 Majida Charif1,2
Majida Charif1,2 Céline Bris1,3
Céline Bris1,3 David Goudenège1,3Valérie Desquiret-Dumas1,3Estelle Colin1,3
David Goudenège1,3Valérie Desquiret-Dumas1,3Estelle Colin1,3 Alban Ziegler1,3Vincent Procaccio1,3
Alban Ziegler1,3Vincent Procaccio1,3 Pascal Reynier1,3
Pascal Reynier1,3 Dominique Bonneau1,3
Dominique Bonneau1,3 Guy Lenaers1*
Guy Lenaers1* Patrizia Amati-Bonneau1,3
Patrizia Amati-Bonneau1,3- 1University Angers, MitoLab team, UMR CNRS 6015—INSERM U1083, Unité MitoVasc, SFR ICAT, Angers, France
- 2Genetics and Immuno-Cell Therapy Team, Mohammed First University, Oujda, Morocco
- 3Departments of Biochemistry and Genetics, University Hospital Angers, Angers, France
Advances in next-generation sequencing (NGS) facilitate the diagnosis of genetic disorders. To evaluate its use for the molecular diagnosis of inherited optic neuropathy (ION), a blinding disease caused by the degeneration of retinal ganglion cells, we performed genetic analysis using targeted NGS of 22 already known and candidate genes in a cohort of 1,102 affected individuals. The panel design, library preparation, and sequencing reactions were performed using the Ion AmpliSeq technology. Pathogenic variants were detected in 16 genes in 245 patients (22%), including 186 (17%) and 59 (5%) dominant and recessive cases, respectively. Results confirmed that OPA1 variants are responsible for the majority of dominant IONs, whereas ACO2 and WFS1 variants are also frequently involved in both dominant and recessive forms of ION. All pathogenic variants were found in genes encoding proteins involved in the mitochondrial function, highlighting the importance of mitochondria in the survival of retinal ganglion cells.
Introduction
Inherited optic neuropathies (IONs), the most common cause of underlying blindness of genetic origin, are mitochondrial diseases that cause degeneration of retinal ganglion cells (RGCs) and optic nerves with varying levels of vision loss. Their clinical presentation can be isolated, without extraocular symptoms, or syndromic, in which the optic atrophy is the primary symptom associated with a wide variety of secondary symptoms, mainly of neuromuscular origin (1–3). IONs may be caused either by autosomal dominant variants, leading to dominant optic atrophy (DOA MIM#165500), or by autosomal recessive variants, leading to recessive optic atrophy (ROA), or finally by mitochondrial DNA (mtDNA) variants, causing Leber's hereditary optic neuropathy (LHON, MIM #535000).
The diagnosis of ION is based on three main criteria: (i) reduced visual acuity with a central or cecocentral scotoma; (ii) the presence of a pallor of the optic disk on funduscopic examination, while the retina remains normal; and (iii) the loss of the layer of retinal nerve fibers objectified by optical coherence tomography (4–6). In general, DOA begins in childhood or in young adults, leading to a chronic and slow progression of changes in vision, whereas recessive forms affect younger children more severely (3, 7). Conversely, maternally transmitted LHON leads, in general, to acute loss of vision in one eye, followed by the other eye on average 2 months later.
DOA is mainly caused by pathogenic OPA1 variants (8–10) and less frequently by mutations in other genes including OPA3, MFN2, SPG7, AFG3L2, DNM1L, and SSBP1 (11–17). These genes are all involved in controlling the mitochondrial dynamics, with the exception of SSBP1, which is involved in mtDNA replication. This suggests that the equilibrium between mitochondrial fusion and fission is a key process for the maintenance of RGC physiology. Another gene involved in dominant ION is WFS1, the gene responsible for Wolfram syndrome, a recessive condition. WFS1 is also responsible for DOA associated with neurosensorial deafness (18) and for isolated recessive isolated ION (19). Importantly, WFS1 encodes a protein of the endoplasmic reticulum that is involved in mitochondrial calcium homeostasis (20).
