 Tommaso Bocci
Tommaso Bocci Davide Baloscio2
Davide Baloscio2 Roberta Ferrucci
Roberta Ferrucci Ferdinando Sartucci
Ferdinando Sartucci Alberto Priori
Alberto Priori- 1“Aldo Ravelli” Center for Neurotechnology and Experimental Brain Therapeutics, Department of Health Sciences, University of Milan & Azienda Socio-Sanitaria Territoriale Santi Paolo e Carlo, Milan, Italy
- 2Section of Neurophysiopathology, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
Introduction: In recent years, a growing body of literature has investigated the use of non-invasive brain stimulation (NIBS) techniques as a putative treatment in Huntington's Disease (HD). Our aim was to evaluate the effects of cerebellar transcranial Direct Current Simulation (ctDCS) on the motor outcome in patients affected by HD, encompassing at the same time the current knowledge about the effects of NIBS both on motor and non-motor dysfunctions in HD.
Materials and Methods: Four patients (two females) were enrolled and underwent ctDCS (both anodal or sham, elapsed by at least 3 months: 2.0 mA, 20 min per day, 5 days a week). Clinical scores were assessed by using the Unified Huntington's Disease Rating Scale – part I (UHDRS-I), immediately before ctDCS (T0), at the end of the 5-days treatment (T1) and 4 weeks later (T2).
Results: Anodal ctDCS improved motor scores compared to baseline (p = 0.0046), whereas sham stimulation left them unchanged (p = 0.33, Friedman test). In particular, following anodal ctDCS, UHDRS-I score significantly improved, especially regarding the subitem “dystonia,” both at T1 and T2 compared to sham condition (p < 0.05; Wilcoxon matched-pairs signed test).
Conclusions: ctDCS improved motor scores in HD, with effects lasting for about 4 weeks after tDCS completion. This is the first study discussing the putative role of cerebellar non-invasive simulation for the treatment of HD.
Introduction
Huntington's disease (HD) is a neurodegenerative, progressive and fatal disorder, clinically characterized by cognitive impairment, behavioral dysfunctions and hyperkinetic movements. Its prevalence is higher than previously thought, geographically variable and increasing worldwide (1, 2). The disease is caused by an expanded CAG trinucleotide repeat (of variable length) in HTT, the gene encoding the protein huntingtin; it is inherited in an autosomal dominant manner, with age-dependent penetrance, where longer CAG repeats predicting earlier onset (3). HD belongs to the so-called “poly-glutamine disorders,” a heterogeneous group of diseases caused by an abnormal expansion of a glutamine encoding CAG repeat in the affected genes. To date, disease modifying therapies are not yet available and the pharmacological treatment is only symptomatic. Also non-invasive brain stimulation techniques (NIBS), as repetitive Transcranial Magnetic Stimulation (rTMS) and transcranial Direct Current Stimulation (tDCS), have reported conflicting results in the treatment of severe choreic and dystonic movements, failing to provide a significant, possibly long-lasting, clinical improvement in HD patients (4–8).
The multisystem character of HD is emphasized by a distribution pattern of neurodegeneration which includes not only the striatum, but also the cerebral neo-and allocortex, thalamus, pallidum, brainstem and the cerebellum, thus sharing more similarities with polyglutamine spinocerebellar ataxias than previously described (9). A possible clinical and pathogenetic overlap has been recently supported also by neurophysiological findings (10, 11).
Recently, a growing attention has been focused on the cerebellar involvement in the pathogenesis of HD (12–14). Clinically, several symptoms of HD could be attributed to cerebellar damage, comprising dysarthria, ataxia, gait disturbances and abnormal oculomotor function. Moreover, animal studies have strengthened the possibility of a key cerebellar involvement in HD models (15–17). Magnetic resonance imaging (MRI) studies have further corroborated these data in humans, showing that aberrant cerebellar diffusion and smaller cerebellar volumes are associated both with a worst motor performance and increased psychiatric symptoms at early stages (12).
To date, only few papers have investigated the role of non-invasive brain stimulation in HD and no studies targeted the cerebellum. In this pilot trial, our purpose was to assess the possibility to improve motor symptoms with cerebellar transcranial Direct Current Stimulation (ctDCS) in clinical manifest HD, encompassing the current literature about the use of cerebellar NIBS for the treatment of HD. Patients underwent anodal and sham (placebo) ctDCS; although there are different factors driving the direction of ctDCS after-effects, as the electrode size and the montage, anodal stimulation is known to exert an overall excitatory effect on cerebellar functions, both motor and non-motor (18–21).
