 Xuewen Xiao1
Xuewen Xiao1 Bin Jiao
Bin Jiao- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Central South University, Changsha, China
- 3Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
Objective: This review summarizes recent findings on the epigenetics of Alzheimer's disease (AD) and provides therapeutic strategies for AD.
Methods: We searched the following keywords: “genetics,” “epigenetics,” “Alzheimer's disease,” “DNA methylation,” “DNA hydroxymethylation,” “histone modifications,” “non-coding RNAs,” and “therapeutic strategies” in PubMed.
Results: In this review, we summarizes recent studies of epigenetics in AD, including DNA methylation/hydroxymethylation, histone modifications, and non-coding RNAs. There are no consistent results of global DNA methylation/hydroxymethylation in AD. Epigenetic genome-wide association studies show that many differentially methylated sites exist in AD. Several studies investigate the role of histone modifications in AD; for example, histone acetylation decreases, whereas H3 phosphorylation increases significantly in AD. In addition, non-coding RNAs, such as microRNA-16 and BACE1-antisense transcript (BACE1-AS), are associated with the pathology of AD. These epigenetic changes provide us with novel insights into the pathogenesis of AD and may be potential therapeutic strategies for AD.
Conclusion: Epigenetics is associated with the pathogenesis of AD, including DNA methylation/hydroxymethylation, histone modifications, and non-coding RNAs, which provide potential therapeutic strategies for AD.
Introduction
Alzheimer's disease (AD) is a devastating neurological disease characterized by progressive cognitive impairments. As the most common form of dementia in the world, AD accounts for an estimated 60–80% of dementia cases worldwide (1). It is estimated that 50 million people are living with dementia worldwide currently, and the figure will increase to 152 million by 2050 (2). The common hypotheses of AD include amyloid cascade hypothesis, tau propagation, neuroimmune activation, mitochondrial cascade hypotheses, and infectious hypothesis (3).
AD can be classified into familial and sporadic AD based on family history. Three causative genes, including presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) are involved in the pathogenesis of familial AD in an autosomal-dominant trait (4, 5). PSEN1 is the most common genetic cause in familial AD, and the second involved gene is PSEN2. PSEN1 and PSEN2 encode the presenilins, which are catalytic subunits of BACE1. APP is the third involved gene that encodes the APP from which amyloid-β peptide is cleaved. These genetic mutations trigger the cascade of amyloid-β deposition, resulting in cognitive impairments in patients with AD (6, 7).
The genetics of sporadic AD are much more complex than that of familial AD. With the developments of genetic sequencing technology, particularly the Genome-Wide Association Study (GWAS), scientists have identified a number of loci containing susceptibility alleles in sporadic AD. One of the most important loci is the apolipoprotein E gene (APOE), and the three major isoforms are APOE ε2, ε3, and ε4 based on two-point mutations (rs429358 and rs7412). The APOE ε4 haplotype increases the likelihood of the onset of AD (8). Since 2009, tens of GWAS studies on AD have identified more than 20 novel loci including ABCA7, BIN1, CD33, CLU, CR1, CD2AP, EPHA1, MS4A6A-MS4A4E, PICALM, and SORL1, among others (9, 10).
Currently, much attention has been drawn to the nongenetic factors. Nongenetic factors are of paramount significance in the etiology of AD. Increasing evidence identifies multiple nongenetic factors for AD, most of which are related to lifestyle. A systematic review and meta-analysis based on the current evidence proposes 21 nongenetic factors for the prevention of AD, such as low level of education, hypertension, hyperhomocysteinemia, diabetes, obesity in late life, depression, and stress (11). The underlying mechanisms of how these nongenetic factors affect AD are not fully understood. The common hypotheses include oxidative stress, inflammation, brain reserve theory, the hypoperfusion hypothesis, and hypomethylation theory (12). In the 5xFAD mouse model, environmental enrichment, a combination of cognitive and sensory stimulation and social interaction, improves cognitive impairment via altering epigenetic markers (13).
Epigenetics may explain the roles of non-genetic factors involved in AD and help us to better understand the etiology of AD. Epigenetics was so named first by Conrad Waddington in the 1940's, and it is now defined as the study of molecules and mechanisms that perpetuate alternative gene activity states without changing the DNA sequence (14). Specifically, epigenetics includes DNA methylation/hydroxymethylation, histone modifications, and non-coding RNA regulation. These modifications play a crucial role in the gene readout, such as gene silencing, transcription, and post-transcriptional RNA processing. Therefore, epigenetics makes a significant impact on the disease (15, 16). Evidence shows that epigenetics is involved in the pathogenesis of AD (Figure 1). Here, we review recent findings on the epigenetics of AD and its potential therapeutic strategies.
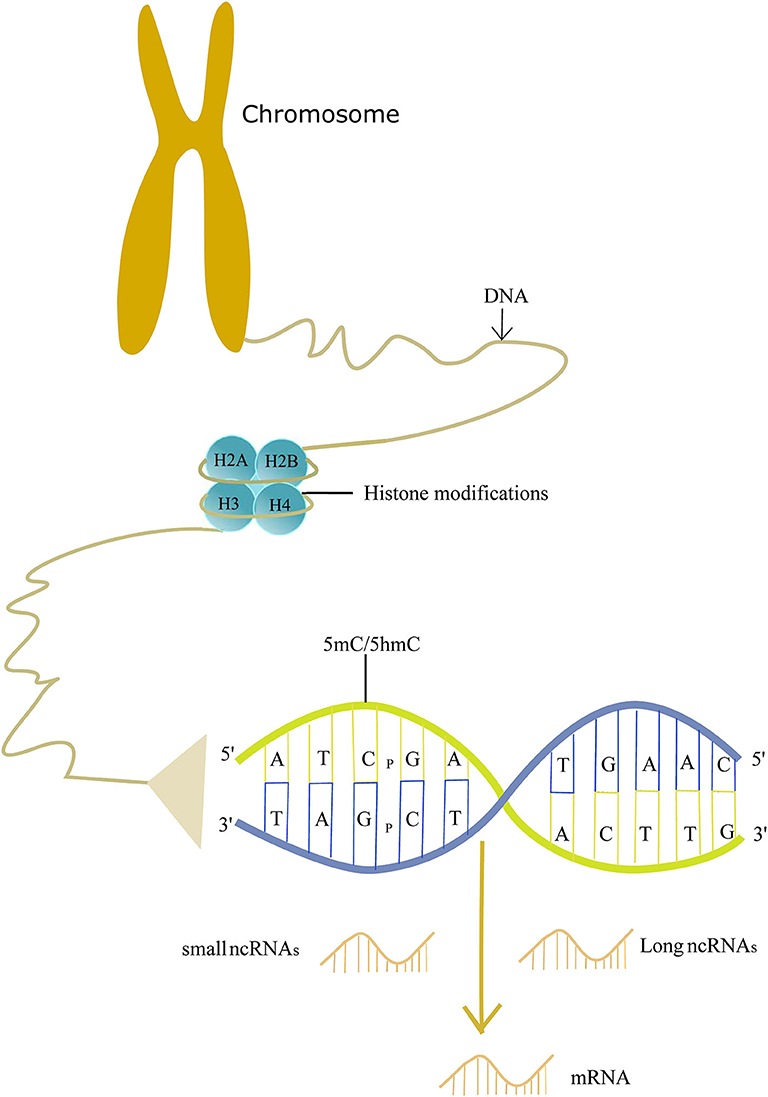
Figure 1. Schematic picture of epigenetics in the pathogenesis of AD.
DNA Methylation/Hydroxymethylation in AD
DNA methylation refers to methylation of the nucleobases in DNA by a set of proteins called DNA methyltransferases (DNMTs). Most of the methylated nucleobases are cytosines in CpGs and CpHs (H = A, T, C), forming 5-methylcytosine (5 mC). Five mC is mostly enriched in CpG dinucleotides (17). Although a lot of attention on DNA methylation focuses on CpGs, CpHs methylation is found to be associated with gene expression in neurons (18). In addition to 5 mC, methylation of another nucleobase is identified. In mouse prefrontal cortical neurons, deoxyadenosine methylation on N6 (m6dA) is correlated with gene expression and the formation of memory (19). DNMTs include DNA methylTransferase 1, DNA methylTransferase 3A, and DNA methylTransferase 3B (20). DNA hydroxymethylation is the hydroxymethylation of the cytosine to develop 5-hydroxymethylcytosine (5 hmC), and 5 hmC is converted from 5 mC by ten-eleven translocase (TET) isoforms (21). CpG and CpH are abbreviations for the cytosine and other nucleobases divided by a phosphate. The methyl/hydroxymethyl groups are located in the major groove of the DNA helix where many DNA-binding proteins make contact. Therefore, DNA methylation can lead to gene silencing, X-chromosome inactivation, and genomic imprinting. The exact functions of DNA hydroxymethylation remain unclear, and it may promote or repress gene expression by binding to regulatory regions of a gene (22, 23).
To date, a variety of studies have analyzed the role of DNA methylation in AD. One of the most studied genes of DNA methylation is the APP gene. CpG dinucleotides are enriched in the 5′ region of the APP gene, and alteration of its methylation levels can affect APP expression. Some studies identify hypomethylation of the APP gene in patients with AD compared with normal controls by analyzing postmortem brains or peripheral blood leucocytes in vitro (24–26), whereas, two other studies show no differences between AD and normal controls (27, 28). The reasons for the different results are not well-understood. Possible explanations include different test methods, different tissues examined, and relatively small sample sizes (29). Specifically, these studies examine the APP methylation levels in postmortem brain tissues or blood from a few AD patients and normal controls. Large-scale studies on methylation of the APP gene may address such different results in the future. Meanwhile, by analyzing genomic DNA, there are markedly different methylation levels of the APP gene in different human tissues (30). Therefore, the different tissues examined may also account for the different findings in the APP methylation levels. Furthermore, methylcytosines are reduced in the APP gene with age, which may be associated with Aβ deposition in AD (31). In addition to age, gender affects the methylation levels of the APP gene. Hypermethylation of the APP gene is identified in the female mouse cerebral cortex compared to male mice (32).
PSEN1 and PSEN2 genetic methylation patterns do not differ significantly between AD samples and normal controls (27, 28). After adjusting for gender and APOE, there are no significant methylation levels of PSEN1 and PSEN2 between AD patients and normal controls (33). No difference in PSEN1 methylation levels is observed in the AD mouse model, suggesting that Aβ production is not associated with PSEN1 methylation (34). Nevertheless, reduced methylation of the PSEN1 gene is identified in the human cerebral cortex (35, 36). In blood DNA, PSEN1 methylation is significantly downregulated in AD patients compared to normal controls, which is associated with higher PSEN1 expression during the progression of AD (37). The inconsistent findings may be associated with significant interindividual methylation variance of the PSEN1 gene (38). For APOE, two studies show that there is no significant difference in DNA methylation levels in postmortem brain tissues between AD patients and normal controls (39, 40). However, Foraker et al. observe a significant decrease in DNA methylation levels of APOE by evaluating methylation profiles of AD postmortem brains in vitro (41). The reason for this inconsistency may result from the detection platform. The previous two studies mainly focus on APOE promoter regions lacking CpG islands based on bead-chip. In the subsequent study, CpG methylation of APOE is obtained by pyrosequencing, which is more reliable in reflecting APOE methylation levels (41). Another study demonstrates that non-neuronal cells mainly contribute to the low levels of APOE DNA methylation in AD patients (42).
Further, a lot of studies examine methylation sites of candidate susceptibility genes for AD (Figure 2). An et al. show that the 2′,5′-oligoadenylate (2-5A) synthetase gene is hypomethylated in AD cells in vitro (43). Sanchez-Mut et al. show that TBXA2R, SORBS3, and SPTBN4 are hypermethylated in 12 distinct AD mouse brain regions, suggesting that the axon initial segment and cAMP response element-binding protein (CREB) activation pathway is involved in AD pathogenesis (44). Lei Yu et al. demonstrate that DNA methylation levels in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 are associated with greater odds for pathological AD by examining 28 reported AD loci discovered from recent GWAS reports on AD, which indicate that the change of DNA methylation of AD risk genes contribute to the etiology of AD (45). The hypermethylation of BDNF is repeatedly observed in AD patients' blood samples, which might be a diagnostic marker of AD (46, 47). Nevertheless, Carboni et al. find that BDNF, SIRT1, and PSEN1 exhibit no different methylation patterns in AD compared with controls using peripheral blood samples in vitro. The inconsistency among the studies may result from methodological differences. Bisulphite pyrosequencing technology and methylation-specific primer real-time PCR cover different CpGs when evaluating CpG sites in BDNF (48).