In addition to WFS1, six genes have been identified as responsible for ROA, namely, TMEM126A, ACO2, RTN4IP1, NDUFS2, MCAT, and SLC25A46 (21–26). All these genes encode proteins with a mitochondrial function, but only SLC25A46 is involved in mitochondrial dynamics (21).
Angers University Hospital (CHU) has provided a national genetic diagnosis for ION for the last 20 years. More than 4,000 DNA samples have now been collected, and an ongoing collection of 800 additional samples is collected each year. Sanger sequencing of OPA1, OPA3, and WFS1, as well as all LHON mtDNA mutation, was performed for each sample until 2015. Currently, in order to improve the molecular diagnosis of ION, we have designed a resequencing chip targeting 22 ascertained or candidate genes.
Here, we report the results obtained with this ION chip on more than 1,000 DNA samples from individuals affected with ION. This procedure allowed a molecular diagnosis for more than 22% of the cases.
Materials and Methods
Patients
The Department of Biochemistry and Genetics at Angers University Hospital (France) is a national center for the genetic diagnosis of ION. DNA samples from 1,102 individuals affected with autosomal ION were collected in this center between 2015 and 2018. All patients had a proven case of clinical optic atrophy, investigated by an ophthalmologist before genetic testing. Where possible, DNA samples were obtained from affected and healthy relatives. Written informed consent was obtained from every individual involved in this diagnosis procedure.
ION Panel Design, Library Preparation, and Sequencing
The patients were initially screened for the three primary LHON mutations or for all mtDNA genome.
An amplicon library of the target exons was designed with the Ion AmpliSeq Designer (http://ampliseq.com) for the most genes reported to be responsible for ION, or candidate genes identified by whole-exome sequencing. These candidate genes do not have a direct mitochondrial function, but these are selected after analysis of the WES data of other NOH patients by filtering with a list of 1,600 mitochondrial genes. Four primer pools (125- to 275-bp amplicon target sizes) were designed to amplify 313 and 274 amplicons, covering 22 genes (Supplementary Table 1) (12) of a total length of 55.28 and 50.99 kb.
Genomic DNA of patients was extracted from peripheral blood. The Qubit dsDNA High Sensitivity Assay Kit (Thermo Fisher Scientific) was used to quantify DNA for next-generation sequencing (NGS) library construction. Library preparation for each sample was performed using Ion AmpliSeq technologies, and all sequencing data were processed using a dedicated bioinformatics pipeline, which includes three steps: variant calling, annotation, and prioritization (12, 27). The calling module uses a consensus-based approach and combines the prediction of six callers (VariantCaller included with the Torrent Suite, GATK Unified Genotyper, VarScan2, SNVer, LoFreq, and Platypus). All the generated variant calling formats are normalized and decomposed before launching the annotation–prioritization module, which combines NCBI Variant Reporter and ANNOVAR. These tools allow including genomic databases, clinical databases such as CLINVAR, and precomputed results of several prioritization tools (e.g., SIFT, PolyPhen2, LRT, MutationTaster).
Causative Variant Prioritization
For each patient, we looked for causative variants using the following prioritization strategy: first, we checked for reported pathogenic variants in ION genes, and then we looked for novel loss-of-function (stop–gain, frameshift, and splicing) or novel missense variants in these genes, and eventually, we selected new variants in candidate genes, which should be classified as being of “uncertain significance” according to the American College of Medical Genetics and Genomics, until additional evidence is supporting their pathogenicity.
All dominant variants were absent or had a minor allele frequency (MAF) threshold of <0.0001 in public databases, whereas recessive variants were considered with an MAF threshold <0.005. For familial cases, we specifically selected the variants that matched the inheritance pattern predicted from the pedigrees.
Sanger Sequencing Validation and Family Segregation Assessment
All putative variants identified by NGS were confirmed by Sanger sequencing, and their segregation was studied in all available members of the family. Primers were designed using the Primer3 software (28). To ensure the quality of Sanger sequencing, amplicons were designed to have a boundary of around 100 bp away from the variant.
Results
NGS Quality Results
DNA samples were primarily screened for the presence of one of the three primary LHON mutations: m.3460G>A, m.11778G>A, and m.14484T>C, and excluded from this study if the positive. Ion AmpliSeq NGS technology using autosomal ION panel was performed in a cohort of 1,102 individuals. Only one affected individual was studied per family. Within the design region, 95% of bases had coverage of >25×, >50×, and >100×, indicating that a sufficient coverage was achieved to enable high variant detection sensitivity.
