 Rui Dong
Rui Dong Renxuan Huang
Renxuan Huang Jiaoqi Wang
Jiaoqi Wang Hongyu Liu1
Hongyu Liu1 Zhongxin Xu
Zhongxin Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 01 July 2021
Sec. Stroke
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.620948
This article is part of the Research TopicModulation of Microglia Transition Activity in Neuroinflammation following CNS InjuryView all 12 articles
Stroke is one of the most common causes of death worldwide. The subsequent development of neuroinflammation and brain edema dramatically increases the risks associated with stroke, leading to a substantial increase in mortality. Although considerable progress has been made in improving cerebral perfusion in the acute phase of stroke, effective treatment options for the subacute and chronic phases associated with cerebral infarction are limited. Microglia, the innate immune cells of the central nervous system (CNS), can be activated and polarized to take on different phenotypes in response to stimulations associated with stroke, including pro-inflammatory and anti-inflammatory phenotypes, which affect the prognosis of stroke. Therefore, investigation of the activation and polarizing mechanisms of microglia plays a critical role in treating stroke. The aim of this article was to investigate the significance of microglial phenotype regulation in stroke treatment by summarizing the activation, polarizing mechanisms, and general microglia characteristics.
Recent data from the World Health organization (WHO) shows that stroke is a leading cause of death worldwide (1–4). Post-ischemic neuroinflammation and cerebral edema secondary to stroke aggravate the damage caused by stroke to varying degrees (5, 6). Therefore, improvement in therapeutic protocols targeting the regulation of the cerebral microenvironment during stroke, and the repair of neural and tissue damage after stroke, are essential.
Microglia, the resident immune cells in CNS, continuously monitor the brain parenchyma. Microglia can become activated and switch their phenotype in response to changes in the local CNS microenvironment. They can have either a pro- or anti-inflammatory role and influence the prognosis of ischemic stroke (7–9). The M1 phenotype can release cytotoxic factors, such as inflammatory cytokines and nitric oxide (NO). M2-type microglia secrete anti-inflammatory factors, remove cell debris, promote angiogenesis, and promote the repair of injured peripheral nerves (10). Microglia, as an integral part of the immune system, produce inflammatory factors, alter the permeability of the blood-brain barrier (BBB), phagocytose vascular endothelial cells, and lead to BBB breakdown after stroke, which can result in reduced brain recovery following infarction. M1-type microglia also remove necrotic tissue, which promotes neurogenesis, secrete anti-inflammatory factors that regulate inflammation, and produce other neuroprotective effects (11–14). Thus, microglia have a “double-edged sword” function to maintain the homeostasis of the CNS microenvironment after stroke through two opposed pathophysiological effects, both of which play essential roles in recovery after brain injury. Therefore, studying the activation and polarization mechanisms of microglia and exploring possible means of regulating the M1/M2 phenotype transition might provide new ways to improve brain injury treatment after stroke.
This review summarizes the mechanisms associated with microglial activation and polarization, as well as the impact of microglia on stroke prognosis. Also, the therapeutic directions for recovery after a brain injury caused by stroke are discussed. We theorize that limiting microglial hyperactivation, inhibition of the M1 phenotype, and promoting the M2 phenotype through a combination of endogenous mechanisms and exogenous drugs would be beneficial for repairing brain injury following stroke. However, specific mechanisms need further investigation.
Ginhoux has reported that microglia are derived from yolk sac macrophages before embryonic day eight in mice, and contribute to primitive hematopoiesis (15). The microglial stages progress from embryonic to early postnatal then adult CNS (16). Under different conditions of activation, microglia of the developing can display different morphologies (17). Microglia are activated in the embryo and early postnatal brains and display a “generic macrophage”-like and mobile amoeboid morphology, while adult microglia exhibit characteristic ramified extensions that survey the surrounding areas (18).
Early after Middle Cerebral Artery Occlusion (MCAO), activated microglia were observed to be concentrated in the lesion area, and fewer microglia were located inside the ischemic center (19). Over time, the number of microglia in the ischemic center gradually increased, with round and amoeboid characteristics as the predominant morphologies. Highly branched microglia were observed at the borders of the ischemic region during reperfusion for up to 22 h (20). So it is clearly to see that microglia have specific region- and time- dependent features.
Moreover, during embryonic development, the phenotypes and functions of microglia are similar to those of yolk sac macrophages (15). Both cell types share the common characteristics of performing surveillance of the immunological microenvironment, mediating inflammation, and removing debris from dead cells (21). However, these two cells may play different roles during brain injury and can respond in opposite ways to acute inflammatory stimuli (22). According to the results from Yamasaki et al., microglia can protect the injured brain while macrophages concomitantly damage the brain under the same circumstances (21), which may be related to different types of gene expression patterns.
Microglia are the first line of defense against brain injury that takes place following stroke. They respond rapidly to alterations in the brain microenvironment. Early after stroke onset, microglia become activated rapidly, initiate migration, and activate downstream cell signaling (23, 24). Microglia activation is mainly characterized by changes in morphology, phagocytosis, migration to the injured area, and secretion of cytokines and oxidative metabolites such as NO and reactive oxygen species (ROS) (25), the excessive expression of which can lead to neuroinflammation after brain injury (26). In several pathological conditions, including exposure to lipopolysaccharide (LPS), inflammatory factors, hypoxia and other injury- related moleculars, microglia become over activated, and could be induced to exhibit a pro-inflammatory phenotype, where numerous pro-inflammatory cytokines are replicated, leading to neuronal cell damage (27). In fact, not only do microglia aggravate the damage that occurs in the brain and inhibit recovery, but they also remove necrotic cells and debris that promotes nerve regeneration (14). Their phagocytic functions can remove tissue fragments and foreign substances, however, the hyperactivation of microglia and mobilized macrophages caused by stroke reduces their clearance capacity, which could eventually result in damage to neurons and breakdown of the BBB (11, 12).
It's already known that stimulation of microglia with amyloid-β peptide (Aβ) activates transcription factors (e.g., nuclear factor kappa-B, NF-κB) involved in the expression of pro-inflammatory genes (28). On the other hand, nerve growth factor (NGF) promotes TrkA-mediated microglial phagocytosis of Aβ and induces the degradation of Aβ. NGF counteracts the pro-inflammatory activation induced by exposure to Aβ (29). NADPH oxidase (NOX) is a multi-subunit enzyme that produces peroxides and ROS in microglia. The inhibition of NOX significantly reduces pro-inflammatory activation of microglia and produces anti-inflammatory effects (30, 31). Recently, evidence has suggested that MCAO and OGD can induce biphasic microglia activation (32, 33). After ischemia occurs, microglia become activated, and produce detrimental and neuroprotective mediators (34). Thus, microglia carry out dual roles in determining the prognosis of brain injury.
Microglial polarization follows two pathways, the classical activation (M1) pathway and the alternate activation (M2) pathway. When the M1 phenotype is induced by exposure to LPS, microglia exhibit destructive effects in the CNS. However, the M2 phenotype, which can be induced by IL-4, exhibits neuroprotective effects (14, 35). The M1 phenotype is a pro-inflammatory cell state that releases inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), interleukin 1β (II-1β), NO, and so on. The M1 phenotype is also associated with increased production of ROS, the synthesis of extracellular matrix protein hydrolases (MMP3 and MMP9), and the cell surface marker of M1 phenotype, CD68 and CD16/32 (36–38). M2 microglia express an anti-inflammatory state in which cells release anti-inflammatory mediators, including cellular interleukin-10 (IL-10), cellular interleukin-4 (IL-4), Arginase-1(Arg1), chitinase-like protein-1 (Ym1), transforming growth factor-β (TGF-β). In addition, the cell surface marker of M2 phenotype, CD206, can promote reductions in inflammation, increased clearance, and enhanced expression of homeostasis-associated genes (24, 35, 39).
LPS is a critical component of the cell wall of gram-negative bacteria that induces the release of pro-inflammatory cytokines, produces neuroinflammatory responses, and promotes M1-type microglial polarization1 (40). Lipocalin 2 (LCN2) also promotes microglial M1-type activation. The expression of LCN2 in microglia was significantly increased after LPS treatment, indicating increased gene expression related to inflammatory processes in microglia (41).
Microglia and macrophages exhibit a protective M2 phenotype in the early stage after ischemic stroke, then gradually transition to a more classically activated M1 phenotype associated with nerve injury in the later stages after stroke. This transition demonstrates that microglia can exhibit dynamic changes in response to injury at different stages after stroke (23). Therefore, regulation of the microglial M1 and M2 phenotype balance is of great significance for the treatment of stroke.
