A). She was clinically diagnosed with bvFTD but died 2 years after diagnosis at the age of 47.
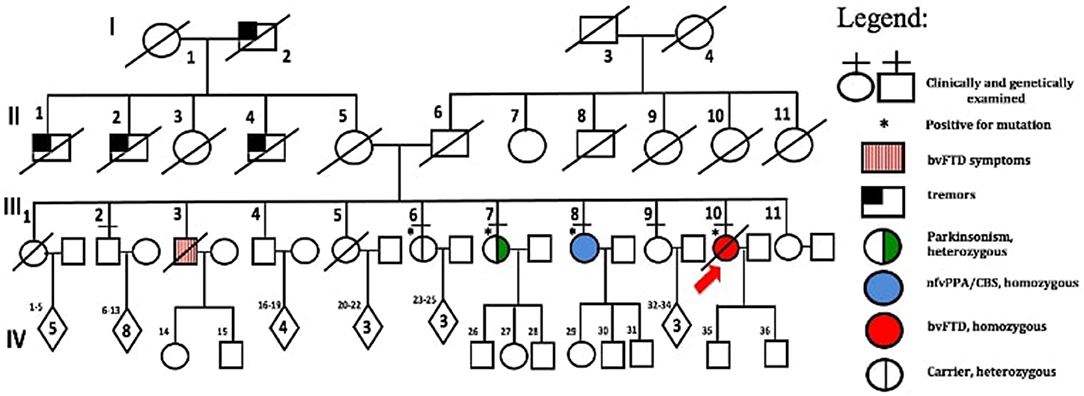
Figure 2. Family pedigree with generations and subjects labeled. Standard symbols were used.
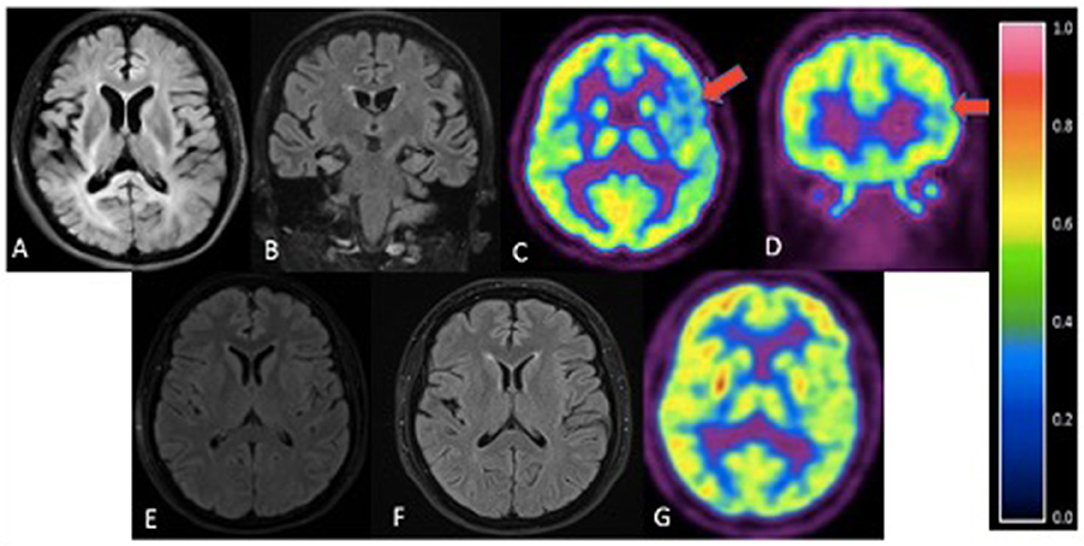
Figure 3. Cranial imaging of family members. (A) - III-10. Axial FLAIR. Mild atrophy of both temporal lobes. (B) - III-8. Coronal FLAIR MRI. Mild atrophy of bilateral parietotemporal areas. (C,D) III-8. Axial (C) and coronal (D) FDG-PET. Mild diffuse FDG hypometabolism in the left parietotemporal cortex (red arrows) with reference color scale to signify FDG uptake. (E) - III-6. Axial FLAIR. Unremarkable MRI. (F) - III-7. Axial FLAIR. No significant atrophy or hyperintensities. (G) - III-7. FDG-PET scan revealed no definite focus of abnormally increased or decreased FDG uptake.
