
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 17 September 2020
Sec. Epilepsy
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.01028
This article is part of the Research Topic Glial Dysfunction in Epileptogenesis View all 16 articles
 Till S. Zimmer1
Till S. Zimmer1 Diede W. M. Broekaart1Victoria-Elisabeth Gruber2
Diede W. M. Broekaart1Victoria-Elisabeth Gruber2 Erwin A. van Vliet1,3Angelika Mühlebner1Eleonora Aronica1,4*
Erwin A. van Vliet1,3Angelika Mühlebner1Eleonora Aronica1,4*Tuberous sclerosis complex (TSC) represents the prototypic monogenic disorder of the mammalian target of rapamycin (mTOR) pathway dysregulation. It provides the rational mechanistic basis of a direct link between gene mutation and brain pathology (structural and functional abnormalities) associated with a complex clinical phenotype including epilepsy, autism, and intellectual disability. So far, research conducted in TSC has been largely neuron-oriented. However, the neuropathological hallmarks of TSC and other malformations of cortical development also include major morphological and functional changes in glial cells involving astrocytes, oligodendrocytes, NG2 glia, and microglia. These cells and their interglial crosstalk may offer new insights into the common neurobiological mechanisms underlying epilepsy and the complex cognitive and behavioral comorbidities that are characteristic of the spectrum of mTOR-associated neurodevelopmental disorders. This review will focus on the role of glial dysfunction, the interaction between glia related to mTOR hyperactivity, and its contribution to epileptogenesis in TSC. Moreover, we will discuss how understanding glial abnormalities in TSC might give valuable insight into the pathophysiological mechanisms that could help to develop novel therapeutic approaches for TSC or other pathologies characterized by glial dysfunction and acquired mTOR hyperactivation.
Tuberous sclerosis complex (TSC) is a rare, genetic multisystem disorder with a prevalence of ~1:6,000 newborns. Common symptoms in TSC include benign tumor growth in kidney, heart, lung, eyes, skin, and brain (1). Characteristic lesions in the brain are cortical tubers and ventricular subependymal nodules, which may progress into subependymal giant cell astrocytomas (SEGAs) (2–4). Neurological manifestations include epilepsy, neurodevelopmental delay, and TSC-associated neuropsychiatric disorders (TANDs), such as intellectual disability and autism spectrum disorder (ASD) (5–7). Moreover, as one of the most debilitating symptoms, TSC represents the most common genetic cause for pediatric epilepsy, with roughly 85% of cases developing seizures, predominantly within the first year of life, and 60% eventually presenting with refractory epilepsy (8, 9). Because uncontrolled seizure activity aggravates cognitive comorbidities, immediate seizure management after or ideally before epilepsy onset is crucial for normal cognitive development of patients (10–12). Currently, the most effective long-term treatment for epilepsy in TSC is vigabatrin, a highly effective drug against infantile spasms in TSC patients (13–15), whereas a subgroup of eligible patients benefits from adjunctive everolimus [mammalian target of rapamycin (mTOR) inhibitor] treatment or surgical resection of the suspected epileptogenic lesion (14, 16–19).
TSC is caused by loss-of-function mutations in the tumor suppressors TSC1 or TSC2, both of which are negative regulators of the mTOR (20, 21). Purely heterozygous germline mutations, as well as mosaic mutations, have been detected in TSC patients (21, 22). mTOR is a serine/threonine protein kinase and the catalytic subunit of mTOR complex 1 (mTORC1) and mTORC2. Under normal conditions, mTOR activity is tightly controlled by upstream regulators and acts as important sensor of cellular energy status and homeostasis. Environmental stimuli, such as cytokines or growth factors can stimulate mTOR, enabling cells to dynamically respond to various extracellular cues via adaptation in metabolism or cellular growth (23, 24). Mutations in either TSC1 or TSC2 lead to uncoupling from upstream regulators and abnormal hyperactivation of mTORC1, causing growth of the characteristic lesions during brain development. While TSC represents the prototypic monogenic disorder of mTOR hyperactivation, other malformations of cortical development, such as megalencephaly, hemimegalencephaly, and focal cortical dysplasia (FCD) are also characterized by aberrant mTOR activation due to acquired mutations in various mTOR regulators (25). Importantly, all share histopathological and clinical characteristics with TSC; hence, this spectrum of diseases is collectively referred to as mTORopathies [reviewed in (26, 27)].
Importantly, mTOR hyperactivity seems to be directly linked to epileptogenesis as mTOR inhibitors can suppress seizures in preclinical TSC models (28, 29), as well as in clinical studies aimed at treating TSC and SEGAs (16–19, 30, 31). Current consensus is that mTOR inhibitors induce a temporary anticonvulsant effect as do currently available antiepileptic drugs, but may also possess disease-modifying potential (15, 32). The clear causative role of mTOR as epileptogenic driver, as well as implications of mTOR activation in acquired epilepsies (33–36), makes TSC an attractive disease model to utilize as translational prototype for epilepsy in general. Despite the progress in understanding the role of the mTOR signaling pathway, there is still a lack in pinpointing the precise cellular substrates responsible for producing seizures. Interestingly, although the neuropathological hallmarks of TSC are primarily found in tubers, some studies showed that the seizure focus in TSC brains could also originate from the surrounding normal-appearing cortex, based on seizure freedom after resection of the perituberal zone, tissue analysis, and electrocorticographic recordings (37–40). However, further progress in the careful examination and advances in the identification of novel histopathological markers may make a discrimination between tuber and perituber obsolete, eventually. Nevertheless, surgical resection of the tuber leads to seizure relief in 50% to 60% of cases, suggesting an important role in epileptogenesis in at least a subset of patients with a clear-cut epileptogenic “driver” lesion (41–44).
In the brain of TSC patients, mTOR hyperactivity promotes development of often multifocal brain lesions characterized by aberrant glioneuronal proliferation, cortical dyslamination, and hypomyelination, along with the presence of dysplastic neurons and improperly developed giant cells (4, 27, 45–47). TSC tissue obtained from surgery due to refractory epilepsy usually presents with a heterogeneous frequency of the aforementioned histopathological hallmarks between patients (27, 46). TSC lesions are thought to arise by the Knudson hypothesis, also known as the “two-hit” hypothesis (48). Accordingly, somatic mutations in either TSC1 or TSC2, resulting in the loss of wild-type alleles, have been detected in different types of TSC neoplastic lesions and to a lesser extent in cortical tubers (21, 49, 50). Thus, it is still an ongoing matter of discussion whether monoallelic inactivation of TSC1/TSC2 is sufficient for tuber development or if the second hit occurs in a specific cellular component complicating its identification (21, 49, 50). Cell specificity, mutation load, and mutation timing during brain development likely give rise to the diverse neuropathological presentations. Recent evidence from in vitro cell cultures and organoid models of TSC revealed that mTORC1 activity during cortical development is tightly controlled, and mTORC1 suppression is required for proper neurogenesis (51). Of note, mTORC1 hyperactivity promotes gliogenesis, likely explaining the increased number of glia in tubers (52–56). More specifically, mTORC1 was shown to activate STAT3 signaling, which represents a major driver of gliogenesis during development (53, 57–60) Furthermore, gliosis and activation of inflammatory signaling pathways are histopathological hallmarks of TSC (46, 52, 61–64). Accordingly, although dysfunctional neuronal circuitry is ultimately required for the development of epilepsy and mTOR can directly regulate neuronal structure, function, and plasticity (65–68), accumulating evidence shows that glial cells represent a crucial element in the pathogenesis of TSC and might pose novel therapeutic strategies (64). This review will focus on the role of glial dysfunction related to mTOR hyperactivity and its contribution to comorbidities, such as epilepsy and TANDs in TSC. In this context, while many studies primarily focused on neuroglial crosstalk, we will emphasize aberrant interglial communication as an essential aspect of TSC. Finally, studying glial abnormalities in TSC might give valuable insight into pathophysiological mechanisms, which could help to develop novel therapeutic approaches for TSC or other pathologies characterized by gliopathic changes and acquired mTOR hyperactivation (summarized in Figure 1).
