
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 04 October 2021
Sec. Dementia and Neurodegenerative Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.619388
 Ting-Bin Chen1,2,3
Ting-Bin Chen1,2,3 Kun-Ju Lin4,5
Kun-Ju Lin4,5 Szu-Ying Lin6
Szu-Ying Lin6 Yi-Jung Lee7Yi-Cheng Lin6,8Chen-Yu Wang9Jun-Peng Chen10
Yi-Jung Lee7Yi-Cheng Lin6,8Chen-Yu Wang9Jun-Peng Chen10 Pei-Ning Wang9,11,12*
Pei-Ning Wang9,11,12*Background and Purpose: Pyroglutamate-modified β-amyloid peptide (AβpE) is crucial for AD pathophysiological process. The potential associations of plasma AβpE and total tau (t-tau) with brain Aβ burden and cognitive performance remain to be clarified.
Methods: Forty-six subjects with unimpaired cognition, mild cognitive impairment, or very mild dementia were enrolled. Plasma levels of AβpE3−40, t-tau, and Aβ42 were quantified by immunomagnetic reduction (IMR) assays. We analyzed individual and combined biomarker correlations with neuropsychological scores and Aβ positivity determined by 18F-florbetapir positron emission tomography (PET).
Results: Both plasma AβpE3−40 levels and AβpE3−40/t-tau ratios correlated negatively with short-term memory and global cognition scores, while correlating positively with PET standardized uptake value ratios (SUVRs). Among the biomarkers analyzed, the combination of AβpE3−40 in a ratio with t-tau had the best discriminatory ability for Aβ PET positivity. Likewise, logistic regression analysis showed that AβpE3−40/t-tau was a highly robust predictor of Aβ PET positivity after controlling for relevant demographic covariates.
Conclusion: Plasma AβpE3−40/t-tau ratios correlate with cognitive function and cerebral Aβ burden. The suitability of AβpE3−40/t-tau as a candidate clinical biomarker of AD pathology in the brain should be examined further in larger studies.
Alzheimer's disease (AD) underlies a major unmet medical need in routine clinical practice and casts a considerable burden on patients, caregivers, and societies. The neuropathological characteristics of AD include neuronal loss, β-amyloid (Aβ) plaques, neurofibrillary tangles, and synaptic loss (1, 2).
The main Aβ variants detected in the human brain are Aβ40 and Aβ42. They are found together with N-terminal truncated forms of these variants (Aβn-40/42), which have been shown to have pyroglutamate modifications, yielding a distinct pyroglutamate-modified Aβ peptide (AβpE) (3–6). The AβpE peptide has been identified in early-stage AD, even before clinical symptoms are apparent, implicating it as a potential seeding molecule that may enable pathological Aβ aggregate formation and triggering hyperphosphorylation of tau (6–11). AβpE is formed when full-length Aβ peptides undergo truncation at the N-terminal glutamate and subsequent dehydration catalyzed by glutaminyl cyclase (12, 13). Compared to full-length peptides, AβpE displays greater β-sheet content, hydrophobicity, aggregation propensity, deposition, amyloidogeneity, resistance to enzymatic degradation, and neuronal toxicity (9, 14–17). In addition, AβpE has been reported to be closely associated with cognitive decline, disease progression, and cerebral amyloidosis in AD (8, 10, 18, 19), suggesting that it may be a key driving force in AD pathogenesis.
Amyloidosis, a primary characteristic of AD pathology, can be detected by positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) measures (20, 21). Both measures possess high diagnostic and prognostic value and may reveal changes many years prior to clinical onset of AD (22–25). Because PET scans are costly and have limited availability, they cannot be incorporated broadly into routine clinical assessments of cognitive impairment before initiation of AD treatment therapies. By contrast, although CSF sampling is less costly and more readily available, it is quite invasive. Neither the PET nor the CSF test is appropriate for population-based screening aimed at identifying high-risk individuals before symptom onset. Moreover, approximately 70% of study participants with clinical syndromes of AD dementia in AD prevention trials and around 25% of participants in AD drug trials did not have brain amyloidosis that was detectable by PET (26, 27). Thus, a minimally invasive measure, such as a blood test, that can reflect cerebral AD pathology accurately and reliably would have critical advantages for supporting clinical decisions regarding AD treatment planning and monitoring of patients during therapeutic trials.
Immunomagnetic reduction (IMR) assay is a relatively new ultrasensitive detection technology capable of quantifying biomarkers down to pg/mL levels. Previously, we observed a positive correlation of IMR-quantified plasma levels of a pyroglutamate cyclization variant of N-terminally truncated Aβ3–40 (AβpE3−40) with cerebral amyloidosis detected by PET imaging (18). In the present study, IMR was implemented to measure plasma levels of AβpE3−40, t-tau, and Aβ42. The primary aim of this work was to investigate associations of these plasma biomarkers, individually and in combination, with cognitive performance and to assess their utility for predicting Aβ PET positivity (+).
Eligible subjects at Taipei Veterans General Hospital, Linkou Chang Gung Memorial Hospital, and Kaohsiung Chang Gung Memorial Hospital were enrolled through the Alzheimer's Disease Neuroimaging Initiative in Taiwan (T-ADNI). The local institutional review boards and the ethics committee of the three hospitals approved the data collection protocol. T-ADNI study was approved by the ethics committees of the three hospitals. The inclusion criteria were an age of 55 to 90 years and educational attainment of at least 6 years. Prior to testing, written informed consent was obtained from all participants and/or their legal guardians. All methods were performed in accordance with the relevant guidelines and regulations.
All participants were interviewed by experienced neurologists to collect information about their demographics, family history, and physical/neurological condition. Additionally, for each participant, neurologists obtained Hachinski ischemic score and vital signs. Blood was drawn for a hematology/chemistry panel and for vitamin B12s, thyroid-stimulating hormone, free thyroxine, and rapid plasma reagin syphilis tests.