Sibling III-8 presented with word-finding difficulty at age 52. She had frequent pauses and prolonged retrieval, but memory was preserved. She found it easier to express her thoughts via written instead of verbal means and frequently interchanged syllables during verbal expression. She also complained of stiffness of her upper back and left arm. The following year, she reported difficulty comprehending written text. There were no hallucinations or delusions. By 55 years of age, she was almost mute, communicating via gestures. There was decreased arm swing on the right; deep tendon reflexes were exaggerated in the right upper and lower extremities but there were no fasciculations, myoclonus, or dystonia. She had a positive snout reflex and positive Babinski sign bilaterally. She had a MoCA-P score of 23/30 and was clinically diagnosed with the non-fluent variant PPA with a corticobasal syndrome (CBS) phenotype. Serial follow-up showed progression of asymmetric parkinsonism. Cranial MRI showed bilateral temporal lobe atrophy (Figure 3B). FDG-PET showed mild diffuse FDG hypometabolism in the left parietotemporal cortex (Figures 3C,D). EMG and muscle ultrasound showed no fasciculations.
Sibling III-7 developed symptoms at 52 years of age with a parkinsonian phenotype consisting of bradykinesia and a stooped posture. Her language and cognition were intact, and she had no delusions, hallucinations, visual perceptual dysfunction or falls. She sought consult at our center 3 years after initial presentation of symptoms. She had a MoCA-P score of 27/30. Extraocular movements were full; she had a stooped posture with slight rigidity and asymmetric cogwheeling with tremors, more prominent on the right. She had a Unified Parkinson Disease Rating Scale (UPDRS) score of 71. There was no limb apraxia, dystonia, fasciculations, or myoclonus. Levodopa/Carbidopa was started but she remained poorly responsive to dopamine replacement therapy. She became wheelchair bound and dependent 7 years after disease onset. There were no hallucinations, delusions, visual perceptual dysfunction, sleep disorders or behavioral changes. Cranial MRI of this sister (Figure 3F) and PET scan findings (Figure 3G) were unremarkable. Surface EMG showed tremors. Muscle ultrasound studies revealed no fasciculations.
The fourth sibling carrying the same OPTN frameshift insertion (III-6) is a female who was 57 years of age during the time of examination and has remained asymptomatic at the time of writing. MRI and FDG-PET imaging were normal (Figure 3E) and there was no evidence of ALS clinically or on neurophysiological testing. Apart from her, further history revealed that the proband's older brother, III-3, presented with behavioral changes at 54 years old suggestive of bvFTD, but was not formally diagnosed. He would jump off a moving jeepney and paid no attention to street signs, crossing the street carelessly. He had delusions, claiming he had a girlfriend abroad. Eventually he had reduced interest in social interaction. He died at age 57. Other family members (I-1, II-1, II-2, II-4), already deceased, had tremors with an unspecified diagnosis. These tremors were termed “kiriw” which described tremulousness or losing control of the hand in local parlance. These affected individuals were occupationally functional but drank alcohol almost daily which worsened the tremors. They did not develop any clinical manifestations of dementia. No blood specimens were collected from these deceased individuals. There is no history of consanguinity in this family.
Discussion
In this study, we screened the proband along with both affected and unaffected family members for FTD and parkinsonism-related genes using a custom targeted exome sequencing panel. We found a novel OPTN frameshift insertion in all three affected family members and one unaffected sibling. Two homozygous carriers demonstrated an FTD phenotype, including bvFTD and nfvPPA, while the heterozygous sibling had a parkinsonism/CBS syndrome. None of the tested family members without the mutation had FTD or parkinsonism. All carriers had no evidence of ALS at present, and were negative for mutations in MAPT, GRN and C9orf72. Interestingly, one heterozygous carrier remains unaffected at age 57 which is past the age of onset of affected siblings. It remains to be seen whether such heterozygous mutation is pathogenic and associated with parkinsonism. This is the first reported family harboring this novel OPTN mutation and it presents without evidence of ALS thus far but with a predominant FTD phenotype. FTLD-TDP cases without motor neuron disease have been reported in association with OPTN and TBK1 (Tank-binding kinase (1) mutations (11). Mutations in TBK1werealso excluded in our affected family members. A possibility remains that we cannot rule out the presence of subclinical motor neuron degeneration, or that symptoms of ALS have not yet manifested in these affected persons.