Sanger sequencing was also performed for regions that are not or are poorly covered by the chip sequencing. This concerned specific regions of the following genes: AFG3L2 (exon1), SPG7 (exon 1, 13, and 17), ACO2 (exon 3), MFN2 (exon 9), RTN4IP1 (exon 2), WFS1 (exon 8), and OPA1 (exon 24).
NGS Results of the ION Panel
After filtering, annotation steps, and variant prioritization procedures, we identified pathogenic variants in 245 individuals (22%) (Supplementary Table 2), including 186 cases (17%) with a dominant mode of inheritance and 59 cases (5%) with a recessive mode of inheritance (Figure 1).
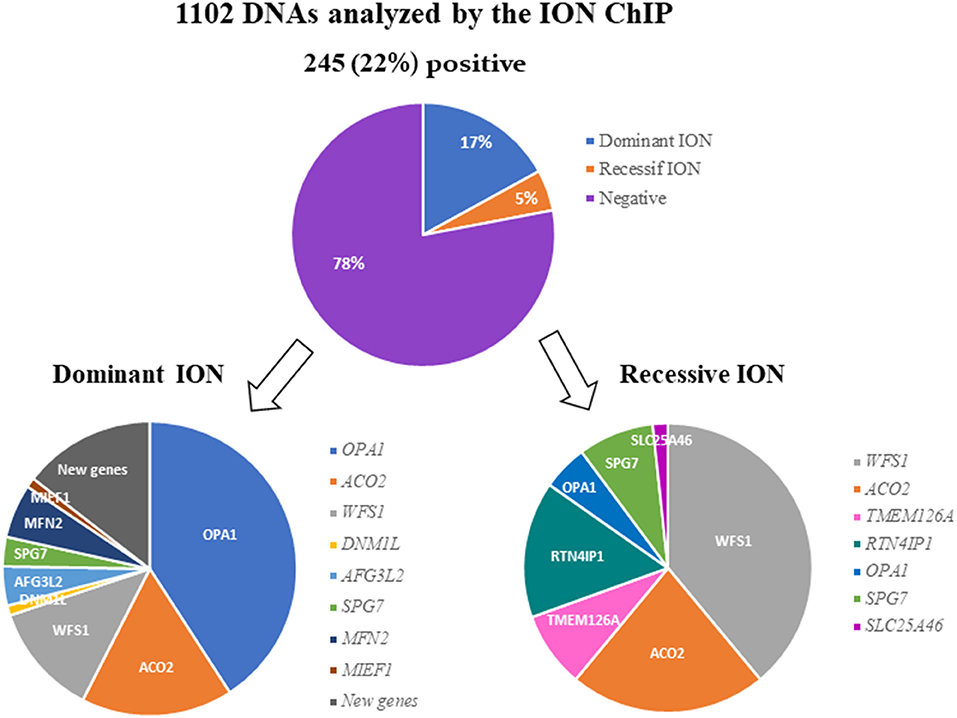
Figure 1. Results of the molecular analysis of 1,102 individuals with ION, using the 22-targeted-gene ION panel. A pathogenic variant was found in 245 (22%) of the DNA samples analyzed. The variants were inherited dominantly or recessively in 186 and 59 cases, respectively. The genes involved in dominant and recessive inheritance are indicated.
Dominant Genes
Out of the 186 individuals affected with DOA, 76 (40.86%) harbored a pathogenic variant in OPA1. The two other genes most involved in DOA were ACO2 and WFS1, with frequencies of 16.67% (31 cases) and 12.37% (23 cases), respectively.
Among the other genes, we identified variants in AFG3L2 in 8 individuals (4.3%), in SPG7 in 6 individuals (3.23%), in MFN2 in 11 individuals (5.91%), and in DNM1L and MIEF1 in 2 individuals each (1.08%). Lastly, we identified five new candidate genes in 27 families (data not shown), for which dominant variants were found to segregate with the disease (Figure 1).