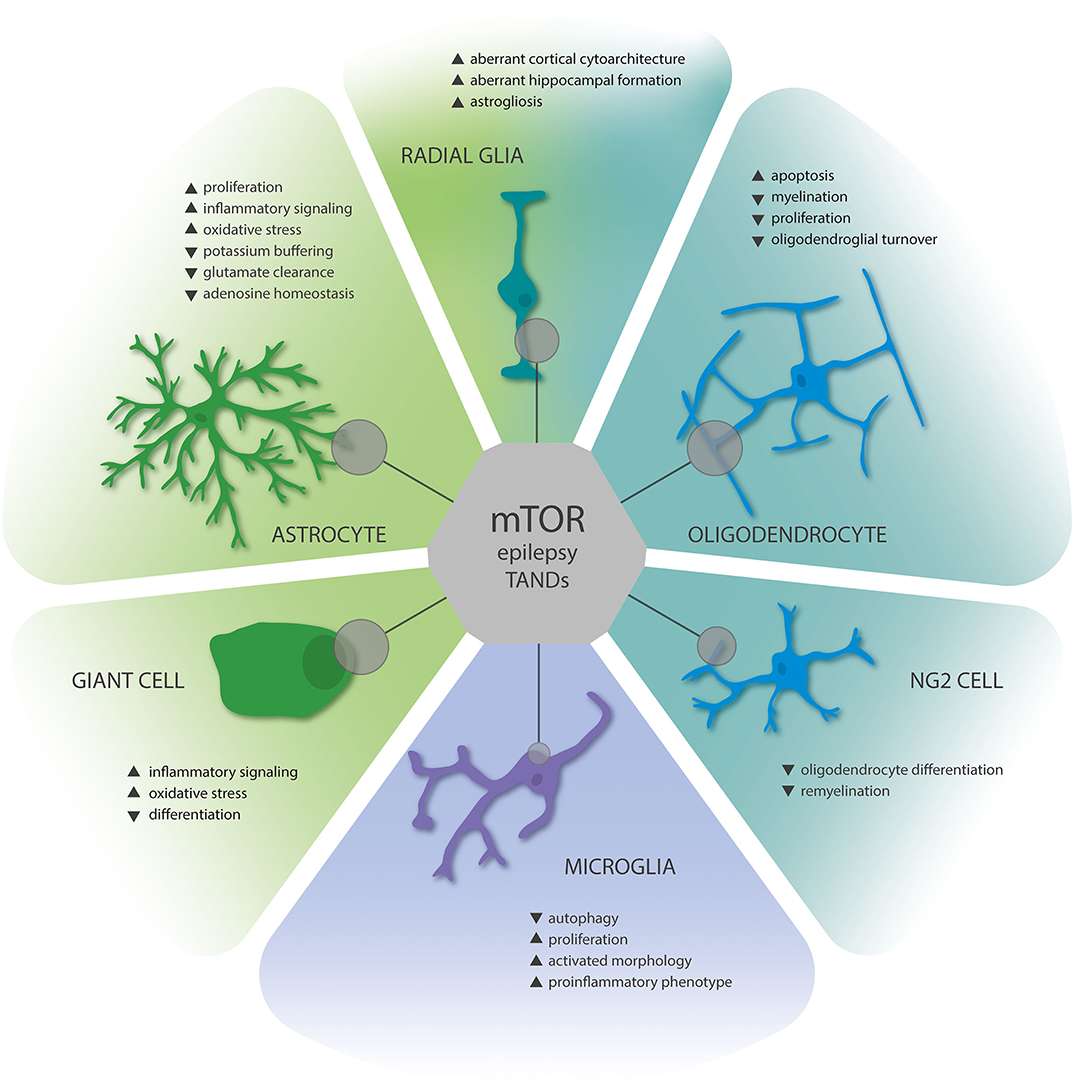
Figure 1. Summary of gliopathic changes due to mTOR hyperactivation in TSC brain lesions. Astrocytes display increased proliferation, activation, and enhanced expression of proinflammatory mediators. Moreover, astrocytes are characterized by decreased homeostatic functions related to ion homeostasis and neurotransmitter metabolism. Radial glia, the neuroglial precursors of astrocytes, oligodendrocytes, and neurons, contribute to malformations of cortical development and aberrant gliogenesis, as well as the formation of giant cells, which display characteristics of proinflammatory glia. Oligodendrocyte dysfunction leads to hypomyelination and disturbed remyelination, and their proliferation is reduced. While dysfunction of NG2 glia in TSC deserves further investigation, they are crucially involved in myelination and crosstalk with neurons, thus representing an essential component of TSC gliopathology. Finally, as for astrocytes, microglia are characterized by enhanced proliferation, activation, and expression of proinflammatory mediators; however, these changes are likely secondary to mTOR activation in the TSC brain. Collectively, these changes contribute to epilepsy and neuropsychiatric comorbidities in TSC. The influence of mTOR signaling on the individual cell types is indicated by the size of gray circles.
Astrocytes display distinct functional changes in a variety of epilepsies with different etiologies, and it becomes increasingly clear that they play crucial roles in the process of epileptogenesis, including TSC (69, 70). Neuropathological hallmarks in resected cortical tubers of TSC patients include increased expression of glial fibrillary acidic protein (GFAP), vimentin, and S100β, as well as higher numbers of astrocytes. Moreover, these astrocytes often present dysplastic and reactive phenotypes compared to the perituberal area and control brain tissue (71, 72). While most studies characterize the total population of astrocytes, some report different subpopulations of astrocytes in TSC (52, 72). One study characterized two subpopulations of astrocytes: “reactive” cells, which are large and vimentin positive and reveal mTOR activation, and “gliotic” astrocytes, which are smaller, do not show mTOR activation, and resemble gliotic astrocytes found in hippocampal sclerosis (HS) (52). Gliotic astrocytes, as in HS, present with decreased expression of inwardly rectifying potassium (Kir) channel subunit Kir4.1, a decrease in the glutamate transporters excitatory amino acid transporter 1 (EAAT1) and EAAT2, and a decrease in glutamine synthetase, all of which represent proepileptogenic changes (70). The authors of this study concluded that the gradual transformation from reactive to gliotic astrocytes might represent a major driving force for the morphological dynamics of tubers over time (52). Another study discriminated between normal astrocytes (no mTOR activation, vimentin-negative, and GFAP-positive), reactive astrocytes (no mTOR activation, vimentin-positive, and GFAP-positive), and dysplastic astroglia (mTOR activation, vimentin-positive, and GFAP-negative), the latter representing an expression pattern common to immature astrocytes and radial glia (72). Taken together, both studies support the notion that populations of improperly differentiated astrocytes with mTOR activation, as well as properly developed, reactive astrocytes without mTOR activation, contribute to TSC pathology. Here, the aforementioned continuum of pathological changes in astrocytes and the precise cellular composition of the tissue might reflect the intrinsic epileptogenicity of the tuber. Importantly, the functional changes in TSC astrocytes are likely caused by a combination of the reactive state in response to seizures known from other diseases, such as mesial temporal lobe epilepsy (TLE), which could induce secondary mTOR activation (33), but also general disturbance in protein translation caused by sustained mTOR activation in mutation-carrying cells. Ultimately, both astrocyte subpopulations could end up having different pathogenic origins, but similar functional outcomes in terms of expression of Kirs, EAATs, or glutamine synthetase, further increasing the epileptogenic potential of the tuber. Whether the different degrees of mTOR activation underlie the wide diversity of astrocyte functions and phenotypes in TSC deserves further investigation. However, for neurons, it has already been shown that extent of mTOR hyperactivity correlates with seizure severity and associated neuropathology (73). Finally, in addition to intrinsic astrocytic properties, maintenance of a non-reactive state in astrocytes was also shown to depend on neuronal mTORC1 signaling, adding yet another level to altered astrocyte function in TSC (74).
The most striking evidence for astrocytic contribution to epileptogenesis in TSC comes from a conditional Tsc1 knockout mouse model (referred to as Tsc1GFAP mice), in which Tsc1 is specifically deleted in GFAP-expressing cells during embryonic development, leading to mTOR hyperactivity in these cells (75, 76). Notably, Tsc1 deletion is also induced in GFAP-positive neural progenitor cells and can be found in neurons, thereby blurring the specific contribution of astrocytes to some extent (77). While this model does not recapitulate all pathological hallmarks of human TSC (most notably lacking tuber formation and giant cells), development of spontaneous recurrent seizures arises in all animals at 1 month of age. This occurs likely via diffuse astrocyte proliferation and dispersion of neurons, causing altered neuronal circuitry. Interestingly, even post-natal deletion of Tsc1 at 2 weeks of age leads to development of epilepsy in half of the animals, although in a less severe form (77). Consequently, TSC1 deletion appears to be the initial insult followed by a latent stage of epileptogenesis, which in TSC patients might be even prenatally. Notably, treatment of Tsc1GFAP mice with the mTOR inhibitor rapamycin suppressed seizures, whereas vigabatrin reduced seizures and partially inhibited mTOR activity, astrogliosis, and neuronal disorganization (29, 78). Interestingly, TSC patients present with differences in disease severity, depending on the underlying mutation, with TSC2 mutations causing a more severe neurological and cognitive phenotype (22, 79–81). In conjunction with this, Tsc2GFAP mice present with more severe epilepsy than Tsc1GFAP mice (82).
While the growth advantage of astrocytes plays an apparent role in disruption of neuronal circuits, astrocytes in this model also display functional changes. A pathological hallmark of acquired epilepsy is impaired potassium buffering by astrocytes (83). Its implication in epileptogenesis is based on increased extracellular potassium upon neuronal depolarization, reduced astrocytic clearance of excess potassium, and consequently neuronal hyperexcitability and seizures. Key players in astrocytic potassium buffering represent aquaporins, Kirs, and connexins, which all display dysregulation in TSC-null astrocytes, Tsc1GFAP mice, and surgically resected TSC tissue (84–87). Another well-established player in neuronal hyperexcitability is impaired astrocyte-mediated clearance of glutamate, which can predispose neurons to sustained excitability, excitotoxicity, and epileptiform activity. Astrocytes in human TSC display altered glutamate receptor expression, whereas Tsc1GFAP mice present with decreased expression of glutamate transporters, implying altered extracellular glutamate metabolism (72, 88, 89). Pharmacological upregulation of glutamate transporters in astrocytes of Tsc1GFAP could reduce seizure frequency and some of the pathological changes, exemplifying the likely importance of extracellular glutamate clearance in TSC (88). Lastly, increased astrogliosis and consequent enhanced astrocytic adenosine kinase activity in epilepsy models and various epileptogenic pathologies, including TSC, result in a deficient homeostatic adenosine tone at the synapse and reveal a direct link between astrocyte activation and network excitability (90, 91).
Besides the reported changes in potassium buffering, glutamate clearance, and adenosine homeostasis, TSC is also characterized by inflammatory changes, and astrocytes are supposed to be both source and target of inflammatory signaling therein (92–95). Indeed, human tuber and SEGA tissue also display activation of inflammation in astrocytes, in particular, the toll-like receptor 4 (TLR-4), interleukin 1β (IL-1β), and complement pathways, as well as increased expression of IL-17, intercellular adhesion molecule 1, tumor necrosis factor α (TNF-α), and nuclear factor κB (NF-κB) (61–63, 96–98). In particular, several large-scale RNA-sequencing studies confirmed that neuroinflammation is a hallmark of TSC-associated lesions, and the retrieved data were enriched for both astrocyte and microglial specific genes (21, 63, 99, 100). Additionally, microRNAs (miRNAs) involved in the regulation of astrocytic inflammatory responses are upregulated in TSC (101). In comparison, astrocytes in Tsc1GFAP mice also present with increased IL-1β and C-X-C motif chemokine 10 expression, most notably, preceding the development of seizures, and are also characterized by increased microglial proliferation (74). Collectively, proinflammatory changes represent an important pathogenic mechanism by further activating astrocytes and could also reinforce mTOR-related dysfunctional processes, e.g., the immunoproteasome pathway, which might represent a direct molecular link between inflammation, mTOR activation, and epilepsy in TSC and other mTORopathies (102).