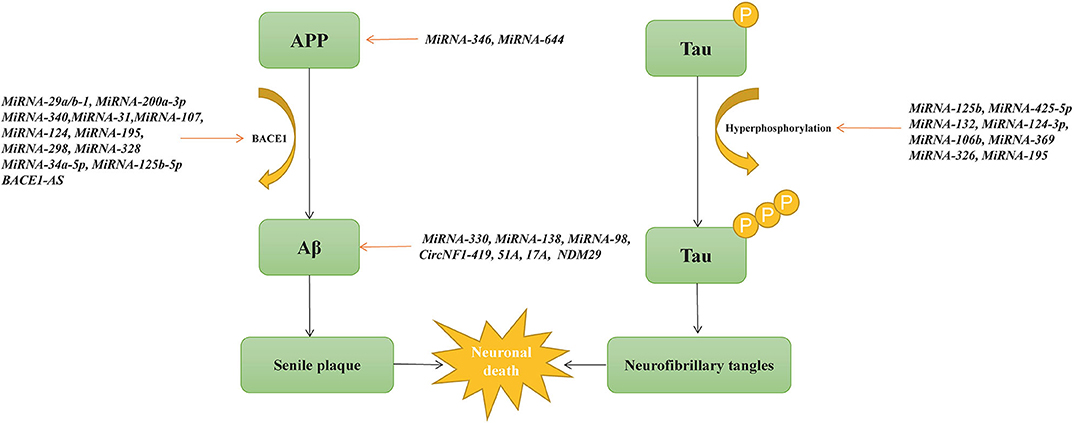
Figure 2. Specific gene methylation in AD: candidate gene approach.
Fabio Coppedè et al. show that there are no methylation differences in DNA repair genes between patients with AD and non-affected individuals, including OGG1, PARP1, MRE11A, BRCA1, MLH1, and MGMT (49). Ma et al. find that UQCRC1 is highly methylated in patients with AD by studying peripheral blood samples of AD, suggesting that inflammation and oxidative stress may contribute to AD (50). The DNA methylation level of PLD3 is increased and correlated with hippocampal Aβ in AD hippocampus compared to controls (51). A lower DNA methylation level at TREM2 is observed in AD patients and associated with TREM2 mRNA expression, which may be a biomarker for AD (52). CRTC1 is hypomethylated in the AD hippocampus and associated with p-tau deposition (53). Hypomethylation of CpGs in BIN1 is identified in participants with AD and confers risk to AD by studying peripheral blood, indicating it may be a biomarker for AD (54). The methylation level of BIN1 is associated with neuritic plaque pathology in the peripheral blood (55). Furthermore, the methylation levels of OPRM1 and OPRL1 are significantly increased in AD compared to controls, suggesting that opioid receptor genes may be potential biomarkers for diagnosing AD (56). The DNA methylation level of PICALM is decreased and linked to cognitive decline in AD (57). Elevated ANK1 DNA methylation exists in the entorhinal cortex of AD and is associated with AD pathology (58). Similarly, another study also identifies that hypermethylated ANK1 is observed in the entorhinal cortex, superior temporal gyrus, and prefrontal cortex (39). Methylation of ABCA2 is negatively associated with AD risk and may be a therapeutic target of AD (59). Furthermore, other studies identify many genes without methylation changes between AD and normal controls, such as SORL1, SIRT1, SST, SSTR4, HSPA8, HSPA9, SIRT3, and ABCA7 (40, 60–63) (Table 1).
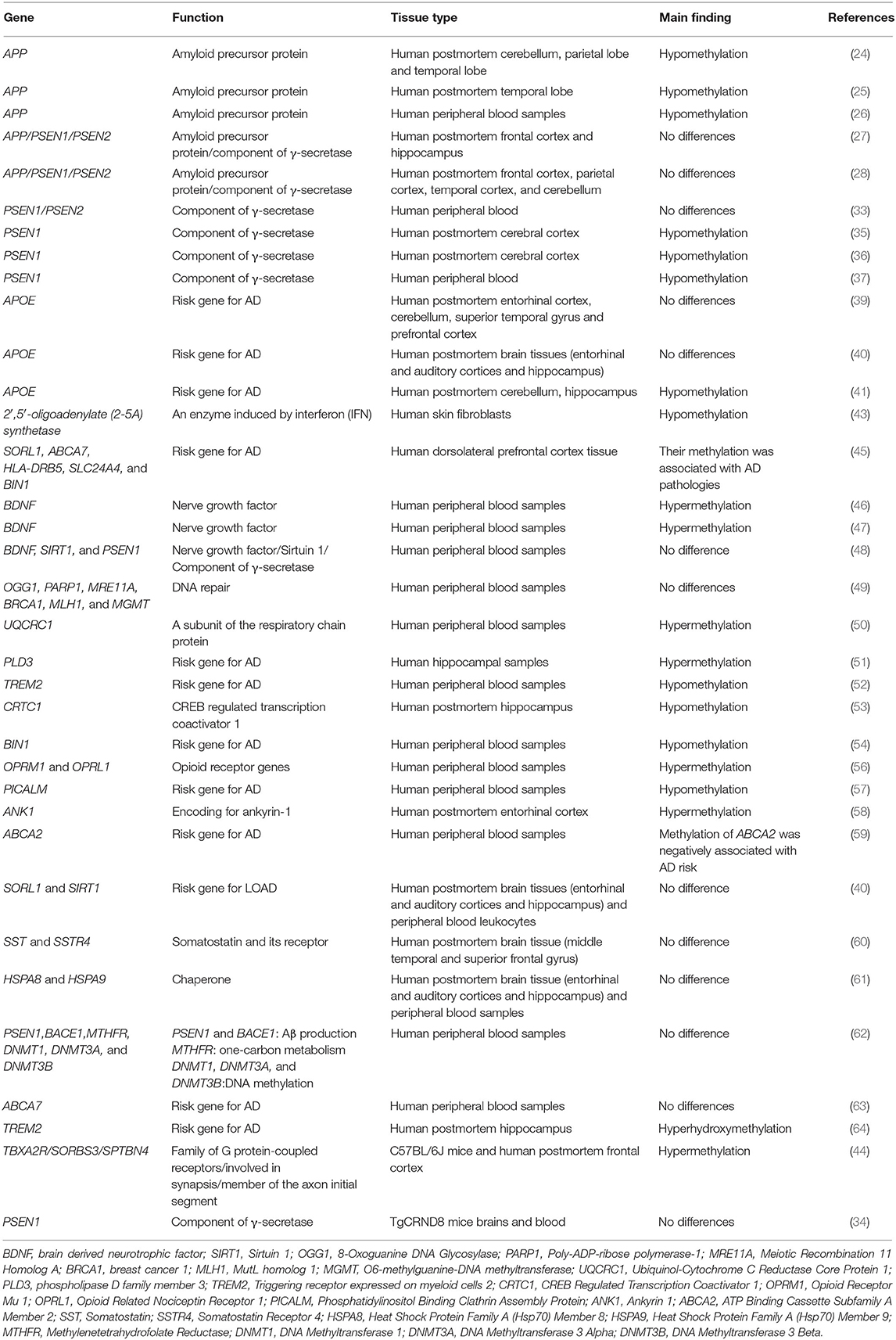
Table 1. Specific gene methylation and hydroxymethylation in Alzheimer's disease: candidate gene approach.
Recently, there were several genome-wide methylation analyses of AD. By examining genome-wide enhancer methylation levels in the prefrontal cortex of AD patients, Li et al. identify 1,224 enhancer regions methylated differentially and most of their methylation levels are decreased in AD neurons, which are associated with Aβ, tau, and cognitive impairment (65). Humphries et al. show that 1,106 of 5,147 CpG sites differ between LOAD patients and controls, 87.3% of which are hypomethylated and related to the myelination network in LOAD (66). Moreover, Watson et al. identify 479 differentially methylated regions in patients with AD compared to normal controls by performing a genome-wide screen of DNA methylation in the temporal gyrus of AD, and 475 of these regions are involved in neuron function and development (67). De Jager et al. show that 11 of 415,848 interrogated CpGs are significantly associated with the AD pathological burden by studying AD autopsied brains, including CpGs in ABCA7 and BIN1 regions (68). Moreover, an epigenome-wide study identifies that DNA methylation of OXT is associated with the risk of AD, indicating it may be a novel promising biomarker or therapeutic target in AD (69). In another epigenome-wide DNA methylation study, among 17,895 differentially methylated CpG sites, there are 11,822 hypermethylated CpGs and 6,073 hypomethylated CpGs in the superior temporal gyrus of AD patients (70). When examining 420,852 DNA methylation sites from four brain regions in late-onset AD and neurotypical controls, 858 sites show differential methylation patterns, indicating that DNA methylation may contribute to AD (71). Altuna et al. profile genome-wide DNA methylation levels in the hippocampus of AD patients in which 118 AD-related differentially methylated positions are found, and these positions are linked to phosphorylated tau burden (72). The first integrated base-resolution genome-wide study finds 39 CpG site-specific and 27 AD region-specific epigenetic changes in AD, providing reliable epigenetic signatures for the diagnosis and treatment of AD (73) (Table 2).

Table 2. Specific gene methylation and hydroxymethylation in AD: Genome-wide approach.
Global DNA methylation/hydroxymethylation is the percentage of methylcytosine/hydroxymethylcytosine of total cytosine. The percentage of methylation of CCGG sites exhibits no significant difference by investigating DNA methylation in the human brain (78). Moreover, no significant global 5mC and 5hmC changes are found in the entorhinal cortex of AD patients (79). Some studies find that the levels of global 5 mC and 5 hmC is decreased in patients with AD (80–82). However, in the postmortem AD cortex, global levels of 5 mC and 5 hmC are significantly increased in AD samples and correlated with biomarkers of AD (83). Global increased 5 mC levels are identified in the postmortem frontal cortex from AD patients (84). By analyzing DNA isolated from peripheral blood, global DNA methylation levels are upregulated in AD patients and correlated with cognitive impairments (85). The reasons for the difference are far from being understood. One possible reason is that they investigated different brain regions. Nevertheless, some findings are contradictory even for the same test method and brain region. The differences in test method procedures and sample size may account for the different findings. In addition to studies in postmortem brain regions, global DNA methylation/hydroxymethylation was also investigated in the AD mouse model. The 5xFAD mouse model is composed of two APP and two PSEN1 mutations. Global high levels of 5-mC and a reduction of 5-hmC were observed in the 5xFAD mouse model, which are related to cognition and paralleled with Aβ deposition (86). In the 3xTg-AD mouse model, global methylation and hydroxymethylation levels are reduced (87). In the SAMP8 mouse model, global DNA methylation levels are decreased while 5-hmC levels are upregulated (88).
Compared to DNA methylation, relatively few studies analyze the role of DNA hydroxymethylation in AD. As we mentioned previously, 5 hmC is the specific product of DNA hydroxymethylation. Five hmC is highly concentrated in the central nervous system and plays an important role in neurodevelopment and neurological function (89, 90). The triggering receptor expressed on myeloid cells (TREM2) is a receptor in brain microglia and contributes to the pathogenesis of AD (91). When examining DNA hydroxymethylation levels in TREM2, Celarain et al. find that the 5 hmC levels were increased in AD patients and involved in TREM2 mRNA expression (64) (Table 1). Shu et al. demonstrate that the 5 hmC levels are decreased in the hippocampus in a mouse model of AD, whereas another study finds that the 5 hmC levels are significantly increased in it in AD patients. Different tissue types may explain the contradictory findings (92, 93). The genome-wide distribution of 5 hmC finds that 517 differentially hydroxymethylated regions (DhMRs) are significantly associated with neuritic plaques, whereas 60 DhMRs are associated with neurofibrillary tangles (74). Moreover, there are 325 genes containing differentially hydroxymethylated loci in AD from genome-wide analyses of 5 hmC in the prefrontal cortex of postmortem AD patients, involving neuronal projection development and neurogenesis (75). By studying genome-wide profiles of 5 mC and 5 hmC profiles in frontal cortex tissues from Chinese AD patients, two significant transcription factor-binding motifs, hypoxia-inducible factor 2α, and hypoxia-inducible factor 1α are enriched in the differentially hydroxymethylated regions (76). As mentioned previously, DNA hypermethylation in ANK1 is identified in AD (58). Hypohydroxymethylation in ANK1 is also found in the entorhinal cortex of AD patients in an epigenome-wide association study, further highlighting the significant role of ANK1 in the development of AD (77) (Table 2).
