 Irina Erchova1
Irina Erchova1 Marcela Votruba
Marcela Votruba
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurol. , 13 April 2021
Sec. Neuro-Ophthalmology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.641259
This article is part of the Research Topic Hereditary Optic Neuropathies: A New Perspective View all 11 articles
Autosomal Dominant Optic Atrophy (ADOA) is an ophthalmological condition associated primarily with mutations in the OPA1 gene. It has variable onset, sometimes juvenile, but in other patients, the disease does not manifest until adult middle age despite the presence of a pathological mutation. Thus, individuals carrying mutations are considered healthy before the onset of clinical symptoms. Our research, nonetheless, indicates that on the cellular level pathology is evident from birth and mutant cells are different from controls. We argue that the adaptation and early recruitment of cytoprotective responses allows normal development and functioning but leads to an exhaustion of cellular reserves, leading to premature cellular aging, especially in neurons and skeletal muscle cells. The appearance of clinical symptoms, thus, indicates the overwhelming of natural cellular defenses and break-down of native protective mechanisms.
Autosomal Dominant Optic Atrophy (ADOA), is a progressive ophthalmological condition caused by degeneration of retinal ganglion cells (RGCs) that leads to visual loss (1). It is associated predominantly with mutations in the OPA1 gene and has variable onset and severity. The pathophysiology of OPA1-related ADOA is believed to be mainly due to haploinsufficiency in OPA1, due to the preponderance of OPA1 mutations that lead to premature translation termination and null mutations (2). This leads to a ~50% reduction in OPA1 protein in most tissues tested (3). In addition, some mutations lead to unstable OPA1 transcripts, due to a premature stop codon, and these appear to be degraded by non-sense mediated mRNA decay, and also lead to haploinsufficiency (4). Thesecond mechanism of disease is linked to missense mutations in the GTPase domain of OPA1, where a dominant-negative effect is postulated to lead to severe “plus” forms of the disease (5).
Affected individuals are usually identified early, as juveniles or adolescents. However, clinical symptoms may appear later in some individuals, such as loss of visual acuity and deficits in color vision (6). Currently, based on the fact that before the onset of symptoms individuals frequently have normal development and good health, supportive treatments are not instituted before the appearance of the clinical symptoms, and disease onset is thus considered age-dependent. As a part of the aging process, the amount of OPA1 protein is believed to decrease, potentially contributing to age-related deterioration of vision, muscle, and memory (7, 8). Moreover, the levels of proteins governing mitochondrial dynamics, which include OPA1, have been found to be significantly altered in Alzheimer Disease (AD) mice and patients (9). On the other hand, there is a mounting body of evidence that suggests that on the cellular level OPA1 mutations cause abnormalities demonstrable from birth, though many are successfully ameliorated (or compensated for) by cellular adaptive mechanisms. In turn, cellular adaptation, though beneficial as it allows normal development, exhausts the antioxidant system, reduces the control of inflammation, and the supply of adult stem cells, thus depleting natural defenses and therefore potentially accelerates the aging process.
OPA1 protein is part of the cellular control of cellular energy production and distribution and thus is essential for development, especially in neurons with long neurites (10). All fetuses carrying homozygous mutations in the Opa1 gene in murine models die during embryonic development (11, 12). As a result, systematic evidence of homozygous pathology in mammals is rare (13). It has however been possible to investigate cellular changes using artificially created mosaics of homozygous cells in non-mammalian experimental models like Drosophila (14, 15), and stress adaptation and life-span changes using nematode, C. elegans (16). In humans, homozygous OPA1 mutations are rarely seen due to presumed fetal loss and when they do occur are associated with very severe developmental conditions, such as encephalomyopathy, muscle weakness, cardiomyopathy, hypertonia, sensory deficits, and more general failure to thrive leading to early death (17). Severe developmental delays and early-onset optic atrophy are also typical for heterozygous mutations causing Behr syndrome, accompanied by spinocerebellar degeneration, ataxia, and sensory deficits (18–21).
Cellular deficits caused by faulty mitochondrial fusion are well-documented. In budding yeast, the tubular mitochondrial network breaks into small spherical segments (22, 23). In Drosophila, a similar process affects the motility of the sperm cells and results in male sterility (17). Similar fragmentation is documented in primary cultures of various animal cells (24, 25) and patient-derived induced pluripotent stem (iPS) cells (26), as well as murine retinal ganglion cells from the B6;C3-Opa1Q285STOP mouse, Opa1+/, (Figure 1) in which there is also accelerated mitochondrial movement (27). Defects in Opa1 primarily affect mitochondrial fusion and motility (28).
