 Stephen Tisch
Stephen Tisch Kishore Raj Kumar
Kishore Raj Kumar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 08 January 2021
Sec. Movement Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.630391
This article is part of the Research Topic Advances in Invasive and Non-Invasive Brain Stimulation for Dystonia and Other Hyperkinetic Movement Disorders View all 8 articles
Globus pallidus internus deep brain stimulation (GPi DBS) is the most effective intervention for medically refractory segmental and generalized dystonia in both children and adults. Predictive factors for the degree of improvement after GPi DBS include shorter disease duration and dystonia subtype with idiopathic isolated dystonia usually responding better than acquired combined dystonias. Other factors contributing to variability in outcome may include body distribution, pattern of dystonia and DBS related factors such as lead placement and stimulation parameters. The responsiveness to DBS appears to vary between different monogenic forms of dystonia, with some improving more than others. The first observation in this regard was reports of superior DBS outcomes in DYT-TOR1A (DYT1) dystonia, although other studies have found no difference. Recently a subgroup with young onset DYT-TOR1A, more rapid progression and secondary worsening after effective GPi DBS, has been described. Myoclonus dystonia due to DYT-SCGE (DYT11) usually responds well to GPi DBS. Good outcomes following GPi DBS have also been documented in X-linked dystonia Parkinsonism (DYT3). In contrast, poorer, more variable DBS outcomes have been reported in DYT-THAP1 (DYT6) including a recent larger series. The outcome of GPi DBS in other monogenic isolated and combined dystonias including DYT-GNAL (DYT25), DYT-KMT2B (DYT28), DYT-ATP1A3 (DYT12), and DYT-ANO3 (DYT24) have been reported with varying results in smaller numbers of patients. In this article the available evidence for long term GPi DBS outcome between different genetic dystonias is reviewed to reappraise popular perceptions of expected outcomes and revisit whether genetic diagnosis may assist in predicting DBS outcome.
Dystonia is a chronic neurological condition characterized by sustained or intermittent muscle contractions resulting in abnormal movements, postures and tremor (1, 2). Genetic dystonias can be defined as those in which an underlying gene is identified as the cause. The genetic understanding of dystonia has expanded with an increasing number of dystonia genes identified. The identification of causative genes has reduced the number of truly idiopathic cases, as many patients previously classified as having idiopathic isolated dystonia can now be attributed to a specific gene. Some genetic dystonias have neurological features beyond pure dystonia and are classified as combined phenotypes. Conceptual shifts in dystonia classification, improved genetic diagnosis, and recognition of phenotypic spectrums for individual genes have allowed for better characterization of dystonic syndromes and their response to treatment both medical and surgical. Globus pallidus internus (GPi) deep brain stimulation (DBS) is the most effective known treatment for medically refractory dystonia with established efficacy in segmental and generalized dystonia of idiopathic, genetic or acquired causes. The potency and versatility of GPi DBS has encouraged its use in an expanding range of medically refractory genetic dystonias with varying degrees of success. In this article we review available information for the effectiveness of GPi DBS in dystonia due to various causative genes in an effort to stratify whether genetic diagnosis may offer some assistance in predicting the DBS treatment outcome.
GPi-DBS is effective in medically refractory dystonia (3–8). Improvement in dystonia after GPi DBS is progressive over months (5, 9–11). Mobile components of dystonia tend to respond more quickly than the tonic elements (12, 13). Isolated dystonia either idiopathic or genetic (primary) dystonia improves to a greater degree than acquired combined (secondary) dystonia (14–17). However, exceptions include acquired tardive dystonia secondary to neuroleptic exposure, (18) and dystonia in neurodegeneration with brain iron accumulation (19, 20), which respond well to GPi DBS (see section on NBIA/DYT-PANK2). Conversely idiopathic isolated craniofacial and laryngeal dystonia may show a poorer than expected response to DBS (21), emphasizing the importance of not only etiology but also body distribution on DBS dystonia outcome. Shorter disease duration has also been correlated with improved GPi DBS outcome (22). Technical aspects of GPi DBS including electrode placement and stimulation parameters also have a significant effect on DBS outcome. Chronic stimulation in the most posteroventral portion of GPi is most effective (23). Dystonia DBS typically requires higher electrical parameters than STN DBS for Parkinson's disease, however longer stimulation pulse widths above 60 ms are not beneficial and are less energy efficient (24–26), while lower frequency stimulation below 100 Hz may be useful in selected patients (27). Rare treatment failures after GPi DBS may occur in all categories of dystonia including genetic isolated generalized dystonia and appear independent of technical reasons such poor lead placement (28).
We review the evidence for GPi-DBS in individual genetic forms of dystonia (Supplementary Table 1).
DYT-TOR1A (DYT1) is the most common cause of young onset familial isolated dystonia and typically begins between 9 and 12 years in a limb then spreads to become generalized with relative sparing of cervical and bulbar segments (29, 30). Among the earliest reports of GPi DBS for generalized dystonia was the study of Coubes et al. reporting 6 children and 1 adult with medically refractory DYT1 generalized dystonia with remarkable improvement after bilateral GPi DBS, resulting in a mean improvement in Burke-Fahn-Marsden Dystonia rating scale (BFMDS) motor score of 90% at 12 months (3). The same group reported a tendency for superior GPi DBS outcome in DYT1 dystonia in 15 patients compared with 17 idiopathic non-DYT1 patients (31). Krause et al. reported superior GPi DBS outcome in 4 DYT1 dystonia patients compared with 6 non-DYT1 patients, with improvements in BFMDS of 55.6 and 35.1% respectively, although one DYT1 patient showed secondary worsening 3 years after DBS (12). Starr et al. also reported marked but variable benefit in DYT1 dystonia following GPi DBS with improvement approaching 100% in two patients, 51% in another and 14% in a patient with long disease duration and fixed orthopedic deformity, factors known to limit overall benefit (32, 33). Longer term studies of GPi DBS in DYT1 have shown sustained benefit for up to 10 years (34) including a large study of 47 DYT1 patients treated with GPi DBS which reported average long-term improvement approaching 80% (35). Some studies have reported a statistically superior outcome of GPi DBS in DYT1 dystonia compared with non-DYT1 dystonia (23), however larger studies with rater-blinded methodology have shown no significant difference (5, 6). A meta regression study by Andrews et al. reviewed individual patient outcomes of GPi DBS in 466 patients including 91 DYT1 and 108 non-DYT generalized dystonia patients. They found DYT1 patients improved by 67.5% in BFMDS compared with 55.8% for non-DYT, and in multivariate analysis DYT1 was in independent predictor of superior outcome, along with shorter disease duration and lower baseline severity score (16). A subsequent larger meta regression study identified DYT1 as a predictor of improved outcome in univariate analysis but found only higher baseline severity score as a significant predictor of superior outcome in multivariate analysis, however, DYT1 status was excluded from the multivariate analysis owing to some studies not reporting DYT1 status (36). A systematic review of GPi DBS outcomes in children found a higher probability of >50% improvement in DYT1 vs. non-DYT1- patients (65 vs 29%) and higher percentage improvement among DYT1 patients compared with non-DYT1 (66 vs. 43%) but not reaching statistical significance (37).
While DYT1 dystonia usually responds well to GPi DBS, especially in patients with shorter disease duration and without orthopedic deformity, there are important exceptions where secondary worsening of dystonia may occur. Cif et al. described secondary worsening after several years in a subgroup of DYT1 dystonia patients with good initial response at 12 months (34). A recent multicentre study reported secondary worsening in DYT1 dystonia after GPi DBS in 11 of 132 patients at 6 months to 3 years after DBS and was associated with younger age of onset, faster disease progression and cranial involvement (38).