A standard battery of neuropsychological tests was administered, including the Geriatric Depression Scale, Mini-Mental State Examination (MMSE) (28), Chinese Version Verbal Learning Test (29), Logical Memory subscale (story A) of the Chinese version of the Wechsler Memory Scale-III (30), 30-item Boston Naming Test (31), categorical Verbal Fluency Test (29), Trail-Making Test A and B (line) (32), and Clinical Dementia Rating Scale (CDR) (33). Only subjects with non-impaired cognition, defined by CDR score of 0, and patients in the prodromal stage or very mild dementia stage, defined by CDR score of 0.5, were invited to participate in this study.
Signs of cognitive impairment were assessed and non-AD etiologies (e.g., tumors, strokes, severe white matter disease, or inflammation) were excluded through a series of clinical interviews, a physical examination, screening laboratory tests, neuropsychological assessments, and brain magnetic resonance (MR) imaging. Exclusionary criteria included any history of major brain trauma, brain tumor, stroke, epilepsy, alcoholism, major psychiatric illness, or any other systemic diseases that might affect cognitive function. Diagnosis of amnestic mild cognitive impairment and probable AD were made on the basis of the core clinical criteria developed by the National Institute on Aging and the Alzheimer's Association's 2011 workgroup (NIA-AA) (34). The demographic characteristics of the study cohort, dichotomized according to Aβ PET results, are summarized in Table 1.
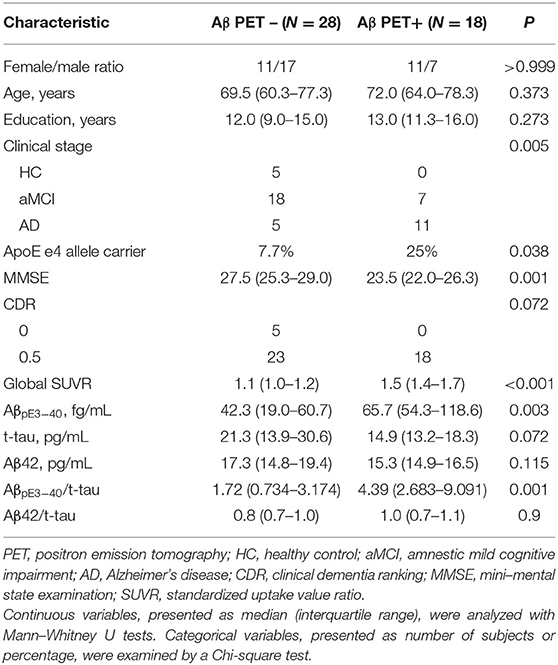
Table 1. Demographic and clinical characteristics of enrolled subjects (N = 46).
An 8-ml non-fasting venous blood sample (K3 EDTA, lavender-top tube) was collected from each subject and then centrifuged (1,500–2,500 ×g for 15 min) within 1 h of the draw. The plasma was then aliquoted into cryotubes and stored at −20°C.
Detailed descriptions of the IMR platform and validation of its accuracy have been published previously (18, 35). IMR reagents were selected based on epitope antigen/antibody affinity, ability to conjugate with MagQu magnetic Fe3O4 nanobeads, and ability to produce linear standard curves of quantitated magnetic signal reduction. Each type of reagent consists of magnetic nanoparticles dispersed in phosphate buffered saline (pH 7.2). By immobilizing functionalized monoclonal antibodies against Aβ37–42 (ABCAM, Cambridge, UK; ab34376) and t-tau protein (Sigma Aldrich; T9450) on the magnetic nanoparticles, two types of reagents were obtained. The antibody against AβpE3−40 was developed by Biogen Inc. The mean diameter of the antibody-functionalized magnetic nanoparticles was 50–60 nm. The magnetic concentration of each type of reagent was 12 mg-Fe/mL. Duplicated/paired measurements of Aβ42, AβpE3−40, and t-tau were performed for each plasma sample. We mixed 60-μl plasma samples with 60 μl of reagent (MF-AB2-0060 or MF-DEX-0060, respectively; MagQu) at room temperature in the Aβ42 and AβpE3−40 assays; for the t-tau assay, 40 μl of plasma with 80 μl of reagent (MF-TAU-0060; MagQu) were used. Magnetic signal changes during the course of interactions between antigens and antibody-functionalized magnetic nanoparticles were detected by an IMR reader (XacPro-S, MagQu) and expressed as percentage reductions in immunomagnetic signal, which were then converted to a sample concentration based on reference to standard curves of the respective analytes. The ratio of the reduction to the alternative-current (ac) magnetic signal of reagent before incubation is expressed as follows:
where χac,o and χac,ϕ are the ac magnetic signals of reagent before and after incubation. For each reported IMR (%) in this study, an averaged value of duplicated IMR measurements was calculated. The standard deviations of all duplicated plasma analyte measurements were <15%. The reported analyte concentrations for each sample are means of the paired measurements.
ApoE genotyping was determined by polymerase chain reaction amplification and DNA sequencing (36). Participants with one or two ε4 alleles were defined as ε4 carriers.
All Aβ PET imaging scans were performed on a Biograph mCT PET/CT scanner (GE Healthcare, Milwaukee, WI) at a single site (Linkou Chang Gung Memorial Hospital) as described in detail elsewhere (37, 38). Briefly, the scan commenced with a 10-min acquisition period (two 5-min frames) beginning 50-min after a 10-mCi injection of 18F-florbetapir tracer. Each image was obtained with the application of a three-dimensional ordered subset expectation maximization reconstruction algorithm (four iterations, 24 subsets; Gaussian filter: 2 mm; zoom: three) with computed tomography-based attenuation correction, as well as scatter and random corrections, with a matrix size of 400 × 400 × 148 and a voxel size of 0.68 × 0.68 × 1.5 mm3. To achieve useful anatomical information and facilitate co-registration with PET images, structural MR scans were obtained for all subjects with a uniform scanning protocol that minimizes and accounts for between-site differences in MR imaging systems.