The OPTN gene is an ubiquitin-binding protein responsible for cell trafficking leading to NF-kappa-B activation and its mechanism is believed to be a loss of function or haploinsufficiency. OPTN is a primary receptor required for the selective autophagy of damaged mitochondria; this process is also regulated via OPTN phosphorylation by TBK1 (31). Deletions, nonsense and frameshift mutations in OPTN have been found in either homozygous or heterozygous state, suggesting that complete loss of function or haploinsufficiency of optineurin is enough to cause or contribute to ALS (32). Most OPTN mutations are heterozygous, except for seven homozygous mutations: p.D127Rfs*21 (33), p.G291fs* (34), p.K359fs* (34), p.E135X (35), p.S174X (36), p.Q398X (37), and Ex5del (13). Homozygous loss-of-function mutations in OPTN were first reported in Japanese families with autosomal recessive ALS (13), while heterozygous OPTN mutations may confer risk for or be causative for ALS (33, 38, 39). The proband (III-10) and sibling III-8 were both homozygous carriers of the frameshift insertion. While the proband had an earlier age at disease onset (45 years), sibling III-8 had a similar age at onset (AAO) of 52 years as sibling III-7, who was heterozygous.III-6 is a heterozygous carrier and without any symptoms at age 57. There is limited information on whether homozygous OPTN carriers have greater loss-of-function and/or variances in clinical presentation including earlier AAO.
While dominant acting variants in OPTN appeared to make a substantial contribution to sporadic ALS, Pottier et al. first reported OPTN mutations in FTD, with their findings supporting a more complex mode of inheritance: sporadic individuals with heterozygous OPTN mutations who develop symptoms may do so because of compound heterozygote mutations (with the second hit not “qualifying” as a pathogenic variant) or because they also carry a mutation(s) in another gene (11). Pathological examination showed typical FTLD pathology without clinical motor neuron disease in 2 cases carrying OPTN mutations, a case of early onset dementia with prominent behavioral symptoms and a male with primary progressive aphasia carrying OPTN and TBK1 double mutation, extending the spectrum of OPTN and TBK1 mutations to pure FTLD (11). To our knowledge, however, our family is the first report of a CBS/parkinsonism phenotype in association with OPTN mutations.
Aside from ALS, other forms of parkinsonism including the 4-repeat tauopathies of corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) remain part of the FTD spectrum of disorders (40–43). At present, the most studied forms of autosomal dominant forms of FTD associated with parkinsonism are those linked to Chromosome 17 i.e., mutations in MAPT and GRN, followed by hexanucleotide repeat expansions in C9 or f72 (2, 44, 45). As mentioned, none of these mutations were reported in siblings III-7 and III-8, who had an asymmetric parkinsonism suggestive of a corticobasal syndrome. Sibling III-7 manifested with rapidly progressive parkinsonism at 52 years but has not demonstrated features of FTD with serial clinical examination thus far. Parkinsonism in FTD may present before, during or after the development of behavioral or language disturbances (45). These symptoms can precede the cognitive and behavioral symptoms of FTD by several years (46) in up to 27% of patients (47). Studies by Baizabal-Carvallo and Jankovic (46) and Gasca-Salas et al. (47) suggest that there is a high probability that parkinsonism among these individuals may precede future onset of FTD and should be evaluated in the serial follow-up of these patients.