It is increasingly becoming accepted that mitochondria play a significant role in the pathogenesis of AD (94). Blanch et al. show that mitochondrial 5 mC levels increase, whereas no significant differences in 5 hmC levels are identified in AD (95). However, another study finds that mitochondrial 5 hmC is significantly increased in the superior temporal gyrus of AD subjects (93). The different test methods among these studies may account for the different findings of mitochondrial 5 hmC in brain tissue. In APP/PS1 transgenic mice, the displacement loop mitochondrial methylation level is reduced while 12 S rRNA gene mitochondrial methylation is increased, indicating that mitochondrial DNA methylation may play a role in the AD development (96).
Histone Modifications in AD
Histone is a kind of octamer consisting of pairs of H2A, H2B, H3, and H4, which form the nucleosome with DNA. Histone can be modified at the N-terminal tails, and the modifications can affect the three-dimensional structure of the chromatin, leading to the changes in the transcription of genes. The common histone modifications include acetylation, methylation, and ubiquitination. The modifications are controlled by a specific set of enzymes, such as acetyltransferases and deacetylases (97).
To date, there have been several studies of histone modifications in AD. A genome-wide study of H3K27 acetylation in AD observed 4,162 differential acetylomic variation peaks between AD patients and normal controls, which were associated with Aβ and tau pathology (98). Zhang et al. show that histone acetylation is significantly decreased in the temporal lobe of patients with AD compared with that of controls, which is consistent with the previous finding in an APP/PS1 mouse model of AD, highlighting that histone acetylation is involved in AD (99, 100). Narayan et al. demonstrate that acetyl histone H3 and H4 levels are significantly increased in postmortem AD brain tissue compared with normal controls by investigating global acetyl histone levels (101). Another study shows a significant increase of monocytic H4K12 acetylation in transgenic AD mouse models and MCI patients (102). Global histone H3 acetylation levels exhibit a significant increase in the frontal cortex in end-stage AD patients, supporting that histone acetylation plays an important role in AD (103). These studies highlight that histone acetylation levels are increased on a global scale in the AD brain. Anderson et al. observe notable decreases in methylation of H2B and H4 residues, whereas, the ubiquitination of H2B residue increases. These post-translational histone modifications are related to AD pathology and of great significance in the development of AD (104). With regard to the regulators of histone modifications, in APP/PS1 mice, age is associated with the global levels of histone modifications, suggesting that age is one of the main risk factors in the histone modifications of AD (105). An epigenome-wide association study demonstrates that tau protein affects histone acetylation changes and an altered chromatin structure in AD prefrontal cortices (106) (Table 3).
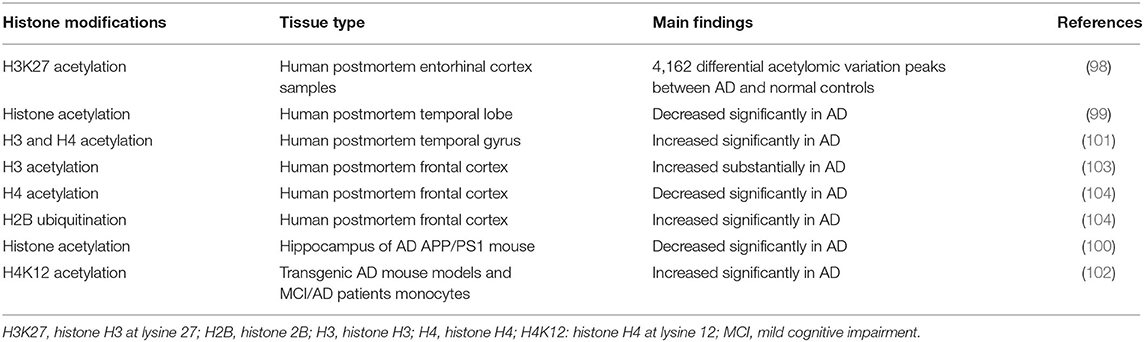
Table 3. Histone modifications in AD.
Non-Coding RNAs in AD
Non-coding RNAs (ncRNAs) are defined as RNA molecules that are not translated into a protein. Less than 2% of the human genome encodes proteins, and the rest produces thousands of ncRNAs, including small ncRNAs (smaller than 200 nucleotides in length), such as microRNAs, small interfering RNAs, piwi-interacting RNAs, and a variety of long ncRNAs (longer than 200 nucleotides) (107).
One of the best characterized small ncRNAs in AD is microRNA. Many microRNAs are surrounded by the CpG island and located within exons, intergenic regions, or introns of genes (108). MicroRNAs can enhance or repress messenger RNA transcription by binding to the 3′-untranslated region or the promoter of the target gene (109). In sporadic AD, microRNAs play an important role in Aβ production and neurofibrillary tangle formation (Figure 3). Two studies demonstrate that microRNA-16 inhibits the expression of APP both in vitro and in vivo, which may be a therapeutic target of AD (110, 111). As mentioned previously, BACE1 is one of the APP-cleaving enzymes in the production of Aβ. By investigating changes in 328 microRNA expression profiles, Hébert et al. find that microRNA cluster microRNA-29a/b-1 suppressed endogenous BACE1 expression, and it is significantly decreased in sporadic AD patients (112). MicroRNA-200a-3p also reduces the expression of BACE1 and is confirmed to be decreased in the hippocampus of APP/PS1 and SAMP8 mice as well as in blood plasma from AD patients (113). MicroRNA-340 is decreased in the hippocampus of an AD mouse model and associated with the overproduction of Aβ by targeting BACE1 (114). MicroRNA-31 reduces the mRNA levels of BACE1 and improves memory deficits in AD triple-transgenic (3xTg-AD) female mice (115). Moreover, microRNA-107, microRNA-124, microRNA-195, microRNA-298, and microRNA-328 are also associated with the expression of BACE1 (116–119). Another recent study shows that microRNA-298 represses the expression of BACE1, APP, Aβ40, and Aβ42 in the cell model, suggesting that microRNA-298 may be a therapeutic target for AD (120). Meanwhile, the addition of microRNA-34a-5p and microRNA-125b-5p reduces Aβ by targeting BACE1 (121). Abnormally phosphorylated tau protein is another key pathological hallmark of AD. Li et al. find that microRNA−219-5p is significantly upregulated in brain tissues of AD patients, contributing to tau phosphorylation and AD progression (122). MicroRNA-125b promotes the phosphorylation of tau by activating cyclin-dependent kinase 5 (CDK5) and p35/25, and microRNA-125b is increased in AD patients (123). Similarly, overexpression of microRNA-125b results in tau hyperphosphorylation by targeting phosphatases DUSP6 and PPP1CA (124). In HEK293/tau cells, microRNA-425-5p overexpression promotes tau phosphorylation through targeting HSPB8 in AD (125). MicroRNA-132, the most significantly downregulated microRNA in AD, is associated with lower levels of tau phosphorylation by regulating EP300, GSK3b, Rbfox1, proteases Calpain2, and caspases 3/7 (126). MicroRNA-124-3p inhibits abnormal tau hyperphosphorylation through targeting the Caveolin-1-PI3K/Akt/GSK3β pathway in AD (127). MicroRNA-106b reduces tau phosphorylation induced by Aβ42 by involving the expression of Fyn (128). In 3xTg-AD mice, knocking out microRNA-369 is associated with tau hyperphosphorylation via regulating Fyn and SRPK2 signaling pathways (129). MicroRNA-326 decreases tau hyperphosphorylation and improves cognitive functions of AD through the JNK signaling pathway (130). Elevating the levels of microRNA-195 diminishes tau hyperphosphorylation and Aβ burden in ApoE4+/+ mice (131).
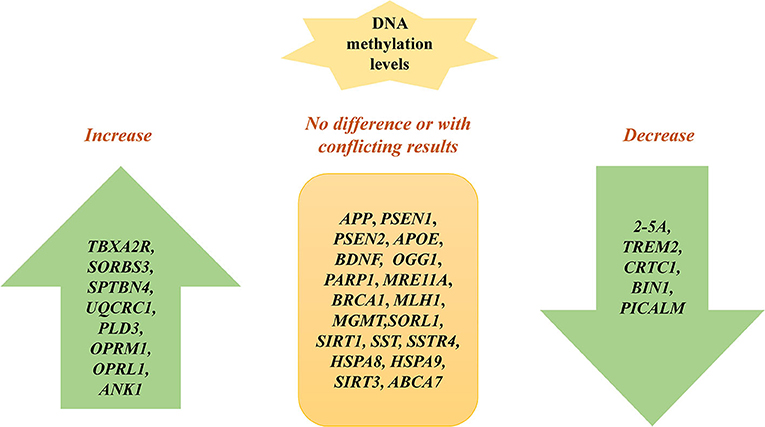
Figure 3. Non-coding RNAs implicated in Aβ and tau protein metabolism.
Furthermore, some altered microRNAs are directly related to APP or Aβ processing in AD. MicroRNA-346 upregulates APP translation and Aβ production by binding to APP 5′UTR, and its levels change in late-stage AD patients (132). MicroRNA-644 downregulate the formation of Aβ via targeting APP 3'UTR (133). Overexpression of microRNA-330 reduces Aβ production and alleviates oxidative stress through the MAPK signaling pathway (134). In APP/PS1 mice, microRNA-138 enhances Aβ production and improves cognitive impairment by decreasing the expression of sirtuin 1 protein (135). MicroRNA-98 improves oxidative stress and downregulates Aβ production by activating the Notch signaling pathway (136). In addition to microRNAs, cortical circular RNAs (circRNAs) are associated with AD diagnosis, clinical dementia severity, and neuropathological severity, suggesting the potentially significant role of circRNA in the pathogenesis of AD (137). CircRNA KIAA1586 is significantly enriched in AD-associated biological processes and may be a novel risk factor in the pathogenesis of AD (138). CircRNA circ_0000950 promotes neuron apoptosis, inhibits the production of neurite outgrowth, and increases the levels of inflammatory cytokine levels via sponging microRNA-103 in AD (139). CircNF1-419 upregulates autophagy and reduces Aβ and tau expression by binding the proteins Dynamin-1 and adaptor protein 2 B1 (140). Circ-AXL, circ-GPHN, and circ-PCCA differ significantly between AD patients and normal controls by studying the circRNA expression profile in cerebrospinal fluid, which may be potential biomarkers in AD (141). In addition, 147 circRNAs are differentially expressed in different AD brain regions, most of which are found in the parahippocampal gyrus, supporting that circRNAs in the parahippocampal gyrus may be biomarkers in AD (142).
In the central nervous system, long ncRNAs (lncRNAs) are prevalent and play a critical role in the pathogenesis of AD (143). A conserved lncRNA called BACE1-antisense transcript (BACE1-AS) increases BACE1 mRNA stability and generates additional Aβ in AD patients as well as in APP transgenic mice, and knockdown of BACE1-AS improves memory (144, 145). The BACE1-AS level differs significantly between pre-AD and healthy controls as well as full-AD and healthy controls, indicating that BACE1-AS may be a potential biomarker of AD (146). LncRNAs, including 51A, 17A, and NDM29, increase the formation of Aβ and contribute to the pathogenesis of AD (147). LncRNA SOX21-AS1 is upregulated in the AD mouse model, and its inhibition reduces neuronal oxidative stress and suppresses neuronal apoptosis via the Wnt signaling pathway (148). LncRNA BC200 levels are enhanced significantly in AD brains and involved in dendritic loss by regulating local protein synthesis (149). Brain-derived neurotrophic factor antisense RNA(BDNF-AS) is an lncRNA that represses BDNF expression. The inhibition of BDNF-AS results in neuronal growth and differentiation, which may be the novel pharmacological target in AD (150). LncRNA Sox2OT represses Sox2 gene expression, involving neurogenesis and neuronal differentiation (151). In APP/PS1 mice, lncRNA EBF3-AS is upregulated in the hippocampus and related to neuron apoptosis by regulating EBF3 expression (152). LncRNA NAT-Rad18 increases the likelihood of apoptosis under DNA damage-related stress via targeting RAD18 (153). LncRNA TUG1 promotes neuronal apoptosis in the hippocampus by increasing microRNA-15a levels and suppressing ROCK1 expression (154). In the SAMP8 mouse model, lifestyle, including diet, exercise, and environmental enrichment, results in epigenetic changes (20). In addition, 3112 differentially expressed lncRNAs are identified in the SAMP8 mouse model, most of which are intergenic and exon sense-overlapping (155). In the human AD brain, 16 age-associated and 13 gender-associated lncRNAs are identified; among them, lncRNAs SNHG19, LINC00672, RNF144A-AS1, LY86-AS1, and LINC00639 are associated with the pathology of AD (156). Nuclear paraspeckle assembly transcript 1 (NEAT1), a lncRNA that is widely expressed in cells, is of great importance in various biological and pathological processes by mediating target genes' expression (157). NEAT1 is involved in Aβ clearance by regulating the expression of endocytosis-related genes in AD (158). Additionally, in an APP/PS1 transgenic mouse model, NEAT1 is increased and promotes the pathogenesis of AD via upregulating PTEN-induced putative kinase 1 (PINK1)'s ubiquitination and degradation, which provided a potential therapeutic strategy in AD (159) (Table 4).