DYT-THAP1 (DYT6) is an early onset dystonia syndrome, with prominent oromandibular and laryngeal involvement at disease onset that typically spreads to the limbs, becoming more generalized (39, 40). The outcomes of GPi DBS in THAP1 dystonia appear poorer and more variable than idiopathic or DYT1 isolated dystonia with average improvements in BFMDS of around 35% and ranging from 16 to 72% with a tendency for limited or no improvement in speech or bulbar function (41–44). However, a few reports have described better outcomes including improvement in speech and swallowing (45, 46). There is some evidence that THAP1 dystonia may take longer to improve after GPi DBS than DYT1 or non-DYT1 dystonia but eventually responds to a similar degree albeit with more variable outcomes (47). A recent larger study of 14 THAP1 dystonia patients treated with GPi DBS with median follow up of 4 years found an average BFMDS improvement of 49% with limited improvements in speech noted, two non-responders and four patients with delayed worsening (48). Delayed worsening in THAP1 with improvement after lead repositioning has been reported in several patients (41, 43, 49). Concerns for effectiveness of GPi DBS in THAP1 dystonia have led to alternative DBS targets being explored. Mure et al. reported 80% improvement in BFMDS at 2 years following bilateral ventral lateral anterior (VLa) thalamic nucleus DBS in a single adult patient with THAP1 dystonia (50). The reasons for poorer and more variable DBS response in THAP1 dystonia are still not fully understood but may in part relate to prominent bulbar involvement which is a body region usually less responsive to DBS. Another possibility is genetic heterogeneity in DYT-THAP1 where many different pathogenic mutations have been described whereas DYT1 dystonia is usually due to a single common GAG deletion.
X-linked dystonia parkinsonism (XDP or “Lubag”) is associated with a SINE-VNTR-Alu retrotransposon insertion within an intron of the TATA box-binding protein–associated factor 1 (TAF1) gene (51, 52). It affects individuals with maternal origin from the island of Panay, in the Philippines (53). In this disorder, affected individuals develop focal or segmental dystonia in adulthood; the dystonia rapidly becomes generalized over several years (51). About 5–10 years after the onset of disease, dystonia features become less prominent, and parkinsonian features predominate (51). The dystonia in XDP is severe and disabling, oral medications have inconsistent benefit and neuroablative therapy targeting the bilateral pallidus and/or thalamus has resulted in major adverse events; thus GPi-DBS has been used as a therapeutic option (51, 54–56).
Perhaps the most comprehensive study of GPi-DBS to date involved 16 males with XDP from the Philippines with predominant dystonia (51). There was an improvement in dystonia post-operatively as well as the Unified Parkinson's Disease Rating Scale Part III (UPDRS III). Additionally, T1-based basal ganglia volumetry showed that caudate atrophy was a predictor of a less beneficial outcome.
Improvements in both BFMDRS and UPDRS measures suggests that GPi-DBS can be beneficial for both dystonia and parkinsonian features in XDP (51). However, there may be a differential effect of bilateral GPi-DBS, with an marked, immediate and sustained improvement in dystonia but with a lesser benefit for parkinsonism (56–58).
Mutations in TUBB4A have been found to cause whispering dysphonia (DYT4)–spasmodic dysphonia combined with other focal or generalized dystonia, and a characteristic “hobby horse” gait. (59–61). Mutations in TUBB4A can also cause other neurological phenotypes such as hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome (62) or hereditary spastic paraplegia (63).
There is a single case report documenting the response to DBS in TUBB4A-related dystonia. A 44 year old man with a p.Arg2Gly variant in TUBB4A was found to have an improvement in dystonia (55% reduction in BFMDRS) with a more prominent improvement in cervical and facial dystonia with bilateral GPi-DBS (64).
DYT11 is an autosomal dominantly inherited condition due to a heterozygous mutation in the SGCE gene, with paternal expression and reduced penetrance with maternal transmission (65). It results in early onset myoclonus dystonia usually presenting in childhood with upper body myoclonus and dystonia frequently with writer's cramp, cervical dystonia, and associated psychiatric problems including anxiety and obsessive compulsive disorder (66–68). The initial reports of beneficial effects of DBS in myoclonus dystonia were in patients without confirmation of SCGE mutation, where suppression of myoclonus without improvement in dystonia following staged bilateral ventralis intermediate nucleus (VIM) DBS was reported (69) and improvement in both myoclonus and dystonia following GPi DBS (70, 71). The first report of GPi DBS for genetically confirmed SGCE myoclonus dystonia was in an 8-year-old boy with marked improvement of both myoclonus and dystonia (72). Subsequently a number of studies reported good response and improvement in both dystonia and myoclonus in DYT11 dystonia following GPi DBS (73–78).
Kosutzka et al. reported long-term outcomes in 9 DYT-SCGE patients treated with bilateral GPi DBS who were followed up for minimum of 5 years. In this study motor improvement was marked with myoclonus improving by 94%, dystonia by 71%, 88% improvement in disability score and significantly improved function and social adjustment (79). A recent meta analysis of individual patient outcomes in 71 patients with myoclonus dystonia and GPi DBS found an average improvement in unified myoclonus rating scale of 79.5% and significant improvements in dystonia motor and disability scores with possible predictive factors for superior myoclonus outcome including shorter disease duration (80). Thalamic stimulation targeting the VIM nucleus appears to be an effective alternative to GPi DBS (73, 81), however pallidal stimulation is generally considered the preferred target (82). Patients with isolated myoclonus SCGE without dystonia also appear to benefit significantly from GPi DBS (83, 84).
While DBS can benefit the motor features of DYT-SGCE, it should be noted that there may not be a parallel improvement in psychiatric comorbidities (85).
Mutations in ATP1A3 can causes a wide spectrum of clinical phenotypes including alternating hemiplegia of childhood type 2, cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome, rapid-onset dystonia-parkinsonism (RDP, DYT12), as well as numerous “non-classical” phenotypes (86). There are case reports of DBS use in ATP1A3-related disorders, providing insights into the likely beneficial response.
One report of a 21 year old woman with presumed RDP who had bilateral GPi-DBS described no improvement in symptoms (87). A 12 year old boy with a novel, de novo, ATP1A3 mutation had onset of dysphagia and dysarthria followed by severe generalized dystonia resulting in inability to walk (88). Following bilateral GPi-DBS he was temporarily able to walk with a temporal improvement in BFMDRS but only a marginal improvement in UPDRS. Two further reports suggested that GPi-DBS was ineffective in ATP1A3-related dystonia (89, 90). However, in a kindred with a novel ATP1A3 variant causing generalized dystonia and paroxysmal dystonic attacks, bilateral GPi-DBS resulted in a remission of paroxysmal episodes and an improvement of interictal dystonia in one family member (91), suggesting that the response to DBS in ATP1A3-related dystonia is not uniformly poor but instead variable.
Mutations in PRKRA cause young onset autosomal recessive dystonia parkinsonism (92). A response to GPi-DBS has been shown in a small number of cases of DYT16; a study of 2 patients showed an improvement in BFMDRS and walking times on gait analysis (93), and GPI-DBS produced a sustained improvement in cranial and limb dystonia over 10 years in one patient with a homozygous PRKRA mutation (94).
Mutations in ANO3 have been associated with autosomal dominant, adult-onset craniocervical dystonia and dystonic tremor as well as myoclonic jerks (95).
To date, there have been at least 5 reported cases documenting a response to bilateral GPi-DBS in ANO3-related dystonia (96–100). For example, one patient with a de novo variant in ANO3 (p.Val561Glu) and early onset, generalized dystonia was found to improve with bilateral GPi-DBS, resulting in a sustained benefit allowing her to walk with assistance (99). Another patient with an ANO3 mutation (p.Glu510Lys) had a substantial improvement in dystonia and tremor but in this case myoclonus persisted (96). A further patient with ANO3-related dystonia had a successful response to GPi-DBS but continued to have episodes of dystonic storms (97). We conclude that DBS is likely to result in a successful but partial response in dystonia due to ANO3 mutations, but large case series are needed.
Autosomal dominant mutations in the GNAL gene cause primary torsion dystonia, typically with a craniocervical onset, although progression to other sites and generalized dystonia can occur, with a phenotype resembling THAP1-associated dystonia (101–103).
There are several case reports or case series documenting a response to GPi-DBS in GNAL mutation carriers (104–108). One patient with a novel missense variant (p.Met97Val) in GNAL with an associated phenotype of late-onset cervical and truncal dystonia had excellent long term and continuous benefit from bilateral GPi-DBS at 5 years follow-up (104).
In a family with a novel GNAL mutation (p.Asp210Asn), the index patient with cervical dystonia and tremor derived a 67% improvement in BFMDRS following bilateral GPi-DBS (105). The index patient's younger sister also obtained a benefit from dystonia in terms of an improvement in cervical and laryngeal dystonia (105).