For OPA1, variants were more frequently evidenced in exons 2, 10, 13, and 19 (Figure 2A). The most frequent OPA1 variants were splice variants (36%), followed by nonsense (26%), missense (24%), and frameshift variants (14%) (Figure 2B).
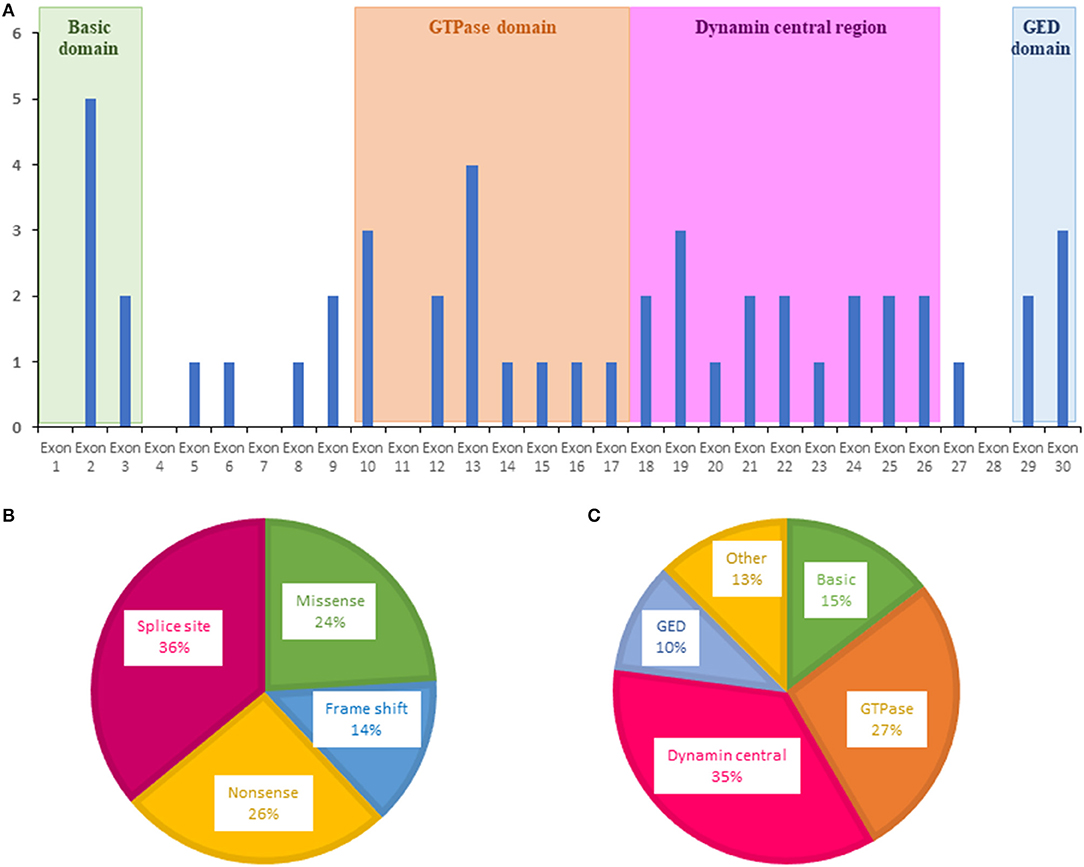
Figure 2. Distribution of variants identified in OPA1 by the ION chip. (A) Exons in which were located the variants are shown as blue bars. (B) Types of variants. (C) Affected domain of the protein. GED, GTPase effector domain.
Regarding localization in the OPA1 protein, most of the variants were localized in the central GTPase (27%) and dynamin (35%) domains, highlighting the important role of these domains in OPA1 functions (Figure 2C).
Recessive Genes
Twenty-three individuals (38.98%) out of 59 ROA cases were harboring biallelic pathogenic variants in WFS1. Regarding the other recessive genes, we found variants in ACO2 in 13 families (22.03%), in RTN4IP1 in 9 families (15.25%), and in TMEM126A or SPG7 in 5 families each (8.47%). Finally, three families (5.08%) harbored OPA1 biallelic variants, and one family, SLC25A46 variants (Figure 1).