An additional pathogenic mechanism frequently encountered and closely linked to inflammation in epilepsy is oxidative stress (OS) (103–105). OS is defined as disturbance in the cell's redox state and was shown to be highly correlated with inflammation in dysmorphic neurons, giant cells, and glia of TSC and other mTORopathies (98). Glial cells in TSC displayed higher expression of the enzymes inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2) (98). Both enzymes produce mediators that contribute to OS and inflammation, thereby supporting the notion that glia are mediators of these pathogenic processes in TSC. In addition, giant cells revealed strong expression of OS (iNOS, cysteine/glutamate antiporter) and inflammation (COX-2, TLR-4) markers, as well as accumulation of NF-κB in the nucleus, supporting the strong correlation between these two processes (98). Another article pointed at the critical role of OS promoting an environment that favors the positive selection of cells with higher antioxidant capacity due to aberrant mTOR activation (106). Further research into OS in TSC revealed that the proinflammatory miRNA-155 might contribute to this sustained activation of antioxidant pathways in giant cells and astrocytes, exemplifying the link between OS and brain inflammation (107). Furthermore, the induction of sustained, miR155-mediated antioxidant signaling in astrocytes led to dysregulation of iron metabolism, which could result in the potentiation of OS in TSC (107).
A final pathological hallmark of TSC is the disruption of the blood–brain barrier (BBB) (108), with implications for a modulatory role of matrix metalloproteinases in BBB remodeling (62, 109–113). In this context, chronic BBB dysfunction and epileptogenesis after status epilepticus (SE)–induced epilepsy could be reduced via treatment with rapamycin, pointing toward a more general role of mTOR-dependent BBB remodeling during epileptogenesis in epilepsy (34, 35, 114).
In the context of gliopathy in TSC, it is noteworthy that giant cells represent a cell type with features of immaturity, highlighting the absence of differentiation to macroglial or neuronal lineage cells prior to migration into the developing cortex (72, 115–117). While the exact precursor of giant cells is unclear, the differential expression of glial (GFAP and S100 protein), neuronal (neurofilament, synaptophysin, MAP2), and neuroglial progenitor (SOX2, nestin, vimentin, CD133, β1-integrin) markers suggests that these cells reflect intermediary, undifferentiated stages of cellular development (45, 62, 115, 116, 118, 119). Many of the expression changes in astrocytes mentioned before are recapitulated in giant cells in tubers; however, on average, they display high heterogeneity, likely due to variation in the frequency of mutations based on the “two-hit” hypothesis (21, 49). Accordingly, balloon cells in FCD, a cell type histologically resembling giant cells in TSC, have been shown to also carry pathogenic somatic, second-hit variants of mTOR regulatory genes, and their density correlates with genetic findings (120). Moreover, non–cell-autonomous effects of the mutation influencing both the interglial and neuroglial crosstalk during cortical development may also contribute to the histological phenotype of malformed cells. Thus, giant cells and balloon cells might represent an important glioneuronal cell type in the generation of disturbed cellular architecture in developmental malformations related to mTOR dysregulation. Functionally, giant cells might contribute to brain inflammation by expressing complement factors and attracting immune cells already very early in development (62, 121). Moreover, they might be actively involved in the aberrant neuronal circuitry leading to the neurological manifestations of TSC by expressing glutamate and γ-aminobutyric acid (GABA) receptors and transporters (72, 122, 123).
One proposed precursor for giant cells are radial glia, progenitor cells of astrocytes and neocortical neurons, and oligodendrocyte progenitors cells (OPCs) (124, 125). Radial glia are localized in the subventricular zone of the developing brain, giving rise to the proliferative, astrogliogenic/neurogenic niche in the developing brain, as well as providing the physical substrate for neurons to migrate along toward their cortical destination (126). In light of this, radial glia perform vital functions in the formation of the cortex, and their malfunction is hypothesized to give rise to improperly differentiated cells, i.e., giant cells and dysmorphic neurons, and malformed cortical cytoarchitecture. Studies on radial glia-specific Tsc1 or Tsc2 knockout mice displayed characteristic features of human TSC, such as aberrant cortical architecture, hippocampal disturbances, astrogliosis, and spontaneous seizures (127–129). Importantly, these alterations displayed specific phenotypic differences between Tsc1 and Tsc2 knockout mice. Moreover, organoid model systems revealed that higher mTOR baseline activation in outer radial glia, a feature linked to the stemness of progenitor cells (130), is specific to primate corticogenesis, suggesting that this cell niche is highly susceptible to perturbations due to germline or somatic mutations in the mTOR pathway and thereby could induce aberrant formation of giant cells in the TSC brain (131, 132). This primate-specific feature could also explain the limitations of most TSC model organisms to reflect the histopathological features of TSC, such as tubers and giant cells. The aforementioned studies imply strong phenotypic effects, depending on the timing of the mutation, as well as the cell type affected, potentially explaining the phenotypic heterogeneity of dysmorphic cells and in particular astrocytes in human TSC (27). Another highly intriguing finding from these studies on brain organoid development revealed that cellular subtype differentiation of progenitor cells might be perturbed in vitro due to enhanced mTOR-dependent glycolysis and endoplasmic reticulum (ER) stress (132, 133), features also implicated in TSC (134, 135).
The central nervous system (CNS) pathology of TSC comprises a range of white matter abnormalities, detectable in presurgical magnetic resonance imaging (MRI) (136, 137), as well as in resected lesional tissue (138). While cortical tubers have classically been the neuropathological hallmark feature observed in these patients, the widespread hypomyelination/dysmyelination has emerged as a synonymous and prominent indication for clinical phenotypes in TSC patients. The cells responsible for the development and maintenance of an intact white matter of the brain are specialized cells called oligodendrocytes. They undergo a complex and precisely timed cycle of proliferation, migration, and differentiation to finally generate myelin by concentrically wrapping axons with multilamellar sheets of plasma membrane consisting of specific proteins and lipids (139). Two distinct terms in regard to white matter pathologies have been established, namely, demyelination and hypomyelination. The term demyelination is generally used if there is loss of myelin, occurring after a normal myelin development. This pathology has been studied accurately in patients suffering from multiple sclerosis (27, 140). Hypomyelination, on the other hand, may emerge if myelin production is disturbed or was never initiated, as seen in TSC patients (27).
Recent technological advances in MRI including diffusion tensor imaging (DTI) and fractional anisotropy (FA) have further emphasized hypomyelination in TSC (141, 142). Data revealed that regions involved in the processing of visual auditory and social stimuli contain more dysmyelinated axons in patients, hence supporting behavioral and cognitive characteristics (142). In addition, a major neuropathological aspect is the limited myelination in resected lesions of TSC patients. A recent study has reported a possible involvement of oligodendroglial turnover, indicating that the inhibition of oligodendroglial cell maturation, supposedly due to constitutive activation of mTOR, may lead to insufficient myelination in TSC patients (138). A principal feature of diseases with abnormal white matter is an oligodendroglial pathology that is frequently associated with cognitive impairments (64). The hypothesis that a dysfunctional white matter and hence abnormal neural circuitry account for the neurological manifestations in TSC has been further investigated by a plethora of studies. Interestingly, TSC patients with ASD have more crucial white matter abnormalities compared to patients without ASD (143, 144).
Oligodendroglial development, from an OPC (also called NG2 glia) to the maintenance of an intact myelin sheath, is tightly controlled by a myriad of both extracellular and intracellular factors, with two regulatory pathways in focus: the mitogen-activated protein kinase kinase/extracellular signal-regulated kinases 1 and 2/mitogen-activated protein kinase (Mek/ERK1/2-MAPK), and the mTOR signaling pathway (145, 146). Specifically, during oligodendrocyte lineage progression and initiation of myelination, the mTOR pathway via mTORC1 has emerged as a key player involved in this process (146). In a recent study, the involvement of mTOR signaling in cytoskeletal reorganization during oligodendrocyte development, as well as in initiation of myelination, was demonstrated. Moreover, the importance of the mTOR pathway on oligodendroglial branching complexity was observed (147). One study demonstrated a decrease in both myelin content and oligodendrocytes in and around cortical lesions of mTORopathy specimens (138). This decrease was linked to elevated mTOR expression and a possible impairment of oligodendroglial turnover, suggesting that mTOR pathway mutations cause a defect in oligodendrocytes (138). Thus, high lesional-specific mTOR activation combined with a decreased number of oligodendrocytes may further strengthen the hypothesis of mTOR pathway-dependent modulation of oligodendroglial differentiation and myelination properties. A plethora of studies have proven the essential role of mTOR signaling on the complex differentiation of oligodendrocytes to the maintenance of an intact myelin sheath (148–150).
Animal models have delved further into the causal relationship between mTOR pathway signaling and proper CNS myelin formation and maintenance. However, there is still considerable uncertainty with regard to the cell autonomous effects of TSC ablation in oligodendrocytes or aberrant signaling from TSC-deficient neurons or astrocytes that may indirectly influence myelination processes in the brain. It has now been suggested that CNS myelination, specifically myelin-associated lipogenesis, and protein gene regulation are mainly dependent on mTORC1 function (151). The same authors demonstrated that oligodendrocyte-specific enhancement of mTORC1, via loss of TSC1, results in abnormal myelination in mice (151). Remarkably, brains of Tsc2Olig2 KO mice reveal distinct hypomyelination, further supporting a cell-autonomous effect of TSC2 inactivation on oligodendrocytes (152). Grier et al. (153) drew attention to the important but more transient contribution of mTORC2 signaling in myelinogenesis by utilizing a mouse model lacking the rapamycin-insensitive companion of mTOR (Rictor), a functional component of the mTORC2, in NG2 glia. They were able to observe that loss of Rictor in these cells decreases and delays the expression of myelin related proteins and causes a developmental hypomyelination (153).