Limitations of this study include the absence of histopathological findings, functional validation of the OPTN frameshift mutation and lack of DNA samples from other affected and unaffected siblings from other generations. Another limitation is the lack of RNA availability which would have provided information in comparing expression levels between homozygous and heterozygous mutation carriers. However, this is the first report of a novel frameshift insertion at OPTN (Chr 10:13166090 G>GA) p.Lys328GluTer11in 4 members of a family from the Philippineswith FTD-related phenotypes. Affected members harboring homozygous mutation presented with FTD-related phenotypes. It should be noted however, that we cannot confer pathogenicity in the heterozygous cases and that parkinsonism may be unrelated. It remains to be determined if this frameshift mutation acts in a recessive and or dominant fashion or whether it is associated with parkinsonism. As such, further close monitoring and follow up of clinical symptoms will be important. We aim for future histopathologic studies in this family to possibly elucidate disease mechanism, unfortunately now, Filipino practice does not allow tampering of the brain post-mortem.
Overall, we identified a novel OPTN frameshift insertion in a family with frontotemporal dementia and parkinsonism/CBS but without clinical and neurophysiological evidence of ALS, expanding the phenotypic spectrum of OPTN mutations. The absence of TBK1 gene mutation in individuals without features of ALS and the other genes commonly implicated in FTD-ALS overlap offer these possibilities: (1) subclinical or, (2) early stage of motor neuron degeneration, (3) presence of other ALS-modifying genes. Given the findings in this family, close follow-up of mutation carriers for the development of new clinical features and wider investigation of additional family members with collection of DNA samples and further genetic analyses will be conducted to investigate the possibility of multiple interacting genetic variants/genetic modifier sin this family which could explain the heterogenous disease phenotype.
Data Availability Statement
Patient WES (whole-exome sequencing) data are deposited in the European Genome-phenome Archive (EGA, https://ega-archive.org, study accession number EGAS00001005220) and are available upon request to Data Access Committee (DAC).
Ethics Statement
The studies involving human participants were reviewed and approved by St. Luke's Medical Center. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JD, JTY, AN, MFD, BN, JC, JY, and MLD contributed to the acquisition, interpretation of clinical, and imaging data. YT, ML, LZ, WL, JF, and ASLN contributed to the collection and processing and analysis of the genetic samples. JD, JTY, and ASLN prepared the initial paper which was reviewed and revised and approved by all authors. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Philippine Council for Health Research and Development of the Philippine Department of Science and Technology [FP 15026], the St. Luke's Medical Center (No. 15-020), Singapore's Ministry of Health National Medical Research Council [ASLN under the NMRC Transition Award (MOH-TA18may-0003)], and National Research Foundation Singapore Fellowship (JF under NRF-NRFF2016-03).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the family's willingness to participate and their cooperation throughout the study. The authors acknowledge Dr. Joven Cuanang, Dr. Ma. Socorro Martinez, and Dr. Raymond Rosales for their comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.645913/full#supplementary-material
Abbreviations
ALS, Amyotrophic Lateral Sclerosis; bvFTD, behavioral variant FTD; CBS, corticobasal syndrome; CDR, Clinical Dementia Rating; C9orf72, chromosome 9 open reading frame 72- SMCR8complex subunit; EMG, electromyography; FTD, Frontotemporal dementia; fFTD, familial frontotemporal dementia; MAPT, microtubule associated protein tau; MMSE, Mini Mental State Exam; MoCA-P, Montreal Cognitive Assessment- Philippines; MRI, magnetic resonance imaging; nfvPPA, non-fluent primary progressive aphasia; OPTN, optineurin; GRN, granulin precursor; svPPA, semantic variant primary progressive aphasia; sALS, sporadic ALS; TBK1, tank-binding kinase 1; UPDRS, Unified Parkinson's Disease Rating Scale.