Altogether, we identified 190 different variants, including 120 missenses, 22 nonsenses, 20 frameshifts, and 28 splice variants. The distribution of the different types of variants according to their dominant or recessive mode of inheritance is shown on Figure 3.
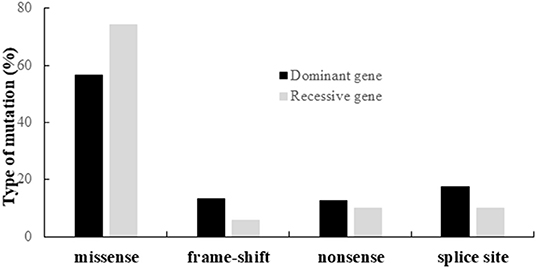
Figure 3. Distribution of the different types of variants identified in the genes responsible for dominant or recessive IONs.
Discussion
NGS is an effective approach that has dramatically improved the molecular diagnosis of genetic disorders, allowing a one-step identification of pathogenic variants in candidate genes. In this study, we have developed a panel for the molecular diagnosis of IONs, allowing the resequencing of 22 targeted genes, based on the Ion AmpliSeq method.
The development of this ION chip has improved the genetic dismemberment of IONs by allowing molecular diagnosis in a large number of cases and by contributing to the identification of new genes responsible for optic nerve degeneration.
Our results revealed that 22% of affected individuals (245 out of 1,102) harbored pathogenic variants in the targeted genes, 17% in the dominant form and 5% in the recessive form. The rather low ratio of success in inferring a molecular diagnosis from our study is most probably related to the fact that we provide a nationwide service to clinicians that might not always be familiar with the clinical diagnosis of ION.
Nevertheless, these results are in agreement with those of the literature indicating that DOAs are the most frequent forms of ION with an estimated frequency of 1/30,000 in the world (29). Conversely, the frequency of autosomal recessive optic neuropathies, which was initially described as very low, is now progressing, thanks to NGS, with an increasing number of genes involved (30).
In our cohort, we found that only 40.9% of the individuals with DOA presented a pathogenic heterozygous OPA1 variant, which is lower than the involvement of this gene in 60–80% of individuals with DOA in the literature (31), which were collected among well-characterized family cases, whereas in our study most patients were isolated cases.
Most of these variants were located in the GTPase (27%) and the central dynamin (35%) domains, in accordance with data from the OPA1 database, which includes 414 pathogenic variants (31).
Among the OPA1 variants, 36% were associated with a splicing defect, 26% were nonsense, 14% were frameshift, and 24% were missense. This confirms that haploinsufficiency is more frequently involved than dominant negative variants in the pathomechanism of DOA (31, 32).
With the exception of WFS1 and SSBP1, all proteins associated with DOA are closely related to mitochondrial dynamics, pointing to a key pathway that is essential for the maintenance and survival of RGC (17, 18, 33, 34). However, while excluding OPA1, the frequency of mutations in DOA that affect the proteins involved in mitochondrial dynamics is low. For example, AFG3L2 is involved in only 4.3% of cases, SPG7 in 3.23%, MFN2 in 5.91%, and DNM1L or MIEF1 in 1.08% of cases with a molecular diagnosis.
On the other hand, the results of ION chip sequencing revealed that ACO2, encoding the mitochondrial aconitase 2 involved in the Krebs cycle, is the second gene most frequently involved in DOA (16.67% of the cases). Variants in ACO2 have until now been mostly involved in recessive IONs (25), but our results show that dominant cases are far more frequent than the recessive ones1. ACO2 is therefore the third gene in order of frequency involved in autosomal IONs, after OPA1 and WFS11.
In this study, we also discovered dominant variants in five novel candidate genes (data not shown), suggesting that other mechanisms a priori complementary to mitochondrial dynamics are also involved in the pathophysiology of DOA.
As mentioned earlier, autosomal recessive IONs are less frequent. The first gene found to be involved in recessive ION was TMEM126A, which encodes a mitochondrial inner membrane protein (23). The most frequent mutation in TMEM126A, namely, p.R55X, was evidenced in five families (8.47%) of our series.