The largest case series so far documented the response of 3 unrelated individuals with isolated dystonia due to a mutation in GNAL (108). All patients improved with bilateral GPi-DBS, with a predominant improvement in cervical dystonia compared to other regions. However, a case report of an individual with a GNAL variant (p.Cys429Tyr) did report improvement in other anatomical regions following bilateral GPi-DBS (107).
In summary, according to the limited patients reported, GNAL-related dystonia is likely to result in a favorable though incomplete response to GPi-DBS.
Heterozygous variants in the KMT2B gene are emerging as one of the commonest causes of early onset dystonia, with a caudocranial pattern evolving into generalized dystonia (109–111). There may be additional features, such as a characteristic facies, microcephaly, short stature, developmental delay, mild psychomotor impairment, and superimposed choreoathetosis or myoclonus (111, 112). Furthermore, MRI findings show characteristic changes including bilateral pallidal hypointensity that may serve as a clue to this disorder (110, 113).
While initially thought to be unresponsive to medical therapy (110), pharmacotherapy with trihexyphenidyl alone or in combination with clonazepam has been reported to reduce dystonia in some cases (114, 115). The reported response to bilateral GPi-DBS is more consistent, with improvements of motor function and gait, and restoration of walking in some patients (110).
A very recent study described the largest sample of KMT2B-related dystonia to date, including 18 individuals with medication-refractory dystonia (111). Significant improvement of motor function and disability [BFMDRS movement (BFMDRS-M) and BFMDRS disability (BFMDRS-D)] was evident at 6 months, 1 year and last follow-up (up to 22 years). Therefore, one of the characteristic features of dystonia due to KMT2B mutations is a response to DBS, which may be the preferred option in severely affected patients (116). The effect of DBS is seen in all anatomical regions, apart from perhaps a less beneficial effect upon laryngeal dystonia (111, 114).
ADCY5-dyskinesia is inherited in an autosomal dominant manner and causes a spectrum of clinical features including chorea, athetosis, myoclonus, dystonia, and ballistic bouts, with an onset in infancy to late-adolescence (117, 118).
In the largest case series to date, 3 patients with ADCY5 mutations has bilateral GPi-DBS (119). There was a subjective general improvement and a reduction in nocturnal episodic dyskinesias. However, on objective measures there was only a mild decrease in involuntary movements, and dystonia improved in 1 of 3 patients.
De novo heterozygous mutations in GNAO1 cause a spectrum of disorders including a neurodevelopmental delay, epileptic encephalopathy, and involuntary movements (120). There are numerous case reports and case series supporting DBS in GNAO1-associated movement disorders (120–124). Of note, emergency GPi-DBS in a severely ill patient produced a dramatic, life-saving response with almost complete remission of the hyperkinesia despite persistence of generalized dystonia (120). Furthermore, GPi-DBS was shown to be effective in two cases of status dystonicus due to GNAO1 mutations.
Biallelic mutations in PANK2 cause neurodegeneration with brain iron accumulation-1 also known as pantothenate kinase-associated neurodegeneration (PKAN), and previously known as Hallervorden-Spatz disease (125). The phenotype may be classic (early onset dystonia, choreoathetosis, dysarthria and rigidity) or atypical (late onset or slowly progressive forms) (19, 126). The MRI findings including iron deposition in the basal ganglia which may result in the characteristic “eye of the tiger sign” (126).
There are several case reports of dystonia due to PKAN treated with GPi-DBS, with most documenting a good response (127–131), although with one instance of no benefit (132). It is notable that genetic testing was not always performed in these cases, making it difficult to be sure of the genotype specific outcome.
A study of 6 individuals with PANK2 mutation-positive PKAN showed a major and sustained improvement in painful spasms, dystonia, and ambulation (19). A further study from Korea demonstrated a benefit in 2 patients with atypical PKAN but a variable benefit in 2 individuals with typical PKAN (133).
The largest study of NBIA to date was of 16 centers contributing 23 individuals who underwent GPi-DBS (20). Fifteen of those had genetic testing, and 14 were found to have PANK2 mutations. They reported an improvement in dystonia severity, disability, and quality of life. However, the improvement was not as great as those with primary generalized dystonia or other secondary dystonias. Additionally, patients with more severe dystonia appeared to derive greater benefit.
There are rarer forms of monogenic dystonia in whom a response to DBS has been reported but the evidence is limited so far. STN-DBS was found to be efficacious in two patients with GCH1 variants for parkinsonism and motor fluctuations following long-term treatment with levodopa (134). A case report of GNB1-related myoclonus dystonia showed an initial marked response to GPi-DBS (135). KCTD17-related myoclonus dystonia is known to have an excellent response to GPi-DBS (136), including an improvement in orolingual dyskinesia and speech (137). In a patient with deafness dystonia syndrome due to an ACTB mutation (p.Arg183Trp), GPi-DBS was also found to be effective (138).
A further case study reported 2 patients with double mutations in DYT1 and DYT11, who a successful response to bilateral VIM DBS (followed by GPi-DBS in one patient) (139).
Heterozygous loss of function mutations in VPS16 have recently been found to cause dystonia with prominent craniocervical and upper limb involvement (140). It is notable that some patients experience an improvement from DBS (140), although further data is required.
GM1 gangliosidosis is one several neurometabolic causes of dystonia (141). A patient with GM1 type 3 gangliosidosis was reported was reported to have a significant functional benefit but no change to disease progression with bilateral GPi-DBS (142).
A very recent systematic review and meta-analysis of GPi-DBS for monogenic dystonia found robust support for DYT1, modest support for DYT6 and PANK2-related dystonia, and promising results for SGCE, DYT3, ACTB and GNAO1-related dystonia, supporting the concept of a differential outcomes for the individual monogenic forms (143). An early age at onset (DYT1 and SGCE) was associated with better outcomes. Moreover, a shorter duration prior to GPi-DBS (DYT1 and DYT3) was associated with a better outcome, suggesting that perhaps earlier intervention would be beneficial in these individuals (143).
A recent systematic review demonstrated an improvement in physical quality of life, but the improvement in mental quality of life was less robust (144).
Pallidal DBS is effective across a range of monogenic forms of DBS, with a suggestion of a gene-specific differential effect (Figure 1). However, the evidence is limited by small cohort sizes or case reports, particularly for the rarer subtypes. Within these limitations, patterns of response between different monogenic dystonias may assist in patient selection for DBS and determining treatment prognosis, particularly if additional factors such as body distribution, disease duration or the presence of orthopedic deformity are included in the overall assessment. Patients should still be warned of the small risks of treatment failure or secondary worsening, even those with monogenic dystonias considered highly responsive to DBS. In this context the genetic diagnosis forms one part of the decision-making algorithm and perhaps with the exception of DYT-ATP1A3, finding a particular gene should not discourage consideration of GPi DBS if alleviation of a disabling dystonic syndrome is clinically imperative. Systematic reviews can be helpful as described (143), and international registries may address this issue further (145), particularly if the collection of clinical data is uniform, allowing for comparison between centers.
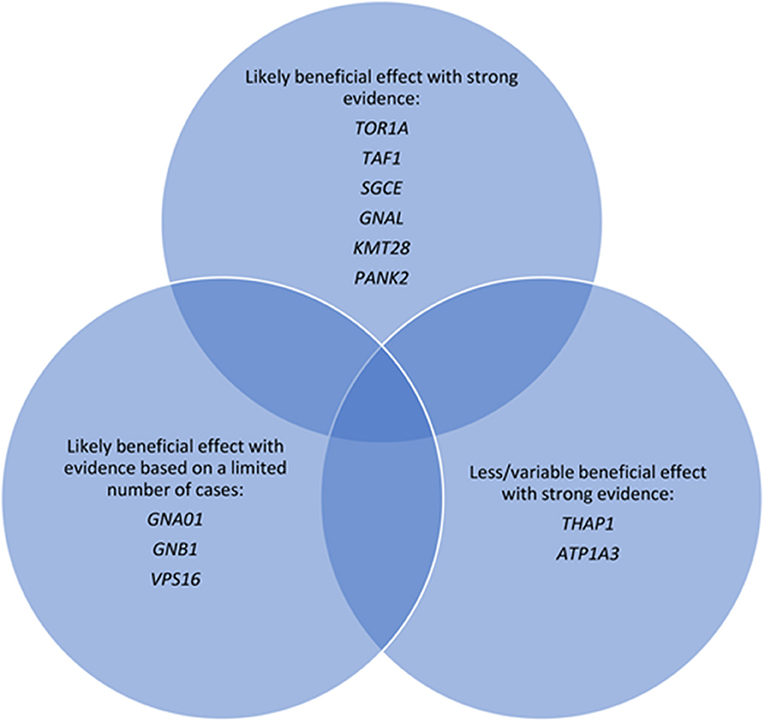
Figure 1. Major monogenic forms of dystonia categorized according to evidence of response to deep brain stimulation.