Besides cell-autonomous effects, a recent study supports the role of abnormal neuron–oligodendroglia communication causing hypomyelination employing induced pluripotent stem cell–derived neuronal and oligodendroglial cultures from TSC patients. Interestingly, neuron–oligodendrocyte cocultures from these patients revealed increased oligodendrocyte proliferation but a decrease in maturation (154). In terms of neuron–glia interaction, it was shown that Tsc1 mutant mice display a striking delay in myelination supporting the hypothesis of an underlying abnormal neuron–oligodendrocyte communication that causes white matter pathologies (155). Further, neuron-specific ablation of Tsc1 in a mouse model results in an increase in connective tissue growth factor secretion that leads to a decrease in the number of oligodendrocytes (156).
In conclusion, there is evidence that mTOR signaling is indeed fundamental to oligodendrocyte differentiation and myelination, as well as critical indications that both cell-autonomous effects and interactions between neurons and oligodendrocytes cause hypomyelination in mTORopathies. Because the outcome of the mTOR pathway hyperactivation observed in TSC patients as well as in animal models is hypomyelination and not the expected hypermyelination, at least five mechanisms were hypothesized to be responsible for this paradoxical impact on myelinogenesis. The constitutive mTORC1 signaling might account for (1) a delayed onset of myelination, (2) triggering non-physiological toxic effects, such as ER stress or apoptosis of oligodendrocytes, (3) TSC subunits exerting non-canonical functions that are independent of mTORC1, (4) suppressing mTORC2 functions, and (5) a negative feedback on mTORC1-independent targets, such as Mek-Erk 1/2 and/or PI3K-Akt pathways [for a detailed review, see (157)].
Because of the emerging evidence for a link between decreased myelin content and the development of neurological deficits, achieving remyelination of axons takes center stage in multiple sclerosis research, implying that it might be beneficial for mTORopathy patients as well (158, 159). As far as disease control is concerned, an important question is whether the observed hypomyelination in TSC patients may be reversible by reducing the constitutive activation of the mTOR signaling pathway. Latest research emphasizes the use of rapamycin or the rapamycin analog, everolimus. Only few researchers have addressed the question if and how the white matter is altered after treatment with an mTOR inhibitor. A pharmacological therapy administering everolimus was able to decrease mean diffusivity and increase FA during DTI measurements in TSC patients (160). In terms of everolimus treatment period, recent data support the hypothesis that longer periods improve the white matter microstructural integrity even more (161). In summary, evidence from experimental and human studies indicates that hypomyelination could be reversed by treatment with everolimus; however, the mechanism of action needs to be studied in more detail.
Apart from astrocytes and oligodendrocytes, NG2 glial cells represent a third macroglial subtype in the CNS, which has received much attention in the past decade [for a detailed review, see (162)]. In the literature, these cells are primarily referred to as OPCs, but also as complex cells, synantocytes, polydendrocytes, and GluR cells, as they depict glial and neuronal functions (163, 164). NG2 glial cells are substantially spread in both gray and white matter of the developing as well as the adult brain (165, 166). A remarkable feature found in cells expressing the proteoglycan NG2 is their proliferative and differentiation potential throughout life (166, 167). Interestingly, in post-natal white matter, these cells mainly differentiate into myelinating oligodendrocytes (168–170), and especially following demyelination, this process is amplified (171). In contrast, NG2 glia in the gray matter retain their neuronal–glial properties post-natally (11). These cell populations receive direct neuronal glutamatergic and GABAergic synaptic input and express voltage-gated ion channels for K+, Na+, and Ca2+ that can trigger long-term potentiation; however, they do not generate action potentials themselves (172–175). The precise functional changes of these cells in response to synaptic input remain largely unknown except some evidence on modulation of inward rectifying potassium channels (176) and proliferation (177, 178). Interestingly, cleaved NG2 was shown to rescue diminished neuronal α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) currents in NG2 knockout mice, establishing a reciprocal signaling loop between NG2 and neurons (179). Apart from these neuronal properties, NG2 glia in the human hippocampus do not couple to other glia via gap junctions, such as astrocytes and lack glutamate transporters, while expressing Kirs (180). Moreover, NG2 ablation was shown to induce microglia-mediated neuroinflammation and neuronal death in the hippocampus. The authors concluded that reduced NG2-derived trophic support via hepatocyte growth factor was responsible for this loss of neurons (181). Furthermore, NG2-derived transforming growth factor β2 (TGF-β2) signaling to TGF-β receptor 2 on microglia was shown to be a key regulator of microglial CX3CR1-mediated immune responses, and deficiency of this signaling axis via NG2 ablation led to neuronal loss and inflammation (182). Hence, the ability of NG2 glia to respond to neuronal inputs, as well as retaining a high proliferative potential in the human brain, makes this cell type another interesting glial cell in the context of TSC.
So far, only one study directly characterized NG2 cells in TSC tubers (52). Although these authors concluded that there were no detectable morphological alterations in oligodendrocytes and NG2 cells, they also acknowledged the lack of knowledge concerning specific activation markers for these cell types. Moreover, this study mainly evaluated morphological changes from an astrocyte perspective and did not convincingly rule out functional changes in NG2 glia (52). Because mTOR is an essential regulator of oligodendrocyte differentiation during development (157), its therapeutic potential was investigated by several studies. Moreover, the OPC-specific NG2 proteoglycan appears be directly linked to mTOR and to positively regulate its activity (183). Deletion of either TSC1 or phosphatase and tensin homolog (PTEN) in NG2 cells, both negatively regulating mTOR, induced an increase in mTORC1 activity. Interestingly, whereas TSC1 deletion in these cells led to the expected hypomyelination and impaired oligodendrocyte development, ablation of PTEN resulted in enhanced NG2 glia proliferation and oligodendrocyte lineage progression. This suggests the involvement of an mTORC1-independent PTEN-downstream signaling process. Further, also deletion of the PTEN-AKT downstream target glycogen synthase kinase 3β (GSK3ß) resulted in a comparable increase in differentiation of oligodendrocytes (184). These results may indicate a possible remyelination mechanism via inhibition of the PTEN-AKT-GSK3β pathway. McLane et al. (185) further revealed that an ablation of TSC1 affects oligodendroglia differently, depending on the olig odendroglial lineage stage. A deletion of TSC1 from NG2 glia speeds up the remyelination process, although TSC1-deficient proteolipid protein–positive oligodendrocytes decelerate remyelination.
Although most research on NG2 glia focused on their function as OPCs, there are also emerging lines of evidence that link them to neuronal function and microglia-mediated neuroinflammation. In the context of TSC, it would be interesting to study models of mTOR activation specifically in gray matter NG2 cells, where they reportedly serve more diverse functions.
Opposed to other neuroglia that are brain-borne, microglia arise from yolk sac–primitive macrophages and invade the brain during development before the BBB is fully formed (186–190). This early migration is a well-preserved mechanism among species, thereby emphasizing the important role of microglia during brain development (188, 191–193). Indeed, the phagocytic function of microglia is most prominent during development as they are capable of phagocytosing newly formed neurons and synapses in the developing brain (194–196). In the adult brain, ramified microglia surveil the brain environment with their processes and migrate to areas of need in response to activation cues, such as chemokine signaling (197). In response to distress signals, microglia can become activated, which is accompanied by a variety of morphological and molecular changes (198, 199). In general, two states of activation can be distinguished: a proinflammatory state (classically M1) that allows immune responses against pathogens and dysfunctional cells, which at the same time might exert damage on surrounding healthy tissue; and an anti-inflammatory state (classically M2) that is thought to be central in repair processes (200–202). However, thanks to sequencing data, microglia activation was identified to be a continuum in which many subtypes can be distinguished (203–206). In addition to their classical role as resident immune-competent cells and noteworthy in the context of TSC, microglia were also shown to modulate neuronal activity directly (207) and can be activated in response to excessive neuronal activity in epilepsy (208).
Several studies have shown increased density and activation of microglia cells in the brains of TSC patients (45, 62, 110, 121, 209). In cortical tubers, microglia with an activated morphology were found throughout the lesional tissue, mostly localized in close proximity to dysmorphic neurons and giant cells with mTOR activation, indicated by phosphorylation of the mTOR target ribosomal protein S6 kinase (pS6K), as well as around blood vessels (62). Similarly, in other epilepsies characterized by mTOR activation, such as FCD, TLE, and Rasmussen encephalitis, increased expression of microglial markers has been found in the respective brain lesions (33, 209–213). Moreover, in TLE patients with glial scarring due to drug-resistant epilepsy, mTOR activation was mostly found in microglia and to a lesser extent in astrocytes (33). Functionally, these microglia have been suggested to have a damaging role as they were shown to colocalize with several proinflammatory markers and surrounded cells expressing caspase 3, indicating that they might be involved in apoptosis (62). Moreover, in FCD lesions and glioneuronal tumors, the number of microglia has been correlated with seizure frequency of the patients (210, 214). Although it remains difficult to pinpoint whether microglia activation is causative or consequential of neurological deficits in these pathologies, the colocalization with pS6-positive cells indicates that microglia respond to mTOR hyperactivation in TSC. Sun et al. (215) showed that microglia activation in FCD and TSC might be partially caused by reductions in the immune modulatory molecules CD47 and CD200 on neurons and their respective receptors on microglia. When exogenously introduced, these molecules could potentially exert anti-inflammatory effects on microglia by suppressing proinflammatory cytokines, such as IL-6 (216).