Table 4. Non-coding RNAs in AD.
The mechanisms underlying epigenetic regulation of non-coding RNAs in AD are complex. As we mention, circRNAs mediate the effect of microRNAs (139). MicroRNAs are regulated by chromatin modifications and DNA methylation, which are implicated in AD by targeting messenger RNA (mRNA) expression (182). Dysregulated circRNAs are associated with the increased number of downstream target mRNAs in the Tg2576 AD mouse model, indicating that circRNA-microRNA-mRNA may play a significant role in the pathogenesis of AD (183). Emerging evidence shows that lncRNAs are involved in multiple epigenetic processes, such as DNA methylation, via regulating the interactions of target genes with chromatin-remodeling enzymes. Most lncRNAs are located in the nucleus, in which they work as scaffolds for chromatin modifiers or transcriptional co-regulators to exhibit regulatory functions (184). In addition, lncRNAs can alter transcription, mRNA stability, alternative splicing, and translational activity in AD, resulting in aberrant gene expression (14). Overall, the deregulated and complex non-coding RNAs are closely associated with core pathophysiological processes of AD via regulating gene expression at different levels, including transcription, RNA processing, and translation (185).
Epigenetic Therapeutic Strategies for AD
DNA hypomethylation of pathogenetic genes is associated with the overproduction of Aβ (186). Oliveira et al. find that DNA methyltransferase Dnmt3a2 is decreased in the hippocampus of mice, and the restoration of Dnmt3a2 recovered the cognitive functions (160, 187). Further, betaine, a methyl donor, ameliorates the memory deficits in mice (161). Another methyl donor, S-adenosylmethionine (SAM), is decreased in the cerebrospinal fluid of AD patients (188). SAM reduces the production of Aβ and tau phosphorylation by upregulating PSEN1 and BACE1 expression in vivo, which improves the cognitive status in AD mouse models (162). The treatment with alcohol extracts from G. lucidum increases methylation regulators and improves memory in APP/PS1 AD model mice (163). In AD and mild cognitive impairment patients, B vitamin intake results in hypermethylation of NUDT15 and TXNRD1, which is associated with better cognitive performance (164). The supplementation of folic acid, a methyl donor, improves cognitive functions in participants who tend to decline with age (165). Interestingly, maternal supplementation of resveratrol promotes cognitive decline in the SAMP8 mice offspring via increasing global methylation levels and decreasing hydroxymethylation levels (166). Consequently, the DNA methyltransferase or the methyl donor may be potential treatments for patients with AD.
Meanwhile, hypermethylation is involved in the development of AD as well; thus, the decrease of methylation levels in some genes may also be a promising therapeutic strategy (109). DNMT inhibitors are used in the treatment of hematopoietic malignancy (189). Also, the administration of DNMT inhibitors is used in some neurodegenerative diseases, such as Friedreich's ataxia (190). Although DNMT inhibitors possess the potential for the therapy of AD, the lack of gene-specificity and security is the main difficulty needed to resolve before its use in AD patients (177). As we previously mentioned, the epigenetic markers are altered in the 5xFAD mouse model. Treatment of UNC0642 inhibits the methyltransferase activity G9a/GLP and restores cognition by reducing 5mC and increasing 5hmC in the 5xFAD mouse model (167). The knockout of the Tet1 gene enhances cognitive function by oxidizing 5 mC to 5 hmC and reducing methylation levels of the brain in mice (191).
Histone deacetylases (HDACs) inhibit gene expression and are associated with memory impairment by restricting access of transcription factors to memory storage-related genes (192). HDAC inhibitors (HDACi) are considered to be the potential therapeutic strategy in AD. Trichostatin A, an HDACi, restores contextual freezing performance and H4 acetylation levels in the APP/PS1 mouse model of AD (100). Valproic acid (VPA) is one of the first discovered HDACi and beneficial in memory enhancement in the mouse model of AD (168). Histone deacetylase 2 (HDAC2) inhibitors improve memory by promoting the formation and growth of dendritic spines in mice (193, 194). Sodium phenylbutyrate, an HDACi, also alleviates memory impairment in transgenic AD mice by inducing neurotrophin expression via the protein kinase C (PKC)-cAMP-response element-binding protein (CREB) pathway (169). M344, an HDACi, lowers the expression of Aβ and prevents cognitive decline with the normalization of several pathogenic pathways in triple transgenic (APPsw/PS1M146V/TauP301L) mice in vivo (170). A mercaptoacetamide-based class II HDACi and a hydroxamide-based class I and II HDACi reduce Aβ levels in vitro and rescue memory loss in AD mice (171). The downregulated PU.1 expression is associated with lower AD risk in a genome-wide association study (195). High-throughput screening of FDA-approved drugs reveals a HDACi, vorinostat, decreases PU.1 expression and may be a useful therapeutic approach in AD (172). Moreover, the administration of RGFP-966, a selective HDAC3 inhibitor, improves cognitive function in the AD mouse model and decreases Aβ and tau in neurons from AD patients, further supporting the significant role of HDACi in patients beyond the AD mouse model (173). Therefore, HDACi can be seen as a potential therapeutic agent for AD. However, HDACi is widely targeted and inevitably causes various side effects, including apoptosis and arrest of the cell cycle; therefore, successful experiments in AD animal models are seldom feasible for clinical application in humans. To increase the sensitivity of HDACi is a critical issue in the future (196). Moreover, histone acetyltransferase (HAT) is involved in the formation of CREB binding protein (CBP), which has an important role in memory (197). The expression of CBP could help transgenic AD mice recover memory impairment. The activator of histone acetyltransferases CBP/p300 is capable of passing the blood–brain barrier and extending the recent memory duration in B57BL6/6J male mice in vivo, which may be a potential treatment target in AD (174). In the late-stage FAD mouse model, the inhibitors of euchromatic histone methyltransferases decrease histone hypermethylation and improve synaptic deficits and cognitive functions, providing a possible novel therapeutic strategy for AD (175).
Noncoding RNAs are also involved in the pathogenesis of AD. The decreased expression of BACE1 is obtained by short-interfering RNA, resulting in the reduction of Aβ and tau phosphorylation levels in AD transgenic mice (176). The expression of APP, PSEN1, and PSEN2 was downregulated by RNA interference, such as short-interfering RNA and short hairpin RNA, which provide a promising therapeutic strategy in the future (177). MicroRNA mimics and anti-microRNAs are being developed by decreasing target protein expression in AD. MicroRNA-384 mimic downregulating the expression of APP and BACE1 in SH-SY5Y cells, demonstrating that microRNA-384 may be a potential target in AD (178). Anti-microRNAs complement respective microRNAs and reduce their levels to restore homeostasis. In the AD mouse model, microRNA-146a is upregulated, and anti-microRNA-146a-base treatment improves cognitive functions and regulates the inflammatory response by using the viral vector delivery system (179). In addition, a number of other microRNAs are the potential treatment targets in AD. MicroRNA-34c increases in the hippocampus and blood of patients with AD, and its inhibitor enhances memory in AD mice models in vivo (180). LncRNA BACE1-AS is positively associated with BACE1 protein expression in vitro and in vivo, and knockdown of BACE1-AS by short interfering RNA improves cognitive function in a mouse model of AD (145). MicroRNA-339-5p, microRNA-29c, and microRNA-124 decrease BACE1 expression in vitro (181, 198, 199) (Table 5). MicroRNA-101 suppresses APP and Aβ expression in hippocampal neurons in vitro (200). MicroRNA-153 decreases APP expression in primary human fetal brain cultures (201). Consequently, non-coding RNAs may be potential targets for AD therapy in the future. Currently, there are several problems in the treatment of AD through non-coding RNAs, including altering such targets, off-target effects, and delivery methods (202). However, it is worthwhile to investigate non-coding RNAs in AD by appropriate understanding and safe manipulation (203).
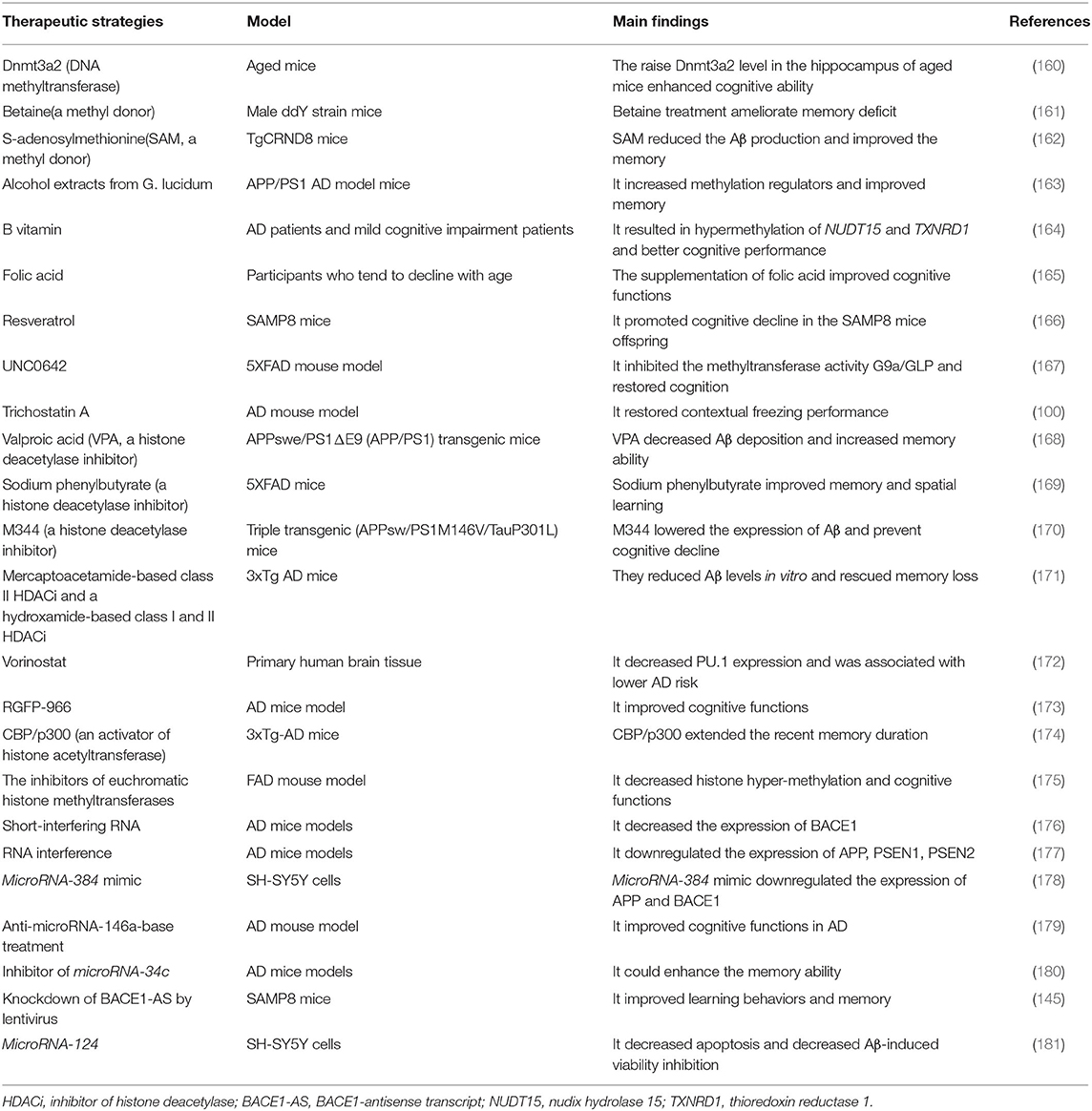
Table 5. Epigenetic therapeutic strategies in AD.