We highlight the importance of identifying a genetic diagnosis in dystonia, which, aside from multiple benefits including assistance with genetic counseling and family planning, may help guide expectations as to the likely outcome of DBS, for both patients and clinicians. This supports the concept of routine genetic testing of patients with dystonia prior to DBS (146). In this case, the options would be targeted gene panel sequencing, whole exome sequencing, or whole genome sequencing (96, 113)–the most cost effective approach is yet to be determined. An additional advantage of a genetic diagnosis in dystonia patients being considered for DBS is that it effectively excludes an unrecognized acquired combined (secondary) etiology where a poorer DBS outcome would be predicted.
It is uncertain whether GPi-DBS should be reserved for severe, medication-resistant cases, or whether it should be instituted at a much earlier point. What is well-accepted is that DBS should be considered in medically refractory dystonia before orthopedic deformity has developed. DBS responses may be variable and depend on other factors such as age (146). Furthermore, DBS may have a differential effect for certain phenotypic manifestations (e.g., improvement in dystonia greater than parkinsonism for DYT/PARK-TAF1 or limited improvement in speech and swallowing in DYT-THAP1).
While genetic testing may have a role with guiding expectations in GPi-DBS for genetic forms of dystonia, ultimately to decision to proceed should be based on the clinical phenotype.
ST and KK contributed to the first draft and revision of the manuscript. Both authors contributed to the article and approved the submitted version.
KK receives funding to study dystonia from the Paul Ainsworth Family Foundation, and receives a Working Group Co-Lead Award from the Michael J. Fox Foundation, Aligning Science Across Parkinson's (ASAP) initiative.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.630391/full#supplementary-material
1. Fahn S. Concept and classification of dystonia. Adv Neurol. (1988) 50:1–8. doi: 10.1212/WNL.50.5_Suppl_5.S1
2. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. (2013) 28:863–73. doi: 10.1002/mds.25475
3. Coubes P, Roubertie A, Vayssiere N, Hemm S, Echenne B. Treatment of DYT1-generalised dystonia by stimulation of the internal globus pallidus. Lancet. (2000) 355:2220–1. doi: 10.1016/S0140-6736(00)02410-7
4. Coubes P, Cif L, El Fertit H, Hemm S, Vayssiere N, Serrat S, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long-term results. J Neurosurg. (2004) 101:189–94. doi: 10.3171/jns.2004.101.2.0189
5. Vidailhet M, Vercueil L, Houeto JL, Krystkowiak P, Benabid AL, Cornu P, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. (2005) 352:459–67. doi: 10.1056/NEJMoa042187
6. Kupsch A, Benecke R, Muller J, Trottenberg T, Schneider GH, Poewe W, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med. (2006) 355:1978–90. doi: 10.1056/NEJMoa063618
7. Volkmann J, Wolters A, Kupsch A, Muller J, Kuhn AA, Schneider GH, et al. Pallidal deep brain stimulation in patients with primary generalised or segmental dystonia: 5-year follow-up of a randomised trial. Lancet Neurol. (2012) 11:1029–38. doi: 10.1016/S1474-4422(12)70257-0
8. Volkmann J, Mueller J, Deuschl G, Kuhn AA, Krauss JK, Poewe W, et al. Pallidal neurostimulation in patients with medication-refractory cervical dystonia: a randomised, sham-controlled trial. Lancet Neurol. (2014) 13:875–84. doi: 10.1016/S1474-4422(14)70143-7
9. Yianni J, Bain PG, Gregory RP, Nandi D, Joint C, Scott RB, et al. Post-operative progress of dystonia patients following globus pallidus internus deep brain stimulation. Eur J Neurol. (2003) 10:239–47. doi: 10.1046/j.1468-1331.2003.00592.x
10. Bittar RG, Yianni J, Wang S, Liu X, Nandi D, Joint C, et al. Stereotactic and functional neurosurgery resident award: deep brain stimulation for generalized dystonia and spasmodic torticollis: rate and extent of postoperative improvement. Clin Neurosurg. (2005) 52:379–83.
11. Tisch S, Limousin P, Rothwell JC, Asselman P, Quinn N, Jahanshahi M, et al. Changes in blink reflex excitability after globus pallidus internus stimulation for dystonia. Mov Disord. (2006) 21:1650–5. doi: 10.1002/mds.20899
12. Krause M, Fogel W, Kloss M, Rasche D, Volkmann J, Tronnier V. Pallidal stimulation for dystonia. Neurosurgery. (2004) 55:1361–8. doi: 10.1227/01.NEU.0000143331.86101.5E
13. Chung M, Huh R. Different clinical course of pallidal deep brain stimulation for phasic- and tonic-type cervical dystonia. Acta Neurochir. (2016) 158:171–80. doi: 10.1007/s00701-015-2646-7
14. Eltahawy HA, Saint-Cyr J, Giladi N, Lang AE, Lozano AM. Primary dystonia is more responsive than secondary dystonia to pallidal interventions: outcome after pallidotomy or pallidal deep brain stimulation. Neurosurgery. (2004) 54:613–19. doi: 10.1227/01.NEU.0000108643.94730.21
15. Vidailhet M, Yelnik J, Lagrange C, Fraix V, Grabli D, Thobois S, et al. Bilateral pallidal deep brain stimulation for the treatment of patients with dystonia-choreoathetosis cerebral palsy: a prospective pilot study. Lancet Neurol. (2009) 8:709–17. doi: 10.1016/S1474-4422(09)70151-6
16. Andrews C, Aviles-Olmos I, Hariz M, Foltynie T. Which patients with dystonia benefit from deep brain stimulation? A metaregression of individual patient outcomes. J Neurol Neurosurg Psychiatry. (2010) 81:1383–9. doi: 10.1136/jnnp.2010.207993
17. Vidailhet M, Jutras MF, Grabli D, Roze E. Deep brain stimulation for dystonia. J Neurol Neurosurg Psychiatr. (2013) 84:1029–42. doi: 10.1136/jnnp-2011-301714
18. Gruber D, Trottenberg T, Kivi A, Schoenecker T, Kopp UA, Hoffmann KT, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology. (2009) 73:53–8. doi: 10.1212/WNL.0b013e3181aaea01
19. Castelnau P, Cif L, Valente EM, Vayssiere N, Hemm S, Gannau A, et al. Pallidal stimulation improves pantothenate kinase-associated neurodegeneration. Ann Neurol. (2005) 57:738–41. doi: 10.1002/ana.20457
20. Timmermann L, Pauls KA, Wieland K, Jech R, Kurlemann G, Sharma N, et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain. (2010) 133:701–12. doi: 10.1093/brain/awq022
21. Limotai N, Go C, Oyama G, Hwynn N, Zesiewicz T, Foote K, et al. Mixed results for GPi-DBS in the treatment of cranio-facial and cranio-cervical dystonia symptoms. J Neurol. (2011) 258:2069–74. doi: 10.1007/s00415-011-6075-0
22. Isaias IU, Alterman RL, Tagliati M. Outcome predictors of pallidal stimulation in patients with primary dystonia: the role of disease duration. Brain. (2008) 131:1895–902. doi: 10.1093/brain/awn120
23. Tisch S, Zrinzo L, Limousin P, Bhatia KP, Quinn N, Ashkan K, et al. Effect of electrode contact location on clinical efficacy of pallidal deep brain stimulation in primary generalised dystonia. J Neurol Neurosurg Psychiatr. (2007) 78:1314–9. doi: 10.1136/jnnp.2006.109694
24. Vercueil L, Houeto JL, Krystkowiak P, Lagrange C, Cassim F, Benazzouz A, et al. Effects of pulse width variations in pallidal stimulation for primary generalized dystonia. J Neurol. (2007) 254:1533–7. doi: 10.1007/s00415-007-0578-8
25. Moro E, Piboolnurak P, Arenovich T, Hung SW, Poon YY, Lozano AM. Pallidal stimulation in cervical dystonia: clinical implications of acute changes in stimulation parameters. Eur J Neurol. (2009) 16:506–12. doi: 10.1111/j.1468-1331.2008.02520.x
26. Agren R, Bartek J Jr, Johansson A, Blomstedt P, Fytagoridis A. Pulse width and implantable pulse generator longevity in pallidal deep brain stimulation for dystonia: a population-based comparative effectiveness study. Stereotact Funct Neurosurg. (2020) 98:331–6. doi: 10.1159/000508794
27. Alterman RL, Miravite J, Weisz D, Shils JL, Bressman SB, Tagliati M. Sixty hertz pallidal deep brain stimulation for primary torsion dystonia. Neurology. (2007) 69:681–8. doi: 10.1212/01.wnl.0000267430.95106.ff
28. Zittel S, Hidding U, Trumpfheller M, Baltzer VL, Gulberti A, Schaper M, et al. Pallidal lead placement in dystonia: leads of non-responders are contained within an anatomical range defined by responders. J Neurol. (2020) 267:1663–71. doi: 10.1007/s00415-020-09753-z
29. Valente EM, Warner TT, Jarman PR, Mathen D, Fletcher NA, Marsden CD, et al. The role of DYT1 in primary torsion dystonia in Europe. Brain. (1998) 121:2335–9. doi: 10.1093/brain/121.12.2335
30. Bressman SB, Sabatti C, Raymond D, De Leon D, Klein C, Kramer PL, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. (2000) 54:1746–52. doi: 10.1212/WNL.54.9.1746
31. Cif L, El Fertit H, Vayssiere N, Hemm S, Hardouin E, Gannau A, et al. Treatment of dystonic syndromes by chronic electrical stimulation of the internal globus pallidus. J Neurosurg Sci. (2003) 47:52–5.