PET imaging data were processed and analyzed in PMOD software (version 3.7, PMOD Technologies Ltd., Zurich, Switzerland), including MR-based spatial normalization to the Montreal Neurological Institute MRI template. We selected seven volumes of interest: frontal, anterior cingulate, posterior cingulate, precuneus, parietal, occipital, and temporal cortical areas. We calculated regional standardized uptake value ratios (SUVRs) for each volume of interest, using the whole cerebellum as a reference region, and then averaged the SUVRs for the seven volumes of interest to yield an estimated global cortical SUVR value for further analysis.
All the PET images were interpreted by an experienced nuclear medicine physician (Kun-Ju Lin) who did not have access to clinical data. Aβ burden was graded on a five-point visual scale, from 0, indicating no tracer retention in cortical gray matter, to 4, denoting high levels of cortical amyloid accumulation. Scores of 0 or 1 were categorized as Aβ PET negativity (–) and scores of 2–4 were categorized as Aβ PET+ (39).
All statistical analyses were performed in SPSS version 22.0 for Windows (SPSS Inc., Chicago, IL, USA). A P < 0.05 was considered significant. All variables were analyzed by non-parametric methods. For continuous variables, differences between Aβ PET- group and Aβ PET+ group were detected with Mann-Whitney U tests. For categorical variables, Chi-square tests were used. We generated neurocognitive numeric composite z-scores by calculating individual z-scores for each test and then averaging them across the cognitive test set. The constituents of the composite z-scores were as follows: short-term memory [Chinese Version Verbal Learning Test and Logical Memory subscale (story A) of the Chinese Version Wechsler Memory Scale-III], semantic memory [Boston Naming Test (total)]; executive function [Trail-Making Test-A/B (line) and categorical Verbal Fluency Test (animal)]; global cognition [short-term memory test, semantic memory test, executive function test, and the MMSE]. Spearman's rank coefficients were calculated to determine correlation of plasma biomarker levels with estimated global cortical SUVR and domains of cognitive performance. Receiver operating characteristic (ROC) and area under the curve (AUC) analyses were performed to define cut-off points for each biomarker analyte or their ratios to further characterize discriminatory properties between the Aβ PET- and Aβ PET+ groups. Finally, logistic regression modeling was performed to investigate the predictive power of biomarker levels for Aβ PET+, in terms of odds ratios (ORs) and 95% confidence interval (CIs), with and without adjusting for age, sex, and ApoE ε4 carrier status.
A total of 46 subjects were enrolled in the study and divided into Aβ PET- and Aβ PET+ groups. Their demographic characteristics and neurocognitive scores are presented in Table 1. There were no significant between-group differences in age, sex, years of education, clinical stage or CDR scores, nor in plasma biomarker levels of t-tau, Aβ42, or Aβ42/t-tau. Compared to the Aβ PET- group, the Aβ PET+ group had more ApoE ε4 carriers (25% vs. 7.7%, p = 0.038), poorer performance on MMSE (23.5 vs. 27.5, p = 0.001), higher Aβ PET SUVR (1.5 vs. 1.1, p < 0.001), and higher levels of plasma AβpE3−40 (65.7 vs. 42.3, p = 0.003) and AβpE3−40/t-tau ratio (4.39 vs. 1.72, p = 0.001).
AβpE3−40 level (r = 0.343, p < 0.005), and AβpE3−40/t-tau ratio values (r = 0.305, p < 0.005) correlated directly with Aβ PET SUVRs (Table 2; Figure 1). Short-term memory scores correlated negatively with AβpE3−40 (r = −0.481, p < 0.001) and AβpE3−40/t-tau values (r = −0.483, p < 0.001), but positively with t-tau (r = 0.3, p < 0.005) and Aβ42 levels (r = 0.391, p < 0.001). Semantic memory scores were found to correlate positively with Aβ42 levels (r = 0.353, p < 0.005), while executive function scores correlated positively with AβpE3−40/t-tau values (r = 0.359, p < 0.005). Global cognition scores correlated negatively with AβpE3−40 (r = −0.337, p < 0.005) and AβpE3−40/t-tau values (r = −0.343, p < 0.005) while correlating positively with Aβ42 levels (r = 0.379, p < 0.005).
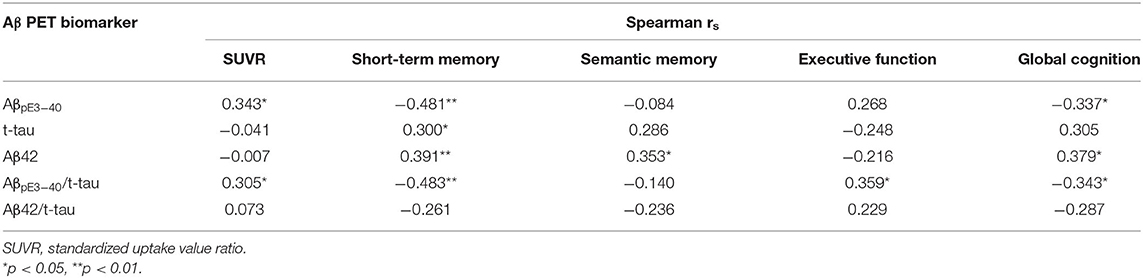
Table 2. Correlation of plasma biomarkers with Aβ burden and cognitive performance (N = 46).
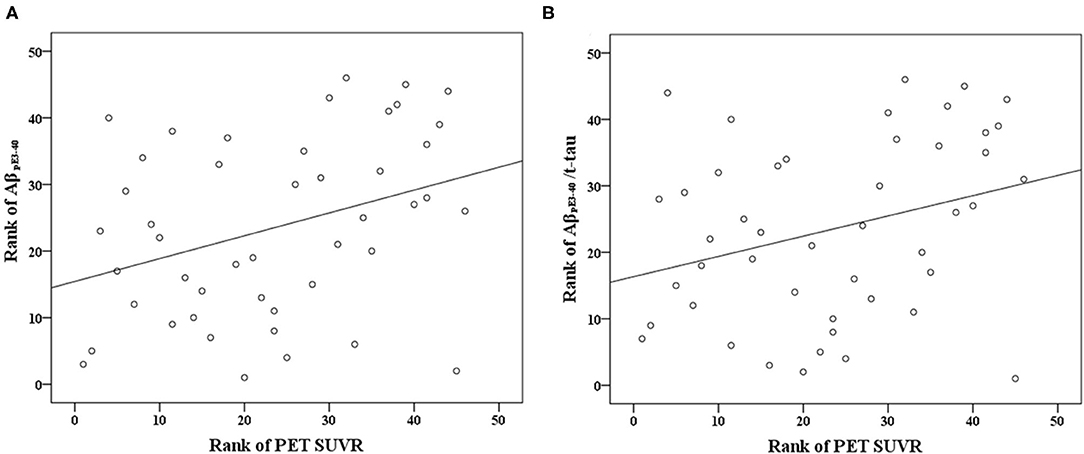
Figure 1. Scatterplots of the associations between Aβ PET SUVR and plasma biomarker levels (pg/mL) of (A) AβpE3−40 and (B) AβpE3−40/t-tau.