BBB permeability increases with aging, as does the release of pro-inflammatory mediators in the nervous system. When the pro- and anti-inflammatory cytokine levels become unbalanced, this leads to an increased inflammatory state in the brain environment. Also, microglial activation and polarization change with aging (42, 43). It has been reported that decreased expression of chemokines is accompanied by increased microglial activation in the brains of aged rats. Additionally, the treatment of aged rats with chemokines can attenuate microglial activation (13). The data from Shoucai Zhao et al. showed an age-related difference in microglial activation after stroke. Indeed, the young stroke brains had higher IRF4 expression, corresponding to a stronger M2 microglial phenotype, as indicated by an up-regulated membrane CD206 level. In contrast, a stronger M1 phenotype was probably induced by up-regulated expression of IRF5 (44).
Microglia exhibit some differences between sexes. It is widely known that estrogens have anti-inflammatory activity, which may be a determining factor in the gender-dependent manifestations observed in brain lesions. Estrogen and progesterone could have neuroprotective roles in ischemic stroke by regulating the expression of chemokines and enhancing the effect of vascular endothelial growth factor (VEGF) (45). Villa et al. described significant differences in the transcriptomes of microglia from adult male and female that may arise from perinatal exposure to sex steroids (46). They also discovered that microglia from female were neuroprotective because they limited the injury caused by acute focal cerebral ischaemia (46). Bodhankar et al. reported that the expression of microglial M2 markers was higher in female mice than in male mice after ischemic stroke. In contrast, the expression of M1 markers, such as TNF-α and IL-1β, were significantly lower (47).
A recent study revealed that hypothermia has a regulatory effect on brain injury after stroke. Specifically, using a MCAO model, hypothermia decreased the number of M1 microglia and the expression of M1 markers and increased the expression of M2 markers. Thus, hypothermia can transform microglia from the M1 state into the M2 state. This supports the supposition that hypothermia has neuroprotective effects on ischemic stroke (48).
The results discussed above demonstrate that the activation and polarization of microglia differ based on age, sex, time after insult, and location. Microglia also are affected by temperature, and the effects of microglial cell production vary according to their environment. These observations suggest that the types of treatments that are effective and the prognostic results for patients experiencing different types of stroke will be different in clinical settings. These data also provide a range of new ideas to target and design novel treatments for stroke.
Given the importance of microglial responses during brain injury, it is critical to investigate the mechanisms of microglial activation and polarization. This will help to understand the molecular processes that underlie microglial morphological and functional changes caused by changes in the brain microenvironment and also provide direction for developing novel therapies to reduce the damage caused by stroke (Figure 1). According to previous studies, we know that the effects of microglia on cerebral ischemic injury are divided into three processes. First, microglia detect changes in the brain microenvironment after ischemic injury and detect extracellular signals through cell-surface and intracellular receptors. Second, these signals are integrated and transduced, through downstream reactions. Finally, these interactions cause microglia to produce pro-inflammatory or anti-inflammatory effects. Chemokine receptor-ligand pairing, transcription factors, signal transduction, activation of transcription, and other pathways are known to influence microglial activation and polarization through regulation of the activation of the pro-inflammatory transcription factor, NF-κB, which, in turn, regulates cytokine secretion.
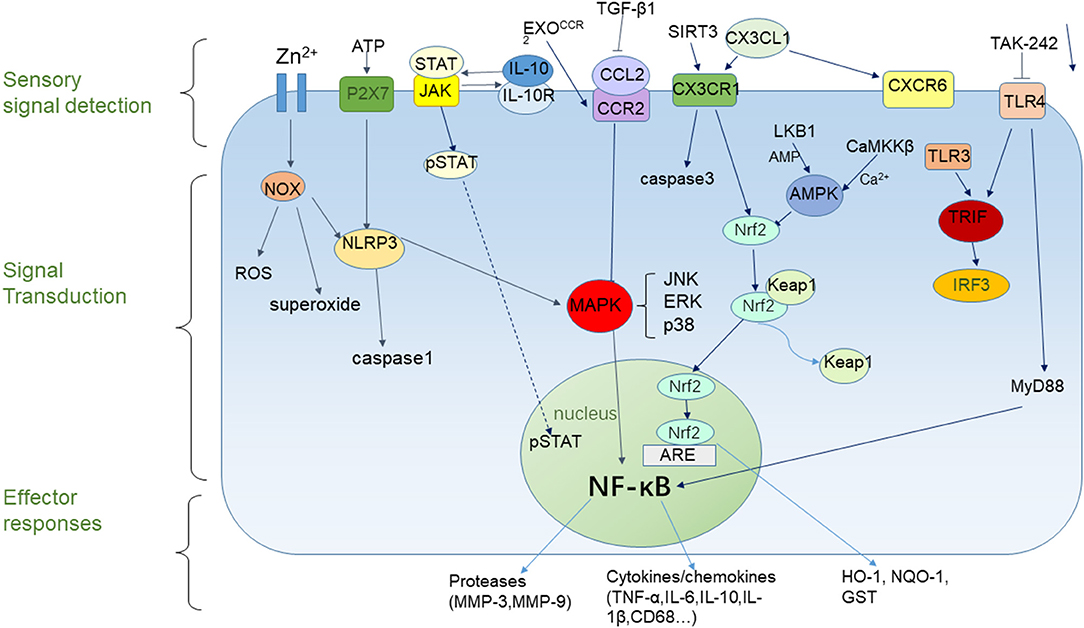
Figure 1. Microglial responses to cerebral ischemia. Microglial responses to cerebral ischemia can be divided into three parts. The sensory component can detect extracellular signals, the signal transduction component can influence gene expression, and the effector component can active pro-inflammatory or anti- inflammatory responses.
NF-κB regulates the transcription of a range of pro-inflammatory factors. After ischemic stroke, NF-κB is activated in activated microglia and translocates from the cytoplasm into the nucleus, which induces the production of inflammatory cytokines and results in secondary brain injury after stroke (49). NF-κB also induces microglia activation and polarization of the M1 phenotype (50). Recently, it has been shown that zinc induces microglial activation through activation of NOX and NF-κB (51). MAPKs are a family of serine/threonine protein kinases that regulate cell proliferation and survival. This family includes JNK, ERK, and p38, which are involved in ischemia-induced neuroinflammation (49). It is evident that MAPK and NF-κB plays an essential role in regulating neuroinflammation after ischemic stroke, as well as the activation and polarization of microglia. Therefore, targeting inhibitors of NF-κB or the activation of pathways that inhibit NF-κB activity might provide a new direction for the treatment of stroke.
Several recent studies have demonstrated that AMP-activated protein kinase (AMPK) and nuclear factor erythroid-2-related factor 2 (Nrf2) play crucial roles in the transition from a pro-inflammatory phenotype in microglia to an anti-inflammatory phenotype (52–54). AMPK is a trimeric serine/threonine kinase with two in vivo upstream kinases, including liver kinase B1 (LKB1) and calmodulin-dependent protein kinase kinase β (CaMKKβ). These kinases activate AMPK in AMP-dependent and Ca2+-dependent pathways, respectively (55, 56). AMPK, known as a central regulator in the body (57), stimulates energy production by shutting down energy-consuming pathways and phosphorylating antioxidant transcription factors, thereby maintaining metabolic and cellular energy homeostasis (58, 59). Activation of AMPK inhibits NF-κB activation and LPS-mediated pro-inflammatory activation of microglia, promoting M2-type microglial polarization (54). Recently, it has been reported that telmisartan can induce AMPK activation and regulate M2 phenotype polarization in microglia (60).
Nrf2 is a master transcriptional regulator of antioxidant pathways and exerts its anti-inflammatory effects through the activation of antioxidants. The N-terminal domain, Neh2, binds to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm. After oxidative stress, the Keap1-Nrf2 complex dissociates, Nrf2 accumulates in the nucleus and binds to antioxidant response elements (ARE) together with small Maf transcription factors. This complex promotes the generation of antioxidant and anti-inflammatory proteins, including heme oxygenase-1 (HO-1), NADPH quinone oxidoreductase-1 (NQO-1), Glutathione S-transferase (GST), and others (61–63). HO-1 has been reported to result from binding of ARE to a dimer, consisting of Nrf2 and activating transcription factor 4 (ATF4) (64). Similar to AMPK, Nrf2 activation suppresses neuroinflammation by inhibiting LPS-induced pro-inflammatory factor expression, and promotes macrophage M2 polarization, resulting in an anti-inflammatory effect (65). Tae et al. found that plumbagin, an activator of the Nrf2/ARE pathway, reduced the area of brain injury in a mouse model of MCAO and reduced the impairment of neurological function (53). The work of Zhang et al. shows that overexpression of SIRT6 in the brain by in vivo gene transfer potentiated anti-oxidant NRF2 signaling, reduced oxidative stress and the extent of cerebral I/R-induced brain tissue damage and neurological impairments, while in NRF2 knockout mice these neuroprotective effects were abolished (66). Another result shows that L-F001 can increase the expression levels of the M2 microglia marker CD206 via NRF2 signaling pathways activation in vitro (67), which corroborated the protective effect of NRF2 activation.