32. Starr PA, Turner RS, Rau G, Lindsey N, Heath S, Volz M, et al. Microelectrode-guided implantation of deep brain stimulators into the globus pallidus internus for dystonia: techniques, electrode locations, and outcomes. Neurosurg Focus. (2004) 17:E4. doi: 10.3171/foc.2004.17.1.4
33. Starr PA, Turner RS, Rau G, Lindsey N, Heath S, Volz M, et al. Microelectrode-guided implantation of deep brain stimulators into the globus pallidus internus for dystonia: techniques, electrode locations, and outcomes. J Neurosurg. (2006) 104:488–501. doi: 10.3171/jns.2006.104.4.488
34. Cif L, Vasques X, Gonzalez V, Ravel P, Biolsi B, Collod-Beroud G, et al. Long-term follow-up of DYT1 dystonia patients treated by deep brain stimulation: an open-label study. Mov Disord. (2010) 25:289–99. doi: 10.1002/mds.22802
35. Panov F, Gologorsky Y, Connors G, Tagliati M, Miravite J, Alterman RL. Deep brain stimulation in DYT1 dystonia: a 10-year experience. Neurosurgery. (2013) 73:86–93. doi: 10.1227/01.neu.0000429841.84083.c8
36. Moro E, Lereun C, Krauss JK, Albanese A, Lin JP, Walleser Autiero S, et al. Efficacy of pallidal stimulation in isolated dystonia: a systematic review and meta-analysis. Eur J Neurol. (2017) 24:552–60. doi: 10.1111/ene.13255
37. Hale AT, Monsour MA, Rolston JD, Naftel RP, Englot DJ. Deep brain stimulation in pediatric dystonia: a systematic review. Neurosurg Rev. (2020) 43:873–80. doi: 10.1007/s10143-018-1047-9
38. Tsuboi T, Cif L, Coubes P, Ostrem JL, Romero DA, Miyagi Y, et al. Secondary worsening following DYT1 dystonia deep brain stimulation: a multi-country cohort. Front Hum Neurosci. (2020) 14:242. doi: 10.3389/fnhum.2020.00242
39. Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. (2009) 41:286–8. doi: 10.1038/ng.304
40. Houlden H, Schneider SA, Paudel R, Melchers A, Schwingenschuh P, Edwards M, et al. THAP1 mutations (DYT6) are an additional cause of early-onset dystonia. Neurology. (2010) 74:846–50. doi: 10.1212/WNL.0b013e3181d5276d
41. Groen JL, Ritz K, Contarino MF, Van De Warrenburg BP, Aramideh M, Foncke EM, et al. DYT6 dystonia: mutation screening, phenotype, and response to deep brain stimulation. Mov Disord. (2010) 25:2420–7. doi: 10.1002/mds.23285
42. Zittel S, Moll CK, Bruggemann N, Tadic V, Hamel W, Kasten M, et al. Clinical neuroimaging and electrophysiological assessment of three DYT6 dystonia families. Mov Disord. (2010) 25:2405–12. doi: 10.1002/mds.23279
43. Panov F, Tagliati M, Ozelius LJ, Fuchs T, Gologorsky Y, Cheung T, et al. Pallidal deep brain stimulation for DYT6 dystonia. J Neurol Neurosurg Psychiatr. (2012) 83:182–7. doi: 10.1136/jnnp-2011-300979
44. Krause P, Bruggemann N, Volzmann S, Horn A, Kupsch A, Schneider GH, et al. Long-term effect on dystonia after pallidal deep brain stimulation (DBS) in three members of a family with a THAP1 mutation. J Neurol. (2015) 262:2739–44. doi: 10.1007/s00415-015-7908-z
45. Franca S, Massano J, Linhares P, Rosas MJ, Volkmann J. Pallidal deep brain stimulation in DYT6: significant long-term improvement of dystonia and disability. Mov Disord Clin Pract. (2014) 1:118–20. doi: 10.1002/mdc3.12019
46. Miri S, Shahidi GA, Parvaresh M, Rohani M. Pallidal deep brain stimulation for the treatment of DYT6 dystonia: a case report and review of literature. Med J Islam Repub Iran. (2014) 28:39.
47. Bruggemann N, Kuhn A, Schneider SA, Kamm C, Wolters A, Krause P, et al. Short- and long-term outcome of chronic pallidal neurostimulation in monogenic isolated dystonia. Neurology. (2015) 84:895–903. doi: 10.1212/WNL.0000000000001312
48. Danielsson A, Carecchio M, Cif L, Koy A, Lin JP, Solders G, et al. Pallidal deep brain stimulation in DYT6 dystonia: clinical outcome and predictive factors for motor improvement. J Clin Med. (2019) 8:2163. doi: 10.3390/jcm8122163
49. Oterdoom DLM, Van Egmond ME, Ascencao LC, Van Dijk JMC, Saryyeva A, Beudel M, et al. Reversal of status dystonicus after relocation of pallidal electrodes in DYT6 generalized dystonia. Tremor Other Hyperkinet Mov. (2018) 8:530. doi: 10.5334/tohm.409
50. Mure H, Morigaki R, Koizumi H, Okita S, Kawarai T, Miyamoto R, et al. Deep brain stimulation of the thalamic ventral lateral anterior nucleus for DYT6 dystonia. Stereotact Funct Neurosurg. (2014) 92:393–6. doi: 10.1159/000365577
51. Bruggemann N, Domingo A, Rasche D, Moll CKE, Rosales RL, Jamora RDG, et al. Association of pallidal neurostimulation and outcome predictors with X-linked dystonia parkinsonism. JAMA Neurol. (2019) 76:211–6. doi: 10.1001/jamaneurol.2018.3777
52. Westenberger A, Reyes CJ, Saranza G, Dobricic V, Hanssen H, Domingo A, et al. A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann Neurol. (2019) 85:812–22. doi: 10.1002/ana.25488
53. Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci USA. (2017) 114:E11020–8. doi: 10.1073/pnas.1712526114
54. Martinez-Torres I, Limousin P, Tisch S, Page R, Pinto A, Foltynie T, et al. Early and marked benefit with GPi DBS for lubag syndrome presenting with rapidly progressive life-threatening dystonia. Mov Disord. (2009) 24:1710–2. doi: 10.1002/mds.22656
55. Aguilar JA, Vesagas TS, Jamora RD, Teleg RA, Ledesma L, Rosales RL, et al. The promise of deep brain stimulation in X-linked dystonia parkinsonism. Int J Neurosci. (2011) 121 (Suppl. 1):57–63. doi: 10.3109/00207454.2010.541573
56. Abejero JEE, Jamora RDG, Vesagas TS, Teleg RA, Rosales RL, Anlacan JP, et al. Long-term outcomes of pallidal deep brain stimulation in X-linked dystonia parkinsonism (XDP): up to 84 months follow-up and review of literature. Parkinsonism Relat Disord. (2019) 60:81–6. doi: 10.1016/j.parkreldis.2018.09.022
57. Oyama G, Fernandez HH, Foote KD, Zeilman P, Hwynn N, Jacobson CET, et al. Differential response of dystonia and parkinsonism following globus pallidus internus deep brain stimulation in X-linked dystonia-parkinsonism (Lubag). Stereotact Funct Neurosurg. (2010) 88:329–33. doi: 10.1159/000319961
58. Kilbane C, Witt J, Galifianakis NB, Glass GA, Volz M, Heath S, et al. Long-term outcomes of bilateral pallidal deep brain stimulation for X-linked dystonia and parkinsonism. Stereotact Funct Neurosurg. (2018) 96:320–6. doi: 10.1159/000492823
59. Wilcox RA, Winkler S, Lohmann K, Klein C. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Mov Disord. (2011) 26:2404–8. doi: 10.1002/mds.23866
60. Hersheson J, Mencacci NE, Davis M, Macdonald N, Trabzuni D, Ryten M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol. (2013) 73:546–53. doi: 10.1002/ana.23832
61. Lohmann K, Wilcox RA, Winkler S, Ramirez A, Rakovic A, Park JS, et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol. (2013) 73:537–45. doi: 10.1002/ana.23829
62. Simons C, Wolf NI, Mcneil N, Caldovic L, Devaney JM, Takanohashi A, et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. (2013) 92:767–73. doi: 10.1016/j.ajhg.2013.03.018
63. Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L, et al. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov Disord. (2015) 30:854–8. doi: 10.1002/mds.26196
64. Delorme C, Roze E, Karachi C, Vidailhet M, Hainque E. Whispering dysphonia in TUBB4A-related disorders responsive to bipallidal deep brain stimulation. Eur J Neurol. (2020). doi: 10.1111/ene.14602. [Epub ahead of print].