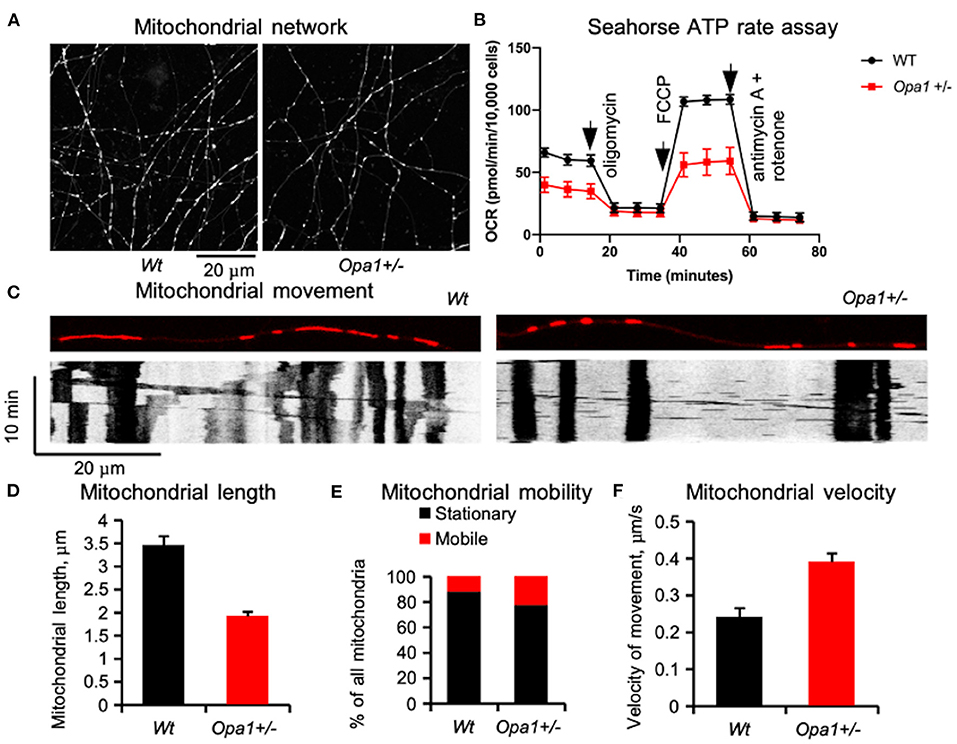
Figure 1. Mitochondrial network in primary retinal ganglion cells (RGCs) in mutant mice (B6;C3-Opa1Q285STOP, Opa1+/−) [adapted from Sun et al. (27)]. (A) Tubular mitochondrial network in control and mutant mice labeled in live culture by MitoTracker®Red and recorded by time-lapse imaging on the 8th day in vitro. (B) Mitochondrial network functionality assessed by Seahorse assay. (C) Mitochondrial movement in RGC dendrites is illustrated by automatically generated kymographs using FIJI software. (D) Average length of a single mitochondrial segment. (E) Distribution of the mitochondrion population classified as stationary and motile. (F) Velocities of mitochondrial movement. The increased mitochondrial mobility and velocity may be a compensatory mechanism for a poor cellular distribution network. The data were obtained from 1280 mitochondria in 91 processes of wild-type RGCs and 1205 mitochondria in 92 processes of Opa1+/- RGCs.
A reduction in mitochondrial quality control, and accelerated mitochondrial movement, are not the only compromises that allow survival. In Drosophila, mitochondrial fusion and fission imbalance is tolerable in young flies that mobilize natural antioxidant protection via Nrf2 and Foxo to up-regulate cytoprotective mechanisms (29). It nonetheless leads to accelerated aging and a much shorter life span (29).