ROC-AUC analyses aimed at determining plasma biomarker discriminatory cut-off values yielded the following optimal significant cut-off values for the differentiation of Aβ PET– and Aβ PET+ groups: 55.45 fg/mL for AβpE3−40; 2.85 for AβpE3−40/t-tau; and 0.8 for Aβ42/t-tau. The combination of AβpE3−40 with t-tau in a AβpE3−40/t-tau ratio had a greater AUC than individual biomarkers (i.e., AβpE3−40, t-tau, and Aβ42) and the composite Aβ42/t-tau ratio. Detailed AUCs, cut-off values, sensitivities, and specificities are reported in Table 3.
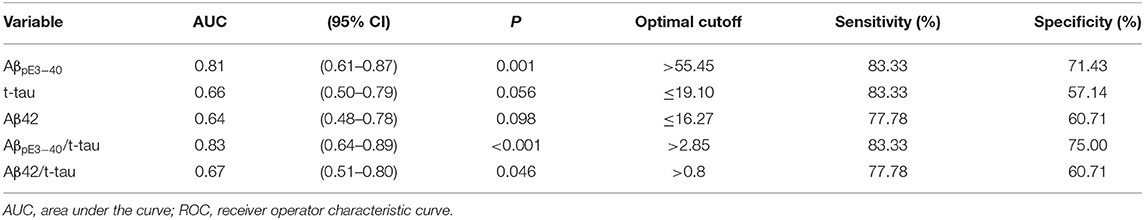
Table 3. ROC analysis for identifying Aβ PET positivity (N = 46).
The relationship between plasma biomarkers and Aβ PET positivity was examined through a logistic regression analysis with the aforementioned optimal cut-offs. All of the analyzed markers were found to be significantly associated with Aβ PET positivity risk in both the unadjusted and adjusted logistic regression modeling results (Table 4). All adjusted ORs (aORs) became more pronounced after controlling for age, sex, and ApoE ε4 carrier status. Participants with AβpE3−40 > 55.45 fg/mL, t-tau ≤ 19.10 pg/mL, Aβ42 ≤ 16.27 pg/mL, AβpE3−40/t-tau > 2.85, or Aβ42/t-tau > 0.8 were at increased risk for Aβ PET positivity (aOR, 95% CI, and p-values are reported in Table 4).
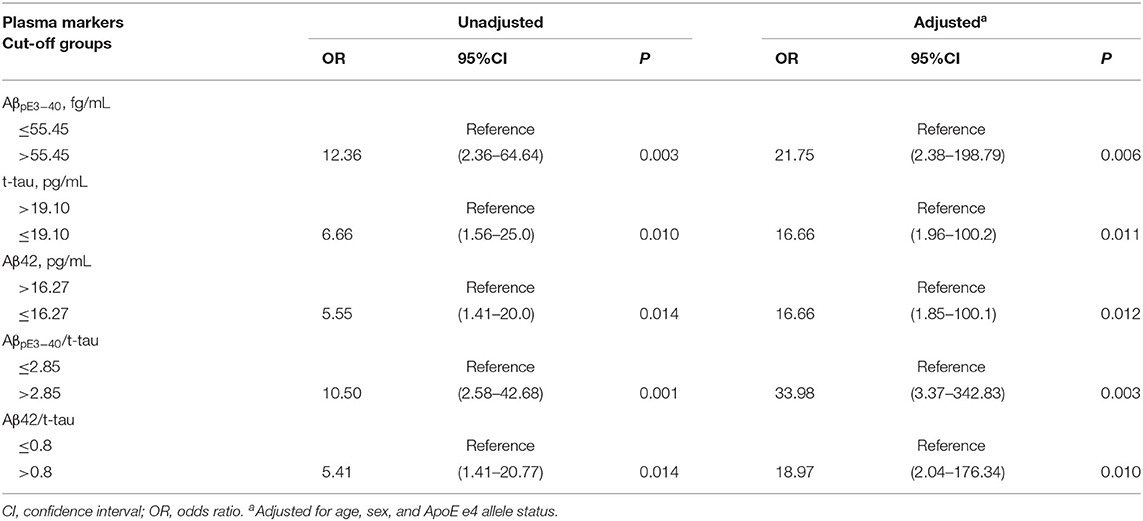
Table 4. Association of plasma biomarkers with Aβ PET positivity (N = 46).
The current study assessed the association of levels of AD-related biomarkers, especially AβpE3−40, quantified by IMR technology with cognitive performance as well as their predictive power in Aβ PET positivity. We found that participants with Aβ PET+ had higher levels of plasma AβpE3−40 and higher AβpE3−40/t-tau ratio values than participants with Aβ PET-. These elevated values correlated positively with PET analysis findings and correlated negatively with short-term memory and global cognition scores. AβpE3−40 alone had a high discriminatory ability, and consideration of AβpE3−40 together with t-tau (i.e., in AβpE3−40/t-tau) provided greater differential value than any of the individual biomarker values examined and the Aβ42/t-tau ratio. After adjusting for demographic covariates (age, sex, and ApoE ε4 carrier status), AβpE3−40/t-tau proved to be the strongest predictive biomarker for Aβ PET positivity. Collectively, these findings suggest that plasma AβpE3−40/t-tau may indeed be reflective of cerebral amyloid pathology. To the best of our knowledge, the current study is the first to report relationships among plasma t-tau level, plasma AβpE3−40 level, and brain Aβ accumulation revealed by in vivo PET.