65. Raymond D, Saunders-Pullman R, Ozelius L. SGCE myoclonus-dystonia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle, WA: University of Washington (1993).
66. Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. (2001) 29:66–9. doi: 10.1038/ng709
67. Peall KJ, Kurian MA, Wardle M, Waite AJ, Hedderly T, Lin JP, et al. SGCE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J Neurol. (2014) 261:2296–304. doi: 10.1007/s00415-014-7488-3
68. Roze E, Lang AE, Vidailhet M. Myoclonus-dystonia: classification, phenomenology, pathogenesis, and treatment. Curr Opin Neurol. (2018) 31:484–90. doi: 10.1097/WCO.0000000000000577
69. Trottenberg T, Meissner W, Kabus C, Arnold G, Funk T, Einhaupl KM, et al. Neurostimulation of the ventral intermediate thalamic nucleus in inherited myoclonus-dystonia syndrome. Mov Disord. (2001) 16:769–71. doi: 10.1002/mds.1119
70. Liu X, Griffin IC, Parkin SG, Miall RC, Rowe JG, Gregory RP, et al. Involvement of the medial pallidum in focal myoclonic dystonia: a clinical and neurophysiological case study. Mov Disord. (2002) 17:346–53. doi: 10.1002/mds.10038
71. Magarinos-Ascone CM, Regidor I, Martinez-Castrillo JC, Gomez-Galan M, Figueiras-Mendez R. Pallidal stimulation relieves myoclonus-dystonia syndrome. J Neurol Neurosurg Psychiatr. (2005) 76:989–91. doi: 10.1136/jnnp.2004.039248
72. Cif L, Valente EM, Hemm S, Coubes C, Vayssiere N, Serrat S, et al. Deep brain stimulation in myoclonus-dystonia syndrome. Mov Disord. (2004) 19:724–7. doi: 10.1002/mds.20030
73. Gruber D, Kuhn AA, Schoenecker T, Kivi A, Trottenberg T, Hoffmann KT, et al. Pallidal and thalamic deep brain stimulation in myoclonus-dystonia. Mov Disord. (2010) 25:1733–43. doi: 10.1002/mds.23312
74. Kurtis MM, San Luciano M, Yu Q, Goodman RR, Ford B, Raymond D, et al. Clinical and neurophysiological improvement of SGCE myoclonus-dystonia with GPi deep brain stimulation. Clin Neurol Neurosurg. (2010) 112:149–52. doi: 10.1016/j.clineuro.2009.10.001
75. Azoulay-Zyss J, Roze E, Welter ML, Navarro S, Yelnik J, Clot F, et al. Bilateral deep brain stimulation of the pallidum for myoclonus-dystonia due to epsilon-sarcoglycan mutations: a pilot study. Arch Neurol. (2011) 68:94–8. doi: 10.1001/archneurol.2010.338
76. Kim JH, Na YC, Lee WH, Chang WS, Jung HH, Chang JW. Bilateral globus pallidus interna deep-brain stimulation in a patient with myoclonus-dystonia: a case report. Neuromodulation. (2014) 17:724–8. doi: 10.1111/ner.12162
77. Fernandez-Pajarin G, Sesar A, Relova JL, Ares B, Jimenez-Martin I, Blanco-Arias P, et al. Bilateral pallidal deep brain stimulation in myoclonus-dystonia: our experience in three cases and their follow-up. Acta Neurochir. (2016) 158:2023–8. doi: 10.1007/s00701-016-2904-3
78. Rocha H, Linhares P, Chamadoira C, Rosas MJ, Vaz R. Early deep brain stimulation in patients with myoclonus-dystonia syndrome. J Clin Neurosci. (2016) 27:17–21. doi: 10.1016/j.jocn.2015.08.034
79. Kosutzka Z, Tisch S, Bonnet C, Ruiz M, Hainque E, Welter ML, et al. Long-term GPi-DBS improves motor features in myoclonus-dystonia and enhances social adjustment. Mov Disord. (2019) 34:87–94. doi: 10.1002/mds.27474
80. Wang X, Yu X. Deep brain stimulation for myoclonus dystonia syndrome: a meta-analysis with individual patient data. Neurosurg Rev. (2020). doi: 10.1007/s10143-019-01233-x. [Epub ahead of print].
81. Zhang YQ, Wang JW, Wang YP, Zhang XH, Li JP. Thalamus stimulation for myoclonus dystonia syndrome: five cases and long-term follow-up. World Neurosurg. (2019) 122:e933–9. doi: 10.1016/j.wneu.2018.10.177
82. Rughani AI, Lozano AM. Surgical treatment of myoclonus dystonia syndrome. Mov Disord. (2013) 28:282–7. doi: 10.1002/mds.25326
83. Ramdhani RA, Frucht SJ, Behnegar A, Kopell BH. Improvement of isolated myoclonus phenotype in myoclonus dystonia after pallidal deep brain stimulation. Tremor Other Hyperkinet Mov. (2016) 6:369. doi: 10.5334/tohm.290
84. Besa Lehmann V, Rosenbaum M, Bulman DE, Read T, Verhagen L. A case report of myoclonus-dystonia with isolated myoclonus phenotype and novel mutation successfully treated with deep brain stimulation. Neurol Ther. (2020) 9:187–91. doi: 10.1007/s40120-020-00186-4
85. Contarino MF, Foncke EM, Cath DC, Schuurman PR, Speelman JD, Tijssen MA. Effect of pallidal deep brain stimulation on psychiatric symptoms in myoclonus-dystonia due to epsilon-sarcoglycan mutations. Arch Neurol. (2011) 68:1087–8. doi: 10.1001/archneurol.2011.180
86. Brashear A, Sweadner KJ, Cook JF, Swoboda KJ, Ozelius L. ATP1A3-related neurologic disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle, WA: University of Washington (1993).