Inflammation is another process that affects cell viability. Mitochondrial fusion and OPA1 protein are directly involved in this process via the TNFα-NF-kB–OPA1 regulatory pathway (29, 30). This pathway works via increasing mitochondrial fusion and improving respiratory chain efficiency in response to cellular stress. TNFα (Tumor Necrosis Factor alpha) is a protein (an inflammatory cytokine) produced by macrophages during acute inflammation. It is a signaling molecule that is passed on to other cells. NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a cytosol-based protein complex that controls transcription of DNA and is involved in cellular responses to stress. While in an inactivated state, NF-κB is in complex with an inhibitory protein, but once activated, it is released from the complex and translocated into the nucleus, where it binds to response elements (specific sequences) of DNA, recruiting co-activators and RNA polymerase. The mRNAs are then translated into proteins changing cell function. Normally, inflammatory stress increases TNFα, which activates NF-kB, and ultimately increases the production of OPA1 protein. The latter part of the pathway NF-kB–OPA1 is also involved in synaptic development (30). The inflammatory response is usually regulated via complex multi-gene signaling adjusting the homeostatic balance of organelles in response to cellular stress (31).
Mitochondrial fusion is essential for normal embryonic development (32). Recently, it has been shown that in human stem cells OPA1 haploinsufficiency causes impairment in neural stem cell self-renewal, thus causing age-dependent depletion, leading to reduced adult neurogenesis and cognitive deficits (33). Similarly, depletion of Opa1 protein affects both stem cell identity and self-renewal, causing age-dependent depletion of adult stem cells and thus possible deficits in adult neurogenesis in a mouse model with a mutation in Dynamin-related Protein gene (DRP) (34). Several recent studies have looked into the molecular mechanisms of this depletion. Sênos Demarco et al. showed that in Drosophila stem cells, depletion of Opa1 leads to activation of Target of Rapamycin (TOR) and a marked accumulation of lipid droplets, thus reducing the capacity of stem cells for self-renewal (35). In Drosophila, Sandoval et al. showed that mitochondrial fusion regulates larval growth and synaptic development via steroid hormone production (36). Moreover, in genetically modified human embryonic and patient-derived induced pluripotent stem cells OPA1 haploinsufficiency leads to aberrant nuclear DNA methylation and thus alters DNA transcription in neural progenitor cells. For instance, the transcription factor needed for GABAergic neuronal development is suppressed, causing reduced generation of GABAergic interneurons, whereas the formation of glutamatergic neurons is not affected (33). The potential reduction in the generation of GABAergic interneurons requires further investigation and exploration, but theoretically, this could result in a more vulnerable neural network, which might require more repair or maintenance. A recent example of the effect of a poorly developed GABA-ergic network has recently been discussed in optic nerve hypoplasia and autism. Clinical observations and recent reports indicate a high frequency of autism spectrum disorders (ASD) in children with optic nerve hypoplasia (ONH) (37, 38). In children with ONH, there are additional characteristics of ASD beyond those attributable to visual impairment alone, such as echolalia and stereotypic motor movements.
In Drosophila, experimental oxidative stress was seen in mitochondrial areas abnormally rich in myelin without cytochrome oxidase activity (39). Oxidative stress and up-regulated production of reactive oxygen species (ROS), mostly by mitochondria, are the main factors causing tissue damage. Though these processes are likely to occur independently of mutation, the early deployment of antioxidant defenses leaves limited capacity to cope with additional stress caused by mitochondrial aging. If tissue damage triggers an inflammatory response, then there is also a potentially limited ability to mobilize cellular resources or suppress inflammation. Reduced mitochondrial quality control, adopted early on to prevent degradation of functional, but fragmented mitochondria, also contribute to accelerated aging, by failing to identify and renew damaged mitochondrial fragments in a timely fashion. It is involved in the early loss of skeletal muscle mass and strength in OPA1 mutation carriers (40). Casuso and Huertas recently reviewed the topic in light of the support and protection offered by regular physical exercise (41). The down-regulation of mitochondrial fusion is linked with age-dependent axonal degeneration, with the visible accumulation of fragmented mitochondria in the axons without mitophagy (42).
Apart from the physical symptoms of aging associated with failing body strength and health, there is cognitive aging, characterized by increased anxiety and reduced working memory. On a cellular level, the symptoms are associated with synaptic loss in pyramidal cells and reduced numbers of inhibitory cells (especially somatostatin neurons) involved in the signaling pathways (43). We recently characterized subtly reduced working memory in Opa1 mutant mouse (B6;C3-Opa1Q285STOP, Opa1+/−) (44). We found that hippocampal CA1 cells showed age-related loss of synapses, developmental abnormalities such as the reduction in the number of GABA-ergic neurons, and defects in adult neurogenesis, which would contribute to the observed effect. Figure 2 illustrates our additional finding in the Dentate Gyrus (DG) region of aged mutant mice. The changes were similar to those observed by Llorens-Martín et al. and are decremental (45).