In a previous study, we observed a positive association of AβpE3−40 levels with Aβ PET SUVRs (18). In this study, we affirmed those prior findings and further showed that AβpE3−40-related biomarkers were more closely related to brain Aβ accumulation revealed by Aβ PET than were Aβ42, t-tau, and Aβ42/t-tau values. These results are discordant with previous data obtained with ultrasensitive analytical assays—such as, xMAP, stable isotope labeling kinetics-mass spectrometry, single-molecule array, and immunoprecipitation mass spectrometry approaches—showing that lower plasma Aβ42 levels and Aβ42/Aβ40 ratios were associated with a higher brain Aβ burden revealed by Aβ PET over the normal–mild cognitive impairment–AD cognitive spectrum (40–48). This divergence of findings could be consequent to the critically discrepant roles of AβpE and Aβ42 in AD pathophysiological processes. AβpE has been considered to be a principal initiator of early-stage AD pathogenesis and has been shown to correlate with tau pathology and cognitive decline in AD (10, 12, 15, 19, 49), whereas plasma Aβ42 was found to be inversely correlated with cortical Aβ burden, likely due to impaired Aβ plaque clearance from the brain or sequestration and deposition of Aβ species within the brain. A recent animal study showed the evidence that impaired meningeal lymphatic drainage can exacerbate the neuroinflammatory response and enhancing meningeal lymphatic function could achieve better ability of monoclonal antibodies to clear Aβ aggregates (50). Together with a phase 2 trial revealing that donanemab, a monoclonal antibody against AβpE epitope, could curb cognitive decline in early AD (51), we speculate that AβpE3−40 is a better plasma biomarker than Aβ42 for detection and monitoring of cerebral amyloidosis.
We found that AβpE3−40, Aβ42, and AβpE3−40/t-tau correlated significantly with short-term memory and global cognition performance measures, whereas t-tau and Aβ42/t-tau did not correlate well with any cognitive performance measures in our study. AβpE3−40 has specifically been found to be more cytotoxic to hippocampal and cortical neurons than Aβ40 and Aβ42 in animal studies (52, 53), highlighting the close relationship to hippocampal-dependent cognitive impairment. In addition, AβpE3−40 and AβpE3−40/t-tau showed modestly positive correlation with Aβ PET SUVRs. In a previous study, Aβ42/t-tau ratios were also reported to have no correlation with Aβ PET SUVRs, but have positive correlation with brain tau accumulation and longitudinal changes in hippocampal volume and cerebral glucose metabolism (54). Unlike Aβ42/Aβ40 and Aβ42/t-tau in other studies, AβpE3−40/t-tau modestly paralleled with the presence of cerebral amyloidosis in this study. Our findings of a positive relationship between Aβ42 level and cognitive function measures are consistent with the findings of prior studies in which ultrasensitive detection methods were used, affirming the supposition that cognitive decline may be associated with low Aβ42 levels (55, 56), but not t-tau levels. There have been discordant findings regarding Aβ42 and t-tau levels correlating closely or not at all with cognitive test scores (35, 55, 57–66), perhaps due to methodological differences related to study design and the quantitative methods used. AβpE has been reported to trigger AD-related neuronal loss, neurodegeneration, neurological deficits, and cognitive decline and has been shown to have differential expression patterns between AD progression and normal aging (19, 67–69). Similar to the finding indicating that a modified version of tau (i.e., ultrasensitive blood immunoassay-detected tau phosphorylated at threonine 181) (70) appears to be a better marker of disease progression than t-tau, AβpE-related marker is a better biomarker of brain Aβ burden and cognitive decline than Aβ42 or t-tau, and may thus provide useful insights into brain functioning in the AD continuum.
Our ROC-AUC analyses indicated that AβpE3−40 (p = 0.001), AβpE3−40/t-tau (p < 0.001), and Aβ42/t-tau (p = 0.046) can be used to differentiate between individuals with Aβ PET+ and individuals with Aβ PET-. Among the singular biomarkers, only AβpE3−40 had a significant and moderate-to-high predictive ability. Between the two aforementioned composite biomarkers, AβpE3−40/t-tau yielded a higher AUC (0.83) as well as greater sensitivity (83.33%) and specificity (75.00%). Among the five biomarkers analyzed, AβpE3−40/t-tau exhibited the best discriminatory power. Combining markers reflective of two distinct underlying pathophysiological derangements did indeed lead to an incremental benefit to between-group differentiation. There are differential time courses of Aβ and tau changes in AD progression (24, 62). The better correspondence to Aβ PET findings for AβpE, relative to other singular markers, may reflect AβpE being an earlier marker than Aβ42 or tau in AD pathogenesis (9). Although subjects with an early disease stage (i.e., healthy controls with Aβ PET+) were not enrolled in this study, it may be reasonable to pursue AβpE3−40/t-tau as a potential predictor of cerebral Aβ deposition in population-based screening for high-risk individuals before AD symptom onset.
After controlling for demographic covariates, all of the presently examined putative biomarkers had some ability to predict Aβ PET positivity, with the combination of AβpE3−40 with t-tau appearing to be particularly useful for reflecting disease state along the AD continuum. Our ROC-AUC analyses indicated that AβpE3−40/t-tau possessed higher AUC, sensitivity, and specificity than Aβ42/t-tau, supporting the notion that utilization of plasma AβpE3−40 and t-tau together is superior to considering absolute levels of individual peptides or Aβ42/t-tau ratio as markers of cerebral amyloidosis. In the face of the high cost and low accessibility of Aβ PET restricting wide use in clinical practice, AβpE3−40/t-tau may help support clinical decisions and be used as a clinical screening tool to rule out individuals who do not need costly Aβ PET scanning in scenarios other than confirmatory diagnosis.