According to existing studies, AMPK phosphorylates Nrf2 at the Ser550 residue, leading to nuclear accumulation of Nrf2. AMPK also induces HO-1 production through the Nrf2/ARE pathway42 (59, 68), indicating that Nrf2 is a downstream signal for AMPK, both of which participate in neuroprotection following brain injury. 3-N-butylphthalate (NBP) has been shown to activate Nrf2 (69, 70). HP-1c, which is composed of telmisartan and the NBP derivative 2-(1-hydroxypentyl)-benzoic acid (HPBA), has been shown to promote M2 microglial polarization and exhibits antioxidant and anti-inflammatory effects through activation of the AMPK/Nrf2 pathway (68).
In summary, activation of AMPK and Nrf2 pathways promotes microglial M2 polarization, which reduces inflammation in the brain after stroke and plays a role in preventing further inflammation.
The chemokine fractalkine (CX3CL1)/CX3CR1 receptor-ligand pair was reported to be a communication link between neurons and microglia. CX3CL1 is an inhibitory factor expressed on neurons, and its receptor, CX3CR1, is primarily expressed on microglia (13, 71–73). The combination of these two components keeps microglia quiescent. CX3CL1 release from neurons is significantly reduced in the presence of nerve injury, which leads to microglial activation (74). The CX3CL1/CX3CR1 signaling axis promotes microglial phagocytic function in the early phase after ischemia (74).
After establishing a MCAO model for local cerebral ischemia in mice, Jolivel et al. observed that serum proteins lead to pooling of activated microglia near blood vessels, and vascular endothelial cells were phagocytosed, which ultimately led to BBB disintegration. On the other hand, the CX3CR1−/− (CX3CR1 loss of function) mouse model resulted in a reduction in BBB breakdown and a decrease in the area of stroke injury (12). Deficiency in CX3CR1 also leads to microglial morphological arborization and reduced expression of M1 phenotypic markers, thereby promoting microglial conversion to the M2 phenotype (74).
Overexpression of SIRT3 after ischemia leads to the upregulation of CX3CR1 expression, which promotes G protein-dependent migration of microglia (75). Thus, activation of the CX3CL1/CX3CR1 signaling axis in the early stage after ischemic stroke promotes microglial activation, migration, intrinsic phagocytosis, the release of pro-inflammatory substances, and exacerbate nerve injury. In contrast, a deficiency of the CX3CR1 receptor results in neuroprotective effects.
Recent experiments have revealed that a deficiency in CX3CR1 leads to significant reductions in the transcription factor, Nrf2 (76). These results suggest a link between CX3CR1 and Nrf2 during inflammation. CX3CL1 overexpression activates the transcription factor, Nrf2, and its target genes. For example, HO-1 limits the excessive activation of microglia, and when Nrf2 and CX3CR1 are knocked out, microglia do not express HO-1 (77). Also, CX3CL1 inhibits LPS-induced microglial activation and reduces the release of inflammatory factors in microglia, including NO, IL-6, and TNF-α through activation of the PI3k/Akt pathway, which effectively inhibits neuronal death (78, 79). The use of exogenous CX3CL1 in a MCAO mouse model reduced the local ischemia-induced cerebral infarct size, neurological deficits, and caspase-3 activation (80). These results indicate that CX3CL1/CX3CR1 signaling also plays a role in post-ischemic neuroprotection.
The studies described above demonstrate that the CX3CL1/CX3CR1 pathway can activate microglia, as well as promote their phagocytosis, migration to regions of brain tissue injury, and the release of inflammatory factors after stroke. Silencing CX3CR1 during the early post-ischemic period inhibited microglial activation, reduced the expression of inflammatory factors, polarized microglial toward the M2 phenotype, and effectively reduced the area of brain injury. Therefore, these data suggest that the lack of CX3CR1 is neuroprotective. However, the expression of CX3CR1 is not necessarily always detrimental to recovery after brain injury. Over time, the deficiency of CX3CR1 might induce other responses in the brain microenvironment, which may exacerbate brain injury in severe cases. For example, a deficiency in CX3CR1 leads to reduced expression of HO-1. Accordingly, we speculated that the functional contradiction exhibited by the CX3CL1/CX3CR1 pathway mightay be related to different times of onset as well as the course of the disease. According to recent reports, when inhibition of CX3CR1 is targeted early, or CX3CL1 expression levels are increased, administration of exogenous CX3CL1 after ischemia can reverse the neurological damage caused by stroke to some extent (81, 82). However, because this pathway contradiction results in both neuroprotective and neurodamaging effects, the specific mechanisms of action and treatment need additional, extensive research.
Monocyte chemoattractant protein-1 (MCP-1), also named CC chemokine ligand 2 (CCL2), can induce recruitment of monocytes or macrophages and increase the production of inflammatory cytokines, such as IL-1β and IL-6 (83, 84). The signaling pathway for MCP-1 is connected with CC chemokine receptor 2 (CCR2), which also plays an important regulatory role during stroke.
Overexpression of CCL2 has been shown to increase macrophage infiltration and the area of cerebral infarction at the injury site (84), suggesting that CCL2 can induce post-stroke inflammation and adversely affect post-stroke recovery. Overexpression of transforming growth factor-β1 (TGF-β1) after ischemia-reperfusion injury down-regulates CCL2, resulting in a significant reduction in the area of cerebral infarction (85), indicating that the neuroprotective effect was due to inhibition of the expression of chemokine CCL2, thus, confirming the neurotoxic effect of CCL2. Meanwhile, deficienc in CCL2 and CCR2 greatly reduce the recruitment of macrophages after stroke, the size of the cerebral infarction area, and the degree of BBB disruption after ischemia-reperfusion injury. Such deficiencies also reduce expression levels of inflammatory cytokines (86–89).
A human umbilical cord mesenchymal stem cell (HUC-MSC)-derived CCR2-overexpressing exosome, ExoCCR2, promotes microglial/macrophage M2-type polarization by competitively binding CCL2 to CCR2. This results in inhibition of nerve injury caused by CCL2-mediated macrophage migration and activation, reduction in the release of inflammatory factors, and decreased NF-κB expression, all of which promote microglial and macrophage M2-type polarization (90).
Therefore, activation of the CCL2/CCR2 pathway can recruit inflammatory cells and release pro-inflammatory factors, which are detrimental to the stroke prognosis. Inhibition of this pathway activity has a significant effect on ischemic stroke and cognitive impairment after stroke. Thus, targeting the inhibition of the CCL2 expression or its receptor CCR2 could improve the prognosis of stroke.
CXCL16 expression is increased in microglia treated with CX3CL1, and cerebral ischemia can result in overexpression of CXCL16 (91). Francesca Lepore proposed that activation of the CXCL16/CXCR6 axis regulated microglial polarization to the M2 phenotype after ischemia, and inhibited LPS- and IFNγ-mediated microglial polarization to the M1 type which, reduced the area of necrosis, promoted neuroprotective mechanisms, and inhibited ischemic neuronal death (92).
Toll-like receptors (TLR) are signaling receptors in the innate immune system that promote microglial activation and polarization (93). Currently, 10 and 12 members of the TLR family have been found in humans and mice, respectively, and exhibit different distributions and functions. For example, TLR2, TLR4, and others are located on the cell surface, while TLR3, TLR7, TLR9, and others are located inside the cell (93). TLR can recognize both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). The dimers that are formed bind to adaptor proteins such as MyD88, TRIF, and other chaperones to activate downstream signaling pathways, ultimately activating NF-κB and inducing the expression of inflammatory factors in microglia (94, 95).
TLR4 recognizes LPS on the cell surface and mediates microglial activation and the production of pro-inflammatory factors after brain injury (40, 96, 97). These functions aid in reducing secondary nerve injury caused by traumatic brain injury through inhibition of TLR4 signaling and regulating microglial polarization to the M2 phenotype (98).
Heme induces NF-κB activation after intracerebral hemorrhage injury by activating microglia and signaling MyD88 and TRIF through the TLR4 pathway, which in turn increases the expression of inflammatory factors and exacerbates inflammatory injury in brain tissue (99). In a model of hemorrhage stroke, the use of the TLR4 inhibitor, TAK-242, reduced the infiltration of peripheral inflammatory cells, expression of pro-inflammatory factors, and resulted in neurological deficits (100). This suggests that TLR4 is involved in the process of neuroinflammation after brain injury, and downregulation of TLR4 expression could control the adverse process to some extent. A recent study revealed that after ischemia-reperfusion treatment, smaller cerebral infarction areas occurred in TLR4 knockout mice. Also, downstream NF-κB and p65 expression were reduced, and there was no significant difference in infarct size or p65 expression levels between TLR3 and TLR9 knockout mice and wild-type mice (101).