References
1. Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. (2005) 65:1817–9. doi: 10.1212/01.wnl.0000187068.92184.63
2. Woollacott IOC, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem. (2016) 138:6–31. doi: 10.1111/jnc.13654
3. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456–77. doi: 10.1093/brain/awr179
4. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
5. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. (2019) 266:2075–86. doi: 10.1007/s00415-019-09363-4
6. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
7. Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. (2012) 11:297–8. doi: 10.1016/S1474-4422(12)70046-7
8. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. (2009) 65:470–3. doi: 10.1002/ana.21612
9. Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. (2012) 79:1556–62. doi: 10.1212/WNL.0b013e31826e25df
10. Barmada SJ, Finkbeiner S. Pathogenic TARDBP mutations in amyotrophic lateral sclerosis and frontotemporal dementia: disease-associated pathways. Rev Neurosci. (2010) 21:251–72. doi: 10.1515/REVNEURO.2010.21.4.251
11. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. (2015) 130:77–92. doi: 10.1007/s00401-015-1436-x
12. Del Bo R, Tiloca C, Pensato V, Corrado L, Ratti A, Ticozzi N, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2011) 82:1239–43. doi: 10.1136/jnnp.2011.242313
13. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. (2010) 465:223–6. doi: 10.1038/nature08971
14. Feng S, Che C, Feng S, Liu C, Li L, Li Y, et al. Novel mutation in optineurin causing aggressive ALS+/–frontotemporal dementia. Ann Clin Transl Neurol. (2019) 6:2377–83. doi: 10.1002/acn3.50928
15. Pottier C, Rampersaud E, Baker M, Wu G, Wuu J, McCauley JL, et al. Identification of compound heterozygous variants in OPTN in an ALS-FTD patient from the CReATe consortium: a case report. Amyotroph Lateral Scler Front Degener. (2018) 19:469–71. doi: 10.1080/21678421.2018.1452947
16. Nishiyama A, Niihori T, Warita H, Izumi R, Akiyama T, Kato M, et al. Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging. (2017) 53:194.e1–194.e8. doi: 10.1016/j.neurobiolaging.2017.01.004
17. Mol MO, van Rooij JGJ, Wong TH, Melhem S, Verkerk AJMH, Kievit AJA, et al. Underlying genetic variation in familial frontotemporal dementia: sequencing of 198 patients. Neurobiol Aging. (2021) 97:148.e9–148.e16. doi: 10.1016/j.neurobiolaging.2020.07.014
18. Folstein MF. The mini-mental state examination. Arch Gen Psychiatry. (1983) 40:812. doi: 10.1001/archpsyc.1983.01790060110016
19. Dominguez JC, Soriano JR, Magpantay CD, Orquiza MGS, Solis WM, Reandelar Jr, MF, et al. Early detection of mild alzheimer's disease in filipino elderly: validation of the montreal cognitive assessment-philippines (MoCA-P). Adv Alzheimer Dis. (2014) 03:160–7. doi: 10.4236/aad.2014.34015
20. The Movement Disorder Society Task Force on Rating Scales for Parkinson's Disease. The Unified Parkinson's Disease Rating Scale (UPDRS): status and recommendations. Mov Disord. (2003) 18:738–50. doi: 10.1002/mds.10473
21. Ng ASL, Tan YJ, Yi Z, Tandiono M, Chew E, Dominguez J, et al. Targeted exome sequencing reveals homozygous TREM2 R47C mutation presenting with behavioral variant frontotemporal dementia without bone involvement. Neurobiol Aging. (2018) 68:160.e15–160.e19. doi: 10.1016/j.neurobiolaging.2018.04.003
22. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
23. Li H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs With BWA-MEM. (2013). Available online at: http://arxiv.org/abs/1303.3997 (accessed September 10, 2020).
24. Auwera GA, Carneiro MO, Hartl C. From Fast Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. (2013). Available online at: https://onlinelibrary.wiley.com/doi/abs/10.1002/0471250953.bi1110s43 (accessed September 10, 2020).
25. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
26. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. (2015) 10:1556–66. doi: 10.1038/nprot.2015.105
27. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. (2016) 11:1–9. doi: 10.1038/nprot.2015.123
28. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using polyphen-2. Curr Protoc Hum Genet. (2013) 76:7. doi: 10.1002/0471142905.hg0720s76
29. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
30. Genome Aggregation Database (2020). Available online at: https://gnomad.broadinstitute.org/ (accessed June 15, 2020).