87. Deutschlander A, Asmus F, Gasser T, Steude U, Botzel K. Sporadic rapid-onset dystonia-parkinsonism syndrome: failure of bilateral pallidal stimulation. Mov Disord. (2005) 20:254–7. doi: 10.1002/mds.20296
88. Kamm C, Fogel W, Wachter T, Schweitzer K, Berg D, Kruger R, et al. Novel ATP1A3 mutation in a sporadic RDP patient with minimal benefit from deep brain stimulation. Neurology. (2008) 70:1501–3. doi: 10.1212/01.wnl.0000310431.41036.e0
89. Brucke C, Horn A, Huppke P, Kupsch A, Schneider GH, Kuhn AA. Failure of pallidal deep brain stimulation in a case of rapid-onset dystonia parkinsonism (DYT12). Mov Disord Clin Pract. (2015) 2:76–8. doi: 10.1002/mdc3.12124
90. Albanese A, Di Giovanni M, Amami P, Lalli S. Failure of pallidal deep brain stimulation in DYT12-ATP1A3 dystonia. Parkinsonism Relat Disord. (2017) 45:99–100. doi: 10.1016/j.parkreldis.2017.09.008
91. Zuniga-Ramirez C, Kramis-Hollands M, Mercado-Pimentel R, Gonzalez-Usigli HA, Saenz-Farret M, Soto-Escageda A, et al. Generalized dystonia and paroxysmal dystonic attacks due to a novel ATP1A3 variant. Tremor Other Hyperkinet Mov. (2019) 9. doi: 10.5334/tohm.490
92. Camargos S, Scholz S, Simon-Sanchez J, Paisan-Ruiz C, Lewis P, Hernandez D, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. (2008) 7:207–15. doi: 10.1016/S1474-4422(08)70022-X
93. Casagrande SCB, Listik C, Coelho DB, Limongi JCP, Teixeira LA, Teixeira MJ, et al. Deep brain stimulation in patients with isolated generalized dystonia caused by PRKRA mutation. Mov Disord Clin Pract. (2019) 6:616–8. doi: 10.1002/mdc3.12811
94. Quadri M, Olgiati S, Sensi M, Gualandi F, Groppo E, Rispoli V, et al. PRKRA mutation causing early-onset generalized dystonia-parkinsonism (DYT16) in an Italian family. Mov Disord. (2016) 31:765–7. doi: 10.1002/mds.26583
95. Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. (2012) 91:1041–50. doi: 10.1016/j.ajhg.2012.10.024
96. Zech M, Boesch S, Jochim A, Weber S, Meindl T, Schormair B, et al. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Mov Disord. (2017) 32:549–59. doi: 10.1002/mds.26808
97. Lasky L, Bliss L, Sidiropoulos C. Successful pallidal deep brain stimulation treatment in a case of generalized dystonia due to a novel ANO3 mutation. Case Rep Neurol Med. (2019) 2019:3154653. doi: 10.1155/2019/3154653
98. Li S, Wang L, Yang Y, Ma J, Wan X. ANO3 mutations in chinese dystonia: a genetic screening study using next-generation sequencing. Front Neurol. (2019) 10:1351. doi: 10.3389/fneur.2019.01351
99. Olschewski L, Jesus S, Kim HJ, Tunc S, Lons S, Junker J, et al. Role of ANO3 mutations in dystonia: a large-scale mutational screening study. Parkinsonism Relat Disord. (2019) 62:196–200. doi: 10.1016/j.parkreldis.2018.12.030
100. Yoo D, Kim HJ, Chae JH, Paek SH, Jeon B. Successful pallidal deep brain stimulation in a patient with childhood-onset generalized dystonia with ANO3 mutation. J Mov Disord. (2019) 12:190–1. doi: 10.14802/jmd.19016
101. Charlesworth G, Bhatia KP, Wood NW. The genetics of dystonia: new twists in an old tale. Brain. (2013) 136:2017–37. doi: 10.1093/brain/awt138
102. Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet. (2013) 45:88–92. doi: 10.1038/ng.2496
103. Kumar KR, Lohmann K, Masuho I, Miyamoto R, Ferbert A, Lohnau T, et al. Mutations in GNAL: a novel cause of craniocervical dystonia. JAMA Neurol. (2014) 71:490–4. doi: 10.1001/jamaneurol.2013.4677
104. Ziegan J, Wittstock M, Westenberger A, Dobricic V, Wolters A, Benecke R, et al. Novel GNAL mutations in two German patients with sporadic dystonia. Mov Disord. (2014) 29:1833–4. doi: 10.1002/mds.26066
105. Carecchio M, Panteghini C, Reale C, Barzaghi C, Monti V, Romito L, et al. Novel GNAL mutation with intra-familial clinical heterogeneity: expanding the phenotype. Parkinsonism Relat Disord. (2016) 23:66–71. doi: 10.1016/j.parkreldis.2015.12.012
106. Ahn JH, Kim AR, Kim NKD, Park WY, Kim JS, Kim M, et al. The effect of globus pallidus interna deep brain stimulation on a dystonia patient with the GNAL mutation compared to patients with DYT1 and DYT6. J Mov Disord. (2019) 12:120–4. doi: 10.14802/jmd.19006
107. Pandey S, Sankhla CS, Ramprasad VL, Geetha TS. Novel GNAL mutation in an Indian patient with generalized dystonia and response to deep brain stimulation. Parkinsonism Relat Disord. (2019) 62:246–7. doi: 10.1016/j.parkreldis.2019.01.011
108. Sarva H, Trosch R, Kiss ZHT, Furtado S, Luciano MS, Glickman A, et al. Deep brain stimulation in isolated dystonia with a GNAL mutation. Mov Disord. (2019) 34:301–3. doi: 10.1002/mds.27585
109. Zech M, Boesch S, Maier EM, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am J Hum Genet. (2016) 99:1377–87. doi: 10.1016/j.ajhg.2016.10.010
110. Meyer E, Carss KJ, Rankin J, Nichols JM, Grozeva D, Joseph AP, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. (2017) 49:223–37. doi: 10.1038/ng.3740
111. Cif L, Demailly D, Lin JP, Barwick KE, Sa M, Abela L, et al. KMT2B-related disorders: expansion of the phenotypic spectrum and long-term efficacy of deep brain stimulation. Brain. (2020) 143:3242–61. doi: 10.1093/brain/awaa304
112. Kawarai T, Miyamoto R, Nakagawa E, Koichihara R, Sakamoto T, Mure H, et al. Phenotype variability and allelic heterogeneity in KMT2B-associated disease. Parkinsonism Relat Disord. (2018) 52:55–61. doi: 10.1016/j.parkreldis.2018.03.022
113. Kumar KR, Davis RL, Tchan MC, Wali GM, Mahant N, Ng K, et al. Whole genome sequencing for the genetic diagnosis of heterogenous dystonia phenotypes. Parkinsonism Relat Disord. (2019) 69:111–8. doi: 10.1016/j.parkreldis.2019.11.004
114. Carecchio M, Invernizzi F, Gonzalez-Latapi P, Panteghini C, Zorzi G, Romito L, et al. Frequency and phenotypic spectrum of KMT2B dystonia in childhood: a single-center cohort study. Mov Disord. (2019) 34:1516–27. doi: 10.1002/mds.27771
115. Dafsari HS, Sprute R, Wunderlich G, Daimaguler HS, Karaca E, Contreras A, et al. Novel mutations in KMT2B offer pathophysiological insights into childhood-onset progressive dystonia. J Hum Genet. (2019) 64:803–13. doi: 10.1038/s10038-019-0625-1
116. Li XY, Dai LF, Wan XH, Guo Y, Dai Y, Li SL, et al. Clinical phenotypes, genotypes and treatment in Chinese dystonia patients with KMT2B variants. Parkinsonism Relat Disord. (2020) 77:76–82. doi: 10.1016/j.parkreldis.2020.06.002
117. Hisama FM, Friedman J, Raskind WH, Bird TD. ADCY5 dyskinesia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle, WA: University of Washington (1993).