Similarly, the use of β-caryophyllene after ischemia-reperfusion injury decreased TLR4 expression levels, reduced the release of pro-inflammatory factors, and inhibited microglial activation as well as M1-type polarization (93). These results suggest that TLR4 expression by microglia is associated with aggravation of stroke-induced secondary brain injury. Thus, targeted inhibition of TLR4 could reduce microglial activation and release of pro-inflammatory factors, coordinate the M1/M2 polarization of microglia, and play a protective role in stroke-induced brain injury.
Beside TLR4, also TLR3 plays a key role in microglia activation. TLR3 induces activation of IFN regulatory factor 3 (IRF3) and NF-κB via the TRIF pathway (96). Pan et al. established an ischemia-reperfusion model after preconditioning rats using the TLR3 agonist, polyinosinic-polycytidylic acid [poly (I:C)]. They reported that the degree of nerve injury, cerebral infarction area, and the expression levels of TNFα and IL-6 were significantly reduced compared with the control group (102). Injecting poly (I:C) into animal models after cerebral ischemia also down-regulated the conduction of the TLR4/MyD88 signaling pathway by activating the TLR3/TRIF pathway. Poly (I:C) significantly reduced the expression levels of TNF-α and IL-1β, indicating it could have a therapeutic role in ischemia-reperfusion injury (103). Based on the results discussed above, we hypothesize that there is an antagonistic effect between the TLR3 and TLR4 pathways. Activation of the TLR3/TRIF pathway could down-regulate the levels of TLR4/MyD88, reducing microglial activation and the expression of inflammatory factors to reverse the adverse effects of TLR4 on post-stroke injury. Therefore, activation of TLR3 should be investigated as a possible therapy for ischemic brain injury. In summary, TLR could be an important target for stroke treatment due to its essential role in regulating the inflammatory response.
The JAK/STAT pathway is one of the signal transduction cascades and plays an essential role in cytokine receptor signaling (104). JAK is a member of the Janus kinase family of protein tyrosine kinases, which is found primarily on cell membranes. It binds to and phosphorylates a JAK-binding site containing domains for cytokine receptors, and subsequently forms STAT-binding sites to recruit signal transducers and activators of transcription (STATs). The STATs are then phosphorylate and translocated to the nucleus to regulate gene expression (105). IL-10 protects against inflammation by inhibiting the release of pro-inflammatory cytokines from monocytes/macrophages and the activity of LPS (106–108). One of the IL-10 signaling pathways is the JAK/STAT pathway. Binding of IL-10 to the IL-10 receptor (IL-10R) activates JAK1 and STAT3, which is necessary to induce IL-10 to inhibit macrophage activation and produce anti-inflammatory effects (109–111).
Recently, it has been reported that the neuroprotective effect induced by the inhibition of NOX may be generated through activation of the IL-10/STAT3 pathway (31). Melatonin has been shown to inhibit microglial M1-type polarization by activating STAT3 and induces M2-type polarization in microglia, which results in neuroprotection (112). Resveratrol up-regulated the expression of the suppressor of cytokine signaling 3 (SOCS3) by promoting the release of IL-10, which activated the JAK1/STAT3 signaling pathway and, in turn, inhibited LPS-induced microglial pro-inflammatory factor release and M1-type polarization (113). On the other hand, STATs also exhibit several negative roles. For example, the activation and phosphorylation of STAT1 is associated with M1 microglia activation in hypoxia-activated BV2 cells, and acts as the increased expression levels of M1 microglia (114). Furthermore, STAT3 was also associated with M1 microglia polarization in both an MCAO-induced and a bilateral common carotid arteries stenosis (BcaS)-induced model of ischemic stroke (115, 116). The results discussed above reveal the regulatory role of STAT on the inflammatory response after stroke. However, due to these contradictory evidence on regulation of microglia polarization, further studies are required.
Extracellular nucleotide receptors (P2 receptors) include two families, ionotropic receptors (P2X) and metabotropic receptors (P2Y). The purinergic ligand-gated ion channel 7 receptor (P2X7) is a trimeric cation channel, which is activated by extracellular ATP. P2X7 activates Pyrin domain-containing 3 (NLRP3). After injury resulting from ischemic stroke, elevated extracellular ATP concentrations activate P2X7, triggering a series of intracellular responses that lead to NLRP3 inflammatory body activation and assembly (117). Botulinum toxin type A (BTX-A) inhibits M1-type microglial polarization and induces M2 polarization in rats through inhibition of P2X7 expression (118). A role of P2Y12 receptor in microglia activation was also shown. Indeeed, extracellular ADP acting on the P2Y12 receptor activated NF-κB and the NLRP3 inflammasome to enhance microglial inflammation (119). Furthermore, in primary cultured microglia, ticagrelor, a direct-acting, reversibly binding P2Y12-receptor antagonist, fully inhibited ADP-induced chemotaxis (120).
NLRP3 is a member of the Nod-like receptor (NLR) family (121). It is a cytosolic complex that activates caspase-1, and its interaction with the adaptor protein, Asc, also can recruit and activate caspase-1 that subsequently leads to the maturation of IL-1β and IL-18 (122, 123). It has been shown that upregulation of NLRP3 expression and increased release of pro-inflammatory cytokines in microglia after ischemia exacerbated the resulting post-ischemic neurological damage. However, after inhibiting the expression of NLRP3 in an ischemic mouse model, the brain injury caused by ischemia was alleviated by reducing the degree of cerebral infarction and the BBB disruption (124). This study also found that NLRP3 expression was down-regulated in the NOX2-deficient model, suggesting that NOX is involved in the activation and expression of NLRP3 in microglia. These results further elucidated the mechanisms by which NOX exerts its toxic effects.
Thus, P2X7, P2Y12, and NLRP3 play essential roles in regulating the occurrence of neuroinflammation after ischemic stroke. Inhibition of their expression reduces the release of pro-inflammatory factors in microglia and can alleviate post-stroke brain injury.
Glutaminase-1 (GLS1) is a mitochondrial enzyme that catalyzes the hydrolysis of glutamine to produce glutamate. Elevated GLS1 expression can be observed in activated microglia, and GLS1 induces inflammatory activation of microglia and the release of exosomes (125). Gao et al. observed that the degree of upregulation of GLS1 expression after ischemia was positively correlated with increased expression of microglial M1-type markers. Also, overexpression of GLS1 induced microglial activation by increasing the release of inflammatory exosomes (126). Meanwhile, inhibition of GLS1 activity by CB839 reduced the release of exosomes, which alleviated neuroinflammation resulting from cerebral ischemia. Thus, this effect was the same as inhibition of exosome release by GW4869, which also alleviated the inflammatory response (126). Therefore, inhibition of GLS1 expression might produce neuroprotective effects and could be a new treatment avenue for ischemic stroke.
Peroxisome proliferator-activator receptor γ (PPARγ) is a member of the nuclear receptor family that binds to PPAR response elements (PPRE) in the promoter region of target genes. Currently three PPAR subtypes have been discovered, including PPAR-γ, PPAR-α, and PPAR-δ (127).
PPARγ is known to play a key role in regulating lipid metabolism, cell apoptosis and inflammation (128, 129). In addition, PPAR-γ activation is reported to reduce neurodegenerative and inflammatory processes in the brain (127). Recent studies have described a close relationship between PPARγ and ischemia injury, suggesting that the effect of PPARγ on ischemic injury is primarily associated with regulation of the inflammatory response (130). Also, PPARγ has been shown to coordinate the switch in the microglia/macrophage phenotype from a pro-inflammatory to an anti-inflammatory phenotype, leading to inhibition of inflammation (131). For example, a current study showed that 10-O-(N,N-dimethylaminoethyl)-ginkgolide B methanesulfonate (XQ-1H) promoted anti-inflammatory microglial polarization through activation of the PPARγ signaling pathway after ischemic stroke (130). Also, rhFGF21 treatment inhibited M1 polarization and pro-inflammatory cytokine expression in microglia by inhibiting NF-κB and upregulating PPAR-γ (132). This evidence suggests a neuroprotective effect of PPAR-γ activation on cerebral ischemic injury.
Based on the analysis described above, we conclude that microglia play a critical role in regulating the repair process that takes place in post-stroke injury. Microglia not only sense the changes in the brain microenvironment and respond through regulation of key factors but microglia, in turn, are regulated by specific signaling pathways and cytokines. Thus, finding treatments that effectively inhibit the activation and polarization of the pro-inflammatory microglial phenotype has important clinical implications for improving stroke prognosis. Some of the proven drugs categorized based on their targets are listed below (Table 1).