Materials and Methods
Patients
Four HD patients were enrolled in a timeline ranging from April 2016 to November 2018. They had undergone genetic testing, which was diagnostic in all (CAG number ⩾ 40). The mean duration of symptoms was about 2 year (23.3 ± 7.8 months). Inclusion criteria for the early manifest HD patients were a CAG repeat ⩾ 40, with a UHDRS motor score ⩾ 5, and a total functional capacity (TFC) score ⩾ 7. Demographic and clinical data are summarized in Table 1. The pharmacological therapy remained the same during the whole experimental protocol.
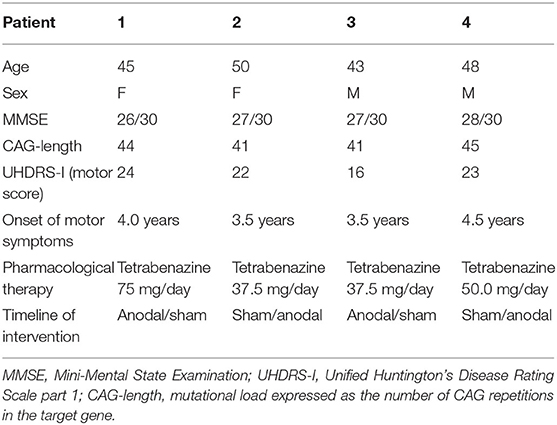
Table 1. Demographic and clinical features of HD patients.
The patients were enrolled and the experimental procedures performed at the Section of Neurophysiopathology, University of Pisa.
Experimental Protocol
In a crossover, double-blind, sham-controlled design, each patient underwent sham and anodal ctDCS. All patients carried out the two experimental conditions, held at least 3 months apart to avoid carry-over effects. Each session, either anodal or sham, lasted 5 days a week (Monday to Friday, 30 min a day).
Clinical scores were assessed at baseline (T0), immediately at the end of the stimulation week (T1), 4 weeks (T2) later.
Patients were enrolled by a physician with expertise in movement disorder medicine (F.S.); clinical scores were administered by a neurologist (T.B.), blinded to the ctDCS condition. A third neurologist with expertise in movement disorders (A.P.) served as blinded video-rater and confirmed the motor outcome.
Informed consent was obtained from all individual participants included in the study. The study was approved by the local ethical committee at the University of Pisa (formally named “Comitato Etico di Area Vasta Nord Ovest della Toscana”), in accordance with the tenets of Helsinki.
Transcranial Direct Current Stimulation
Cerebellar transcutaneous direct current stimulation (ctDCS) was applied using a battery-driven constant current stimulator (HDCStim, Newronika®, Italy) and a pair of electrode in two saline-soaked synthetic sponges with a surface area of 35 cm2. Direct current was transcranially applied for 20 min with an intensity of 2.0 mA and constant current flow was measured by an ampere meter (current density ~0.08 mA/cm2). At the offset of tDCS, the current was decreased in a ramp-like manner, a method shown to achieve a high level of blinding between sessions (18). For anodal ctDCS, the anode was applied on the median line, 2 cm below the inion, with lateral borders about 1 cm medially to the mastoid apophysis, and the cathode over the right shoulder (19, 21–25). For sham ctDCS, the current was turned on for 5 s and then turned off in a ramp-shaped fashion, thus inducing skin sensations similar to those produced by real ctDCS.
We stimulated the cerebellum bilaterally, as previous studies have shown that varying the position of the active electrode with ~1 cm induced negligible changes in the electrical field distribution (26).
Patients were blinded to the tsDCS protocol and did not discriminate between anodal and sham condition. In order to report possible adverse effects, the questionnaire developed by Brunoni et al. was administered to each patient (27).
Clinical Outcome and Statistical Analysis
The clinical outcome was assessed by evaluating the Unified Huntington Disease Rating Scale-part I (UHDRS-I) score, particularly its “dystonia” (cumulative values for trunk and extremities) and “chorea” (face, mouth, trunk and extremities) subscores, at the different time points (T0, T1, and T2) (28). ctDCS-induced changes in UHDRS-I score and related sub-items were assessed with a Friedman test (non-parametric analysis on paired data) with the main factor “time” (three levels: T0, T1, and T2). In order to disclose significant changes at each time point between anodal and sham ctDCS (T0, T1, T2,), a Wilcoxon matched-pairs signed test was then applied. Statistical significance was set at P < 0.05. The data were analyzed using SPSS v. 21.0 for Windows (SPSS Inc.). Raw data are shown as mean values ± 1 standard error (S.E.).