Currently, a few drugs are available for the treatment of AD. No drug can cure or stop disease development. Given the huge number and seriousness of AD, the need for clinical trials is necessary. To date, most of the clinical trials of AD have targeted Aβ, tau protein (204). Several clinical trials investigate epigenetics for the treatment of AD. Oral betaine was given in eight AD patients; however, the efficacy of betaine could not be determined due to the lack of controls and small sample size (205). With the use of S-adenosylmethionine and nutriceutical, almost 30% improvement in the neuropsychiatric inventory and activities of daily living is observed in AD patients compared to normal controls (206). RDN-929, a selective HDAC inhibitor, is being investigated in a phase I clinical trial for the treatment of AD patients. The result is not disclosed at clinicaltrials.gov. EVP-0334, also called FRM-0334, a CNS-penetrant HDACi, phase I testing of AD is completed; however, no result is posted (207). Because increasing evidence shows that epigenetics plays an essential role in AD, the drugs related to epigenetics may be breakthroughs for AD in the future.
Conclusion
To date, although some genetic and non-genetic factors are well-studied, the pathogenesis of AD remains unclear. Epigenetics provides us with an important insight into how AD develops. There is an increasing number of studies about epigenetics in AD patients, including DNA methylation/hydroxymethylation, histone modifications, and non-coding RNAs. Epigenetic genome-wide association (EGWA) studies show that many differentially methylated sites exist in AD compared with normal controls. Several studies investigate the role of histone modifications in AD. Non-coding RNAs play an important role in the pathogenesis of AD. LncRNAs, such as BACE1-AS, increases BACE1 mRNA stability and generates additional Aβ in AD. These studies show us that epigenetics is of great importance in AD, suggesting that epigenetics can be a potential intervention target in treating AD given the reversible nature of epigenetic changes. Therapeutic attempts include the use of inhibitors of HDACs, DNA methyltransferase, and inhibitors of non-coding RNAs, which have shown some exciting results in animal studies. Despite the numerous and exciting findings of epigenetics in AD, the results are less satisfying. The data is often controversial and lacks definite results. There is a need to design some larger longitudinal cohorts to study the epigenetic changes of AD, which may help us better understand the pathogenesis of AD and find novel strategies to treat AD in the future.
Author Contributions
BJ was involved in the review design, modified, and revised the manuscript. XX and XL searched and reviewed the articles. XX wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81701134 to BJ) and Provincial Technology Innovation Guidance Plan Project of Hunan (No. 2108SK52601 to BJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Alzheimer's disease facts and figures. Alzheimers Dement (2016) 12:459–509. doi: 10.1016/j.jalz.2016.03.001
2. Patterson C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. An Analysis of Prevalence, Incidence, Cost and Trends. Alzheimer's Disease International. (2018).
3. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. (2019) 179:312–39. doi: 10.1016/j.cell.2019.09.001
4. Tang M, Ryman DC, McDade E, Jasielec MS, Buckles VD, Cairns NJ, et al. Neurological manifestations of autosomal dominant familial Alzheimer's disease: a comparison of the published literature with the dominantly inherited Alzheimer network observational study (DIAN-OBS). Lancet Neurol. (2016) 15:1317–25. doi: 10.1016/S1474-4422(16)30229-0
5. Wang JC, Alinaghi S, Tafakhori A, Sikora E, Azcona LJ, Karkheiran S, et al. Genetic screening in two Iranian families with early-onset Alzheimer's disease identified a novel PSEN1 mutation. Neurobiol Aging. (2018) 62:244.e215–7. doi: 10.1016/j.neurobiolaging.2017.10.011
6. Hunter S, Brayne C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatr. (2018) 23:81–93. doi: 10.1038/mp.2017.218
7. Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. (2017) 14:e1002270. doi: 10.1371/journal.pmed.1002270
8. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. (1993) 261:921–3. doi: 10.1126/science.8346443
9. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. (2013) 45:1452–8. doi: 10.1038/ng.2802
10. Sims R, Williams J. Defining the genetic architecture of Alzheimer's disease: where next. Neurodegener Dis. (2016) 16:6–11. doi: 10.1159/000440841
11. Yu JT, Xu W, Tan CC, Andrieu S, Suckling J, Evangelou E, et al. Evidence-based prevention of Alzheimer's disease: systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J Neurol Neurosurg Psychiatr. (2020). doi: 10.1136/jnnp-2019-321913. [Epub ahead of print].
12. Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. (2018) 14:653–66. doi: 10.1038/s41582-018-0070-3
13. Griñán-Ferré C, Izquierdo V, Otero E, Puigoriol-Illamola D, Corpas R, Sanfeliu C, et al. Environmental enrichment improves cognitive deficits, AD hallmarks and epigenetic alterations presented in 5xFAD mouse model. Front Cell Neurosci. (2018) 12:224. doi: 10.3389/fncel.2018.00224
14. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. (2019) 571:489–99. doi: 10.1038/s41586-019-1411-0
16. Qureshi IA, Mehler MF. Understanding neurological disease mechanisms in the era of epigenetics. JAMA Neurol. (2013) 70:703–10. doi: 10.1001/jamaneurol.2013.1443
17. Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A. The role of DNA methylation in epigenetics of aging. Pharmacol Ther. (2019) 195:172–85. doi: 10.1016/j.pharmthera.2018.11.001
18. Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. (2014) 17:215–22. doi: 10.1038/nn.3607
19. Li X, Zhao Q, Wei W, Lin Q, Magnan C, Emami MR, et al. The DNA modification N6-methyl-2′-deoxyadenosine (m6dA) drives activity-induced gene expression and is required for fear extinction. Nat Neurosci. (2019) 22:534–44. doi: 10.1038/s41593-019-0339-x
20. Griñán-Ferré C, Corpas R, Puigoriol-Illamola D, Palomera-Ávalos V, Sanfeliu C, Pallàs M. Understanding epigenetics in the neurodegeneration of Alzheimer's disease: SAMP8 mouse model. J Alzheimers Dis. (2018) 62:943–63. doi: 10.3233/JAD-170664
21. Morris-Blanco KC, Kim T, Lopez MS, Bertogliat MJ, Chelluboina B, Vemuganti R. Induction of DNA hydroxymethylation protects the brain after stroke. Stroke. (2019) 50:2513–21. doi: 10.1161/STROKEAHA.119.025665
22. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. (2014) 6:a019133. doi: 10.1101/cshperspect.a019133
23. Seritrakul P, Gross JM. Tet-mediated DNA hydroxymethylation regulates retinal neurogenesis by modulating cell-extrinsic signaling pathways. PLoS Genet. (2017) 13:e1006987. doi: 10.1371/journal.pgen.1006987
24. Iwata A, Nagata K, Hatsuta H, Takuma H, Bundo M, Iwamoto K, et al. Altered CpG methylation in sporadic Alzheimer's disease is associated with APP and MAPT dysregulation. Hum Mol Genet. (2014) 23:648–56. doi: 10.1093/hmg/ddt451
25. West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer's disease patient. J Mol Neurosci. (1995) 6:141–6. doi: 10.1007/BF02736773
26. Hou Y, Chen H, He Q, Jiang W, Luo T, Duan J, et al. Changes in methylation patterns of multiple genes from peripheral blood leucocytes of Alzheimer's disease patients. Acta Neuropsychiatr. (2013) 25:66–76. doi: 10.1111/j.1601-5215.2012.00662.x
27. Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. (2009) 68:880–91. doi: 10.1097/NEN.0b013e3181af2e46
28. Brohede J, Rinde M, Winblad B, Graff C. A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J Neurogenet. (2010) 24:179–81. doi: 10.3109/01677063.2010.503978
29. Li Q, Hermanson PJ, Springer NM. Detection of DNA methylation by whole-genome bisulfite sequencing. Methods Mol Biol. (2018) 1676:185–96. doi: 10.1007/978-1-4939-7315-6_11
30. Rogaev EI, Lukiw WJ, Lavrushina O, St Rogaeva EA. George-Hyslop PH. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics. (1994) 22:340–7. doi: 10.1006/geno.1994.1393
31. Ledoux S, Nalbantoglu J, Cashman NR. Amyloid precursor protein gene expression in neural cell lines: influence of DNA cytosine methylation. Brain Res Mol Brain Res. (1994) 24:140–4. doi: 10.1016/0169-328X(94)90125-2
32. Mani ST, Thakur MK. In the cerebral cortex of female and male mice, amyloid precursor protein (APP) promoter methylation is higher in females and differentially regulated by sex steroids. Brain Res. (2006) 1067:43–7. doi: 10.1016/j.brainres.2005.10.006
33. Piaceri I, Raspanti B, Tedde A, Bagnoli S, Sorbi S, Nacmias B. Epigenetic modifications in Alzheimer's disease: cause or effect? J Alzheimers Dis. (2015) 43:1169–73. doi: 10.3233/JAD-141452
34. Fuso A, Cavallaro RA, Nicolia V, Scarpa S. PSEN1 promoter demethylation in hyperhomocysteinemic TgCRND8 mice is the culprit, not the consequence. Curr Alzheimer Res. (2012) 9:527–35. doi: 10.2174/156720512800618053
35. Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE. (2007) 2:e895. doi: 10.1371/journal.pone.0000895
36. Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, et al. Genome-wide DNA methylation differences between late-onset Alzheimer's disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. (2012) 29:571–88. doi: 10.3233/JAD-2012-111223
37. Monti N, Cavallaro RA, Stoccoro A, Nicolia V, Scarpa S, Kovacs GG, et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics. (2020) 15:781–99. doi: 10.1080/15592294.2020.1722917
38. Flanagan JM, Popendikyte V, Pozdniakovaite N, Sobolev M, Assadzadeh A, Schumacher A, et al. Intra- and interindividual epigenetic variation in human germ cells. Am J Hum Genet. (2006) 79:67–84. doi: 10.1086/504729
39. Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer's disease. Nat Neurosci. (2014) 17:1164–70. doi: 10.1038/nn.3782
40. Furuya TK, da Silva PN, Payao SL, Rasmussen LT, de Labio RW, Bertolucci PH, et al. SORL1 and SIRT1 mRNA expression and promoter methylation levels in aging and Alzheimer's disease. Neurochem Int. (2012) 61:973–5. doi: 10.1016/j.neuint.2012.07.014
41. Foraker J, Millard SP, Leong L, Thomson Z, Chen S, Keene CD, et al. The APOE gene is differentially methylated in Alzheimer's disease. J Alzheimers Dis. (2015) 48:745–55. doi: 10.3233/JAD-143060
42. Tulloch J, Leong L, Thomson Z, Chen S, Lee EG, Keene CD, et al. Glia-specific APOE epigenetic changes in the Alzheimer's disease brain. Brain Res. (2018) 1698:179–86. doi: 10.1016/j.brainres.2018.08.006
43. An S, Khanna KK, Wu JM. mRNA levels and methylation patterns of the 2-5A synthetase gene in control and Alzheimer's disease (AD) fibroblasts. Biochem Mol Biol Int. (1994) 33:835–40.