The other genes involved in recessive IONs are responsible for isolated or syndromic forms of optic atrophy. Recessive variants in ACO2 were found in 13 families (22.03%), whereas nine families (15.25%) harbored RTN4IP1 recessive variants. RTN4IP1 encodes a mitochondrial protein with a quinone oxidoreductase activity, which is involved in complexes I and IV respiratory functions, and neuronal dendritic branching (22). For these three genes, mutated individuals had a clinical presentation that was, in most cases, restricted to isolated optic atrophy.
In contrast, the recessive variants identified in SPG7 (8.47%), OPA1 (5.08%), and SLC25A46 (1.69%) were associated with syndromic optic atrophies.
In our series, variants in WFS1 were found in 12.37 and 38.98% of dominant and recessive IONs, respectively, with most variants localized in exon 8 and leading to truncating and missense changes (18, 35–37). Mutations in WFS1 are responsible for many different clinical presentations with dominant and recessive inheritance, including the Wolfram syndrome, autosomal recessive non-syndromic OA, isolated DOA (19), and DOA associated with hearing loss (18, 38).
The recurrent, non-founder mutation in WFS1, p.A684V, has been identified in six families from this series. This variant is a common cause of isolated autosomal DOA associated with deafness, as is the recurrent p.R500H mutation in OPA1. WFS1 encodes the wolframin, a transmembrane endoplasmic reticulum protein that plays an essential role in calcium homeostasis and interorganelle cross-talk at mitochondria-associated membranes (33, 39), highlighting the important role of these structures in the pathophysiology of IONs.
Conclusion
The NGS approach allowed us to identify the genetic cause of 22% of autosomal IONs. A total of 190 different variants were found in known genes and in five novel candidate genes. Further improvements in NGS technology, with a greater number of targeted genes, or the use of the whole-exome or whole-genome sequencing will undoubtedly increase the success rate of the molecular diagnosis of this group of diseases.
Finally, pathophysiological studies on the new genes involved in IONs will broaden our vision of the mechanisms responsible for the degeneration of CGRs and will eventually lead to new therapeutic strategies.
Data Availability Statement
The datasets presented in this article are not readily available because they have been collected for official clinical molecular diagnosis performed at the University Hospital of Angers. Requests to access the datasets should be directed to GL and PA-B (Z3V5LmxlbmFlcnNAdW5pdi1hbmdlcnMuZnI= and cGFib25uZWF1QGNodS1hbmdlcnMuZnI=).
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board Committee of the University Hospital of Angers, Authorization number: AC-2012-1507. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
GL and PA-B: conception and design of the study. DB, EC, and AZ: recruitment of patients. MC, CB, DG, VD-D, PR, and PA-B: acquisition and data analysis. MC and GL: drafting a significant portion of the manuscript and figures. DB and VP: revised the manuscript for intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the University of Angers, Angers University Hospital, the Région Pays de la Loire, Angers Loire Métropole, the Fondation Maladies Rares, the Fondation VISIO, Kjer-France, Ouvrir Les Yeux, Retina France, UNADEV, the Association Française contre les Myopathies and the Fondation de France.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all clinicians for their participation in patient recruitment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.602979/full#supplementary-material
Footnote
1. ^Charif M, Gueguen N, Ferré M, Elkarhat Z, Khiati S, LeMao M, et al. Dominant ACO2 mutations are a frequent cause of isolated Optic Atrophy. Brain Commun. (in press).