Several attempts have been made to study aberrant mTOR activation in microglia and its impact on their function. Zhao et al. (217) showed that deletion of the Tsc1 gene in CX3CR1-expressing cells (referred to as Tsc1CX3CR1), either congenitally or post-natally, increased microglial mTOR activity and their overall number in the cortex and hippocampus. All of the Tsc1CX3CR1 mice developed spontaneous recurrent seizures around 5 weeks of age, as well as two-thirds of the post-natally induced conditional knockout mice. However, that same year, Zhang et al. (218) reported that CX3CR1-Cre driver lines in Tsc1CX3CR1 animals target not solely the alleged microglia cells, but also NeuN- and rarely GFAP-positive cells. Therefore, they concluded that the effects seen in Tsc1CX3CR1 mice were not exclusively driven by reactive microglia but could also be elicited by affected neurons and astrocytes. Furthermore, post-natal induction of the knockout, which had a higher specificity for microglia showing only 5% of non-microglial cells affected, did not result in spontaneous ictal activity (218), in contrast to the previous study (217). These studies emphasize that it is essential to precisely target and characterize cell types in TSC KO models, as only small percentages of affected neurons can lead to increased neuronal excitability (219). Nevertheless, isolated microglia from Tsc1CX3CR1 animals displayed clear cellular alterations. Thus, these two studies support the role of mTOR-dependent microglial abnormalities, and its role in epileptogenesis, especially in the context of inflammation, cannot be excluded.
In another model, direct manipulation of the mTOR pathway was induced by in utero electroporation of constitutively active Rheb, an mTORC1 activator. With this method, Nguyen et al. (73) observed that mTOR hyperactivity resulted in a global increase in Iba1-positive microglia that were both larger and had a more activated morphology. Furthermore, these Iba1-positive microglia were positively correlated with seizure frequency. However, with this technique, not only microglial cells were targeted. Indeed, mTOR activation also induced hypertrophy and cytoarchitectural misplacement of neurons in these animals, which together with the activated microglia were concluded to be responsible for seizure generation. Finally, in the BV2 microglial cell line direct activation of mTOR by MHY1485 treatment in vitro induced expression of proinflammatory cytokines, such as TNF-α, IL-6, and HMGB1, and decreased anti-inflammatory cytokines, such as TGF-β and IL-10. Furthermore, microglia displayed a shift from an anti-inflammatory toward a proinflammatory subtype, and markers of autophagy were reduced due to mTOR activation (220).
Besides direct genetic or chemical activation of the mTOR signaling pathway, the majority of research on the interaction of microglia and mTOR is supported by experiments that evaluated microglia in disease states characterized by increased mTOR activity through various brain injuries and/or by means of chemical mTOR inhibition. For example, pilocarpine-induced SE resulted in mTOR activation in neurons and microglia, and subsequent rapamycin treatment could alleviate microgliosis and had beneficial effects on cognitive performance of animals (221). Moreover, kainic acid–injected rats treated with rapamycin displayed reduced microglial activation (35). In contrast, rapamycin treatment in an electrically induced post-SE model did not change expression of inflammation markers or number of CD11b/c and CD68-positive cells, indicating that rapamycin did not affect brain inflammation in this model. Other studies have used brain injuries, such as stroke or vascular dementia in combination with mTOR blockage to evaluate microglial changes. Treatment with rapamycin or its derivatives everolimus or sirolimus could revert medial cerebral artery occlusion (MCAO)–induced increases of cytokines and/or chemokines, as well as promote anti-inflammatory microglial polarization (222, 223). In this same study, RaptorCX3CR1 mice, characterized by lacking regulatory-associated protein of mTOR (Raptor) specifically in microglia leading to mTORC1 activation, undergoing MCAO were found to have similar beneficial responses to chemical mTOR inhibition in terms of microglia activation and cytokine induction. Treatment with everolimus in mice with bilateral common carotid artery stenosis, a vascular dementia model that induces mTOR activation, also caused a shift toward anti-inflammatory microglia due to a loss of inhibitory feedback of mTORC1 on PI3K, alternatively activating the prosurvival kinase Akt (224). Likewise, spinal cord injury induced increases in OX42-positive microglia, which could be attenuated by treatment with wortmannin, an inhibitor of the PI3K/Akt/mTOR pathway (225). Interestingly, according to Yang et al. (226), because of its ability to also interact with mTORC2, everolimus is more effective than rapamycin in counteracting lipopolysaccharide (LPS)/kainic acid–induced microglial responses. Of note, some authors argued that the anti-inflammatory effect of mTOR inhibitors might be mediated primarily by other cell types than microglia (222). Despite these claims, in pure microglia cell cultures, such as the BV2 and N9 cell line, inhibition of mTOR activity after oxygen glucose deprivation, LPS, or a cytokine challenge reduced both microglia activation and inflammation (224, 227–229). Furthermore, LPS-stimulated N9 microglia even exerted neuroprotective effects after rapamycin treatment, as conditioned medium could suppress neurotoxicity in a neuronal cell line (229). Lastly, in a kainic acid–induced SE model, the long-term epileptogenic effects of early life seizures could be reduced via treatment with an inhibitor of microglia activation, minocycline, directly linking microglial activation and epileptogenesis (230).
Finally, assuming microglial activation secondary to mTOR-driven malformations of cortical development in TSC, depletion of resident microglia, and repopulation of the brain with fresh microglia might offer a drastic, yet promising therapeutic option to resolve chronic neuroinflammation (231). Importantly, this approach could relieve the neuroinflammatory burden in the post-natal TSC brain even after aberrant brain development. Adjunctive with mTOR inhibitors, this approach could target source (mTOR hyperactivation) and symptom (microglia activation) of TSC brain malformations simultaneously and offer a valuable disease-modifying therapy.
While neuron–glia interactions are the focus in many of the studies discussed here, interactions between glia may offer new therapeutic and diagnostic opportunities (Figure 2). In the context of neuroinflammation, bilateral signaling between microglia and astrocytes likely plays an essential role in brains of TSC patients. For example, Tsc1GFAP mice do not only display alterations in astrocytes, but microglia number and size were abnormally increased in cortex and hippocampus, pointing toward an indirect effect of mTOR hyperactivation in astrocytes on microglia (232). However, the importance of microglia in the induction of a reactive phenotype in astrocytes has been shown (233), and Tsc1CX3CR1 mice also display increased proliferation and reactive changes of astrocytes (217). Together, this reinforcing crosstalk might be crucial for the maintenance of a proinflammatory environment in TSC with contributions from functionally normal glia, as well as glia with cell-autonomous mTOR-related alterations. The effect of microglia is likely contributing to the proinflammatory environment of TSC tubers as their functions involve inflammation initiation and propagation in conjunction with astrocytes (233, 234). Moreover, microglia activation may exert proinflammatory/damaging effects on oligodendrocytes and neurons, contributing to neuronal dysfunction and resulting neurological comorbidities and hyperexcitability (235, 236).
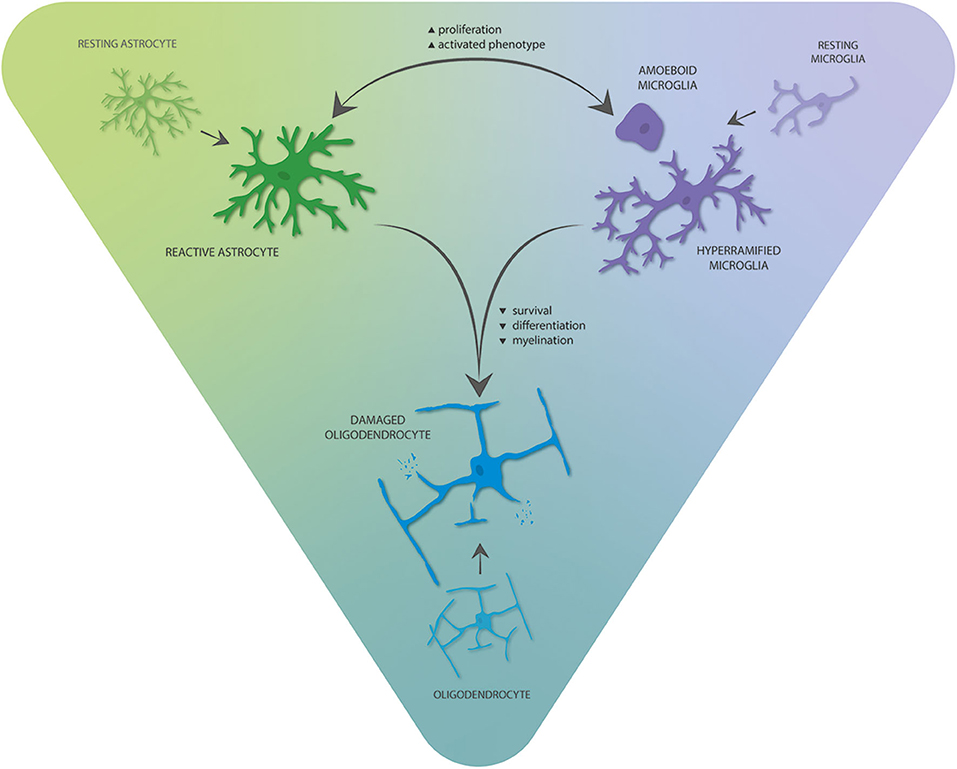
Figure 2. Interglial crosstalk of the three main glial cell types in the TSC brain. Astrocytes and microglia can stimulate and reinforce proliferation and phenotypic activation of each other, thereby promoting proinflammatory gene expression. These alterations mediate negative consequences on oligodendrocyte survival, differentiation, and myelination.