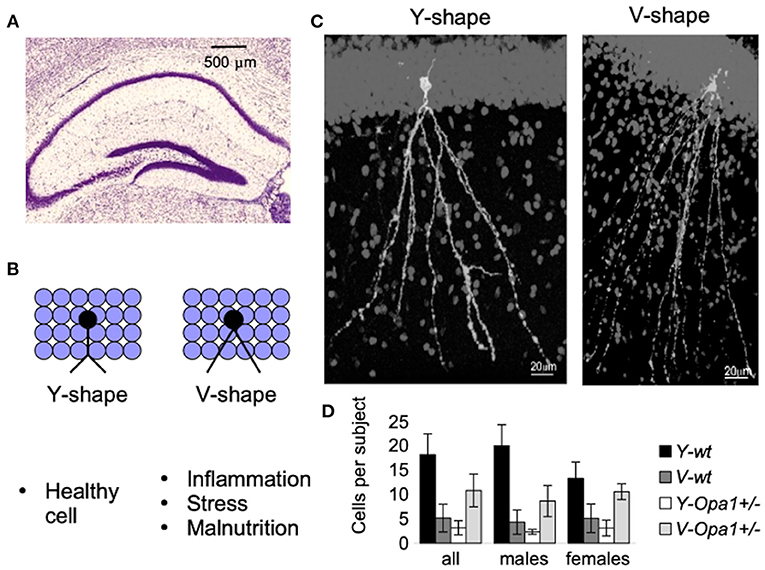
Figure 2. New-born neurons are continuously being added to the hippocampal Dentate Gyrus (DG) throughout adulthood (A). Detrimental factors (such as schizophrenia, stress, Alzheimer's disease, seizures, stroke, inflammation, dietary deficiencies, or the consumption of drugs of abuse or toxic substances) and neuroprotective factors (physical exercise and environmental enrichment) influence maturation and morphology of new-born granular cells [reviewed in Llorens-Martín et al. (45)]. For example, there is a striking difference in the shape of the apical dendrites (B). These morphological alterations may have important consequences (such as change in the pattern of synaptic connections, or change in the pattern of dendritic summation, and ultimately, change in cell excitability and functionality). An example of granular cells with different dendritic shafts are represented in (C). Though both wt and Opa1 +/- animals had both types of granular cell morphology, in aged mice, the Y-type was attributed to 80% of all Diolistically labeled DG granular cells in wt [see Bevan et al. (44) for the details]. By contrast, in age-matched Opa1 +/- animals more cells were of V-type (D). These changes are likely to be induced by a combination of stress factors in conjunction with impaired adult hippocampal neurogenesis.
Recent studies highlight the fact that fusion/fission mitochondrial dynamics and cellular metabolism are coupled: in cultured cell lines, elongated mitochondria are observed in conditions associated with increased ATP requirements (46, 47), whilstin nutrient-restricted conditions, mitochondria form large interconnected networks (48). When fusion is reduced secondary to nutrient excess or genetic abnormality (such as OPA1 deficiency) mitochondrial fragmentation promotes mitochondrial degradation through mitophagy (49). It appears that elongated shape and a fused mitochondrial network protect individual mitochondria from autophagosomal degradation (50, 51). When the tubular network is fragmented not only does energy production decrease but mitochondria themselves are at risk of degradation. To protect mitochondria when the tubular network is compromised, the cellular mechanism responsible for “quality control” is adjusted (52), allowing a larger quantity of disconnected mitochondria to circulate in the cell. This adjustment helps to maintain a sufficient number of fragmented mitochondria to meet the immediate cellular demands in development when most mitochondria are healthy, but with time might lead to accumulation of damaged mitochondria. At present, it is not clear whether mitochondrial fragmentation itself causes any deficit in cellular function, although the mitochondrial network does influence cellular functioning. In cultured HeLa cells, for example, the mitochondrial network interacts with the endoplasmic reticulum (ER) and uses Ca2+ signals to rapidly tune ATP production to the demands of the cell (53). However, in the rat cardiac muscle, the developmentally fragmented mitochondrial network later assumes a tubular structure (54). In post-natal primary RGC culture from the B6;C3-Opa1Q285STOP, Opa1+/−, mouse the mitochondrial network forms tubular structures but remains, nonetheless, more fragmented compared to controls, and levels of ATP production are reduced (Figure 1). Another important function of the mitochondrial network is the rapid spatial distribution of energy resources to distant cellular compartments. This function may be partially compensated via increased mitochondrial movement (27).