All patients in this study were classified into amnestic-type MCI or AD based on the clinical diagnostic criteria proposed by the 2011 NIA-AA workgroup (34). We intended to identify potential plasma biomarkers with proper cutoff values indicating Aβ PET positivity over the normal–mild cognitive impairment–AD cognitive spectrum, rather than to correlate clinical diagnoses with Aβ PET positivity or to differentiate clinical groups by Aβ PET positivity.
The current study had some limitations. First, we could only infer a possible association of plasma AβpE3−40 and t-tau with imminent risk of Aβ PET positivity. Owing to small sample size and lack of Aβ+ healthy controls, further replication, particularly with larger samples and longer follow-up, is warranted to validate the predictive values obtained in this study and to clarify the temporal relationship of these variables with Aβ PET+. Second, the striatum was not included in the SUVR measurements for amyloid PET. Third, we did not account for comorbidity covariates that may affect plasma biomarker levels in our analyses. Finally, the relatively small sample size of our cohort limits the generalizability of the present findings.
In conclusion, a dual-factor biomarker consisting of the ratio between AβpE3−40 and t-tau measures, each determined by an ultrasensitive and easy-to-implement IMR assessment, showed superior performance characteristics and was in concordance with PET analysis and cognitive function assessment results. Given that Aβ and tau have long been considered hallmarks of AD pathogenesis, it may be reasonable to pursue an AβpE3−40 and t-tau composite factor as a predictor of cerebral Aβ deposition. AβpE3−40/t-tau might be an effective and feasible candidate for blood-based screening of cerebral AD-related neuropathology aimed at identifying at-risk individuals that should be recommended for CSF and/or PET studies prior to clinical treatment planning. These findings must be interpreted with caution given the relatively small sample size.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Taipei Veterans General Hospital, Linkou Chang Gung Memorial Hospital, and Kaohsiung Chang Gung Memorial Hospital. The patients/participants provided their written informed consent to participate in this study.
T-BC and P-NW contributed to the conception and design of the study, acquisition of data, interpretation of the data, critical revision of the manuscript, and wrote the first draft of the manuscript. K-JL conducted PET imaging analysis. S-YL, Y-JL, Y-CL, and C-YW were involved in the collection and/or analysis of data. J-PC performed statistical analysis. All authors read and approved the submitted version.
This study was carried out with financial support from the Brain Research Center, National Yang-Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, the Taiwan Alzheimer's Disease Neuroimaging Initiative (T-ADNI) group, the National Science Council and the Ministry of Science and Technology, Taiwan (MOST 105-2325-B-182A-005-, MOST 108-2321-B-010-013-MY2, and MOST 110-2321-B-010-007), Taipei Veterans General Hospital (V107C-090, V108C-060), and Chang Gung Memorial Hospital (CMRPG3D1802).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge the help of our staff and participants for their efforts and contributions.
1. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. (1985) 82:4245–9. doi: 10.1073/pnas.82.12.4245
2. Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. (2004) 44:181–93. doi: 10.1016/j.neuron.2004.09.010
3. Geddes JW, Tekirian TL, Mattson MP. N-terminus truncated beta-amyloid peptides and C-terminus truncated secreted forms of amyloid precursor protein: distinct roles in the pathogenesis of Alzheimer's disease. Neurobiol Aging. (1999) 20:75–9; discussion 87.
4. Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem. (1992) 267:17082–6. doi: 10.1016/S0021-9258(18)41896-0
5. Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. (1992) 359:325–7. doi: 10.1038/359325a0
6. Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3: in senile plaques. Neuron. (1995) 14:457–66. doi: 10.1016/0896-6273(95)90301-1
7. Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimer's disease as new targets for the vaccination approach. J Neurochem. (2003) 85:1581–91. doi: 10.1046/j.1471-4159.2003.01818.x
8. Rijal Upadhaya A, Kosterin I, Kumar S, von Arnim CA, Yamaguchi H, Fändrich M, et al. Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer's disease. Brain. (2014) 137(Pt. 3):887–903. doi: 10.1093/brain/awt362
9. Moro ML, Phillips AS, Gaimster K, Paul C, Mudher A, Nicoll JAR, et al. Pyroglutamate and isoaspartate modified amyloid-beta in ageing and Alzheimer's disease. Acta Neuropathol Commun. (2018) 6:3. doi: 10.1186/s40478-017-0505-x
10. Mandler M, Walker L, Santic R, Hanson P, Upadhaya AR, Colloby SJ, et al. Pyroglutamylated amyloid-β is associated with hyperphosphorylated tau and severity of Alzheimer's disease. Acta Neuropathol. (2014) 128:67–79. doi: 10.1007/s00401-014-1296-9
11. Neddens J, Daurer M, Flunkert S. Correlation of pyroglutamate amyloid β and ptau Ser202/Thr205 levels in Alzheimer's disease and related murine models. PLoS ONE. (2020) 15:e0235543. doi: 10.1371/journal.pone.0235543
12. Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J Biol Chem. (2011) 286:38825–32. doi: 10.1074/jbc.R111.288308
13. Cynis H, Frost JL, Crehan H, Lemere CA. Immunotherapy targeting pyroglutamate-3 Aβ: prospects and challenges. Mol Neurodegen. (2016) 11:48. doi: 10.1186/s13024-016-0115-2