Results
No patient reported adverse effect during stimulation or the follow-up period.
Figures 1A,B shows changes in UHDRS motor score (UHDRS-I) following either sham (gray columns) or anodal ctDCS (red columns).
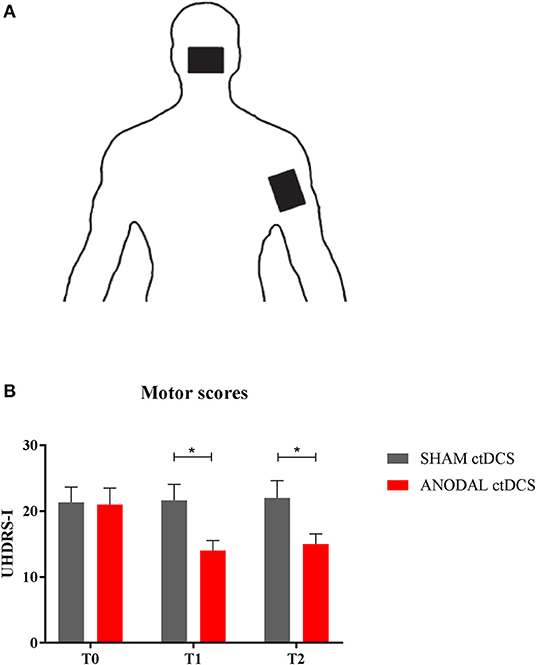
Figure 1. (A) ctDCS montage. The active electrode was placed over the cerebellar area, while the return electrode was positioned over the right shoulder. (B) Motor score changes. The histogram shows UHDRS-I values at different time intervals (T0, T1, and T2), following either sham (gray columns) or anodal ctDCS (red columns). Note that real (anodal) stimulation significantly reduced motor impairment, both at T1 and T2. Data are shown as mean values ± 1 S.E. (*p < 0.05).
Overall, compared to baseline (T0), anodal ctDCS improved motor scores (UHDRS-I: T0 = 22.0 ± 1.9; T1 = 14.5 ± 1.2; T2 = 15.5 ± 1.1: p = 0.0046), whereas sham stimulation left them unchanged (T0 = 21.7 ± 1.7; T1 = 22.1 ± 1.7; T2 = 22.5 ± 2.6: p = 0.33, Friedman test). In particular, following anodal polarization, UHDRS-I score significantly improved both at T1 and T2 compared to sham tDCS (p = 0.46 and p = 0.48, respectively; Wilcoxon matched-pairs signed test).
As regards UHDRS-1 subscores, anodal ctDCS significantly improved dystonia over time (T0 = 8.0 ± 0.7; T1 = 3.7 ± 0.5; T2 = 4.5 ± 0.4: p = 0.037), whereas sham stimulation had no significant effects (T0 = 8.0 ± 0.9; T1 = 7.5 ± 0.9; T2 = 8.3 ± 0.7: p = 0.29, Friedman test; Figure 2). In particular, following anodal polarization, dystonia was reduced both at T1 and T2 compared to sham tDCS (p = 0.46 and p = 0.48, respectively; Wilcoxon matched-pairs signed test).
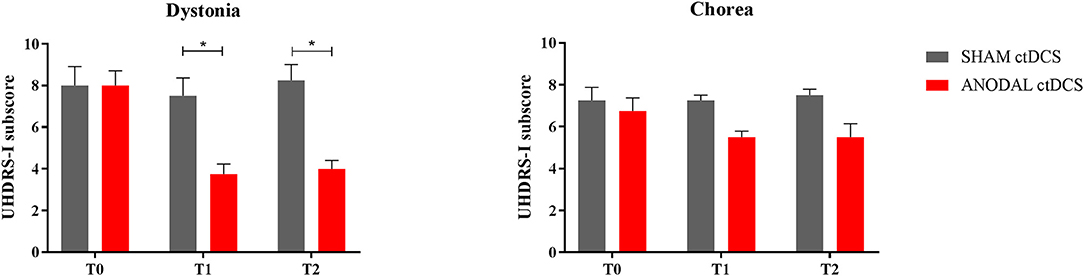
Figure 2. Motor scores changes in the subitems “dystonia” (left) and “chorea” (right). The histogram shows UHDRS-I subscores at different time intervals (T0, T1, and T2), following either sham (gray columns) or anodal ctDCS (red columns). Note that real (anodal) stimulation significantly reduced dystonia, with a smaller effect on choreic movements. Data are shown as mean values ± 1 S.E. (*p < 0.05).