118. Wali GM, Wali G, Kumar KR, Sue CM. Long-term follow-up and evolution of ADCY5—from a ballistic to dystonic phenotype. Mov Disord Clin Pract. (2020) 7:985–6. doi: 10.1002/mdc3.13069
119. De Almeida Marcelino AL, Mainka T, Krause P, Poewe W, Ganos C, Kuhn AA. Deep brain stimulation reduces (nocturnal) dyskinetic exacerbations in patients with ADCY5 mutation: a case series. J Neurol. (2020) 267:3624–31. doi: 10.1007/s00415-020-09871-8
120. Danti FR, Galosi S, Romani M, Montomoli M, Carss KJ, Raymond FL, et al. GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol Genet. (2017) 3:e143. doi: 10.1212/NXG.0000000000000143
121. Kulkarni N, Tang S, Bhardwaj R, Bernes S, Grebe TA. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol. (2016) 31:211–4. doi: 10.1177/0883073815587945
122. Yilmaz S, Turhan T, Ceylaner S, Gokben S, Tekgul H, Serdaroglu G. Excellent response to deep brain stimulation in a young girl with GNAO1-related progressive choreoathetosis. Childs Nerv Syst. (2016) 32:1567–8. doi: 10.1007/s00381-016-3139-6
123. Benato A, Carecchio M, Burlina A, Paoloni F, Sartori S, Nosadini M, et al. Long-term effect of subthalamic and pallidal deep brain stimulation for status dystonicus in children with methylmalonic acidemia and GNAO1 mutation. J Neural Transm. (2019) 126:739–57. doi: 10.1007/s00702-019-02010-2
124. Yamashita Y, Ogawa T, Ogaki K, Kamo H, Sukigara T, Kitahara E, et al. Neuroimaging evaluation and successful treatment by using directional deep brain stimulation and levodopa in a patient with GNAO1-associated movement disorder: a case report. J Neurol Sci. (2020) 411:116710. doi: 10.1016/j.jns.2020.116710
125. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in hallervorden-spatz syndrome. Nat Genet. (2001) 28:345–9. doi: 10.1038/ng572
126. Gregory A, Hayflick SJ. Pantothenate kinase-associated neurodegeneration. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle, WA: University of Washington (1993).
127. Umemura A, Jaggi JL, Dolinskas CA, Stern MB, Baltuch GH. Pallidal deep brain stimulation for longstanding severe generalized dystonia in hallervorden-spatz syndrome. Case Rep J Neurosurg. (2004) 100:706–9. doi: 10.3171/jns.2004.100.4.0706
128. Krause M, Fogel W, Tronnier V, Pohle S, Hortnagel K, Thyen U, et al. Long-term benefit to pallidal deep brain stimulation in a case of dystonia secondary to pantothenate kinase-associated neurodegeneration. Mov Disord. (2006) 21:2255–7. doi: 10.1002/mds.21166
129. Mikati MA, Yehya A, Darwish H, Karam P, Comair Y. Deep brain stimulation as a mode of treatment of early onset pantothenate kinase-associated neurodegeneration. Eur J Paediatr Neurol. (2009) 13:61–4. doi: 10.1016/j.ejpn.2008.01.006
130. Grandas F, Fernandez-Carballal C, Guzman-De-Villoria J, Ampuero I. Treatment of a dystonic storm with pallidal stimulation in a patient with PANK2 mutation. Mov Disord. (2011) 26:921–2. doi: 10.1002/mds.23586
131. Tanrikulu B, Ozen A, Gunal DI, Turkdogan D, Bayrakli F, Bayri Y, et al. Deep brain stimulation as treatment for dystonic storm in pantothenate kinase-associated neurodegeneration syndrome: case report of a patient with homozygous C.628 2 T > G mutation of the PANK2 gene. Acta Neurochir. (2015) 157:1513–16. doi: 10.1007/s00701-015-2514-5
132. Sharma MC, Aggarwal N, Bihari M, Goyal V, Gaikwed S, Vaishya S, et al. Hallervorden spatz disease: MR and pathological findings of a rare case. Neurol India. (2005) 53:102–4. doi: 10.4103/0028-3886.15072
133. Lim BC, Ki CS, Cho A, Hwang H, Kim KJ, Hwang YS, et al. Pantothenate kinase-associated neurodegeneration in Korea: recurrent R440P mutation in PANK2 and outcome of deep brain stimulation. Eur J Neurol. (2012) 19:556–61. doi: 10.1111/j.1468-1331.2011.03589.x
134. Daida K, Nishioka K, Shimo Y, Umemura A, Yoshino H, Hattori N. Deep brain stimulation shows high efficacy in two patients with GCH1 variants. Parkinsonism Relat Disord. (2019) 65:277–8. doi: 10.1016/j.parkreldis.2019.06.002
135. Jones HF, Morales-Briceno H, Barwick K, Lewis J, Sanchis-Juan A, Raymond FL, et al. Myoclonus-dystonia caused by GNB1 mutation responsive to deep brain stimulation. Mov Disord. (2019) 34:1079–80. doi: 10.1002/mds.27708
136. Mencacci NE, Rubio-Agusti I, Zdebik A, Asmus F, Ludtmann MH, Ryten M, et al. A missense mutation in KCTD17 causes autosomal dominant myoclonus-dystonia. Am J Hum Genet. (2015) 96:938–47. doi: 10.1016/j.ajhg.2015.04.008
137. Marce-Grau A, Correa M, Vanegas MI, Munoz-Ruiz T, Ferrer-Aparicio S, Baide H, et al. Childhood onset progressive myoclonic dystonia due to a de novo KCTD17 splicing mutation. Parkinsonism Relat Disord. (2019) 61:7–9. doi: 10.1016/j.parkreldis.2019.01.004
138. Skogseid IM, Rosby O, Konglund A, Connelly JP, Nedregaard B, Jablonski GE, et al. Dystonia-deafness syndrome caused by ACTB p.Arg183Trp heterozygosity shows striatal dopaminergic dysfunction and response to pallidal stimulation. J Neurodev Disord. (2018) 10:17. doi: 10.1186/s11689-018-9235-z
139. Wang JW, Li JP, Wang YP, Zhang XH, Zhang YQ. Deep brain stimulation for myoclonus-dystonia syndrome with double mutations in DYT1 and DYT11. Sci Rep. (2017) 7:41042. doi: 10.1038/srep41042
140. Steel D, Zech M, Zhao C, Barwick KES, Burke D, Demailly D, et al. Loss-of-function variants in HOPS complex genes VPS16 and VPS41 cause early onset dystonia associated with lysosomal abnormalities. Ann Neurol. (2020) 88:867–77. doi: 10.1002/ana.25879
141. Phua CS, Kumar KR, Levy S. Clinical characteristics and diagnostic clues to neurometabolic causes of dystonia. J Neurol Sci. (2020) 419:117167. doi: 10.1016/j.jns.2020.117167
142. Roze E, Navarro S, Cornu P, Welter ML, Vidailhet M. Deep brain stimulation of the globus pallidus for generalized dystonia in GM1 type 3 gangliosidosis: technical case report. Neurosurgery. (2006) 59:E1340. doi: 10.1227/01.NEU.0000245620.24603.1B
143. Artusi CA, Dwivedi A, Romagnolo A, Bortolani S, Marsili L, Imbalzano G, et al. Differential response to pallidal deep brain stimulation among monogenic dystonias: systematic review and meta-analysis. J Neurol Neurosurg Psychiatr. (2020) 91:426–33. doi: 10.1136/jnnp-2019-322169
144. Tsuboi T, Wong JK, Okun MS, Ramirez-Zamora A. Quality of life outcomes after deep brain stimulation in dystonia: a systematic review. Parkinsonism Relat Disord. (2020) 70:82–93. doi: 10.1016/j.parkreldis.2019.11.016
145. Van Coller R, Bardien S, Neethling A, Carr J, Schutte C. A South African family with myoclonus-dystonia syndrome with a novel mutation in the SGCE gene responding to deep brain stimulation. Parkinsonism Relat Disord. (2019) 68:63–4. doi: 10.1016/j.parkreldis.2019.10.001
Keywords: dystonia, deep brain stimulation, globus pallidus internus, pallidal, gene, mutation, monogenic
Citation: Tisch S and Kumar KR (2021) Pallidal Deep Brain Stimulation for Monogenic Dystonia: The Effect of Gene on Outcome. Front. Neurol. 11:630391. doi: 10.3389/fneur.2020.630391
Received: 17 November 2020; Accepted: 09 December 2020;
Published: 08 January 2021.
Edited by:
Anna Sadnicka, University of London, United KingdomReviewed by:
Francesca Morgante, University of Messina, ItalyCopyright © 2021 Tisch and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Tisch, c3RlcGhlbi50aXNjaEBzdmhhLm9yZy5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.