Moreover, a poorly controlled exuberant inflammatory response leads to tissue deterioration and accelerated aging. The chronic inflammatory response in cell lines and skeletal muscle caused by OPA1 deficiency is well-documented in patients and animal models (55–57). In addition, the adequate response to sepsis caused by pulmonary infection, whichoften complicates other chronic conditions, also requires rapid up-regulation of OPA1 (58). In post-mortem analyses of lung tissue obtained from areas with mild and severe emphysema, impaired fusion characterized by a low quantity of OPA1 protein is correlated with disease severity (59). Evidence from our lab suggests that levels of signaling protein NF-kB are already elevated in healthy Opa1 +/- mice compared to controls without changes in the levels of TNFα. This suggests that developmental adaptive mechanisms are likely to recruit a part of the inactive inflammatory pathway to alleviate cellular deficits in OPA1 during synaptic development (60–62). Unfortunately, this early recruitment of the regulatory pathway may compromise normal inflammatory responses, resulting in chronic inflammation or poor outcome in the case of severe acute inflammation (63, 64).
The aging process in mitochondrial networks at the microscopic level is very different from the tubular fragmentation described above. It characterized by mitochondrial swelling, reduced cristae, and damaged membranes (65, 66). Accelerated aging does not only manifest itself in sensory and cognitive deficits. There are numerous subtle changes that do not manifest themselves in everyday life. For example, recent studies showed that there is an elevated risk of cardiovascular conditions and reduced capacity for a successful recovery. By using C.elegans, Machiela et al. showed that disrupted mitochondrial fusion changed the normal pattern of responses to cellular stress. Cells became more resistant to both heat and oxidative stress, but more sensitive to osmotic variations and hypoxia. Sensitivity to hypoxia is critical in recovery from ischaemic stroke (67). Guo et al. showed that the increased risk of cerebral vascular injury in diabetic patients is partially due to chronically reduced levels of OPA1. They also reported more severe damage in this group of patients (68). Accordingly, Lai et al. demonstrated that rapid restoration of OPA1 levels after stroke reduces neuronal death and improved both survival and recovery of functions (69). Similarly, Xin and Lu showed in a murine model, that Opa1 expression was down-regulated in infarcted hearts, but Opa1 overexpression protected cardiomyocytes (70). Simulated ischaemia in the cardiac myogenic cell line H9c2 cells reduced OPA1 protein levels resulting in mitochondria fragmentation and apoptosis (71).
Thus, in this “Perspective” we summarize the evidence that OPA1 haploinsufficiency affects cellular functions from the molecular perspective of natural cellular resistance during development and adulthood. Deficits in OPA1 protein impact mitochondrial fusion, reduce cellular energy supply and thus impair cell survival. From the clinical perspective, this means that patients, identified as having a pathological mutation, may benefit from being monitoredbefore, or in the absence of, any clinical symptoms of disease. This could include careful multi-modal imaging of the retina and optic nerve and functional investigation with electrodiagnostic tests. Pre-symptomatic screening would contribute valuable clinical information allowing for the identification of markers of early disease and putative biomarkers that would be essential in the testing of novel therapeutic interventions. It also adds some weight to the idea that by supporting natural defenses, such as maintaining a healthy diet, avoiding smoking and alcohol consumption, and a regular exercise regime throughout the normal lifespan, it may be feasible to delay the onset of premature aging. Smoking is known to disturb mitochondrial function, and may thus be a factor that helps accelerate the onset and progression of visual loss in patients with mutations that impair mitochondrial function [as for example, in Leber Hereditary Optic Neuropathy and ADOA (72)].
There are many further potentially important research questions, such as why and how mitochondria in different tissues differ and whether this affects the apparent different rates of aging in different body tissues, which we would suggest may be worth addressing in future research.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by UK PPL Home Office PP7147250.
IE: conceptualization, methodology, investigation, writing-original draft preparation, and visualization. SS: investigation, visualization, writing-reviewing, and editing. MV: resources, writing-reviewing and editing, supervision, and funding acquisition. All authors: contributed to the article and approved the submitted version.