Conversely, both active and sham ctDCS left chorea scores unchanged over time, whereas a trend to improve was shown following the anodal stimulation (anodal ctDCS: T0 = 6.7 ± 0.6; T1 = 5.5 ± 0.3; T2 = 5.5 ± 0.6: p = 0.074; sham ctDCS: T0 = 7.3 ± 0.6; T1 = 7.3 ± 0.3; T2 = 7.5 ± 0.3: p = 0.8).
Discussion and a Reappraisal of the Current Literature
Cerebellar direct current polarization improved motor scores in HD patients, especially dystonia, with the effects lasting for 1 month after the end of stimulation. To the best of our knowledge, this is the first report showing a significant effect of cerebellar stimulation on motor scores in HD. Only few papers have evaluated NIBS techniques for the treatment of both motor and non-motor symptoms in HD patients (Table 2). Among these, Brusa and colleagues found an improvement of choreic movements by 1 Hz rTMS, when applied over the supplementary motor area (SMA) (6); conversely, Shukla and co-workers showed that bilateral low-frequency rTMS over the SMA did not reduce choreiform movements in severe HD (5). A third, single-case study has recently proposed the use of “deep” TMS (dTMS, at 1 Hz and 120% of the motor threshold) for the treatment of depression and anxiety in HD (31). To support beneficial effects induced by TMS on HD patients, some Authors have found that Transcranial magnetic stimulation attenuates cell loss, oxidative and nitrosative damage in the striatum of a murine HD model (29). More recently, it has been demonstrated that anodal tDCS enhances working memory in HD, when applied over the left dorsolateral prefrontal cortex (DLPFC), and this improvement is greatest in patients with more severe motor symptoms, thus suggesting that motor scores may help identify patients who are most likely to benefit from tDCS (4).
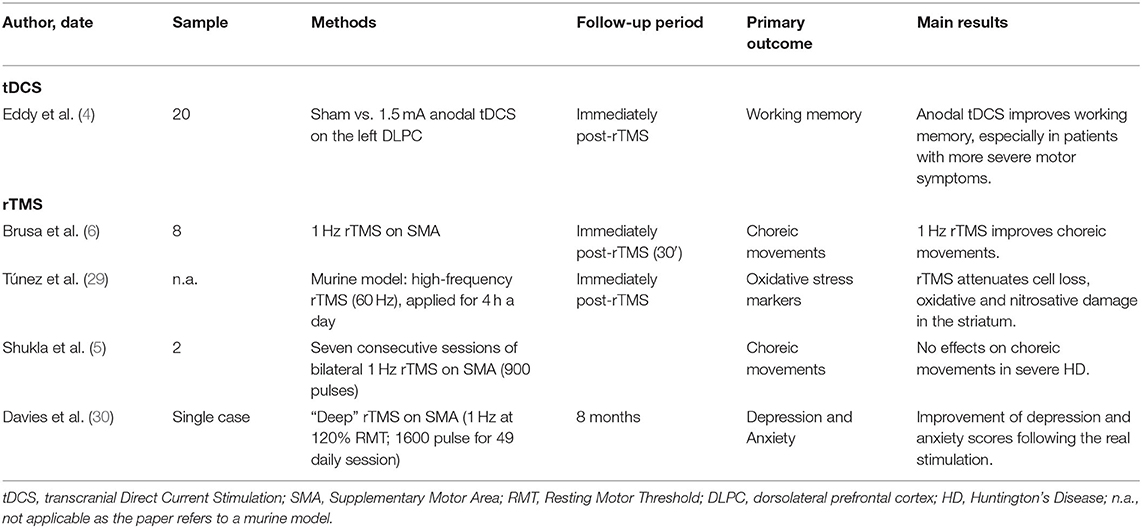
Table 2. Non Invasive Brain Stimulation (NIBS) for the treatment of HD: current literature.