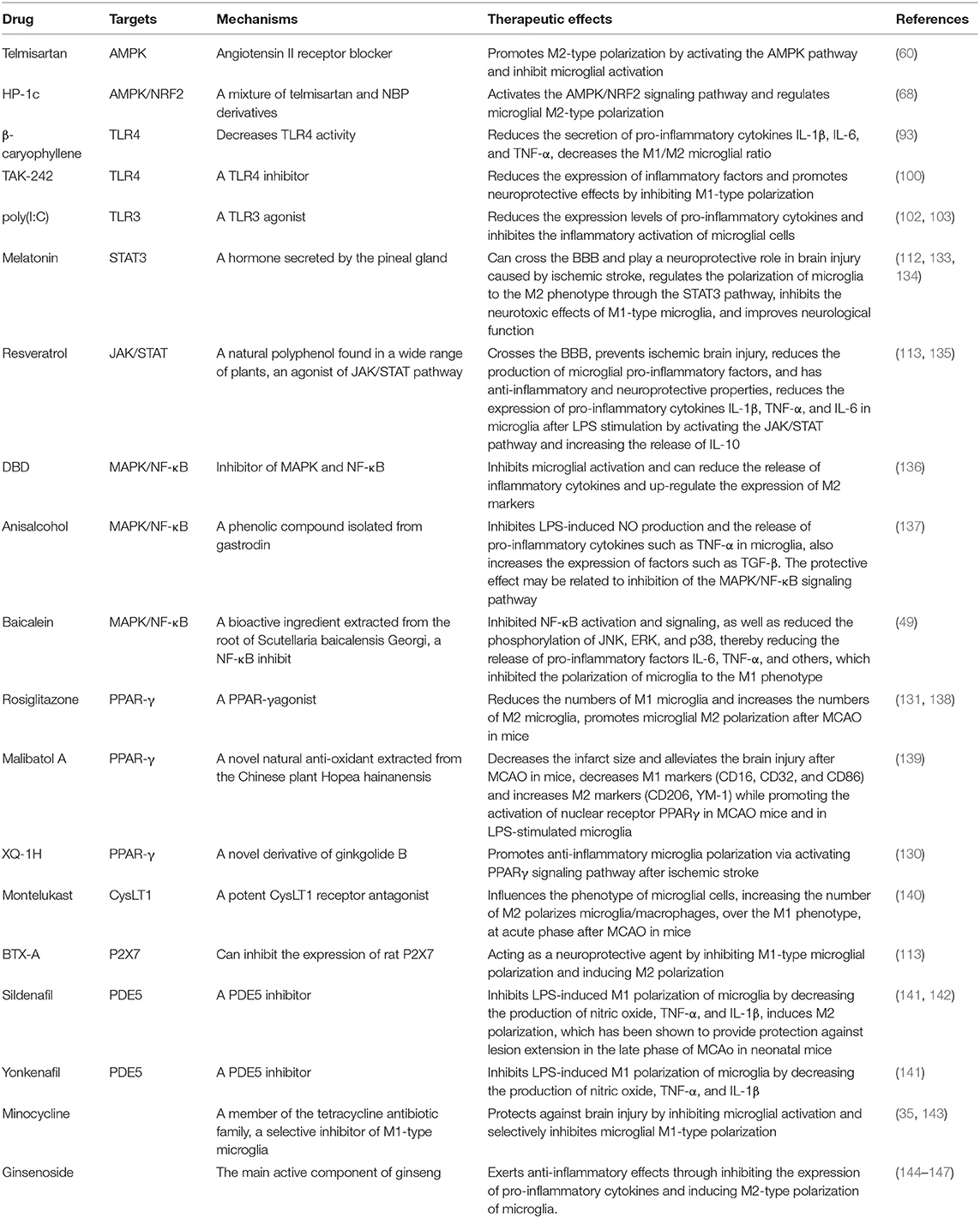
Table 1. Drug factors affecting microglial activation and polarization.
Stroke is a leading cause of death worldwide, and effective treatments are a pressing challenge. Over the years, research into the cellular events and mechanisms associated with stroke has been ongoing. However, there are still unanswered questions, such as the activation of some targets that are protective against stroke and also have adverse effects. There are still disputes concerning how these targets can be regulated to maximize their protective effects, how to use activators of these targets, and whether the timing and dose of these drugs might have different effects on prognosis. These questions need further exploration.
It is known that microglia are rapidly activated and polarized into M1 and M2 phenotypes after stroke, which produces opposite effects on post-stroke brain injury. Based on current experiments, we conclude that inhibiting M1 microglia and promoting M2 microglia is protective against neurological damage. However, some signaling pathways and drugs have been shown to induce both M1 and M2-type polarization in microglia. Does this indicate a dual role for these pathways and drugs, or do microglia possess additional, undiscovered properties? These questions need to be resolved by further experiments. Moreover, microglia can exhibit a mixed phenotype between M1 and M2 (8). Thus, limiting microglia to the two phenotypes representing two polar states after stroke is oversimplified. Therefore, the classification and nomenclature of microglia requir further exploration.
Although successful experimental treatment protocols for stroke have been developed, they have not been entirely successful in humans. We know that the current treatment for stroke in the clinical setting is still challenging. There are two possible reasons for this: (1) In experimental models, the MCAO model is usually used to simulate stroke in vivo as well as the OGD model in vitro, which may not adequately simulate clinical stroke. This is because stroke involves a long process of disease progression, which is influenced by a range of factors, including age, gender, temperature, environment and others. Several cellular events also are involved in the stroke process. For stroke patients experiencing other diseases at the same time, the diseases may interact and could cause increases or decreases the damage caused by the stroke. This is why experimental models have limitations. (2) Species differences exist between humans and experimental models. Also, the expression level of the same targets and effects of drugs are likely to vary in different species. Therefore, experimental results can only provide therapeutic ideas for clinical research, and translational research is particularly important.
In conclusion, we suggest several recommendations: (1) Researchers should search for novel and accurate experimental stroke models. (2) The communication between human microglia and neurons should be observed directly to study the protective effects of human microglia on the nervous system. (3) Stroke-related pathway proteins, targets or markers of M1 and M2 microglia phenotypes should be extracted and evaluated from the cerebrospinal fluid or serum of stroke patients to observe the effects of stroke on different molecular pathways and cell phenotypes in humans. (4) Additional drugs need to be identified that may have protective effects against stroke, e.g., network pharmacology should be used to find herbal medicines that have the same targets as stroke, and identify new nanomaterials. In addition, the mechanisms that influence microglia activation and polarization during stroke need to be studied in greater depth.
RD and JW: conceptualization. RD: writing—original draft preparation. JW, RH, and HL: writing—review and editing. ZX: reading and suggestions. All authors have read and agreed to the published version of the manuscript.
This research was funded by the National Natural Science Foundation of China (81671159), the National Natural Science Foundation of China (81371298), the National Natural Science Foundation of China (31700927), Jilin Province Health Research Talent Project (2019SCZ045), and Jilin Province Science and Technology Development Project (20190103081JH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We appreciated EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
CNS, Central nervous system; NO, Nitric oxide; BBB, Blood-brain barrier; MCAO, Middle Cerebral Artery Occlusion; ROS, Reactive oxygen species; Aβ, Amyloid-β peptide; NF-κB, Nuclear factor kappa-B; NGF, Nerve growth factor; NOX, NADPH oxidase; TNF-α, Tumor necrosis factor-α; IL-6, Interleukin 6; II-1β, Interleukin 1β; IL-10, Interleukin-10; TGF-β, Transforming growth factor-β; LPS, Lipopolysaccharide; LCN2, Lipocalin 2; VEGF, Vascular endothelial growth factor; AMPK, AMP-activated protein kinase; Nrf2, Nuclear factor erythroid-2-related factor 2; LKB1, Liver kinase B1; CaMKKβ, Calmodulin-dependent protein kinase kinase β; Keap1, Kelch-like ECH-associated protein 1; ARE, Antioxidant response elements; HO-1, Heme oxygenase-1; NQO-1, NADPH quinone oxidoreductase-1; GST, Glutathione S-transferase; ATF4, Activating transcription factor 4; MCP-1, Monocyte chemoattractant protein-1; CCL2, CC chemokine ligand 2; CCR2, CC chemokine receptor 2; HUC-MSC, Human umbilical cord mesenchymal stem cell; TLR, Toll-like receptors; STAT, Signal PAMP, Pathogen-associated molecular pattern; DAMP, Damage-associated molecular patterns; STAT, Signal transducers and activator of transcription; SOCS3, Suppressor of cytokine signaling 3; PPAR-γ, Peroxisome proliferator-activator receptor γ; BTX-A, Botulinum toxin type A; NLR, Nod-like receptor; DBD, 3, 4-Dihydroxybenzaldehyde; GLS1, Glutaminase-1; BTX-A, Botulinum toxin type A; PDE5, phosphodiesterase of type 5; ARB, Angiotensin II receptor blocker.
1. Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain. (2018) 11:15. doi: 10.1186/s13041-018-0357-8
2. Rothwell PM, Algra A, Amarenco P. Medical treatment in acute and long-term secondary prevention after transient ischaemic attack and ischaemic stroke. Lancet. (2011) 377:1681–92. doi: 10.1016/S0140-6736(11)60516-3
3. Hachinski V, Azarpazhooh MR. Stroke is a burdensome but preventable brain disorder. Lancet Neurol. (2016) 15:892–3. doi: 10.1016/S1474-4422(16)30120-X
4. WHO. Leading Causes of Death and Disability 2000-2019-A Visual Summary. Available online at: https://www.who.int/data/stories/leading-causes-of-death-and-disability-2000-2019-a-visual-summary.