This work was supported by Cardiff University to IE and MV and by China Scholarship Council (201706100202) to SS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab. (2018) 125:315–21. doi: 10.1016/j.ymgme.2018.10.003
2. Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferré M, Guillet V, et al. OPA1-associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol. (2009) 41:1855–65. doi: 10.1016/j.biocel.2009.0401
3. Marchbank NJ, Craig JE, Leek JP, Toohey M, Churchill AJ, Markham AF, et al. Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet. 2002 39:e47. doi: 10.1136/jmg.39.8e47
4. Schimpf S, Fuhrmann N, Schaich S, Wissinger B. Comprehensive cDNA study and quantitative transcript analysis of mutant OPA1 transcripts containing premature termination codons. Hum Mutat. 2008 29:106–12. doi: 10.1002/humu20607
5. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain. 2008 131:338–51. doi: 10.1093/brain/awm298
6. Jurkute N, Leu C, Pogoda HM, Arno G, Robson AG, Nürnberg G, et al. SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. (2019) 86:368–83. doi: 10.1002/ana.25550
7. Shah SI, Paine JG, Perez C, Ullah G. Mitochondrial fragmentation and network architecture in degenerative diseases. PLoS ONE. (2019) 14:e0223014. doi: 10.1371/journal.pone.0223014
8. Zorov D, Vorobjev I, Popkov V, Babenko V, Zorova L, Pevzner I, et al. Lessons from the discovery of mitochondrial fragmentation (fission): a review and update. Cells. (2019) 8:175. doi: 10.3390/cells8020175
9. Murrell JR, Hake AM, Quaid KA, Farlow MR, Ghetti B. Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch Neurol. (2000) 57:885–7. doi: 10.1001/archneur.57.6.885
10. Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. (2017) 5:25. doi: 10.3390/antiox6020025
11. Baker C, Ebert S. Development of aerobic metabolism in utero: requirement for mitochondrial function during embryonic and fetal periods. OA Biotechnol. (2013) 2:16. doi: 10.13172/2052-0069-2-2-571
12. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
13. Hartmann B, Wai T, Hu H, MacVicar T, Musante L, Fischer-Zirnsak B, et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. Elife. (2016) 5:e16078. doi: 10.7554/eLife.16078
14. Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. (2001) 24:351–254. doi: 10.1016/S0166-2236(00)01791-4
15. Smith GA, Lin TH, Sheehan AE, Van der Goes van Naters W, Neukomm LJ, Graves HK, et al. Glutathione S-transferase regulates mitochondrial populations in axons through increased glutathione oxidation. Neuron. (2019) 103:52–65.e6. doi: 10.1016/j.neuron.2019.04.017
16. Dancy BM, Sedensky MM, Morgan PG. Effects of the mitochondrial respiratory chain on longevity in C. elegans. Exp Gerontol. (2014) 56:245–55. doi: 10.1016/j.exger.2014.03.028
17. Spiegel R, Saada A, Flannery PJ, Burté F, Soiferman D, Khayat M, et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J Med Genet. (2016) 53:127–31. doi: 10.1136/jmedgenet-2015-103361
18. Bonneau D, Colin E, Oca F, Ferré M, Chevrollier A, Guéguen N, et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain. (2014) 137:e301. doi: 10.1093/brain/awu184
19. Carelli V, Sabatelli M, Carrozzo R, Rizza T, Schimpf S, Wissinger B, et al. “Behr syndrome” with OPA1 compound heterozygote mutations. Brain. (2015) 138:e321. doi: 10.1093/brain/awu234
20. Rahman S. Mitochondrial disease in children. J Intern Med. (2020) 287:609–33. doi: 10.1111/joim.13054
21. Schaaf CP, Blazo M, Lewis RA, Tonini RE, Takei H, Wang J, et al. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol Genet Metab. (2011) 103:383–7. doi: 10.1016/j.ymgme.2011.04.018
22. Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, et al. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. (1998) 143:359–73. doi: 10.1083/jcb.143.2.359
23. Rapaport D, Brunner M, Neupert W, Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J Biol Chem. (1998) 273:20150–5. doi: 10.1074/jbc.273.32.20150
24. Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. (2008) 9:505–18. doi: 10.1038/nrn2417