The finding that ctDCS significantly improved specific subscores than others fits with recent data showing a key cerebellar involvement in the pathophysiology of dystonia (32–36), especially in inherited disorders (37–41). In particular, the cerebellum is likely engaged in a hyper-direct, short-delay cerebello-talamo-striatal pathway, projecting from the dentate nucleus to the striatum and the external segment of the globus pallidus (GPe), via the intralaminar nuclei of the thalamus (42, 43). Under pathological conditions, this pathway can bypass the cerebello-thalamo-cortical stream and drive abnormal activity within the basal ganglia, thus inducing dystonic movements (43).
Recently, an increasing body of literature has specifically investigated the relationship between HD pathogenesis and the cerebellum.
Both autoptic and MRI studies have suggested that cerebellar abnormalities are present in HD at early stages, mainly involving the gray matter of the deep cerebellar nuclei, with a relative preservation of the cerebellar cortex (9, 44–47). The cerebellar involvement is particularly marked, both from a clinical and a neuropathological perspective, especially in the Juvenile Huntington's Disease (JHD), a rare HD variant arising before 20 years age and characterized by a variable presentation, including myoclonus, seizures, Parkinsonism, and cognitive decline (48–51).
Nonetheless, how these abnormalities play a contributing role in the pathophysiology of the disease is still a matter of debate. From a neuropathological point of view, HD is characterized by a bilateral and symmetrical neuronal loss in the neostriatum, mainly caused by the extensive demise of GABA-ergic medium spiny stellate projections neurons (52–57). The lack of any inhibitory cerebellar effect in patients with dystonia may contribute to the loss of M1 inhibition and the development of incorrect motor programs and maladaptive behaviors (58, 59). Anodal ctDCS may ultimately interfere with cerebello-thalamo-cortical loops, possibly restoring the physiological inhibition exerted by cerebellar nuclei on cortical processing.
Moreover, as described above, the cerebellum itself directly interferes with striatal networks through a disynaptic pathway leading to the intralaminar nuclei of the thalamus and to the dorsolateral putamen (42).
Limitations
Our study has some limitations. First, the small sample, given that our study has been designed as a pilot research; nonetheless, other studies have enrolled a similar number of patients, probably due to the fact that HD is a quite rare disease. Second, other clinical measures (e.g., quality of life and depression) were not assessed and included as secondary outcomes. Third, a longer observation period should be used in further studies, in order to assess how long the beneficial effects of cerebellar tDCS persist. As a pilot trial, further studies are needed.
Conclusions
This is the first pilot trial about the use of cerebellar tDCS for the treatment of motor symptoms in HD. Further and large-scale studies are needed, possibly including non-motor features as secondary outcomes. Moreover, the impact of a combined cerebello-cortical stimulation may be of interest, in order to improve therapeutic effects on motor scores.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Università di Pisa. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
TB and FS: concept and design. TB and DB: acquisition of data. TB, DB, and RF: acquisition and interpretation of data. TB, RF, AP, and FS: drafting of the manuscript. AP: critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. (2015) 1:15005. doi: 10.1038/nrdp.2015.5
2. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. (2011) 10:83–98. doi: 10.1016/S1474-4422(10)70245-3
3. Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, International Huntington's Disease Collaborative G. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. (2004) 65:267–77. doi: 10.1111/j.1399-0004.2004.00241.x
4. Eddy CM, Shapiro K, Clouter A, Hansen PC, Rickards HE. Transcranial direct current stimulation can enhance working memory in Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatr. (2017) 77:75–82. doi: 10.1016/j.pnpbp.2017.04.002
5. Shukla A, Jayarajan RN, Muralidharan K, Jain S. Repetitive transcranial magnetic stimulation not beneficial in severe choreiform movements of Huntington disease. J ECT. (2013) 29:e16–7. doi: 10.1097/YCT.0b013e3182711dfc
6. Brusa L, Versace V, Koch G, Bernardi G, Iani C, Stanzione P, et al. Improvement of choreic movements by 1 Hz repetitive transcranial magnetic stimulation in Huntington's disease patients. Ann Neurol. (2005) 58:655–6. doi: 10.1002/ana.20613
7. Latorre A, Rocchi L, Berardelli A, Bhatia KP, Rothwell JC. The interindividual variability of transcranial magnetic stimulation effects: implications for diagnostic use in movement disorders. Mov Disord. (2019) 34:936–49. doi: 10.1002/mds.27736
8. Latorre A, Rocchi L, Berardelli A, Bhatia KP, Rothwell JC. The use of transcranial magnetic stimulation as a treatment for movement disorders: a critical review. Mov Disord. (2019) 34:769–82. doi: 10.1002/mds.27705
9. Rub U, Seidel K, Heinsen H, Vonsattel JP, den Dunnen WF, Korf HW. Huntington's disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. (2016) 26:726–40. doi: 10.1111/bpa.12426
10. Bocci T, Baloscio D, Ferrucci R, Briscese L, Priori A, Sartucci F. Interhemispheric connectivity in idiopathic cervical dystonia and spinocerebellar ataxias: a transcranial magnetic stimulation study. Clin EEG Neurosci. (2020). doi: 10.1177/1550059420957487. [Epub ahead of print].