5. Kalaria RN, Akinyemi R, Ihara M. Stroke injury, cognitive impairment and vascular dementia. Biochim Biophys Acta. (2016) 1862:915–25. doi: 10.1016/j.bbadis.2016.01.015
6. Rajan WD, Wojtas B, Gielniewski B, Gieryng A, Zawadzka M, Kaminska, et al. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia. (2019) 67:232–45. doi: 10.1002/glia.23536
7. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. (2017) 13:420–33. doi: 10.1038/nrneurol.2017.69
8. Loane DJ, Kumar A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol. (2016) 275:316–27. doi: 10.1016/j.expneurol.2015.08.018
9. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, et al. dual functions of microglia in ischemic stroke. Neurosci Bull. (2019) 35:921–33. doi: 10.1007/s12264-019-00388-3
10. Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-β. J Neuroimmunol. (2009) 210:3–12. doi: 10.1016/j.jneuroim.2009.02.003
11. Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. (2012) 87:10–20. doi: 10.1016/j.brainresbull.2011.10.004
12. Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. (2015) 129:279–95. doi: 10.1007/s00401-014-1372-1
13. Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int J Mol Sci. (2018) 19:318. doi: 10.3390/ijms19010318
14. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
15. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
16. Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell. (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
17. Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. (1992) 48:405–15. doi: 10.1016/0306-4522(92)90500-2
18. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. (1990) 39:151–70. doi: 10.1016/0306-4522(90)90229-W
19. Wang J, Xing H, Wan L, Jiang X, Wang C, Wu Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed Pharmacother. (2018) 105:518–25. doi: 10.1016/j.biopha.2018.05.143
20. Zhang Z, Chopp M, Powers C. Temporal profile of microglial response following transient (2h) middle cerebral artery occlusion. Brain Res. (1997) 744:189–98. doi: 10.1016/S0006-8993(96)01085-2
21. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. (2016) 142:23–44. doi: 10.1016/j.pneurobio.2016.05.001
22. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
23. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. (2012) 43:3063–70. doi: 10.1161/STROKEAHA.112.659656
24. Jin X, Yamashita T. Microglia in central nervous system repair after injury. J Biochem. (2016) 159:491–6. doi: 10.1093/jb/mvw009
25. Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M, et al. Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav Immunity. (2014) 37:1–14. doi: 10.1016/j.bbi.2013.12.010
26. Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflam. (2012) 9:5. doi: 10.1186/1742-2094-9-5
27. Luan P, Ding X, Xu J, Jiang L, Xu Y, Zhu Y, et al. Salvianolate reduces neuronal apoptosis by suppressing OGD-induced microglial activation. Life Sci. (2020) 260:118393. doi: 10.1016/j.lfs.2020.118393
28. Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. (2001) 21:1179–8. doi: 10.1523/JNEUROSCI.21-04-01179.2001
29. Rizzi C, Tiberi A, Giustizieri M, Marrone MC, Gobbo F, Carucci NM, et al. NGF steers microglia toward a neuroprotective phenotype. Glia. (2018) 66:1395–416. doi: 10.1002/glia.23312
30. Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxidants Redox Signal. (2006) 8:1583–96. doi: 10.1089/ars.2006.8.1583
31. Barrett JP, Henry RJ, Villapol S, Stoica BA, Kumar A, Burns MP, et al. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflam. (2017) 14:65. doi: 10.1186/s12974-017-0843-4
32. Mota M, Porrini V, Parrella E, Benarese M, Bellucci A, Rhein S, et al. Neuroprotective epi-drugs quench the inflammatory response and microglial/macrophage activation in a mouse model of permanent brain ischemia. J Neuroinflammation. (2020) 17:361. doi: 10.1186/s12974-020-02028-4
33. Yao Z, Liu N, Zhu X, Wang L, Zhao Y, Liu Q, et al. Subanesthetic isoflurane abates ROS-activated MAPK/NF-kappaB signaling to repress ischemia-induced microglia inflammation and brain injury. Aging. (2020) 12:26121–39. doi: 10.18632/aging.202349
34. Zhao SC, Ma LS, Chu ZH, Xu H, Wu WQ, Liu F. Regulation of microglial activation in stroke. Acta Pharmacol Sin. (2017) 38:445–58. doi: 10.1038/aps.2016.162
35. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. (2013) 4:e525. doi: 10.1038/cddis.2013.54
36. Zhang J, Zheng Y, Luo Y, Du Y, Zhang X, Fu Jl. Curcumin inhibits LPS-induced neuroinflammation by promoting microglial M2 polarization via TREM2/ TLR4/ NF-kappaB pathways in BV2 cells. Mol Immunol. (2019) 116:29–37. doi: 10.1016/j.molimm.2019.09.020
37. Higashi Y, Aratake T, Shimizu S, Shimizu T, Nakamura K, Tsuda M, et al. Influence of extracellular zinc on M1 microglial activation. Sci Rep. (2017) 7:43778. doi: 10.1038/srep43778
38. Zhao R, Ying M, Gu S, Yin W, Li Y, Yuan H, et al. Cysteinyl leukotriene receptor 2 is involved in inflammation and neuronal damage by mediating microglia M1/M2 polarization through NF-kappaB pathway. Neuroscience. (2019) 422:99–118. doi: 10.1016/j.neuroscience.2019.10.048
39. Collmann FM, Pijnenburg R, Hamzei-Taj S, Minassian A, Folz-Donahue K, Kukat C, et al. Individual in vivo profiles of microglia polarization after stroke, represented by the genes iNOS and Ym1. Front Immunol. (2019) 10:1236. doi: 10.3389/fimmu.2019.01236
40. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
41. Jang E, Lee S, Kim JH, Kim JH, Seo JW, Lee WH, et al. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. (2013) 27:1176–90. doi: 10.1096/fj.12-222257
42. von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. (2015) 7:124. doi: 10.3389/fnagi.2015.00124
43. Hua K, Schindler MK, McQuail JA, Forbes ME, Riddle DR. Regionally distinct responses of microglia and glial progenitor cells to whole brain irradiation in adult and aging rats. PLoS ONE. (2012) 7:e52728. doi: 10.1371/journal.pone.0052728
44. Zhao SC, Wang C, Xu H, Wu WQ, Chu ZH, Ma LS, et al. Age-related differences in interferon regulatory factor-4 and−5 signaling in ischemic brains of mice. Acta Pharmacol Sin. (2017) 38:1425–34. doi: 10.1038/aps.2017.122
45. Dang J, Mitkari B, Kipp M, Beyer C. Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immunity. (2011) 25:715–26. doi: 10.1016/j.bbi.2011.01.013
46. Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-specific features of microglia from adult mice. Cell Rep. (2018) 23:3501–11. doi: 10.1016/j.celrep.2018.05.048
47. Bodhankar S, Lapato A, Chen Y, Vandenbark AA, Saugstad JA, Offner H. Role for microglia in sex differences after ischemic stroke: importance of M2. Metab Brain Dis. (2015) 30:1515–29. doi: 10.1007/s11011-015-9714-9
48. Liu L, Liu X, Wang R, Yan F, Luo Y, Chandra A, et al. Mild focal hypothermia regulates the dynamic polarization of microglia after ischemic stroke in mice. Neurol Res. (2018) 40:508–15. doi: 10.1080/01616412.2018.1454090
49. Yang S, Wang H, Yang Y, Wang R, Wang Y, Wu C, et al. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed Pharmacother. (2019) 117:109102. doi: 10.1016/j.biopha.2019.109102
50. Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol Med Rep. (2020) 21:2006–18. doi: 10.3892/mmr.2020.11003
51. Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci. (2008) 28:5827–35. doi: 10.1523/JNEUROSCI.1236-08.2008
52. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. (2013) 18:251–64. doi: 10.1016/j.cmet.2013.06.017
53. Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, et al. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem. (2010) 112:1316–26. doi: 10.1111/j.1471-4159.2009.06552.x
54. Chu X, Cao L, Yu Z, Xin D, Li T, Ma W, et al. Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J Neuroinflammation. (2019) 16:104. doi: 10.1186/s12974-019-1488-2
55. Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. (2009) 9:407–16. doi: 10.1016/j.cmet.2009.03.012
56. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. (2007) 403:139–48. doi: 10.1042/BJ20061520
57. Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. (2016) 36:1931–42. doi: 10.1128/MCB.00118-16
58. Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
59. Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol. (2011) 300:H84–93. doi: 10.1152/ajpheart.00749.2010
60. Xu Y, Xu Y, Wang Y, Wang Y, He L, Jiang Z, et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain Behav Immun. (2015) 50:298–313. doi: 10.1016/j.bbi.2015.07.015
61. Dinkova-Kostova AT, Kostov RV, Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. (2018) 285:3576–90. doi: 10.1111/febs.14379
62. Satoh T, McKercher SR, Lipton SA. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med. (2013) 65:645–57. doi: 10.1016/j.freeradbiomed.2013.07.022
63. Tkachev VO, Menshchikova EB, Zenkov NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochem Biokhimiia. (2011) 76:407–22. doi: 10.1134/S0006297911040031
64. He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. implication for heme oxygenase-1 gene regulation. J Biol Chem. (2001) 276:20858–65. doi: 10.1074/jbc.M101198200
65. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:11624. doi: 10.1038/ncomms11624
66. Zhang W, Wei R, Zhang L, Tan Y, Qian C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience. (2017) 366:95–104. doi: 10.1016/j.neuroscience.2017.09.035
67. Chen J, Yin W, Tu Y, Wang S, Yang X, Chen Q, et al. L-F001, a novel multifunctional ROCK inhibitor, suppresses neuroinflammation in vitro and in vivo: Involvement of NF-kappaB inhibition and Nrf2 pathway activation. Eur J Pharmacol. (2017) 806:1–9. doi: 10.1016/j.ejphar.2017.03.025
68. Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, et al. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxidants Redox Signal. (2018) 28:141–63. doi: 10.1089/ars.2017.7003
69. Feng X, Peng Y, Liu M, Cui L. dl-3-n-butylphthalide extends survival by attenuating glial activation in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology. (2012) 62:1004–10. doi: 10.1016/j.neuropharm.2011.10.009
70. Hua K, Sheng X, Li TT, Wang LN, Zhang YH, Huang ZJ, et al. The edaravone and 3-n-butylphthalide ring-opening derivative 10b effectively attenuates cerebral ischemia injury in rats. Acta Pharmacol Sin. (2015) 36:917–27. doi: 10.1038/aps.2015.31
71. Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. (2013) 3:130181. doi: 10.1098/rsob.130181
72. Cotter R, Williams C, Ryan L, Erichsen D, Lopez A, Peng H, et al. Fractalkine (CX3CL1) and brain inflammation: implications for HIV-1-associated dementia. J Neurovirol. (2002) 8:585–98. doi: 10.1080/13550280290100950
73. Lauro C, Catalano M, Trettel F, Limatola C. Fractalkine in the nervous system: neuroprotective or neurotoxic molecule? Ann N Y Acad Sci. (2015) 1351:141–8. doi: 10.1111/nyas.12805
74. Fumagalli S, Perego C, Ortolano F, De Simoni MG. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. (2013) 61:827–42. doi: 10.1002/glia.22474
75. Cao R, Li S, Yin J, Guo L, Shi J. Sirtuin 3 promotes microglia migration by upregulating CX3CR1. Cell Adh Migr. (2019) 13:229–35. doi: 10.1080/19336918.2019.1629224
76. Castro-Sánchez S, García-Yagüe ÁJ, Kügler S, Lastres-Becker I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. (2019) 22:101118. doi: 10.1016/j.redox.2019.101118
77. Lastres-Becker I, Innamorato NG, Jaworski T, Rábano A, Kügler S, Van Leuven F, et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. (2014) 137:78–91. doi: 10.1093/brain/awt323
78. Lyons A, Lynch AM, Downer EJ, Hanley R, O'Sullivan JB, Smith A, et al. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. (2009) 110:1547–56. doi: 10.1111/j.1471-4159.2009.06253.x
79. Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. (2003) 979:65–70. doi: 10.1016/S0006-8993(03)02867-1
80. Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. (2011) 31:16327–35. doi: 10.1523/JNEUROSCI.3611-11.2011
81. Cisbani G, Le Behot A, Plante MM, Prefontaine P, Lecordier M, Rivest S. Role of the chemokine receptors CCR2 and CX3CR1 in an experimental model of thrombotic stroke. Brain Behav Immun. (2018) 70:280–92. doi: 10.1016/j.bbi.2018.03.008
82. Yang G, Liu Z, Wang L, Chen X, Wang X, Dong Q, et al. MicroRNA-195 protection against focal cerebral ischemia by targeting CX3CR1. J Neurosurg. (2018) 131:1–10. doi: 10.3171/2018.5.JNS173061
83. Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. (1998) 187:601–8. doi: 10.1084/jem.187.4.601
84. Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. (2003) 23:748–55. doi: 10.1097/01.WCB.0000071885.63724.20
85. Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY. Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke. (2001) 32:544–52. doi: 10.1161/01.STR.32.2.544
86. Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma. (2014) 31:1677–88. doi: 10.1089/neu.2013.3252
87. Morganti JM, Jopson TD, Liu S, Riparip L.-K, Guandique CK, Gupta N. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. (2015) 35:748–60. doi: 10.1523/JNEUROSCI.2405-14.2015
88. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. (2007) 38:1345–53. doi: 10.1161/01.STR.0000259709.16654.8f
89. Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. (2002) 22:308–17. doi: 10.1097/00004647-200203000-00008
90. Yang HC, Zhang M, Wu R, Zheng HQ, Zhang LY, Luo J, et al. C-C chemokine receptor type 2-overexpressing exosomes alleviated experimental post-stroke cognitive impairment by enhancing microglia/macrophage M2 polarization. World J Stem Cells. (2020) 12:152–67. doi: 10.4252/wjsc.v12.i2.152
91. Rosito M, Lauro C, Chece G, Porzia A, Monaco L, Mainiero F, et al. Trasmembrane chemokines CX3CL1 and CXCL16 drive interplay between neurons, microglia and astrocytes to counteract pMCAO and excitotoxic neuronal death. Front Cell Neurosci. (2014) 8:193. doi: 10.3389/fncel.2014.00193
92. Lepore F, D'Alessandro G, Antonangeli F, Santoro A, Esposito V, Limatola C, et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol. (2018) 9:2750. doi: 10.3389/fimmu.2018.02750
93. Tian X, Liu H, Xiang F, Xu L, Dong Z. beta-Caryophyllene protects against ischemic stroke by promoting polarization of microglia toward M2 phenotype via the TLR4 pathway. Life Sci. (2019) 237:116915. doi: 10.1016/j.lfs.2019.116915
94. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
95. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
96. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
97. Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur C, et al. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflam. (2013) 10:23. doi: 10.1186/1742-2094-10-23
98. Yao X, Liu S, Ding W, Yue P, Jiang Q, Zhao M, et al. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J Neuroimmunol. (2017) 310:38–45. doi: 10.1016/j.jneuroim.2017.06.006
99. Lin S, Yin Q, Zhong Q, Lv F.-L, Zhou Y, Li J.-Q, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflamm. (2012) 9:46. doi: 10.1186/1742-2094-9-46
100. Wang YC, Wang PF, Fang H, Chen J, Xiong XY, Yang QW. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke. (2013) 44:2545–52. doi: 10.1161/STROKEAHA.113.001038
101. Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. (2010) 171:258–67. doi: 10.1016/j.neuroscience.2010.08.054
102. Pan LN, Zhu W, Li C, Xu XL, Guo LJ, Lu Q. Toll-like receptor 3 agonist Poly I:C protects against simulated cerebral ischemia in vitro and in vivo. Acta Pharmacol Sin. (2012) 33:1246–53. doi: 10.1038/aps.2012.122
103. Wang PF, Fang H, Chen J, Lin S, Liu Y, Xiong XY, et al. Polyinosinic-polycytidylic acid has therapeutic effects against cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via TLR3. J Immunol. (2014) 192:4783–94. doi: 10.4049/jimmunol.1303108
104. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. (2007) 178:2623–9. doi: 10.4049/jimmunol.178.5.2623
105. Behrmann I, Smyczek T, Heinrich PC, Schmitz-Van de Leur H, Komyod W, Giese B, et al. Janus kinase (Jak) subcellular localization revisited: the exclusive membrane localization of endogenous Janus kinase 1 by cytokine receptor interaction uncovers the Jak.receptor complex to be equivalent to a receptor tyrosine kinase. J Biol Chem. (2004) 279:35486–93. doi: 10.1074/jbc.M404202200
106. de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. (1991) 174:1209–20. doi: 10.1084/jem.174.5.1209
107. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. (1991) 147:3815–22.
108. Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate Aβ(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol. (2001) 113:49–62. doi: 10.1016/S0165-5728(00)00404-5
109. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the Interleukin-10 Receptor. Ann Rev Immunol. (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
110. Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. (2004) 172:567–76. doi: 10.4049/jimmunol.172.1.567
111. Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. (1999) 274:16513–21. doi: 10.1074/jbc.274.23.16513
112. Liu ZJ, Ran YY, Qie SY, Gong WJ, Gao FH, Ding ZT, et al. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci Ther. (2019) 25:1353–62. doi: 10.1111/cns.13261
113. Cianciulli A, Dragone T, Calvello R, Porro C, Trotta T, Lofrumento DD, et al. IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int Immunopharmacol. (2015) 24:369–76. doi: 10.1016/j.intimp.2014.12.035
114. Butturini E, Boriero D, Carcereri de Prati A, Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch Biochem Biophys. (2019) 669:22–30. doi: 10.1016/j.abb.2019.05.011
115. Qin C, Fan WH, Liu Q, Shang K, Murugan M, Wu LJ, et al. Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke. (2017) 48:3336–46. doi: 10.1161/STROKEAHA.117.018505
116. Ding Y, Qian J, Li H, Shen H, Li X, Kong Y, et al. Effects of SC99 on cerebral ischemia-perfusion injury in rats: selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci Res. (2019) 142:58–68. doi: 10.1016/j.neures.2018.05.002
117. Di Virgilio F. Liaisons dangereuses: P2X7 and the inflammasome. Trends Pharmacol Sci. (2007) 28:465–72. doi: 10.1016/j.tips.2007.07.002
118. Gui X, Wang H, Wu L, Tian S, Wang X, Zheng H, et al. Botulinum toxin type A promotes microglial M2 polarization and suppresses chronic constriction injury-induced neuropathic pain through the P2X7 receptor. Cell Biosci. (2020) 10:45. doi: 10.1186/s13578-020-00405-3
119. Suzuki T, Kohyama K, Moriyama K, Ozaki M, Hasegawa S, Ueno T, et al. Extracellular ADP augments microglial inflammasome and NF-kappaB activation via the P2Y12 receptor. Eur J Immunol. (2020) 50:205–19. doi: 10.1002/eji.201848013
120. Gelosa P, Lecca D, Fumagalli M, Wypych D, Pignieri A, Cimino M, et al. Microglia is a key player in the reduction of stroke damage promoted by the new antithrombotic agent ticagrelor. J Cereb Blood Flow Metab. (2014) 34:979–88. doi: 10.1038/jcbfm.2014.45
121. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. (2015) 6:e1954. doi: 10.1038/cddis.2015.277
122. Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. (2014) 35:253–61. doi: 10.1016/j.it.2014.02.007
123. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. (2014) 1319:82–95. doi: 10.1111/nyas.12458
124. Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab. (2014) 34:660–7. doi: 10.1038/jcbfm.2013.242
125. Gao G, Zhao S, Xia X, Li C, Li C, Ji C, et al. Glutaminase C regulates microglial activation and pro-inflammatory exosome release: relevance to the pathogenesis of Alzheimer's disease. Front Cell Neurosci. (2019) 13:264. doi: 10.3389/fncel.2019.00264
126. Gao G, Li C, Zhu J, Wang Y, Huang Y, Zhao S, et al. Glutaminase 1 regulates neuroinflammation after cerebral ischemia through enhancing microglial activation and pro-inflammatory exosome release. Front Immunol. (2020) 11:161. doi: 10.3389/fimmu.2020.00161
127. Culman J, Zhao Y, Gohlke P, Herdegen T. PPAR-gamma: therapeutic target for ischemic stroke. Trends Pharmacol Sci. (2007) 28:244–9. doi: 10.1016/j.tips.2007.03.004
128. Lin CH, Liao LY, Yang TY, Chang YJ, Tung CW, Hsu SL, et al. Microglia-derived adiposomes are potential targets for the treatment of ischemic stroke. Cell Mol Neurobiol. (2019) 39:591–604. doi: 10.1007/s10571-019-00665-9
129. Zhou D, Ji L, Chen Y. TSPO modulates IL-4-induced microglia/macrophage M2 polarization via PPAR-gamma pathway. J Mol Neurosci. (2020) 70:542–9. doi: 10.1007/s12031-019-01454-1
130. Liu R, Diao J, He S, Li B, Fei Y, Li Y, et al. XQ-1H protects against ischemic stroke by regulating microglia polarization through PPARgamma pathway in mice. Int Immunopharmacol. (2018) 57:72–81. doi: 10.1016/j.intimp.2018.02.014
131. Li Y, Zhu ZY, Lu BW, Huang TT, Zhang YM, Zhou NY, et al. Rosiglitazone ameliorates tissue plasminogen activator-induced brain hemorrhage after stroke. CNS Neurosci Ther. (2019) 25:1343–52. doi: 10.1111/cns.13260
132. Wang D, Liu F, Zhu L, Lin P, Han F, Wang X, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. (2020) 17:257. doi: 10.1186/s12974-020-01921-2
133. Alghamdi BS. The neuroprotective role of melatonin in neurological disorders. J Neurosci Res. (2018) 96:1136–49. doi: 10.1002/jnr.24220
134. Kilic E, Ozdemir YG, Bolay H, Keleştimur H, Dalkara T. Pinealectomy aggravates and melatonin administrationattenuates brain damage in focal ischemia. J Cereb Blood Flow Metab. (1999) 19:511–6. doi: 10.1097/00004647-199905000-00005
135. Zhang F, Liu J, Shi J.-S. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in microglial activation. Europ J Pharmacol. (2010) 636:1–7. doi: 10.1016/j.ejphar.2010.03.043
136. Li X, Xiang B, Shen T, Xiao C, Dai R, He F, et al. Anti-neuroinflammatory effect of 3,4-dihydroxybenzaldehyde in ischemic stroke. Int Immunopharmacol. (2020) 82:106353. doi: 10.1016/j.intimp.2020.106353
137. Xiang B, Xiao C, Shen T, Li X. Anti-inflammatory effects of anisalcohol on lipopolysaccharide-stimulated BV2 microglia via selective modulation of microglia polarization and down-regulation of NF-kappaB p65 and JNK activation. Mol Immunol. (2018) 95:39–46. doi: 10.1016/j.molimm.2018.01.011
138. Han L, Cai W, Mao L, Liu J, Li P, Leak RK, et al. Rosiglitazone promotes white matter integrity and long-term functional recovery after focal cerebral ischemia. Stroke. (2015) 46:2628–36. doi: 10.1161/STROKEAHA.115.010091
139. Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ, et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J Neuroinflam. (2015) 12:51. doi: 10.1186/s12974-015-0270-3
140. Gelosa P, Bonfanti E, Castiglioni L, Delgado-Garcia JM, Gruart A, Fontana L, et al. Improvement of fiber connectivity and functional recovery after stroke by montelukast, an available and safe anti-asthmatic drug. Pharmacol Res. (2019) 142:223–36. doi: 10.1016/j.phrs.2019.02.025
141. Zhao S, Yang J, Wang L, Peng S, Yin J, Jia L, et al. NF-kappaB upregulates type 5 phosphodiesterase in N9 microglial cells: inhibition by sildenafil and yonkenafil. Mol Neurobiol. (2016) 53:2647–58. doi: 10.1007/s12035-015-9293-0
142. Moretti R, Leger PL, Besson VC, Csaba Z, Pansiot J, Di Criscio L, et al. Sildenafil, a cyclic GMP phosphodiesterase inhibitor, induces microglial modulation after focal ischemia in the neonatal mouse brain. J Neuroinflam. (2016) 13:95. doi: 10.1186/s12974-016-0560-4
143. Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho, et al. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. (1999) 96:13496–500. doi: 10.1073/pnas.96.23.13496
144. Im DS. Pro-resolving effect of ginsenosides as an anti-inflammatory mechanism of panax ginseng. Biomolecules. (2020) 10:444. doi: 10.3390/biom10030444
145. Yu T, Rhee MH, Lee J, Kim SH, Yang Y, Kim HG, et al. Ginsenoside Rc from Korean Red Ginseng (Panax ginseng C.A. Meyer) attenuates inflammatory symptoms of gastritis, hepatitis and arthritis. Am J Chin Med. (2016) 44:595–615. doi: 10.1142/S0192415X16500336
146. Ahn S, Siddiqi MH, Aceituno VC, Simu SY, Yang DC. Suppression of MAPKs/NF-kappaB activation induces intestinal anti-inflammatory action of ginsenoside Rf in HT-29 and RAW264.7 cells. Immunol Invest. (2016) 45:439–49. doi: 10.3109/08820139.2016.1168830
Keywords: microglia, polarization, stroke, MCAO, phenotype
Citation: Dong R, Huang R, Wang J, Liu H and Xu Z (2021) Effects of Microglial Activation and Polarization on Brain Injury After Stroke. Front. Neurol. 12:620948. doi: 10.3389/fneur.2021.620948
Received: 28 October 2020; Accepted: 03 May 2021;
Published: 01 July 2021.
Edited by:
Jong Eun Lee, Yonsei University, South KoreaReviewed by:
Luigi Sironi, University of Milan, ItalyCopyright © 2021 Dong, Huang, Wang, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongxin Xu, eHV6aG9uZ3hpbkBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.