Next to microglia, particularly interesting in the context of interglial crosstalk in TSC is the role of astrocytes to directly influence the production and survival of cells of the oligodendrocyte lineage (237, 238). Accordingly, reactive and enlarged dysplastic astrocytes with enhanced activation of mTOR and gain of aberrant functions in cortical tubers, including a proinflammatory phenotype, may pose detrimental in the function of other glia in TSC. In support of this, there is a growing body of evidence that supports the concept of astrocytopathies within the field of childhood white matter disorders in which dysfunctional astrocytes have been suggested to drive degeneration of the white matter (239, 240). As mentioned previously, astrocytic gap-junction coupling in TSC models is disturbed (85), and heterotypic gap-junction coupling between astrocytes and oligodendrocytes was shown to be essential for (re-)myelination in animal models (241–243). Moreover, dysregulation of glutamate metabolism by astrocytes in TSC (72, 88, 89) could promote excitotoxic cell death in oligodendrocytes as they express functional N-methyl-D-aspartic acid, AMPA, and kainate-type receptors that mediate toxic effects of excess glutamate (244–247). Moreover, astrocyte-specific NF-κB activation in TSC might also play a role in suppressing myelination (248). Lastly, evidence from the “twitcher” mouse model supports the role of microglial COX-2 in demyelination. Here, secreted microglial prostaglandins (PGDs) could stimulate PGD receptors on astrocytes, inducing astrogliosis as indicated by hypertrophy and a rise in intracellular calcium, and blocking this pathway increased oligodendrocyte survival (249). Because increased COX-2 expression is observed not only in glia, but also giant cells/balloon cells and dysmorphic neurons in TSC and FCD (98), this specific crosstalk might link hypomyelination to COX-2 expression.
As for mature oligodendrocytes, NG2 function in TSC likely depends on other glia. In vitro, it was shown that astrocyte- and microglia-conditioned medium exerts important effects in the development of oligodendrocytes, with astrocytic factors promoting oligodendrocyte survival and microglial factors supporting differentiation and myelination (250). Considering aberrant number and function of both cell types already early in development, this interglial crosstalk might contribute to the hypomyelination observed in TSC. Moreover, NG2 glia survival and differentiation can be impaired by OS and TNF produced by activated microglia (251–253). In essence, astrocytes and microglia could participate in the pathological link between OS, inflammation, and the dysregulated iron metabolism in TSC by inducing aberrant oligodendrocyte maturation and myelination in TSC (252, 254). Importantly, OS-dependent dysregulation of histone–deacetylase activity could promote astrogenesis/neurogenesis over oligodendrogenesis potentially contributing to the disturbed cell ratio observed in TSC brain tissue (252).
While the most debilitating CNS symptoms of TSC, epilepsy, and neurodevelopmental comorbidities ultimately result from neuronal dysfunction, it is also clear that glial alterations contribute and shape the complex mechanisms generating these symptoms. Moreover, glia provide the proliferative precursor of pre-natal and post-natal neurons in the form of radial glia and astrocytes, respectively. It is important to stress that in TSC there is likely a mixture of cells with cell-autonomous mTOR activation because of intrinsic TSC mutations and cells with regular mTOR activity that respond to changes due to this intrinsically dysfunctional cellular substrate. Nevertheless, the major triad of glial cells displays conserved features in response to mTOR activation in TSC, TSC models, and conditions of mTOR hyperactivation.
Although it is likely that increased proliferation of astrocytes and resulting physical disruption of neuronal circuits can impact epileptogenesis in TSC, studies on surgically resected tubers and TSC models suggest that astrocytes also present with epileptogenic functional changes. More importantly, these changes are likely caused by a mixture of primary astrocytic changes during brain development due to mTOR activation and secondary effects that promote reactive states of astrocytes, such as brain inflammation later on. Finally, astrocyte dysfunction in TSC recapitulates findings from other epileptogenic pathologies, thus potentially representing common astrocytopathic mechanisms of epilepsy that could be targeted by novel astrocyte-centered therapies.
For oligodendrocytes, it is of utmost interest to find targets by which the endogenous remyelination in TSC patients might be enhanced. The mTOR signaling pathway has been proposed to be an attractive target to promote remyelination; however, recent results emphasize the importance of correctly applied therapeutics, because what may be beneficial to OPC development might be noxious to myelinating oligodendrocytes.
Lastly, alterations in microglial functions in TSC might be caused by cell-autonomous mTOR activation or secondary to the altered brain environment in TSC. Whether their activation depends on either or both is not clearly defined yet; however, the presence of proinflammatory microglia upon mTOR activation likely contributes to pathology, while a shift toward an anti-inflammatory phenotype via mTOR inhibition might have beneficial effects.
Although challenging, a better understanding of the complexity of the glial pathology in TSC may provide opportunities for novel therapeutic approaches targeting glial-mediated mechanisms. In particular, a combinatorial therapy targeting different glial cell types and their crosstalk might be translated into disease-modifying treatments for various epilepsies associated with a deregulation of mTOR. Considering the evidence for mTOR inhibition not only rescuing neuronal, but also glial dysfunction, in preclinical TSC models (29, 255, 256), mTOR inhibitors, which were recently approved by the US Food and Drug Administration and European Medicines Agency for the treatment of refractory seizures associated to TSC starting from the age of 2 years (257), represent promising candidates to target TSC gliopathy. Finally and most importantly, mTOR inhibition as therapy of TSC could potentially be extrapolated to other genetic and acquired epilepsies (258, 259).
TZ and EA drafted the content of this review. TZ, DB, VG, EV, and AM contributed the specific topics and assisted in the final editing and revision of the manuscript. EA conceived the idea and was invited to participate in the editorial issue. All authors contributed to the article and approved the submitted version.
This work was supported by Stichting TSC Fonds (EA); the European Union's Seventh Framework Program (FP7/2007-2013) under grant agreement 602102 (EPITARGET; EV, EA) and 602391 (EPISTOP; AM, EA); the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no. 722053 (EU-GliaPhD; TZ, EA); the Dutch Epilepsy Foundation, project 16-05 (DB, EV) and 20-02 (AM) and the grant of the Austrian Epilepsy Society dedicated to Dr. Feucht (VG). VG is an affiliated partner of the EpiCARE European reference network.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. (2006) 355:1345–56. doi: 10.1056/NEJMra055323
2. Mizuguchi M, Takashima S. Neuropathology of tuberous sclerosis. Brain Dev. (2001) 23:508–15. doi: 10.1016/s0387-7604(01)00304-7
3. DiMario FJ Jr. Brain abnormalities in tuberous sclerosis complex. J Child Neurol. (2004) 19:650–7. doi: 10.1177/08830738040190090401
4. Curatolo P, Moavero R, van Scheppingen J, Aronica E. mTOR dysregulation and tuberous sclerosis-related epilepsy. Expert Rev Neurother. (2018) 18:185–201. doi: 10.1080/14737175.2018.1428562
5. Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. (2015) 14:733–45. doi: 10.1016/S1474-4422(15)00069-1
6. de Vries PJ, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. TSC-associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet J Rare Dis. (2018) 13:157. doi: 10.1186/s13023-018-0901-8
7. Moavero R, Benvenuto A, Emberti Gialloreti L, Siracusano M, Kotulska K, Weschke B, et al. Early clinical predictors of autism spectrum disorder in infants with tuberous sclerosis complex: results from the EPISTOP study. J Clin Med. (2019) 8:788. doi: 10.3390/jcm8060788
8. Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. (2010) 51:1236–41. doi: 10.1111/j.1528-1167.2009.02474.x
9. Nabbout R, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. Epilepsy in tuberous sclerosis complex: findings from the TOSCA study. Epilepsia Open. (2019) 4:73–84. doi: 10.1002/epi4.12286
10. Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. (2011) 22:735–9. doi: 10.1016/j.yebeh.2011.08.037
11. Gupta A, de Bruyn G, Tousseyn S, Krishnan B, Lagae L, Agarwal N, et al. Epilepsy and Neurodevelopmental comorbidities in tuberous sclerosis complex: a natural history study. Pediatr Neurol. (2020) 106:10–6. doi: 10.1016/j.pediatrneurol.2019.12.016
12. Tye C, McEwen FS, Liang H, Underwood L, Woodhouse E, Barker ED, et al. Long-term cognitive outcomes in tuberous sclerosis complex. Dev Med Child Neurol. (2020) 62:322–9. doi: 10.1111/dmcn.14356
13. Curatolo P, Verdecchia M, Bombardieri R. Vigabatrin for tuberous sclerosis complex. Brain Dev. (2001) 23:649–53. doi: 10.1016/s0387-7604(01)00290-x
14. Curatolo P, Nabbout R, Lagae L, Aronica E, Ferreira JC, Feucht M, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol. (2018) 22:738–48. doi: 10.1016/j.ejpn.2018.05.006