11. Bocci T, Hensghens MJM, Di Roll A, Parenti L, Barloscio D, Rossi S, et al. Impaired interhemispheric processing in early Huntington's disease: a transcranial magnetic stimulation study. Clin Neurophysiol. (2016) 127:1750–2. doi: 10.1016/j.clinph.2015.10.036
12. Rees EM, Farmer R, Cole JH, Haider S, Durr A, Landwehrmeyer B, et al. Cerebellar abnormalities in Huntington's disease: a role in motor and psychiatric impairment? Mov Disord. (2014) 29:1648–54. doi: 10.1002/mds.25984
13. Wolf RC, Thomann PA, Sambataro F, Wolf ND, Vasic N, Landwehrmeyer GB, et al. Abnormal cerebellar volume and corticocerebellar dysfunction in early manifest Huntington's disease. J Neurol. (2015) 262:859–69. doi: 10.1007/s00415-015-7642-6
14. Gellersen HM, Guo CC, O'Callaghan C, Tan RH, Sami S, Hornberger M. Cerebellar atrophy in neurodegeneration-a meta-analysis. J Neurol Neurosurg Psychiatr. (2017) 88:780–8. doi: 10.1136/jnnp-2017-315607
15. Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. (2000) 97:8093–7. doi: 10.1073/pnas.110078997
16. Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, Goldowitz D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci. (2011) 31:14794–9. doi: 10.1523/JNEUROSCI.2774-11.2011
17. Dougherty SE, Reeves JL, Lesort M, Detloff PJ, Cowell RM. Purkinje cell dysfunction and loss in a knock-in mouse model of Huntington disease. Exp Neurol. (2013) 240:96–102. doi: 10.1016/j.expneurol.2012.11.015
18. Galea JM, Jayaram G, Ajagbe L, Celnik P. Modulation of cerebellar excitability by polarity-specific noninvasive direct current stimulation. J Neurosci. (2009) 29:9115–22. doi: 10.1523/JNEUROSCI.2184-09.2009
19. Bocci T, Santarcangelo E, Vannini B, Torzini A, Carli G, Ferrucci R, et al. Cerebellar direct current stimulation modulates pain perception in humans. Restor Neurol Neurosci. (2015) 33:597–609. doi: 10.3233/RNN-140453
20. Priori A, Ciocca M, Parazzini M, Vergari M, Ferrucci R. Transcranial cerebellar direct current stimulation and transcutaneous spinal cord direct current stimulation as innovative tools for neuroscientists. J Physiol. (2014) 592:3345–69. doi: 10.1113/jphysiol.2013.270280
21. Grimaldi G, Argyropoulos GP, Bastian A, Cortes M, Davis NJ, Edwards DJ, et al. Cerebellar Transcranial Direct Current Stimulation (ctDCS): a novel approach to understanding cerebellar function in health and disease. Neuroscientist. (2016) 22:83–97. doi: 10.1177/1073858414559409
22. Bocci T, De Carolis G, Ferrucci R, Paroli M, Mansani F, Priori A, et al. Cerebellar Transcranial Direct Current Stimulation (ctDCS) ameliorates phantom limb pain and non-painful phantom limb sensations. Cerebellum. (2019) 18:527–35. doi: 10.1007/s12311-019-01020-w
23. Ferrucci R, Cortese F, Priori A. Cerebellar tDCS: how to do it. Cerebellum. (2015) 14:27–30. doi: 10.1007/s12311-014-0599-7
24. Ferrucci R, Brunoni AR, Parazzini M, Vergari M, Rossi E, Fumagalli M, et al. Modulating human procedural learning by cerebellar transcranial direct current stimulation. Cerebellum. (2013) 12:485–92. doi: 10.1007/s12311-012-0436-9
25. Ferrucci R, Cortese F, Bianchi M, Pittera D, Turrone R, Bocci T, et al. Cerebellar and motor cortical transcranial stimulation decrease levodopa-induced dyskinesias in Parkinson's disease. Cerebellum. (2016) 15:43–7. doi: 10.1007/s12311-015-0737-x