References
1. Votruba M. Molecular genetic basis of primary inherited optic neuropathies. Eye. (2004) 18:1126–32. doi: 10.1038/sj.eye.6701570
2. Carelli V, La Morgia C, Iommarini L, Carroccia R, Mattiazzi M, Sangiorgi S, et al. Mitochondrial optic neuropathies: how two genomes may kill the same cell type? Biosci Rep. (2007) 27:173–84. doi: 10.1007/s10540-007-9045-0
3. Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, et al. Dominant optic atrophy. Orphanet J Rare Dis. (2012) 7:46. doi: 10.1186/1750-1172-7-46
4. La Morgia C, Carbonelli M, Barboni P, Sadun AA, Carelli V. Medical management of hereditary optic neuropathies. Front Neurol. (2014) 5:114. doi: 10.3389/fneur.2014.00141
5. Leruez S, Amati-Bonneau P, Verny C, Reynier P, Procaccio V, Bonneau D, et al. Mitochondrial dysfunction affecting visual pathways. Rev Neurol (Paris). (2014) 170:344–54. doi: 10.1016/j.neurol.2014.03.009
6. Yu-Wai-Man P, Chinnery PF. Leber hereditary optic neuropathy - Therapeutic challenges and early promise. Taiwan J Ophthalmol. (2011) 1:12–5. doi: 10.1016/j.tjo.2011.09.001
7. Meunier I, Lenaers G, Hamel C, Defoort-Dhellemmes S. Hereditary optic neuropathies: From clinical signs to diagnosis. J Fr Ophtalmol. (2013) 36:886–900. doi: 10.1016/j.jfo.2013.05.007
8. Alexander C, Votruba M, Pesch UEA, Thiselton DL, Mayer S, Rodriguez M, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. (2000) 26:211–5. doi: 10.1038/79944
9. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. (2000) 26:207–10 doi: 10.1038/79936
10. Ferré M, Caignard A, Milea D, Leruez S, Cassereau J, Chevrollier A, et al. Improved locus-specific database for OPA1 mutations allows inclusion of advanced clinical data. Hum Mutat. (2015) 36:20–5. doi: 10.1002/humu.22703
11. Charif M, Roubertie A, Salime S, Mamouni S, Goizet C, Hamel CP, et al. A novel mutation of AFG3L2 might cause dominant optic atrophy in patients with mild intellectual disability. Front Genet. (2015) 6:311. doi: 10.3389/fgene.2015.00311
12. Charif M, Chevrollier A, Gueguen N, Bris C, Goudenège D, Desquiret-Dumas V, et al. Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy. Neurol Genet. (2020) 6:e428. doi: 10.1212/nxg.0000000000000428
13. Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain. (2012) 135:2980–93. doi: 10.1093/brain/aws240
14. Piro-Mégy C, Sarzi E, Tarrés-Solé A, Péquignot M, Hensen F, Quilès M, et al. Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy. J Clin Invest. (2020) 130:143–56. doi: 10.1172/JCI128513
15. Reynier P, Amati-Bonneau P, Verny C, Olichon A, Simard G, Guichet A, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. (2004) 41:1–6. doi: 10.1136/jmg.2003.016576
16. Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain. (2012) 135(Pt 1):23–34. doi: 10.1093/brain/awr323
17. Gerber S, Charif M, Chevrollier A, Chaumette T, Angebault C, Kane MS, et al. Mutations in DNM1L, as in OPA1, result indominant optic atrophy despite opposite effectson mitochondrial fusion and fission. Brain. (2017) 140:2586–96. doi: 10.1093/brain/awx219
18. Rendtorff ND, Lodahl M, Boulahbel H, Johansen IR, Pandya A, Welch KO, et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy hearing impairment. Am J Med Genet Part A. (2011) 155:1298–313. doi: 10.1002/ajmg.a.33970
19. Grenier J, Meunier I, Daien V, Baudoin C, Halloy F, Bocquet B, et al. WFS1 in optic neuropathies: mutation findings in nonsyndromic optic atrophy and assessment of clinical severity. Ophthalmology. (2016) 123:1989–98. doi: 10.1016/j.ophtha.2016.05.036
20. Takeda K. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet. (2001) 10:477–84. doi: 10.1093/hmg/10.5.477
21. Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. (2015) 47:926–32. doi: 10.1038/ng.3354
22. Angebault C, Guichet PO, Talmat-Amar Y, Charif M, Gerber S, Fares-Taie L, et al. Recessive mutations in RTN4IP1 cause isolated and syndromic optic neuropathies. Am J Hum Genet. (2015) 97:754–60. doi: 10.1016/j.ajhg.2015.09.012