44. Sanchez-Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A, et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer's disease. Brain. (2013) 136:3018–27. doi: 10.1093/brain/awt237
45. Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J, et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. (2015) 72:15–24. doi: 10.1001/jamaneurol.2014.3049
46. Nagata T, Kobayashi N, Ishii J, Shinagawa S, Nakayama R, Shibata N, et al. Association between DNA methylation of the BDNF promoter region and clinical presentation in Alzheimer's disease. Dement Geriatr Cogn Dis Extra. (2015) 5:64–73. doi: 10.1159/000375367
47. Chang L, Wang Y, Ji H, Dai D, Xu X, Jiang D, et al. Elevation of peripheral BDNF promoter methylation links to the risk of Alzheimer's disease. PLoS ONE. (2014) 9:e110773. doi: 10.1371/journal.pone.0110773
48. Carboni L, Lattanzio F, Candeletti S, Porcellini E, Raschi E, Licastro F, et al. Peripheral leukocyte expression of the potential biomarker proteins Bdnf, Sirt1, and Psen1 is not regulated by promoter methylation in Alzheimer's disease patients. Neurosci Lett. (2015) 605:44–8. doi: 10.1016/j.neulet.2015.08.012
49. Coppede F, Tannorella P, Stoccoro A, Chico L, Siciliano G, Bonuccelli U, et al. Methylation analysis of DNA repair genes in Alzheimer's disease. Mech Ageing Dev. (2017) 161:105–11. doi: 10.1016/j.mad.2016.04.003
50. Ma SL, Tang NL, Lam LC. Association of gene expression and methylation of UQCRC1 to the predisposition of Alzheimer's disease in a Chinese population. J Psychiatr Res. (2016) 76:143–7. doi: 10.1016/j.jpsychires.2016.02.010
51. Blanco-Luquin I, Altuna M, Sanchez-Ruiz de Gordoa J, Urdanoz-Casado A, Roldan M, Camara M, et al. PLD3 epigenetic changes in the hippocampus of Alzheimer's disease. Clin Epigenetics. (2018) 10:116. doi: 10.1186/s13148-018-0547-3
52. Ozaki Y, Yoshino Y, Yamazaki K, Sao T, Mori Y, Ochi S, et al. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer's disease. J Psychiatr Res. (2017) 92:74–80. doi: 10.1016/j.jpsychires.2017.04.003
53. Mendioroz M, Celarain N, Altuna M, Sanchez-Ruiz de Gordoa J, Zelaya MV, Roldan M, et al. CRTC1 gene is differentially methylated in the human hippocampus in Alzheimer's disease. Alzheimers Res Ther. (2016) 8:15. doi: 10.1186/s13195-016-0183-0
54. Salcedo-Tacuma D, Melgarejo JD, Mahecha MF, Ortega-Rojas J, Arboleda-Bustos CE, Pardo-Turriago R, et al. Differential methylation levels in CpGs of the BIN1 gene in individuals with Alzheimer disease. Alzheimer Dis Assoc Disord. (2019) 33:321–6. doi: 10.1097/WAD.0000000000000329
55. Yu L, Chibnik LB, Yang J, McCabe C, Xu J, Schneider JA, et al. Methylation profiles in peripheral blood CD4+ lymphocytes versus brain: the relation to Alzheimer's disease pathology. Alzheimers Dement. (2016) 12:942–51. doi: 10.1016/j.jalz.2016.02.009
56. Xu C, Liu G, Ji H, Chen W, Dai D, Chen Z, et al. Elevated methylation of OPRM1 and OPRL1 genes in Alzheimer's disease. Mol Med Rep. (2018) 18:4297–302. doi: 10.3892/mmr.2018.9424
57. Mercorio R, Pergoli L, Galimberti D, Favero C, Carugno M, Dalla Valle E, et al. PICALM gene methylation in blood of Alzheimer's disease patients is associated with cognitive decline. J Alzheimers Dis. (2018) 65:283–92. doi: 10.3233/JAD-180242
58. Smith AR, Smith RG, Burrage J, Troakes C, Al-Sarraj S, Kalaria RN, et al. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol Aging. (2019) 74:70–76. doi: 10.1016/j.neurobiolaging.2018.09.024
59. Hu W, Lin X, Zhang H, Zhao N. ATP binding cassette subfamily A member 2 (ABCA2) expression and methylation are associated with Alzheimer's disease. Med Sci Monit. (2017) 23:5851–61. doi: 10.12659/MSM.905524
60. Grosser C, Neumann L, Horsthemke B, Zeschnigk M, van de Nes J. Methylation analysis of SST and SSTR4 promoters in the neocortex of Alzheimer's disease patients. Neurosci Lett. (2014) 566:241–6. doi: 10.1016/j.neulet.2014.02.046
61. Silva PN, Furuya TK, Braga IL, Rasmussen LT, Labio RW, Bertolucci PH, et al. Analysis of HSPA8 and HSPA9 mRNA expression and promoter methylation in the brain and blood of Alzheimer's disease patients. J Alzheimers Dis. (2014) 38:165–70. doi: 10.3233/JAD-130428
62. Tannorella P, Stoccoro A, Tognoni G, Petrozzi L, Salluzzo MG, Ragalmuto A, et al. Methylation analysis of multiple genes in blood DNA of Alzheimer's disease and healthy individuals. Neurosci Lett. (2015) 600:143–7. doi: 10.1016/j.neulet.2015.06.009
63. Yamazaki K, Yoshino Y, Mori T, Yoshida T, Ozaki Y, Sao T, et al. Gene expression and methylation analysis of ABCA7 in patients with Alzheimer's disease. J Alzheimers Dis. (2017) 57:171–81. doi: 10.3233/JAD-161195
64. Celarain N, Sanchez-Ruiz de Gordoa J, Zelaya MV, Roldan M, Larumbe R, Pulido L, et al. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer's disease hippocampus. Clin Epigenetics. (2016) 8:37. doi: 10.1186/s13148-016-0202-9
65. Li P, Marshall L, Oh G, Jakubowski JL, Groot D, He Y, et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer's disease pathology and cognitive symptoms. Nat Commun. (2019) 10:2246. doi: 10.1038/s41467-019-10101-7
66. Humphries CE, Kohli MA, Nathanson L, Whitehead P, Beecham G, Martin E, et al. Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer's disease. J Alzheimers Dis. (2015) 44:977–87. doi: 10.3233/JAD-141989
67. Watson CT, Roussos P, Garg P, Ho DJ, Azam N, Katsel PL, et al. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer's disease. Genome Med. (2016) 8:5. doi: 10.1186/s13073-015-0258-8
68. De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. (2014) 17:1156–63. doi: 10.1038/nn.3786
69. Lardenoije R, Roubroeks JAY, Pishva E, Leber M, Wagner H, Iatrou A, et al. Alzheimer's disease-associated (hydroxy)methylomic changes in the brain and blood. Clin Epigenetics. (2019) 11:164. doi: 10.1186/s13148-019-0755-5
70. Gao Z, Fu HJ, Zhao LB, Sun ZY, Yang YF, Zhu HY. Aberrant DNA methylation associated with Alzheimer's disease in the superior temporal gyrus. Exp Ther Med. (2018) 15:103–8. doi: 10.3892/etm.2017.5394
71. Semick SA, Bharadwaj RA, Collado-Torres L, Tao R, Shin JH, Deep-Soboslay A, et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease. Acta Neuropathol. (2019) 137:557–69. doi: 10.1007/s00401-019-01966-5
72. Altuna M, Urdánoz-Casado A, Sánchez-Ruiz de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ, et al. DNA methylation signature of human hippocampus in Alzheimer's disease is linked to neurogenesis. Clin Epigenetics. (2019) 11:91. doi: 10.1186/s13148-019-0672-7
73. Fetahu IS, Ma D, Rabidou K, Argueta C, Smith M, Liu H. Epigenetic signatures of methylated DNA cytosine in Alzheimer's disease. Sci Adv. (2019) 5:eaaw2880. doi: 10.1126/sciadv.aaw2880
74. Zhao J, Zhu Y, Yang J, Li L, Wu H, De Jager PL, et al. A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer's disease. Alzheimers Dement. (2017) 13:674–88. doi: 10.1016/j.jalz.2016.10.004
75. Bernstein AI, Lin Y, Street RC, Lin L, Dai Q, Yu L, et al. 5-Hydroxymethylation-associated epigenetic modifiers of Alzheimer's disease modulate Tau-induced neurotoxicity. Hum Mol Genet. (2016) 25:2437–50. doi: 10.1093/hmg/ddw109
76. Qin L, Xu Q, Li Z, Chen L, Li Y, Yang N, et al. Ethnicity-specific and overlapping alterations of brain hydroxymethylome in Alzheimer's disease. Hum Mol Genet. (2020) 29:149–58. doi: 10.1093/hmg/ddz273
77. Smith AR, Smith RG, Pishva E, Hannon E, Roubroeks JAY, Burrage J, et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer's disease. Clin Epigenetics. (2019) 11:52. doi: 10.1186/s13148-019-0636-y
78. Schwob NG, Nalbantoglu J, Hastings KE, Mikkelsen T, Cashman NR. DNA cytosine methylation in brain of patients with Alzheimer's disease. Ann Neurol. (1990) 28:91–4. doi: 10.1002/ana.410280117
79. Lashley T, Gami P, Valizadeh N, Li A, Revesz T, Balazs R. Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer's disease. Neuropathol Appl Neurobiol. (2015) 41:497–506. doi: 10.1111/nan.12183
80. Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol Aging. (2010) 31:2025–37. doi: 10.1016/j.neurobiolaging.2008.12.005
81. Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol Aging. (2013) 34:2091–9. doi: 10.1016/j.neurobiolaging.2013.02.021
82. Condliffe D, Wong A, Troakes C, Proitsi P, Patel Y, Chouliaras L, et al. Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer's disease brain. Neurobiol Aging. (2014) 35:1850–4. doi: 10.1016/j.neurobiolaging.2014.02.002
83. Coppieters N, Dieriks BV, Lill C, Faull RL, Curtis MA, Dragunow M. Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol Aging. (2014) 35:1334–44. doi: 10.1016/j.neurobiolaging.2013.11.031
84. Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients. Transl Psychiatry. (2012) 2:e132. doi: 10.1038/tp.2012.55
85. Di Francesco A, Arosio B, Falconi A, Micioni Di Bonaventura MV, Karimi M, Mari D, et al. Global changes in DNA methylation in Alzheimer's disease peripheral blood mononuclear cells. Brain Behav Immun. (2015) 45:139–44. doi: 10.1016/j.bbi.2014.11.002
86. Griñán-Ferré C, Sarroca S, Ivanova A, Puigoriol-Illamola D, Aguado F, Camins A, et al. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY). (2016) 8:664–84. doi: 10.18632/aging.100906
87. Martínez-Iglesias O, Carrera I, Carril JC, Fernández-Novoa L, Cacabelos N, Cacabelos R. DNA methylation in neurodegenerative and cerebrovascular disorders. Int J Mol Sci. (2020) 21:2220. doi: 10.3390/ijms21062220
88. Cosín-Tomás M, Álvarez-López MJ, Companys-Alemany J, Kaliman P, González-Castillo C, Ortuño-Sahagún D, et al. Temporal integrative analysis of mRNA and microRNAs expression profiles and epigenetic alterations in female SAMP8, a model of age-related cognitive decline. Front Genet. (2018) 9:596. doi: 10.3389/fgene.2018.00596
89. Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. (2010) 5:e15367. doi: 10.1371/journal.pone.0015367
90. Cheng Y, Bernstein A, Chen D, Jin P. 5-Hydroxymethylcytosine: a new player in brain disorders? Exp Neurol. (2015) 268:3–9. doi: 10.1016/j.expneurol.2014.05.008
91. Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. (2016) 17:201–7. doi: 10.1038/nrn.2016.7