25. Kushnareva Y, Seong Y, Andreyev AY, Kuwana T, Kiosses W, Votruba M, et al. Mitochondrial dysfunction in an Opa1Q285STOP mouse model of dominant optic atrophy results from Opa1 haploinsufficiency. Cell Death Dis. (2016) 7:e2309. doi: 10.1038/cddis.2016.160
26. Prieto J, León M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. (2016) 7:1–13. doi: 10.1038/ncomms11124
27. Sun S, Erchova I, Sengpiel F, Votruba M. Opa1 deficiency leads to diminished mitochondrial bioenergetics with compensatory increased mitochondrial motility. Investig Ophthalmol Vis Sci. (2020) 61:42. doi: 10.1167/IOVS.61.6.42
28. Chen H, Chan DC. Mitochondrial dynamics in mammals. Curr Top Dev Biol. (2004) 59:119–44. doi: 10.1016/S0070-2153(04)59005-1
29. Gumeni S, Evangelakou Z, Tsakiri EN, Scorrano L, Trougakos IP. Functional wiring of proteostatic and mitostatic modules ensures transient organismal survival during imbalanced mitochondrial dynamics. Redox Biol. (2019) 24:101219. doi: 10.1016/j.redox.2019.101219
30. Shen DN, Zhang LH, Wei EQ, Yang Y. Autophagy in synaptic development, function, and pathology. Neurosci Bull. (2015) 31:416–26. doi: 10.1007/s12264-015-1536-6
31. Müller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. (2013) 49:908–21. doi: 10.1016/j.molcel.2013.01036
32. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
33. Caglayan S, Hashim A, Cieslar-Pobuda A, Jensen V, Behringer S, Talug B, et al. Optic Atrophy 1 Controls human neuronal development by preventing aberrant nuclear DNA methylation. iScience. (2020) 23:101154. doi: 10.1016/j.isci.2020.101154
34. Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. (2016) 19:232–47. doi: 10.1016/j.stem.2016.04.015
35. Sênos Demarco R, Uyemura BS, D'Alterio C, Jones DL. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat Cell Biol. (2019) 21:710–20. doi: 10.1038/s41556-019-0332-3
36. Sandoval H, Yao CK, Chen K, Jaiswal M, Donti T, Lin YQ, et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. Elife. (2014) 3:1–23. doi: 10.7554/eLife.0355
37. Ek U, Fernell E, Jacobson L. Cognitive and behavioural characteristics in blind children with bilateral optic nerve hypoplasia. Acta Paediatr. (2005) 94:1421–6. doi: 10.1111/j.1651-2227.2005.tb01814x
38. Parr JR, Dale NJ, Shaffer LM, Salt AT. Social communication difficulties and autism spectrum disorder in young children with optic nerve hypoplasia and/or septo-optic dysplasia. Dev Med Child Neurol. (2010) 52:917–21. doi: 10.1111/j.1469-8749.2010.03664x
39. Walker DW, Benzer S. Mitochondrial “swirls” induced by oxygen stress and in the Drosophila mutant hyperswirl. Proc Natl Acad Sci USA. (2004) 101:10290–5. doi: 10.1073/pnas.0403767101
40. Liu HW, Chang YC, Chan YC, Hu SH, Liu MY, et al. Dysregulations of mitochondrial quality control and autophagic flux at an early age lead to progression of sarcopenia in SAMP8 mice. Biogerontology. (2020) 21:367–80. doi: 10.1007/s10522-020-09867-x
41. Casuso RA, Huertas JR. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing Res Rev. (2020) 58:101025. doi: 10.1016/j.arr.2020.101025
42. Cao X, Wang H, Wang Z, Wang Q, Zhang S, Deng Y, et al. In vivo imaging reveals mitophagy independence in the maintenance of axonal mitochondria during normal aging. Aging Cell. (2017) 16:1180–90. doi: 10.1111/acel.12654
43. Shukla R, Prevot TD, French L, Isserlin R, Rocco BR, Banasr M, et al. The relative contributions of cell-dependent cortical microcircuit aging to cognition and anxiety. Biol Psychiatry. (2019) 85:257–67. doi: 10.1016/j.biopsych.2018.09.019
44. Bevan RJ, Williams PA, Waters CT, Thirgood R, Mui A, Seto S, et al. OPA1 deficiency accelerates hippocampal synaptic remodelling and age-related deficits in learning and memory. Brain Commun. (2020) 2:fcaa101. doi: 10.1093/braincomms/fcaa101
45. Llorens-Martín M, Rábano A, Ávila J. The ever-changing morphology of hippocampal granule neurons in physiology and pathology. Front Neurosci. (2016) 9:526. doi: 10.3389/fnins.2015.00526
46. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz JA hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. (20098) 106:11960–5. doi: 10.1073/pnas.0904875106
47. Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SlP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. (2009) 28:1589–600. doi: 10.1038/emboj.2009.89
48. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. (2015) 32:678–92. doi: 10.1016/j.devcel.2015.01.029
49. Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science. (2012) 337:1062–5. doi: 10.1126/science.1219855
50. Gomes LC, Benedetto G. Di, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. (2011) 13:589–8. doi: 10.1038/ncb2220
51. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal deadation during nutrient starvation. Proc Natl Acad Sci USA. (2011) 108:10190–5. doi: 10.1073/pnas.1107402108
52. Alavi MV, Fuhrmann N. Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol Neurodegener. (2013) 8:1–12. doi: 10.1186/1750-1326-8-32
53. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. (1998) 280:1763–6. doi: 10.1126/science.280.5370.1763
54. Bakeeva LE, Chentsov YS, Skulachev VP. Intermitochondrial contacts in myocardiocytes. J Mol Cell. Cardiol. (1983) 15:413–20. doi: 10.1016/0022-2828(83)90261-4
55. Baltrusch S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia. (2016) 59:683–7. doi: 10.1007/s00125-016-3891-x
56. Rodríguez-Nuevo A, Díaz-Ramos A, Noguera E, Díaz-Sáez F, Duran X, Muñoz JP, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. (2018) 37:e96553. doi: 10.15252/embj.201796553
57. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-Associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. (2017) 25: 374–89.e6. doi: 10.1016/j.cmet.2017.04.021
58. Shi J, Yu J, Zhang Y, Wu L, Dong S, Wu L, et al. PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab Investig. (2019) 99:1795–809. doi: 10.1038/s41374-019-0286-x
59. Kosmider B, Lin CR, Karim L, Tomar D, Vlasenko L, Marchetti N, et al. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. E Bio Med. (2019) 46:305–16. doi: 10.1016/j.ebiom.2019.07.063
60. Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse. (2013) 35:151–9. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P
61. Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-κB functions in synaptic signaling and behavior. Nat Neurosci. (2003) 6:1072–8. doi: 10.1038/nn1110
62. Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. (2007) 8:49–62. doi: 10.1038/nrm2083
63. Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. (2004) 25:280–8. doi: 10.1016/j.it.2004.03008
64. Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-κB. Cell Death Differ. (2006) 13:759–72. doi: 10.1038/sj.cdd4401838
65. Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. (2002) 269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x
66. Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. (2006) 27:983–93. doi: 10.1016/j.neurobiolaging.2005.05.012
67. Machiela E, Liontis T, Dues DJ, Rudich PD, Traa A, Wyman L, et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. (2020) 34:8475–92. doi: 10.1096/fj.201903235R
68. Guo Y, Wang S, Liu Y, Fan L, Booz GW, Roman RJ, et al. Accelerated cerebral vascular injury in diabetes is associated with vascular smooth muscle cell dysfunction. Gero Sci. (2020) 42:547–61. doi: 10.1007/s11357-020-00179-z
69. Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M, et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. (2020) 34:101503. doi: 10.1016/j.redox.2020.101503
70. Xin T, Lu C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging. (2020). 12:4474–88. doi: 10.18632/aging.102899
71. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. (2009) 84:91–9. doi: 10.1093/cvr/cvp181
Keywords: mitochondria, OPA1, mitochondrial dynamics, aging, cellular adaptation
Citation: Erchova I, Sun S and Votruba M (2021) A Perspective on Accelerated Aging Caused by the Genetic Deficiency of the Metabolic Protein, OPA1. Front. Neurol. 12:641259. doi: 10.3389/fneur.2021.641259
Received: 13 December 2020; Accepted: 19 March 2021;
Published: 13 April 2021.
Edited by:
Rustum Karanjia, University of Ottawa, CanadaReviewed by:
Devin Dean Mackay, Indiana University, United StatesCopyright © 2021 Erchova, Sun and Votruba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela Votruba, dm90cnViYW1AY2FyZGlmZi5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.