26. Parazzini M, Rossi E, Ferrucci R, Liorni I, Priori A, Ravazzani P. Modelling the electric field and the current density generated by cerebellar transcranial DC stimulation in humans. Clin Neurophysiol. (2014) 125:577–84. doi: 10.1016/j.clinph.2013.09.039
27. Brunoni AR, Amadera J, Berbel B, Volz MS, Rizzerio BG, Fregni F. A systematic review on reporting and assessment of adverse effects associated with transcranial direct current stimulation. Int J Neuropsychopharmacol. (2011) 14:1133–45. doi: 10.1017/S1461145710001690
28. Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord. (1996) 11:136–42. doi: 10.1002/mds.870110204
29. Tunez I, Drucker-Colin R, Jimena I, Medina FJ, Munoz Mdel C, Pena J, et al. Transcranial magnetic stimulation attenuates cell loss and oxidative damage in the striatum induced in the 3-nitropropionic model of Huntington's disease. J Neurochem. (2006) 97:619–30. doi: 10.1111/j.1471-4159.2006.03724.x
30. Davis M, Phillips A, Tendler A, Oberdeck A. Deep rTMS for neuropsychiatric symptoms of huntington's disease: case report. Brain Stimul. (2016) 9:960–1. doi: 10.1016/j.brs.2016.09.002
31. Davis M, Phillips A, Tendler A, Oberdeck A. Deep rTMS for neuropsychiatric symptoms of Huntington's disease: case report. Brain Stimul. (2016) 9:960–1. doi: 10.1016/j.brs.2016.09.002
32. Shakkottai VG, Batla A, Bhatia K, Dauer WT, Dresel C, Niethammer M, et al. Current opinions and areas of consensus on the role of the cerebellum in Dystonia. Cerebellum. (2017) 16:577–94. doi: 10.1007/s12311-016-0825-6
33. Fung WKW, Peall KJ. What is the role of the cerebellum in the pathophysiology of dystonia? J Neurol. (2019) 266:1549–51. doi: 10.1007/s00415-019-09344-7
34. Filip P, Lungu OV, Bares M. Dystonia and the cerebellum: a new field of interest in movement disorders? Clin Neurophysiol. (2013) 124:1269–76. doi: 10.1016/j.clinph.2013.01.003
35. Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. (2002) 22:7825–33. doi: 10.1523/JNEUROSCI.22-17-07825.2002
36. Schirinzi T, Sciamanna G, Mercuri NB, Pisani A. Dystonia as a network disorder: a concept in evolution. Curr Opin Neurol. (2018) 31:498–503. doi: 10.1097/WCO.0000000000000580
37. Fremont R, Tewari A, Angueyra C, Khodakhah K. A role for cerebellum in the hereditary dystonia DYT1. Elife. (2017) 6:e2275. doi: 10.7554/eLife.22775
38. Hanssen H, Heldmann M, Prasuhn J, Tronnier V, Rasche D, Diesta CC, et al. Basal ganglia and cerebellar pathology in X-linked dystonia-parkinsonism. Brain. (2018) 141:2995–3008. doi: 10.1093/brain/awy222
39. Sadnicka A, Teo JT, Kojovic M, Parees I, Saifee TA, Kassavetis P, et al. All in the blink of an eye: new insight into cerebellar and brainstem function in DYT1 and DYT6 dystonia. Eur J Neurol. (2015) 22:762–7. doi: 10.1111/ene.12521
40. Liu Y, Xing H, Wilkes BJ, Yokoi F, Chen H, Vaillancourt DE, et al. The abnormal firing of Purkinje cells in the knockin mouse model of DYT1 dystonia. Brain Res Bull. (2020) 165:14–22. doi: 10.1016/j.brainresbull.2020.09.011
41. Tewari A, Fremont R, Khodakhah K. It's not just the basal ganglia: cerebellum as a target for dystonia therapeutics. Mov Disord. (2017) 32:1537–45. doi: 10.1002/mds.27123
42. Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nat Neurosci. (2005) 8:1491–3. doi: 10.1038/nn1544
43. Chen CH, Fremont R, Arteaga-Bracho EE, Khodakhah K. Short latency cerebellar modulation of the basal ganglia. Nat Neurosci. (2014) 17:1767–75. doi: 10.1038/nn.3868