92. Shu L, Sun W, Li L, Xu Z, Lin L, Xie P, et al. Genome-wide alteration of 5-hydroxymenthylcytosine in a mouse model of Alzheimer's disease. BMC Genomics. (2016) 17:381. doi: 10.1186/s12864-016-2731-1
93. Bradley-Whitman MA, Lovell MA. Epigenetic changes in the progression of Alzheimer's disease. Mech Ageing Dev. (2013) 134:486–95. doi: 10.1016/j.mad.2013.08.005
94. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. (2010) 20(Suppl. 2):S265–79. doi: 10.3233/JAD-2010-100339
95. Blanch M, Mosquera JL, Ansoleaga B, Ferrer I, Barrachina M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol. (2016) 186:385–97. doi: 10.1016/j.ajpath.2015.10.004
96. Xu Y, Xu L, Han M, Liu X, Li F, Zhou X, et al. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an APP/PS1 transgenic mouse model of Alzheimer disease. Biochem Biophys Res Commun. (2019) 520:41–6. doi: 10.1016/j.bbrc.2019.09.094
97. Christopher MA, Kyle SM, Katz DJ. Neuroepigenetic mechanisms in disease. Epigenetics Chromatin. (2017) 10:47. doi: 10.1186/s13072-017-0150-4
98. Marzi SJ, Leung SK, Ribarska T, Hannon E, Smith AR, Pishva E, et al. A histone acetylome-wide association study of Alzheimer's disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat Neurosci. (2018) 21:1618–27. doi: 10.1038/s41593-018-0253-7
99. Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics. (2012) 12:1261–8. doi: 10.1002/pmic.201200010
100. Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. (2009) 18:131–9. doi: 10.3233/JAD-2009-1134
101. Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis. (2015) 74:281–94. doi: 10.1016/j.nbd.2014.11.023
102. Plagg B, Ehrlich D, Kniewallner KM, Marksteiner J, Humpel C. Increased acetylation of histone H4 at lysine 12 (H4K12) in monocytes of transgenic alzheimer's mice and in human patients. Curr Alzheimer Res. (2015) 12:752–60. doi: 10.2174/1567205012666150710114256
103. Schueller E, Paiva I, Blanc F, Wang XL, Cassel JC, Boutillier AL, et al. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer's disease patients. Eur Neuropsychopharmacol. (2020) 33:101–16. doi: 10.1016/j.euroneuro.2020.01.015
104. Anderson KW, Turko IV. Histone post-translational modifications in frontal cortex from human donors with Alzheimer's disease. Clin Proteomics. (2015) 12:26. doi: 10.1186/s12014-015-9098-1
105. Dyer M, Phipps AJ, Mitew S, Taberlay PC, Woodhouse A. Age, but not amyloidosis, induced changes in global levels of histone modifications in susceptible and disease-resistant neurons in Alzheimer's disease model mice. Front Aging Neurosci. (2019) 11:68. doi: 10.3389/fnagi.2019.00068
106. Klein HU, McCabe C, Gjoneska E, Sullivan SE, Kaskow BJ, Tang A, et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer's human brains. Nat Neurosci. (2019) 22:37–46. doi: 10.1038/s41593-018-0291-1
107. Zhou X, Xu J. Identification of Alzheimer's disease-associated long noncoding RNAs. Neurobiol Aging. (2015) 36:2925–31. doi: 10.1016/j.neurobiolaging.2015.07.015
108. Moutinho C, Esteller M. MicroRNAs and epigenetics. Adv Cancer Res. (2017) 135:189–220. doi: 10.1016/bs.acr.2017.06.003
109. Wang J, Yu JT, Tan MS, Jiang T, Tan L. Epigenetic mechanisms in Alzheimer's disease: implications for pathogenesis and therapy. Ageing Res Rev. (2013) 12:1024–41. doi: 10.1016/j.arr.2013.05.003
110. Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. (2009) 32:199–206. doi: 10.1016/j.tins.2008.12.003
111. Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer's-associated pathogenesis in SAMP8 mice. Neurobiol Aging. (2012) 33:522–34. doi: 10.1016/j.neurobiolaging.2010.04.034
112. Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA. (2008) 105:6415–20. doi: 10.1073/pnas.0710263105
113. Wang L, Liu J, Wang Q, Jiang H, Zeng L, Li Z, et al. MicroRNA-200a-3p mediates neuroprotection in Alzheimer-related deficits and attenuates amyloid-beta overproduction and tau hyperphosphorylation via coregulating BACE1 and PRKACB. Front Pharmacol. (2019) 10:806. doi: 10.3389/fphar.2019.00806
114. Tan X, Luo Y, Pi D, Xia L, Li Z, Tu Q. miR-340 reduces accumulation of amyloid-beta through targeting BACE1 (beta-site amyloid precursor protein cleaving enzyme 1) in Alzheimer' s disease. Curr Neurovasc Res. (2020) 114:86–92. doi: 10.2174/1567202617666200117103931
115. Barros-Viegas AT, Carmona V, Ferreiro E, Guedes J, Cardoso AM, Cunha P, et al. miRNA-31 improves cognition and abolishes amyloid-β pathology by targeting APP and BACE1 in an animal model of Alzheimer's disease. Mol Ther Nucleic Acids. (2020) 19:1219–36. doi: 10.1016/j.omtn.2020.01.010
116. Nelson PT, Wang WX. MiR-107 is reduced in Alzheimer's disease brain neocortex: validation study. J Alzheimers Dis. (2010) 21:75–9. doi: 10.3233/JAD-2010-091603
117. Fang M, Wang J, Zhang X, Geng Y, Hu Z, Rudd JA, et al. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer's disease. Toxicol Lett. (2012) 209:94–105. doi: 10.1016/j.toxlet.2011.11.032
118. Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. (2009) 284:1971–81. doi: 10.1074/jbc.M807530200
119. Zhu HC, Wang LM, Wang M, Song B, Tan S, Teng JF, et al. MicroRNA-195 downregulates Alzheimer's disease amyloid-beta production by targeting BACE1. Brain Res Bull. (2012) 88:596–601. doi: 10.1016/j.brainresbull.2012.05.018
120. Chopra N, Wang R, Maloney B, Nho K, Beck JS, Pourshafie N, et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol Psychiatr. (2020). doi: 10.1038/s41380-019-0610-2
121. Li P, Xu Y, Wang B, Huang J, Li Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J Neurol Sci. (2020) 413:116793. doi: 10.1016/j.jns.2020.116793
122. Li J, Chen W, Yi Y, Tong Q. miR-219-5p inhibits tau phosphorylation by targeting TTBK1 and GSK-3beta in Alzheimer's disease. J Cell Biochem. (2019) 120:9936–46. doi: 10.1002/jcb.28276
123. Ma X, Liu L, Meng J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer's disease. Neurosci Lett. (2017) 661:57–62. doi: 10.1016/j.neulet.2017.09.043
124. Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer's disease. Embo J. (2014) 33:1667–80. doi: 10.15252/embj.201387576
125. Yuan J, Wu Y, Li L, Liu C. MicroRNA-425-5p promotes tau phosphorylation and cell apoptosis in Alzheimer's disease by targeting heat shock protein B8. J Neural Trans. (2020) 127:339–46. doi: 10.1007/s00702-019-02134-5
126. El Fatimy R, Li S, Chen Z, Mushannen T, Gongala S, Wei Z, et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. (2018) 136:537–55. doi: 10.1007/s00401-018-1880-5
127. Kang Q, Xiang Y, Li D, Liang J, Zhang X, Zhou F, et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget. (2017) 8:24314–26. doi: 10.18632/oncotarget.15149
128. Liu W, Zhao J, Lu G. miR-106b inhibits tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer's disease. Biochem Biophys Res Commun. (2016) 478:852–7. doi: 10.1016/j.bbrc.2016.08.037
129. Yao X, Xian X, Fang M, Fan S, Li W. Loss of miR-369 promotes Tau phosphorylation by targeting the Fyn and serine/threonine-protein kinase 2 signaling pathways in Alzheimer's disease mice. Front Aging Neurosci. (2019) 11:365. doi: 10.3389/fnagi.2019.00365
130. He B, Chen W, Zeng J, Tong W, Zheng P. MicroRNA-326 decreases tau phosphorylation and neuron apoptosis through inhibition of the JNK signaling pathway by targeting VAV1 in Alzheimer's disease. J Cell Physiol. (2020) 235:480–93. doi: 10.1002/jcp.28988
131. Cao J, Huang M, Guo L, Zhu L, Hou J, Zhang L, et al. MicroRNA-195 rescues ApoE4-induced cognitive deficits and lysosomal defects in Alzheimer's disease pathogenesis. Mol Psychiatr. (2020). doi: 10.1038/s41380-020-0824-3
132. Long JM, Maloney B, Rogers JT, Lahiri DK. Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer's disease. Mol Psychiatr. (2019) 24:345–63. doi: 10.1038/s41380-018-0266-3
133. Delay C, Calon F, Mathews P, Hébert SS. Alzheimer-specific variants in the 3'UTR of amyloid precursor protein affect microRNA function. Mol Neurodegener. (2011) 6:70. doi: 10.1186/1750-1326-6-70
134. Zhou Y, Wang ZF, Li W, Hong H, Chen J, Tian Y, et al. Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer's disease by targeting VAV1 via the MAPK signaling pathway. J Cell Biochem. (2018) 119:5437–48. doi: 10.1002/jcb.26700
135. Lu Y, Tan L, Wang X. Circular HDAC9/microRNA-138/sirtuin-1 pathway mediates synaptic and amyloid precursor protein processing deficits in Alzheimer's disease. Neurosci Bull. (2019) 35:877–88. doi: 10.1007/s12264-019-00361-0
136. Chen FZ, Zhao Y, Chen HZ. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer's disease mice. Int J Mol Med. (2019) 43:91–102. doi: 10.3892/ijmm.2018.3957
137. Dube U, Del-Aguila JL, Li Z, Budde JP, Jiang S, Hsu S, et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat Neurosci. (2019) 22:1903–12. doi: 10.1038/s41593-019-0501-5
138. Zhang Y, Yu F, Bao S, Sun J. Systematic characterization of circular RNA-associated CeRNA network identified novel circRNA biomarkers in Alzheimer's disease. Front Bioeng Biotechnol. (2019) 7:222. doi: 10.3389/fbioe.2019.00222
139. Yang H, Wang H, Shang H, Chen X, Yang S, Qu Y, et al. Circular RNA circ_0000950 promotes neuron apoptosis, suppresses neurite outgrowth and elevates inflammatory cytokines levels via directly sponging miR-103 in Alzheimer's disease. Cell Cycle. (2019) 18:2197–214. doi: 10.1080/15384101.2019.1629773
140. Diling C, Yinrui G, Longkai Q, Xiaocui T, Yadi L, Xin Y, et al. Circular RNA NF1-419 enhances autophagy to ameliorate senile dementia by binding Dynamin-1 and adaptor protein 2 B1 in AD-like mice. Aging. (2019) 11:12002–31. doi: 10.18632/aging.102529
141. Li Y, Fan H, Sun J, Ni M, Zhang L, Chen C, et al. Circular RNA expression profile of Alzheimer's disease and its clinical significance as biomarkers for the disease risk and progression. Int J Biochem Cell Biol. (2020) 123:105747. doi: 10.1016/j.biocel.2020.105747
142. Lo I, Hill J, Vilhjálmsson BJ, Kjems J. Linking the association between circRNAs and Alzheimer's disease progression by multi-tissue circular RNA characterization. RNA Biol. (2020). doi: 10.1080/15476286.2020.1783487. [Epub ahead of print].
143. Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, et al. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. (2010) 11:14. doi: 10.1186/1471-2202-11-14
144. Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, et al. Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. (2008) 14:723–30. doi: 10.1038/nm1784
145. Zhang W, Zhao H, Wu Q, Xu W, Xia M. Knockdown of BACE1-AS by siRNA improves memory and learning behaviors in Alzheimer's disease animal model. Exp Ther Med. (2018) 16:2080–86. doi: 10.3892/etm.2018.6359
146. Fotuhi SN, Khalaj-Kondori M, Hoseinpour Feizi MA, Talebi M. Long non-coding RNA BACE1-AS may serve as an Alzheimer's disease blood-based biomarker. J Mol Neurosci. (2019) 69:351–9. doi: 10.1007/s12031-019-01364-2
147. Luo Q, Chen Y. Long noncoding RNAs and Alzheimer's disease. Clin Interv Aging. (2016) 11:867–72. doi: 10.2147/CIA.S107037
148. Zhang L, Fang Y, Cheng X, Lian YJ, Xu HL. Silencing of long noncoding RNA SOX21-AS1 relieves neuronal oxidative stress injury in mice with Alzheimer's disease by upregulating fzd3/5 via the Wnt signaling pathway. Mol Neurobiol. (2019) 56:3522–37. doi: 10.1007/s12035-018-1299-y
149. Mus E, Hof PR, Tiedge H. Dendritic BC200 RNA in aging and in Alzheimer's disease. Proc Natl Acad Sci USA. (2007) 104:10679–84. doi: 10.1073/pnas.0701532104
150. Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. (2012) 30:453–9. doi: 10.1038/nbt.2158
151. Knauss JL, Miao N, Kim SN, Nie Y, Shi Y, Wu T, et al. Long noncoding RNA Sox2ot and transcription factor YY1 co-regulate the differentiation of cortical neural progenitors by repressing Sox2. Cell Death Dis. (2018) 9:799. doi: 10.1038/s41419-018-0840-2
152. Gu C, Chen C, Wu R, Dong T, Hu X, Yao Y, et al. Long noncoding RNA EBF3-AS promotes neuron apoptosis in Alzheimer's disease. DNA Cell Biol. (2018) 37:220–6. doi: 10.1089/dna.2017.4012
153. Parenti R, Paratore S, Torrisi A, Cavallaro S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during beta-amyloid-induced apoptosis. Eur J Neurosci. (2007) 26:2444–57. doi: 10.1111/j.1460-9568.2007.05864.x
154. Li X, Wang SW, Li XL, Yu FY, Cong HM. Knockdown of long non-coding RNA TUG1 depresses apoptosis of hippocampal neurons in Alzheimer's disease by elevating microRNA-15a and repressing ROCK1 expression. Inflamm Res. (2020) 69:897–910. doi: 10.1007/s00011-020-01364-8
155. Hong H, Mo Y, Li D, Xu Z, Liao Y, Yin P, et al. Aberrant expression profiles of lncRNAs and their associated nearby coding genes in the hippocampus of the SAMP8 mouse model with AD. Mol Ther Nucleic Acids. (2020) 20:140–54. doi: 10.1016/j.omtn.2020.02.008
156. Cao M, Li H, Zhao J, Cui J, Hu G. Identification of age- and gender-associated long noncoding RNAs in the human brain with Alzheimer's disease. Neurobiol Aging. (2019) 81:116–26. doi: 10.1016/j.neurobiolaging.2019.05.023
157. Wang Z, Li K, Huang W. Long non-coding RNA NEAT1-centric gene regulation. Cell Mol Life Sci. (2020) 77:3769–79. doi: 10.1007/s00018-020-03503-0
158. Wang Z, Zhao Y, Xu N, Zhang S, Wang S, Mao Y, et al. NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Cell Mol Life Sci. (2019) 76:3005–18. doi: 10.1007/s00018-019-03074-9
159. Huang Z, Zhao J, Wang W, Zhou J, Zhang J. Depletion of LncRNA NEAT1 rescues mitochondrial dysfunction through NEDD4L-dependent PINK1 degradation in animal models of Alzheimer's disease. Front Cell Neurosci. (2020) 14:28. doi: 10.3389/fncel.2020.00028
160. Oliveira AM, Hemstedt TJ, Bading H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat Neurosci. (2012) 15:1111–13. doi: 10.1038/nn.3151
161. Kunisawa K, Nakashima N, Nagao M, Nomura T, Kinoshita S, Hiramatsu M. Betaine prevents homocysteine-induced memory impairment via matrix metalloproteinase-9 in the frontal cortex. Behav Brain Res. (2015) 292:36–43. doi: 10.1016/j.bbr.2015.06.004
162. Fuso A, Nicolia V, Ricceri L, Cavallaro RA, Isopi E, Mangia F, et al. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol Aging. (2012) 33:1482.e1481–1416. doi: 10.1016/j.neurobiolaging.2011.12.013
163. Lai G, Guo Y, Chen D, Tang X, Shuai O, Yong T, et al. Alcohol extracts from ganoderma lucidum delay the progress of Alzheimer's disease by regulating DNA methylation in rodents. Front Pharmacol. (2019) 10:272. doi: 10.3389/fphar.2019.00272
164. An Y, Feng L, Zhang X, Wang Y, Wang Y, Tao L, et al. Dietary intakes and biomarker patterns of folate, vitamin B(6), and vitamin B(12) can be associated with cognitive impairment by hypermethylation of redox-related genes NUDT15 and TXNRD1. Clin Epigenetics. (2019) 11:139. doi: 10.1186/s13148-019-0741-y
165. Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. (2007) 369:208–16. doi: 10.1016/S0140-6736(07)60109-3
166. Izquierdo V, Palomera-Ávalos V, López-Ruiz S, Canudas AM, Pallàs M, Griñán-Ferré C. Maternal resveratrol supplementation prevents cognitive decline in senescent mice offspring. Int J Mol Sci. (2019) 20:1134. doi: 10.3390/ijms20051134
167. Griñán-Ferré C, Marsal-García L, Bellver-Sanchis A, Kondengaden SM, Turga RC, Vázquez S, et al. Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and β-Amyloid plaques in an early-onset Alzheimer's disease mouse model. Aging. (2019) 11:11591–608. doi: 10.18632/aging.102558
168. Xuan AG, Pan XB, Wei P, Ji WD, Zhang WJ, Liu JH, et al. Valproic acid alleviates memory deficits and attenuates amyloid-beta deposition in transgenic mouse model of Alzheimer's disease. Mol Neurobiol. (2015) 51:300–12. doi: 10.1007/s12035-014-8751-4
169. Corbett GT, Roy A, Pahan K. Sodium phenylbutyrate enhances astrocytic neurotrophin synthesis via protein kinase C (PKC)-mediated activation of cAMP-response element-binding protein (CREB): implications for Alzheimer disease therapy. J Biol Chem. (2013) 288:8299–312. doi: 10.1074/jbc.M112.426536
170. Volmar CH, Salah-Uddin H, Janczura KJ, Halley P, Lambert G, Wodrich A, et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing Alzheimer's disease genes and improving memory. Proc Natl Acad Sci USA. (2017) 114:E9135–44. doi: 10.1073/pnas.1707544114
171. Sung YM, Lee T, Yoon H, DiBattista AM, Song JM, Sohn Y, et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer's disease. Exp Neurol. (2013) 239:192–201. doi: 10.1016/j.expneurol.2012.10.005
172. Rustenhoven J, Smith AM, Smyth LC, Jansson D, Scotter EL, Swanson MEV, et al. PU.1 regulates Alzheimer's disease-associated genes in primary human microglia. Mol Neurodegener. (2018) 13:44. doi: 10.1186/s13024-018-0277-1
173. Janczura KJ, Volmar CH, Sartor GC, Rao SJ, Ricciardi NR, Lambert G, et al. Inhibition of HDAC3 reverses Alzheimer's disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc Natl Acad Sci USA. (2018) 115:E11148–e57. doi: 10.1073/pnas.1805436115
174. Chatterjee S, Mizar P, Cassel R, Neidl R, Selvi BR, Mohankrishna DV, et al. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J Neurosci. (2013) 33:10698–712. doi: 10.1523/JNEUROSCI.5772-12.2013
175. Zheng Y, Liu A, Wang ZJ, Cao Q, Wang W, Lin L, et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer's disease. Brain. (2019) 142:787–807. doi: 10.1093/brain/awy354
176. Wang P, Zheng X, Guo Q, Yang P, Pang X, Qian K, et al. Systemic delivery of BACE1 siRNA through neuron-targeted nanocomplexes for treatment of Alzheimer's disease. J Control Release. (2018) 279:220–33. doi: 10.1016/j.jconrel.2018.04.034
177. Qazi TJ, Quan Z, Mir A, Qing H. Epigenetics in Alzheimer's disease: perspective of DNA methylation. Mol Neurobiol. (2018) 55:1026–44. doi: 10.1007/s12035-016-0357-6
178. Liu CG, Wang JL, Li L, Wang PC. MicroRNA-384 regulates both amyloid precursor protein and β-secretase expression and is a potential biomarker for Alzheimer's disease. Int J Mol Med. (2014) 34:160–6. doi: 10.3892/ijmm.2014.1780
179. Jaber VR, Zhao Y, Sharfman NM, Li W, Lukiw WJ. Addressing Alzheimer's disease (AD) neuropathology using anti-microRNA (AM) strategies. Mol Neurobiol. (2019) 56:8101–8. doi: 10.1007/s12035-019-1632-0
180. Bhatnagar S, Chertkow H, Schipper HM, Yuan Z, Shetty V, Jenkins S, et al. Increased microRNA-34c abundance in Alzheimer's disease circulating blood plasma. Front Mol Neurosci. (2014) 7:2. doi: 10.3389/fnmol.2014.00002
181. An F, Gong G, Wang Y, Bian M, Yu L, Wei C. MiR-124 acts as a target for Alzheimer's disease by regulating BACE1. Oncotarget. (2017) 8:114065–71. doi: 10.18632/oncotarget.23119
182. Van den Hove DL, Kompotis K, Lardenoije R, Kenis G, Mill J, Steinbusch HW, et al. Epigenetically regulated microRNAs in Alzheimer's disease. Neurobiol Aging. (2014) 35:731–45. doi: 10.1016/j.neurobiolaging.2013.10.082
183. Lee WJ, Moon J, Jeon D, Shin YW, Yoo JS, Park DK, et al. Possible epigenetic regulatory effect of dysregulated circular RNAs in Alzheimer's disease model. Sci Rep. (2019) 9:11956. doi: 10.1038/s41598-019-48471-z
184. Quan Z, Zheng D, Qing H. Regulatory roles of long non-coding RNAs in the central nervous system and associated neurodegenerative diseases. Front Cell Neurosci. (2017) 11:175. doi: 10.3389/fncel.2017.00175
185. Millan MJ. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer's disease: an integrative review. Prog Neurobiol. (2017) 156:1–68. doi: 10.1016/j.pneurobio.2017.03.004
186. Mulder C, Schoonenboom NS, Jansen EE, Verhoeven NM, van Kamp GJ, Jakobs C, et al. The transmethylation cycle in the brain of Alzheimer patients. Neurosci Lett. (2005) 386:69–71. doi: 10.1016/j.neulet.2005.03.073
187. Oliveira AM, Hemstedt TJ, Freitag HE, Bading H. Dnmt3a2: a hub for enhancing cognitive functions. Mol Psychiatr. (2016) 21:1130–6. doi: 10.1038/mp.2015.175
188. Linnebank M, Popp J, Smulders Y, Smith D, Semmler A, Farkas M, et al. S-adenosylmethionine is decreased in the cerebrospinal fluid of patients with Alzheimer's disease. Neurodegener Dis. (2010) 7:373–8. doi: 10.1159/000309657
189. Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. (2004) 103:1635–40. doi: 10.1182/blood-2003-03-0687
190. Sandi C, Al-Mahdawi S, Pook MA. Epigenetics in friedreich's ataxia: challenges and opportunities for therapy. Genet Res Int. (2013) 2013:852080. doi: 10.1155/2013/852080
191. Kumar D, Aggarwal M, Kaas GA, Lewis J, Wang J, Ross DL, et al. Tet1 oxidase regulates neuronal gene transcription, active DNA hydroxy-methylation, object location memory, and threat recognition memory. Neuroepigenetics. (2015) 4:12–27. doi: 10.1016/j.nepig.2015.10.002
192. Amin SA, Adhikari N, Kotagiri S, Jha T, Ghosh B. Histone deacetylase 3 inhibitors in learning and memory processes with special emphasis on benzamides. Eur J Med Chem. (2019) 166:369–80. doi: 10.1016/j.ejmech.2019.01.077
193. Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. (2012) 483:222–6. doi: 10.1038/nature10849
194. Song JM, Sung YM, Nam JH, Yoon H, Chung A, Moffat E, et al. A mercaptoacetamide-based class II histone deacetylase inhibitor increases dendritic spine density via RasGRF1/ERK pathway. J Alzheimers Dis. (2016) 51:591–604. doi: 10.3233/JAD-150717
195. Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. (2017) 20:1052–61. doi: 10.1038/nn.4587
196. Yang SS, Zhang R, Wang G, Zhang YF. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer's disease. Transl Neurodegener. (2017) 6:19. doi: 10.1186/s40035-017-0089-1
197. Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. (2010) 107:22687–92. doi: 10.1073/pnas.1012851108
198. Long JM, Ray B, Lahiri DK. MicroRNA-339-5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem. (2014) 289:5184–98. doi: 10.1074/jbc.M113.518241
199. Lei X, Lei L, Zhang Z, Zhang Z, Cheng Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer's disease. Int J Clin Exp Pathol. (2015) 8:1565–74.
200. Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. (2010) 285:18344–51. doi: 10.1074/jbc.M110.112664
201. Long JM, Ray B, Lahiri DK. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J Biol Chem. (2012) 287:31298–310. doi: 10.1074/jbc.M112.366336
202. Idda ML, Munk RK, Gorospe M. Noncoding RNAs in Alzheimer's disease. Wiley Interdiscip Rev RNA. (2018) 9:1463. doi: 10.1002/wrna.1463
203. Liu X, Jiao B, Shen L. The epigenetics of Alzheimer's disease: factors and therapeutic implications. Front Genet. (2018) 9:579. doi: 10.3389/fgene.2018.00579
204. Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. (2020) 27:18. doi: 10.1186/s12929-019-0609-7
205. Knopman D, Patterson M. An open-label, 24-week pilot study of the methyl donor betaine in Alzheimer disease patients. Alzheimer Dis Assoc Disord. (2001) 15:162–5. doi: 10.1097/00002093-200107000-00008
206. Remington R, Chan A, Paskavitz J, Shea TB. Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer's disease: a placebo-controlled pilot study. Am J Alzheimers Dis Other Demen. (2009) 24:27–33. doi: 10.1177/1533317508325094
Keywords: Alzheiemer's disease, epigenetic, DNA methylation, DNA hydroxymethylation, histone modifications, non-coding RNA (ncRNA)
Citation: Xiao X, Liu X and Jiao B (2020) Epigenetics: Recent Advances and Its Role in the Treatment of Alzheimer's Disease. Front. Neurol. 11:538301. doi: 10.3389/fneur.2020.538301
Received: 27 February 2020; Accepted: 03 September 2020;
Published: 15 October 2020.
Edited by:
Keith Vossel, Mary S. Easton Center for Alzheimer's Disease Research at UCLA, United StatesReviewed by:
Christian Griñán-Ferré, Bosch i Gimpera Foundation, SpainAnderson A. Butler, University of Alabama at Birmingham, United States
Copyright © 2020 Xiao, Liu and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Jiao, amJpbjA5MTFAMTYzLmNvbQ==