31. Slowicka K, Vereecke L, van Loo G. Cellular functions of optineurin in health and disease. Trends Immunol. (2016) 37:621–33. doi: 10.1016/j.it.2016.07.002
32. Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol. (2017) 154:1–20. doi: 10.1016/j.pneurobio.2017.04.005
33. Goldstein O, Nayshool O, Nefussy B, Traynor BJ, Renton AE, Gana-Weisz M, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology. (2016) 86:446–53. doi: 10.1212/WNL.0000000000002334
34. Özoguz A, Uyan Ö, Birdal G, Iskender C, Kartal E, Lahut S, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging. (2015) 36:1764.e9–1764.e18. doi: 10.1016/j.neurobiolaging.2014.12.032
35. Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. (2018) 89:817–27. doi: 10.1136/jnnp-2017-317611
36. Gotkine C, de Majo M, Wong M, Topp CH, Kanaan S, Michaelson-Cohen M, et al. A novel optineurin truncation mutation identified in a consanguineous palestinian family with amyotrophic lateral sclerosis confirms loss of function as a disease mechanism. E Pub. (2016). doi: 10.7490/f1000research.1113439.1
37. Sundaramoorthy V, Walker AK, Tan V, Fifita JA, Mccann EP, Williams KL, et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet. (2015) 24:3830–46. doi: 10.1093/hmg/ddv126
38. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (80-). (2015) 347:1436–41. doi: 10.1126/science.aaa3650
39. Tümer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, LuCamp, et al. A novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging. (2012) 33:208.e1–208.e5. doi: 10.1016/j.neurobiolaging.2011.07.001
40. Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, et al. Distinct genetic forms of frontotemporal dementia. Neurology. (2008) 71:1220–6. doi: 10.1212/01.wnl.0000319702.37497.72
41. Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AMT, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain. (2008) 131:721–31. doi: 10.1093/brain/awm331
42. Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. (2009) 73:1451–6. doi: 10.1212/WNL.0b013e3181bf997a
43. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci. (2011) 45:343–9. doi: 10.1007/s12031-011-9632-1
44. Giau V, Senanarong V, Bagyinszky E, An S, Kim S. Analysis of 50 neurodegenerative genes in clinically diagnosed early-onset Alzheimer's disease. Int J Mol Sci. (2019) 20:1514. doi: 10.3390/ijms20061514
45. Siuda J, Fujioka S, Wszolek ZK. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord. (2014) 20:957–64. doi: 10.1016/j.parkreldis.2014.06.004
46. Baizabal-Carvallo JF, Jankovic J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat Rev Neurol. (2016) 12:175–85. doi: 10.1038/nrneurol.2016.14
Keywords: familial FTD, novel mutation, parkinsonism, amyotrophic lateral scelerosis, optineurin (OPTN)
Citation: Dominguez J, Yu JT, Tan YJ, Ng A, De Guzman MF, Natividad B, Daroy ML, Cano J, Yu J, Lian MM, Zeng L, Lim WK, Foo JN and Ng ASL (2021) Novel Optineurin Frameshift Insertion in a Family With Frontotemporal Dementia and Parkinsonism Without Amyotrophic Lateral Sclerosis. Front. Neurol. 12:645913. doi: 10.3389/fneur.2021.645913
Received: 15 February 2021; Accepted: 12 April 2021;
Published: 19 May 2021.
Edited by:
Christos Proukakis, University College London, United KingdomReviewed by:
Jack Humphrey, Icahn School of Medicine at Mount Sinai, United StatesMarka van Blitterswijk, Mayo Clinic Florida, United States
Copyright © 2021 Dominguez, Yu, Tan, Ng, De Guzman, Natividad, Daroy, Cano, Yu, Lian, Zeng, Lim, Foo and Ng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adeline S. L. Ng, YWRlbGluZS5uZy5zLmxAc2luZ2hlYWx0aC5jb20uc2c=; Jacqueline Dominguez, amNkb21pbmd1ZXpAc3RsdWtlcy5jb20ucGg=