14. Perez-Garmendia R, Gevorkian G. Pyroglutamate-modified amyloid beta peptides: emerging targets for Alzheimer's disease immunotherapy. Curr Neuropharmacol. (2013) 11:491–8. doi: 10.2174/1570159X11311050004
15. Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. (2012) 485:651–5. doi: 10.1038/nature11060
16. Gunn AP, Masters CL, Cherny RA. Pyroglutamate-Aβ: role in the natural history of Alzheimer's disease. Int J Biochem Cell Biol. (2010) 42:1915–8. doi: 10.1016/j.biocel.2010.08.015
17. Michno W, Nyström S, Wehrli P, Lashley T, Brinkmalm G, Guerard L, et al. Pyroglutamation of amyloid-βx-42 (Aβx-42) followed by Aβ1-40 deposition underlies plaque polymorphism in progressing Alzheimer's disease pathology. J Biol Chem. (2019) 294:6719–32. doi: 10.1074/jbc.RA118.006604
18. Wang PN, Lin KJ, Liu HC, Andreasson U, Blennow K, Zetterberg H, et al. Plasma pyroglutamate-modified amyloid beta differentiates amyloid pathology. Alzheimers Dement. (2020) 12:e12029. doi: 10.1002/dad2.12029
19. Pivtoraiko VN, Abrahamson EE, Leurgans SE, DeKosky ST, Mufson EJ, Ikonomovic MD. Cortical pyroglutamate amyloid-β levels and cognitive decline in Alzheimer's disease. Neurobiol Aging. (2015) 36:12–9. doi: 10.1016/j.neurobiolaging.2014.06.021
20. Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
21. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
22. Shaw LM, Vanderstichele H, Knapik-Czajka M, Figurski M, Coart E, Blennow K, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. (2011) 121:597–609. doi: 10.1007/s00401-011-0808-0
23. Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. (2007) 130(Pt. 11):2837–44. doi: 10.1093/brain/awm238
24. Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron. (2013) 80:1347–58. doi: 10.1016/j.neuron.2013.12.003
25. Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. (2013) 12:357–67. doi: 10.1016/S1474-4422(13)70044-9
26. Sperling R, Donohue M, Raman R, Sun C-K, Yaari R, Siemers E, et al. The anti-amyloid treatment in asymptomatic Alzheimer's disease (A4) study: report of screening data results. Alzheimer's Dementia. (2018) 14:P215–6. doi: 10.1016/j.jalz.2018.06.2337
27. Karran E, Hardy J. Antiamyloid therapy for Alzheimer's disease–are we on the right road? N Engl J Med. (2014) 370:377–8. doi: 10.1056/NEJMe1313943
28. Shyu YI, Yip PK. Factor structure and explanatory variables of the Mini-Mental State Examination (MMSE) for elderly persons in Taiwan. J Formos Med Assoc. (2001) 100:676–83.
29. Chang CC, Kramer JH, Lin KN, Chang WN, Wang YL, Huang CW, et al. Validating the Chinese version of the Verbal Learning Test for screening Alzheimer's disease. J Int Neuropsychol Soc. (2010) 16:244–51. doi: 10.1017/S1355617709991184
30. Gong YX. Manual of Wechsler Memory Scale-Chinese Version: Changsha: Human Medical College Press (1989).
31. Chen TB, Lin CY, Lin KN, Yeh YC, Chen WT, Wang KS, et al. Culture qualitatively but not quantitatively influences performance in the Boston naming test in a chinese-speaking population. Dement Geriatr Cogn Dis Extra. (2014) 4:86–94. doi: 10.1159/000360695
32. Wang PN, Hong CJ, Lin KN, Liu HC, Chen WT. APOE ε4 increases the risk of progression from amnestic mild cognitive impairment to Alzheimer's disease among ethnic Chinese in Taiwan. J Neurol Neurosurg Psychiatry. (2011) 82:165–9. doi: 10.1136/jnnp.2010.209122
33. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. (1982) 140:566–72. doi: 10.1192/bjp.140.6.566
34. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.004
35. Chen TB, Lee YJ, Lin SY, Chen JP, Hu CJ, Wang PN, et al. Plasma Aβ42 and total tau predict cognitive decline in amnestic mild cognitive impairment. Sci Rep. (2019) 9:13984. doi: 10.1038/s41598-019-50315-9
36. Emi M, Wu LL, Robertson MA, Myers RL, Hegele RA, Williams RR, et al. Genotyping and sequence analysis of apolipoprotein E isoforms. Genomics. (1988) 3:373–9. doi: 10.1016/0888-7543(88)90130-9
37. Lin KJ, Hsu WC, Hsiao IT, Wey SP, Jin LW, Skovronsky D, et al. Whole-body biodistribution and brain PET imaging with [18F]AV-45, a novel amyloid imaging agent–a pilot study. Nucl Med Biol. (2010) 37:497–508. doi: 10.1016/j.nucmedbio.2010.02.003
38. Hsiao IT, Huang CC, Hsieh CJ, Wey SP, Kung MP, Yen TC, et al. Perfusion-like template and standardized normalization-based brain image analysis using 18F-florbetapir (AV-45/Amyvid) PET. Eur J Nucl Med Mol Imaging. (2013) 40:908–20. doi: 10.1007/s00259-013-2350-x
39. Johnson KA, Sperling RA, Gidicsin CM, Carmasin JS, Maye JE, Coleman RE, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer's disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. (2013) 9:S72–83. doi: 10.1016/j.jalz.2012.10.007
40. Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. (2017) 13:841–9. doi: 10.1016/j.jalz.2017.06.2266
41. Lui JK, Laws SM, Li QX, Villemagne VL, Ames D, Brown B, et al. Plasma amyloid-beta as a biomarker in Alzheimer's disease: the AIBL study of aging. J Alzheimers Dis. (2010) 20:1233–42. doi: 10.3233/jad-2010-090249
42. Vergallo A, Mégret L, Lista S, Cavedo E, Zetterberg H, Blennow K, et al. Plasma amyloid β 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer's disease. Alzheimers Dement. (2019) 15:764–75. doi: 10.1016/j.jalz.2019.03.009
43. Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma β-amyloid in Alzheimer's disease and vascular disease. Sci Rep. (2016) 6:26801. doi: 10.1038/srep26801
44. Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. (2019) 93:e1647–59. doi: 10.1212/WNL.0000000000008081
45. Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer's disease. Nature. (2018) 554:249–54. doi: 10.1038/nature25456