44. Jeste DV, Barban L, Parisi J. Reduced purkinje cell density in Huntington's disease. Exp Neurol. (1984) 85:78–86. doi: 10.1016/0014-4886(84)90162-6
45. Rodda RA. Cerebellar atrophy in Huntington's disease. J Neurol Sci. (1981) 50:147–57. doi: 10.1016/0022-510X(81)90049-6
46. Fennema-Notestine C, Archibald SL, Jacobson MW, Corey-Bloom J, Paulsen JS, Peavy GM, et al. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. (2004) 63:989–95. doi: 10.1212/01.WNL.0000138434.68093.67
47. Ruocco HH, Lopes-Cendes I, Li LM, Santos-Silva M, Cendes F. Striatal and extrastriatal atrophy in Huntington's disease and its relationship with length of the CAG repeat. Braz J Med Biol Res. (2006) 39:1129–36. doi: 10.1590/S0100-879X2006000800016
48. Latimer CS, Flanagan ME, Cimino PJ, Jayadev S, Davis M, Hoffer ZS, et al. Neuropathological comparison of adult onset and juvenile Huntington's disease with cerebellar atrophy: a report of a father and son. J Huntingtons Dis. (2017) 6:337–48. doi: 10.3233/JHD-170261
49. Patra KC, Shirolkar MS. Childhood-onset (Juvenile) Huntington's disease: a rare case report. J Pediatr Neurosci. (2015) 10:276–9. doi: 10.4103/1817-1745.165709
50. Sakazume S, Yoshinari S, Oguma E, Utsuno E, Ishii T, Narumi Y, et al. A patient with early onset Huntington disease and severe cerebellar atrophy. Am J Med Genet A. (2009) 149A:598–601. doi: 10.1002/ajmg.a.32707
51. Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol. (2006) 21:223–9. doi: 10.2310/7010.2006.00055
52. Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. (2006) 63:2642–60. doi: 10.1007/s00018-006-6242-0
53. Braak H, Braak E. Allocortical involvement in Huntington's disease. Neuropathol Appl Neurobiol. (1992) 18:539–47. doi: 10.1111/j.1365-2990.1992.tb00824.x
54. de la Monte SM, Vonsattel JP, Richardson EP Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol. (1988) 47:516–25. doi: 10.1097/00005072-198809000-00003
55. Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D, et al. Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol. (1994) 88:320–33. doi: 10.1007/BF00310376
56. Rub U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D, et al. Degeneration of the cerebellum in Huntington's disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol. (2013) 23:165–77. doi: 10.1111/j.1750-3639.2012.00629.x
57. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. (1985) 44:559–77. doi: 10.1097/00005072-198511000-00003
58. Hubsch C, Roze E, Popa T, Russo M, Balachandran A, Pradeep S, et al. Defective cerebellar control of cortical plasticity in writer's cramp. Brain. (2013) 136:2050–62. doi: 10.1093/brain/awt147
Keywords: Huntington's disease, neurodegenerative diseases, cerebellum, tDCS, rTMS, non-invasive brain stimulation
Citation: Bocci T, Baloscio D, Ferrucci R, Sartucci F and Priori A (2020) Cerebellar Direct Current Stimulation (ctDCS) in the Treatment of Huntington's Disease: A Pilot Study and a Short Review of the Literature. Front. Neurol. 11:614717. doi: 10.3389/fneur.2020.614717
Received: 06 October 2020; Accepted: 11 November 2020;
Published: 03 December 2020.
Edited by:
Francesco Di Lorenzo, Santa Lucia Foundation (IRCCS), ItalyReviewed by:
Tommaso Schirinzi, University of Rome Tor Vergata, ItalyCarlo Alberto Artusi, University of Turin, Italy
Copyright © 2020 Bocci, Baloscio, Ferrucci, Sartucci and Priori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tommaso Bocci, dG9tbWFzby5ib2NjaUB1bmltaS5pdA==