23. Hanein S, Perrault I, Roche O, Gerber S, Khadom N, Rio M, et al. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am J Hum Genet. (2009) 84:493–8. doi: 10.1016/j.ajhg.2009.03.003
24. Li H, Yuan S, Minegishi Y, Suga A, Yoshitake K, Sheng X, et al. Novel mutations in malonyl-CoA-acyl carrier protein transacylase provoke autosomal recessive optic neuropathy. Hum Mol Genet. (2020) 29:444–58. doi: 10.1093/hmg/ddz311
25. Metodiev MD, Gerber S, Hubert L, Delahodde A, Chretien D, Gérard X, et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J Med Genet. (2014) 51:834–38. doi: 10.1136/jmedgenet-2014-102532
26. Gerber S, Ding MG, Gérard X, Zwicker K, Zanlonghi X, Rio M, et al. Compound heterozygosity for severe and hypomorphic NDUFS2 mutations cause non-syndromic LHON-like optic neuropathy. J Med Genet. (2017) 54:346–56. doi: 10.1136/jmedgenet-2016-104212
27. Felhi R, Sfaihi L, Charif M, Desquiret-Dumas V, Bris C, Goudenège D, et al. Next generation sequencing in family with MNGIE syndrome associated to optic atrophy: Novel homozygous POLG mutation in the C-terminal sub-domain leading to mtDNA depletion. Clin Chim Acta. (2019) 488:104–10. doi: 10.1016/j.cca.2018.11.003
28. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. (2007) 35:W71–4. doi: 10.1093/nar/gkm306
29. Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. (2010) 133:771–86. doi: 10.1093/brain/awq007
30. Yu-Wai-Man P, Votruba M, Burté F, La Morgia C, Barboni P, Carelli V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. (2016) 132:789–806. doi: 10.1007/s00401-016-1625-2
31. Le Roux B, Lenaers G, Zanlonghi X, Amati-Bonneau P, Chabrun F, Foulonneau T, et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J Rare Dis. (2019) 14:1–9. doi: 10.1186/s13023-019-1187-1
32. Ferré M, Bonneau D, Milea D, Chevrollier A, Verny C, Dollfus H, et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum Mutat. (2009) 30:692–705. doi: 10.1002/humu.21025
33. Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. (2015) 11:11–24. doi: 10.1038/nrneurol.2014.228
34. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. (2003) 278:7743–6. doi: 10.1074/jbc.C200677200
35. Bespalova IN, Van Camp G, Bom SJH, Brown DJ, Cryns K, DeWan AT, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet. (2001) 10:2501–8.doi: 10.1093/hmg/10.22.2501
36. Hansen L, Eiberg H, Barrett T, Bek T, Kjærsgaard P, Tranebjærg L, et al. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet. (2005) 13:1275–84. doi: 10.1038/sj.ejhg.5201491
37. Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet. (2001) 10:2509–14. doi: 10.1093/hmg/10.22.2509
38. Hogewind BFT, Pennings RJE, Hol FA, Kunst HPM, Hoefsloot EH, Cruysberg JRM, et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol Vis. (2010) 16:26–35.
39. Strom TM, Hörtnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet. (1998) 7:2021–8. doi: 10.1093/hmg/7.13.2021
Keywords: inherited optic neuropathies, mitochondrial disorders, molecular diagnosis, next generation sequencing, retinal ganglia cells
Citation: Charif M, Bris C, Goudenège D, Desquiret-Dumas V, Colin E, Ziegler A, Procaccio V, Reynier P, Bonneau D, Lenaers G and Amati-Bonneau P (2021) Use of Next-Generation Sequencing for the Molecular Diagnosis of 1,102 Patients With a Autosomal Optic Neuropathy. Front. Neurol. 12:602979. doi: 10.3389/fneur.2021.602979
Received: 04 September 2020; Accepted: 01 February 2021;
Published: 25 March 2021.
Edited by:
Monkol Lek, Yale University, United StatesReviewed by:
Ni-Chung Lee, National Taiwan University, TaiwanNicole J. Lake, Yale University, United States
Copyright © 2021 Charif, Bris, Goudenège, Desquiret-Dumas, Colin, Ziegler, Procaccio, Reynier, Bonneau, Lenaers and Amati-Bonneau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Lenaers, Z3V5LmxlbmFlcnNAaW5zZXJtLmZy