46. Hameed S, Fuh JL, Senanarong V, Ebenezer EGM, Looi I, Dominguez JC, et al. Role of fluid biomarkers and PET imaging in early diagnosis and its clinical implication in the management of Alzheimer's disease. J Alzheimers Dis Rep. (2020) 4:21–37. doi: 10.3233/ADR-190143
47. Lopez OL, Klunk WE, Mathis CA, Snitz BE, Chang Y, Tracy RP, et al. Relationship of amyloid-β1–42 in blood and brain amyloid: Ginkgo Evaluation of Memory Study. Brain Commun. (2019) 2:fcz038. doi: 10.1093/braincomms/fcz038
48. Doecke JD, Pérez-Grijalba V, Fandos N, Fowler C, Villemagne VL, Masters CL, et al. Total Aβ(42)/Aβ(40) ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology. (2020) 94:e1580–91. doi: 10.1212/WNL.0000000000009240
49. Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. (2008) 14:1106–11. doi: 10.1038/nm.1872
50. Da Mesquita S, Papadopoulos Z, Dykstra T, Brase L, Farias FG, Wall M, et al. Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy. Nature. (2021) 593:255–60. doi: 10.1038/s41586-021-03489-0
51. Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, et al. Donanemab in early Alzheimer's disease. N Engl J Med. (2021) 384:1691–704. doi: 10.1056/NEJMoa2100708
52. Russo C, Violani E, Salis S, Venezia V, Dolcini V, Damonte G, et al. Pyroglutamate-modified amyloid beta-peptides–AbetaN3(pE)–strongly affect cultured neuron and astrocyte survival. J Neurochem. (2002) 82:1480–9. doi: 10.1046/j.1471-4159.2002.01107.x
53. Breyhan H, Wirths O, Duan K, Marcello A, Rettig J, Bayer TA. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. (2009) 117:677–85. doi: 10.1007/s00401-009-0539-7
54. Park JC, Han SH, Yi D, Byun MS, Lee JH, Jang S, et al. Plasma tau/amyloid-β1-42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer's disease. Brain. (2019) 142:771–86. doi: 10.1093/brain/awy347
55. Hanon O, Vidal JS, Lehmann S, Bombois S, Allinquant B, Tréluyer JM, et al. Plasma amyloid levels within the Alzheimer's process and correlations with central biomarkers. Alzheimers Dement. (2018) 14:858–68. doi: 10.1016/j.jalz.2018.01.004
56. Yaffe K, Weston A, Graff-Radford NR, Satterfield S, Simonsick EM, Younkin SG, et al. Association of plasma beta-amyloid level and cognitive reserve with subsequent cognitive decline. JAMA. (2011) 305:261–6. doi: 10.1001/jama.2010.1995
57. Cosentino SA, Stern Y, Sokolov E, Scarmeas N, Manly JJ, Tang MX, et al. Plasma ß-amyloid and cognitive decline. Arch Neurol. (2010) 67:1485–90. doi: 10.1001/archneurol.2010.189
58. Llado-Saz S, Atienza M, Cantero JL. Increased levels of plasma amyloid-beta are related to cortical thinning and cognitive decline in cognitively normal elderly subjects. Neurobiol Aging. (2015) 36:2791–7. doi: 10.1016/j.neurobiolaging.2015.06.023
59. Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. (2016) 87:1827–35. doi: 10.1212/WNL.0000000000003246
60. Chiu MJ, Chen YF, Chen TF, Yang SY, Yang FP, Tseng TW, et al. Plasma tau as a window to the brain-negative associations with brain volume and memory function in mild cognitive impairment and early Alzheimer's disease. Hum Brain Mapp. (2014) 35:3132–42. doi: 10.1002/hbm.22390
61. Tsai CL, Liang CS, Lee JT, Su MW, Lin CC, Chu HT, et al. Associations between plasma biomarkers and cognition in patients with Alzheimer's disease and amnestic mild cognitive impairment: a cross-sectional and longitudinal study. J Clin Med. (2019) 8:1893. doi: 10.3390/jcm8111893
62. Chen TB, Lai YH, Ke TL, Chen JP, Lee YJ, Lin SY, et al. Changes in plasma amyloid and tau in a longitudinal study of normal aging, mild cognitive impairment, and Alzheimer's disease. Dement Geriatr Cogn Disord. (2019) 48:180–95. doi: 10.1159/000505435
63. Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology. (2013) 80:1341–8. doi: 10.1212/WNL.0b013e31828ab35d
64. Shekhar S, Kumar R, Rai N, Kumar V, Singh K, Upadhyay AD, et al. Estimation of tau and phosphorylated Tau181 in serum of Alzheimer's disease and mild cognitive impairment patients. PLoS ONE. (2016) 11:e0159099. doi: 10.1371/journal.pone.0159099
65. Pase M, Beiser A, Himali J, Satizabal C, Aparicio HJ, DeCarli C, et al. Plasma tau corresponds to preclinical Alzheimer's disease and is a strong predictor of future dementia (S48.001). Neurology. (2018)90:S48.001.
66. Deters KD, Risacher SL, Kim S, Nho K, West JD, Blennow K, et al. Plasma tau association with brain atrophy in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. (2017) 58:1245–54. doi: 10.3233/JAD-161114
67. Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. (2009) 118:487–96. doi: 10.1007/s00401-009-0557-5
68. Alexandru A, Jagla W, Graubner S, Becker A, Bäuscher C, Kohlmann S, et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J Neurosci. (2011) 31:12790–801. doi: 10.1523/JNEUROSCI.1794-11.2011
69. Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, et al. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. (2005) 280:34186–92. doi: 10.1074/jbc.M501694200
70. Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. (2020) 19:422–33. doi: 10.1016/S1474-4422(20)30071-5
Keywords: pyroglutamate, tau, β-amyloid, Alzheimer's disease, predictor
Citation: Chen T-B, Lin K-J, Lin S-Y, Lee Y-J, Lin Y-C, Wang C-Y, Chen J-P and Wang P-N (2021) Prediction of Cerebral Amyloid Pathology Based on Plasma Amyloid and Tau Related Markers. Front. Neurol. 12:619388. doi: 10.3389/fneur.2021.619388
Received: 20 October 2020; Accepted: 07 September 2021;
Published: 04 October 2021.
Edited by:
Ching-Kuan Liu, Kaohsiung Medical University Hospital, TaiwanReviewed by:
Michael D. Devous, Avid Radiopharmaceuticals, Inc., United StatesCopyright © 2021 Chen, Lin, Lin, Lee, Lin, Wang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-Ning Wang, bGluZGEyODYwQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.