15. van der Poest Clement E, Jansen FE, Braun KPJ, Peters JM. Update on drug management of refractory epilepsy in tuberous sclerosis complex. Paediatr Drugs. (2020) 22:73–84. doi: 10.1007/s40272-019-00376-0
16. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. (2016) 388:2153–63. doi: 10.1016/S0140-6736(16)31419-2
17. Curatolo P, Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. (2018) 2:495–504. doi: 10.1016/S2352-4642(18)30099-3
18. Franz DN, Lawson JA, Yapici Z, Brandt C, Kohrman MH, Wong M, et al. Everolimus dosing recommendations for tuberous sclerosis complex-associated refractory seizures. Epilepsia. (2018) 59:1188–97. doi: 10.1111/epi.14085
19. Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Everolimus for treatment-refractory seizures in TSC: extension of a randomized controlled trial. Neurol Clin Pract. (2018) 8:412–20. doi: 10.1212/CPJ.0000000000000514
20. Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. (2013) 49:255–65. doi: 10.1016/j.pediatrneurol.2013.08.002
21. Martin KR, Zhou W, Bowman MJ, Shih J, Au KS, Dittenhafer-Reed KE, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun. (2017) 8:15816. doi: 10.1038/ncomms15816
22. Ogorek B, Hamieh L, Hulshof HM, Lasseter K, Klonowska K, Kuijf H, et al. TSC2 pathogenic variants are predictive of severe clinical manifestations in TSC infants: results of the EPISTOP study. Genet Med. (2020) 22:1489–97. doi: 10.1038/s41436-020-0823-4
23. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
24. Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience. (2017) 341:112–53. doi: 10.1016/j.neuroscience.2016.11.017
25. Sim NS, Ko A, Kim WK, Kim SH, Kim JS, Shim KW, et al. Precise detection of low-level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. (2019) 138:901–12. doi: 10.1007/s00401-019-02052-6
26. Crino PB. Focal brain malformations: a spectrum of disorders along the mTOR cascade. Novartis Found Symp. (2007) 288:260–72; discussion 272–281. doi: 10.1002/9780470994030.ch18
27. Muhlebner A, Bongaarts A, Sarnat HB, Scholl T, Aronica E. New insights into a spectrum of developmental malformations related to mTOR dysregulations: challenges and perspectives. J Anat. (2019) 235:521–42. doi: 10.1111/joa.12956
28. Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. (2008) 28:5422–32. doi: 10.1523/JNEUROSCI.0955-08.2008
29. Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. (2008) 63:444–53. doi: 10.1002/ana.21331
30. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. (2010) 363:1801–11. doi: 10.1056/NEJMoa1001671
31. Overwater IE, Rietman AB, Bindels-de Heus K, Looman CW, Rizopoulos D, Sibindi TM, et al. Sirolimus for epilepsy in children with tuberous sclerosis complex: a randomized controlled trial. Neurology. (2016) 87:1011–8. doi: 10.1212/WNL.0000000000003077
32. Wong M. Rapamycin for treatment of epilepsy: antiseizure, antiepileptogenic, both, or neither? Epilepsy Curr. (2011) 11:66–8. doi: 10.5698/1535-7511-11.2.66
33. Sosunov AA, Wu X, McGovern RA, Coughlin DG, Mikell CB, Goodman RR, et al. The mTOR pathway is activated in glial cells in mesial temporal sclerosis. Epilepsia. (2012) 53:78–86. doi: 10.1111/j.1528-1167.2012.03478.x
34. van Vliet EA, Forte G, Holtman L, den Burger JC, Sinjewel A, de Vries HE, et al. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. (2012) 53:1254–63. doi: 10.1111/j.1528-1167.2012.03513.x
35. van Vliet EA, Otte WM, Wadman WJ, Aronica E, Kooij G, de Vries HE, et al. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats II: potential mechanisms. Epilepsia. (2016) 57:70–8. doi: 10.1111/epi.13245
36. Crino PB. Mechanistic target of rapamycin (mTOR) signaling in status epilepticus. Epilepsy Behav. (2019) 101:106550. doi: 10.1016/j.yebeh.2019.106550
37. Madhavan D, Weiner HL, Carlson C, Devinsky O, Kuzniecky R. Local epileptogenic networks in tuberous sclerosis complex: a case review. Epilepsy Behav. (2007) 11:140–6. doi: 10.1016/j.yebeh.2007.03.017
38. Major P, Rakowski S, Simon MV, Cheng ML, Eskandar E, Baron J, et al. Are cortical tubers epileptogenic? Evidence from electrocorticography. Epilepsia. (2009) 50:147–54. doi: 10.1111/j.1528-1167.2008.01814.x
39. Marcotte L, Aronica E, Baybis M, Crino PB. Cytoarchitectural alterations are widespread in cerebral cortex in tuberous sclerosis complex. Acta Neuropathol. (2012) 123:685–93. doi: 10.1007/s00401-012-0950-3
40. Ruppe V, Dilsiz P, Reiss CS, Carlson C, Devinsky O, Zagzag D, et al. Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia. (2014) 55:539–50. doi: 10.1111/epi.12545
41. Jansen FE, Van Huffelen AC, Van Rijen PC, Leijten FS, Jennekens-Schinkel A, Gosselaar P, et al. Epilepsy surgery in tuberous sclerosis: the Dutch experience. Seizure. (2007) 16:445–53. doi: 10.1016/j.seizure.2007.03.001
42. Madhavan D, Schaffer S, Yankovsky A, Arzimanoglou A, Renaldo F, Zaroff CM, et al. Surgical outcome in tuberous sclerosis complex: a multicenter survey. Epilepsia. (2007) 48:1625–8. doi: 10.1111/j.1528-1167.2007.01112.x
43. Fallah A, Rodgers SD, Weil AG, Vadera S, Mansouri A, Connolly MB, et al. Resective epilepsy surgery for tuberous sclerosis in children: determining predictors of seizure outcomes in a multicenter retrospective cohort study. Neurosurgery. (2015) 77:517–24; discussion 524. doi: 10.1227/NEU.0000000000000875
44. Neal A, Ostrowsky-Coste K, Jung J, Lagarde S, Maillard L, Kahane P, et al. Epileptogenicity in tuberous sclerosis complex: a stereoelectroencephalographic study. Epilepsia. (2020) 61:81–95. doi: 10.1111/epi.16410
45. Boer K, Troost D, Jansen F, Nellist M, van den Ouweland AM, Geurts JJ, et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology. (2008) 28:577–90. doi: 10.1111/j.1440-1789.2008.00920.x
46. Muhlebner A, van Scheppingen J, Hulshof HM, Scholl T, Iyer AM, Anink JJ, et al. Novel histopathological patterns in cortical tubers of epilepsy surgery patients with tuberous sclerosis complex. PLoS ONE. (2016) 11:e0157396. doi: 10.1371/journal.pone.0157396
47. Cotter JA. An update on the central nervous system manifestations of tuberous sclerosis complex. Acta Neuropathol. (2020) 139:613–24. doi: 10.1007/s00401-019-02003-1
48. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
49. Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology. (2010) 74:1716–23. doi: 10.1212/WNL.0b013e3181e04325
50. Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE, et al. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. (2010) 20:1096–105. doi: 10.1111/j.1750-3639.2010.00416.x
51. Afshar Saber W, Sahin M. Recent advances in human stem cell-based modeling of tuberous sclerosis complex. Mol Autism. (2020) 11:16. doi: 10.1186/s13229-020-0320-2
52. Sosunov AA, Wu X, Weiner HL, Mikell CB, Goodman RR, Crino PD, et al. Tuberous sclerosis: a primary pathology of astrocytes? Epilepsia. (2008) 49:53–62. doi: 10.1111/j.1528-1167.2008.01493.x
53. Cloetta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, et al. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. (2013) 33:7799–810. doi: 10.1523/JNEUROSCI.3294-12.2013
54. Lee DY. Roles of mTOR signaling in brain development. Exp Neurobiol. (2015) 24:177–85. doi: 10.5607/en.2015.24.3.177
55. Grabole N, Zhang JD, Aigner S, Ruderisch N, Costa V, Weber FC, et al. Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med. (2016) 8:94. doi: 10.1186/s13073-016-0347-3
56. Blair JD, Hockemeyer D, Bateup HS. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med. (2018) 24:1568–78. doi: 10.1038/s41591-018-0139-y
57. Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. (1997) 278:477–83. doi: 10.1126/science.278.5337.477
58. Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. (2000) 10:47–50. doi: 10.1016/s0960-9822(99)00268-7
59. Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. (2007) 54:357–69. doi: 10.1016/j.neuron.2007.04.019
60. Wang B, Xiao Z, Chen B, Han J, Gao Y, Zhang J, et al. Nogo-66 promotes the differentiation of neural progenitors into astroglial lineage cells through mTOR-STAT3 pathway. PLoS ONE. (2008) 3:e1856. doi: 10.1371/journal.pone.0001856
61. Maldonado M, Baybis M, Newman D, Kolson DL, Chen W, McKhann G, et al. Expression of ICAM-1, TNF-alpha, NF kappa B, and MAP kinase in tubers of the tuberous sclerosis complex. Neurobiol Dis. (2003) 14:279–90. doi: 10.1016/s0969-9961(03)00127-x
62. Boer K, Jansen F, Nellist M, Redeker S, van den Ouweland AM, Spliet WG, et al. Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res. (2008) 78:7–21. doi: 10.1016/j.eplepsyres.2007.10.002
63. Boer K, Crino PB, Gorter JA, Nellist M, Jansen FE, Spliet WG, et al. Gene expression analysis of tuberous sclerosis complex cortical tubers reveals increased expression of adhesion and inflammatory factors. Brain Pathol. (2010) 20:704–19. doi: 10.1111/j.1750-3639.2009.00341.x
64. Wong M. The role of glia in epilepsy, intellectual disability, and other neurodevelopmental disorders in tuberous sclerosis complex. J Neurodev Disord. (2019) 11:30. doi: 10.1186/s11689-019-9289-6
65. Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. (2002) 99:467–72. doi: 10.1073/pnas.012605299
66. Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. (2005) 25:11300–12. doi: 10.1523/JNEUROSCI.2270-05.2005
67. Raab-Graham KF, Haddick PC, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. (2006) 314:144–8. doi: 10.1126/science.1131693
68. Wang Y, Barbaro MF, Baraban SC. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett. (2006) 401:35–9. doi: 10.1016/j.neulet.2006.03.011
70. Binder DK. Astrocytes: stars of the sacred disease. Epilepsy Curr. (2018) 18:172–9. doi: 10.5698/1535-7597.18.3.172
71. Richardson EP Jr. Pathology of tuberous sclerosis. Neuropathologic aspects. Ann N Y Acad Sci. (1991) 615:128–39. doi: 10.1111/j.1749-6632.1991.tb37755.x
72. Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE. Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol. (2008) 63:454–65. doi: 10.1002/ana.21342
73. Nguyen LH, Mahadeo T, Bordey A. mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J Neurosci. (2019) 39:2762–73. doi: 10.1523/JNEUROSCI.2260-18.2019
74. Zhang Y, Xu S, Liang KY, Li K, Zou ZP, Yang CL, et al. Neuronal mTORC1 is required for maintaining the nonreactive state of astrocytes. J Biol Chem. (2017) 292:100–11. doi: 10.1074/jbc.M116.744482
75. Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. (2002) 52:285–96. doi: 10.1002/ana.10283
76. Erbayat-Altay E, Zeng LH, Xu L, Gutmann DH, Wong M. The natural history and treatment of epilepsy in a murine model of tuberous sclerosis. Epilepsia. (2007) 48:1470–6. doi: 10.1111/j.1528-1167.2007.01110.x
77. Zou J, Zhang B, Gutmann DH, Wong M. Postnatal reduction of tuberous sclerosis complex 1 expression in astrocytes and neurons causes seizures in an age-dependent manner. Epilepsia. (2017) 58:2053–63. doi: 10.1111/epi.13923
78. Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS ONE. (2013) 8:e57445. doi: 10.1371/journal.pone.0057445
79. Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. (2001) 68:64–80. doi: 10.1086/316951
80. Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. (2007) 9:88–100. doi: 10.1097/gim.0b013e31803068c7
81. Jansen FE, Braams O, Vincken KL, Algra A, Anbeek P, Jennekens-Schinkel A, et al. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology. (2008) 70:908–15. doi: 10.1212/01.wnl.0000280578.99900.96
82. Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. (2011) 20:445–54. doi: 10.1093/hmg/ddq491
83. de Curtis M, Uva L, Gnatkovsky V, Librizzi L. Potassium dynamics and seizures: why is potassium ictogenic? Epilepsy Res. (2018) 143:50–9. doi: 10.1016/j.eplepsyres.2018.04.005
84. Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. (2005) 46:1871–80. doi: 10.1111/j.1528-1167.2005.00289.x
85. Xu L, Zeng LH, Wong M. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2009) 34:291–9. doi: 10.1016/j.nbd.2009.01.010
86. Sukigara S, Dai H, Nabatame S, Otsuki T, Hanai S, Honda R, et al. Expression of astrocyte-related receptors in cortical dysplasia with intractable epilepsy. J Neuropathol Exp Neurol. (2014) 73:798–806. doi: 10.1097/NEN.0000000000000099
87. Short B, Kozek L, Harmsen H, Zhang B, Wong M, Ess KC, et al. Cerebral aquaporin-4 expression is independent of seizures in tuberous sclerosis complex. Neurobiol Dis. (2019) 129:93–101. doi: 10.1016/j.nbd.2019.05.003
88. Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2007) 28:184–96. doi: 10.1016/j.nbd.2007.07.015
89. Zeng LH, Bero AW, Zhang B, Holtzman DM, Wong M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2010) 37:764–71. doi: 10.1016/j.nbd.2009.12.020
90. Aronica E, Sandau US, Iyer A, Boison D. Glial adenosine kinase–a neuropathological marker of the epileptic brain. Neurochem Int. (2013) 63:688–95. doi: 10.1016/j.neuint.2013.01.028
91. Boison D, Aronica E. Comorbidities in neurology: is adenosine the common link? Neuropharmacology. (2015) 97:18–34. doi: 10.1016/j.neuropharm.2015.04.031
92. Aronica E, Ravizza T, Zurolo E, Vezzani A. Astrocyte immune responses in epilepsy. Glia. (2012) 60:1258–68. doi: 10.1002/glia.22312
93. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol. (2013) 244:11–21. doi: 10.1016/j.expneurol.2011.09.033
94. Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. (2013) 69:16–24. doi: 10.1016/j.neuropharm.2012.04.004
95. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. (2019) 15:459–72. doi: 10.1038/s41582-019-0217-x
96. Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T, et al. Activation of Toll-like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain. (2011) 134:1015–32. doi: 10.1093/brain/awr032
97. He JJ, Wu KF, Li S, Shu HF, Zhang CQ, Liu SY, et al. Expression of the interleukin 17 in cortical tubers of the tuberous sclerosis complex. J Neuroimmunol. (2013) 262:85–91. doi: 10.1016/j.jneuroim.2013.05.007
98. Arena A, Zimmer TS, van Scheppingen J, Korotkov A, Anink JJ, Muhlebner A, et al. Oxidative stress and inflammation in a spectrum of epileptogenic cortical malformations: molecular insights into their interdependence. Brain Pathol. (2019) 29:351–65. doi: 10.1111/bpa.12661
99. Mills JD, Iyer AM, van Scheppingen J, Bongaarts A, Anink JJ, Janssen B, et al. Coding and small non-coding transcriptional landscape of tuberous sclerosis complex cortical tubers: implications for pathophysiology and treatment. Sci Rep. (2017) 7:8089. doi: 10.1038/s41598-017-06145-8
100. Bongaarts A, van Scheppingen J, Korotkov A, Mijnsbergen C, Anink JJ, Jansen FE, et al. The coding and non-coding transcriptional landscape of subependymal giant cell astrocytomas. Brain. (2020) 143:131–49. doi: 10.1093/brain/awz370
101. van Scheppingen J, Iyer AM, Prabowo AS, Muhlebner A, Anink JJ, Scholl T, et al. Expression of microRNAs miR21, miR146a, and miR155 in tuberous sclerosis complex cortical tubers and their regulation in human astrocytes and SEGA-derived cell cultures. Glia. (2016) 64:1066–82. doi: 10.1002/glia.22983
102. van Scheppingen J, Broekaart DW, Scholl T, Zuidberg MR, Anink JJ, Spliet WG, et al. Dysregulation of the (immuno) proteasome pathway in malformations of cortical development. J Neuroinflamm. (2016) 13:202. doi: 10.1186/s12974-016-0662-z
103. Pauletti A, Terrone G, Shekh-Ahmad T, Salamone A, Ravizza T, Rizzi M, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain. (2019) 142:e39. doi: 10.1093/brain/awz130
104. Shekh-Ahmad T, Kovac S, Abramov AY, Walker MC. Reactive oxygen species in status epilepticus. Epilepsy Behav. (2019) 101:106410. doi: 10.1016/j.yebeh.2019.07.011
105. Terrone G, Balosso S, Pauletti A, Ravizza T, Vezzani A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology. (2020) 167:107742. doi: 10.1016/j.neuropharm.2019.107742
106. Malik AR, Liszewska E, Skalecka A, Urbanska M, Iyer AM, Swiech LJ, et al. Tuberous sclerosis complex neuropathology requires glutamate-cysteine ligase. Acta Neuropathol Commun. (2015) 3:48. doi: 10.1186/s40478-015-0225-z
107. Zimmer TS, Ciriminna G, Arena A, Anink JJ, Korotkov A, Jansen FE, et al. Chronic activation of anti-oxidant pathways and iron accumulation in epileptogenic malformations. Neuropathol. Appl. Neurobiol. (2019). doi: 10.1111/nan.12596. [Epub ahead of print].
108. van Vliet EA, Aronica E, Gorter JA. Blood-brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol. (2015) 38:26–34. doi: 10.1016/j.semcdb.2014.10.003
109. Li S, Yu S, Zhang C, Shu H, Liu S, An N, et al. Increased expression of matrix metalloproteinase 9 in cortical lesions from patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. Brain Res. (2012) 1453:46–55. doi: 10.1016/j.brainres.2012.03.009
110. Prabowo AS, Iyer AM, Anink JJ, Spliet WG, van Rijen PC, Aronica E. Differential expression of major histocompatibility complex class I in developmental glioneuronal lesions. J Neuroinflammation. (2013) 10:12. doi: 10.1186/1742-2094-10-12
111. Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. (2016) 36:1481–507. doi: 10.1177/0271678X16655551
112. Bongaarts A, de Jong JM, Broekaart DWM, van Scheppingen J, Anink JJ, Mijnsbergen C, et al. Dysregulation of the MMP/TIMP proteolytic system in subependymal giant cell astrocytomas in patients with tuberous sclerosis complex: modulation of MMP by MicroRNA-320d in vitro. J Neuropathol Exp Neurol. (2020) 79:777–90. doi: 10.1093/jnen/nlaa040
113. Broekaart DWM, van Scheppingen J, Anink JJ, Wierts L, van Het Hof B, Jansen FE, et al. Increased matrix metalloproteinases expression in tuberous sclerosis complex: modulation by microRNA 146a and 147b in vitro. Neuropathol Appl Neurobiol. (2020) 46:142–59. doi: 10.1111/nan.12572
114. van Vliet EA, Otte WM, Wadman WJ, Aronica E, Kooij G, de Vries HE, et al. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats I: magnetic resonance imaging. Epilepsia. (2016) 57:59–69. doi: 10.1111/epi.13246
115. Crino PB, Trojanowski JQ, Dichter MA, Eberwine J. Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci USA. (1996) 93:14152–7. doi: 10.1073/pnas.93.24.14152
116. Lee A, Maldonado M, Baybis M, Walsh CA, Scheithauer B, Yeung R, et al. Markers of cellular proliferation are expressed in cortical tubers. Ann Neurol. (2003) 53:668–73. doi: 10.1002/ana.10579
117. Boer K, Lucassen PJ, Spliet WG, Vreugdenhil E, van Rijen PC, Troost D, et al. Doublecortin-like (DCL) expression in focal cortical dysplasia and cortical tubers. Epilepsia. (2009) 50:2629–37. doi: 10.1111/j.1528-1167.2009.02191.x
118. Lopes MB, Altermatt HJ, Scheithauer BW, Shepherd CW, VandenBerg SR. Immunohistochemical characterization of subependymal giant cell astrocytomas. Acta Neuropathol. (1996) 91:368–75. doi: 10.1007/s004010050438
119. Yasin SA, Latak K, Becherini F, Ganapathi A, Miller K, Campos O, et al. Balloon cells in human cortical dysplasia and tuberous sclerosis: isolation of a pathological progenitor-like cell. Acta Neuropathol. (2010) 120:85–96. doi: 10.1007/s00401-010-0677-y
120. Baldassari S, Ribierre T, Marsan E, Adle-Biassette H, Ferrand-Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. (2019) 138:885–900. doi: 10.1007/s00401-019-02061-5
121. Prabowo AS, Anink JJ, Lammens M, Nellist M, van den Ouweland AM, Adle-Biassette H, et al. Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol. (2013) 23:45–59. doi: 10.1111/j.1750-3639.2012.00616.x
122. White R, Hua Y, Scheithauer B, Lynch DR, Henske EP, Crino PB. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol. (2001) 49:67–78. doi: 10.1002/1531-8249(200101)49:1<67::aid-ana10>3.0.co;2-l
123. Boer K, Troost D, Timmermans W, Gorter JA, Spliet WG, Nellist M, et al. Cellular localization of metabotropic glutamate receptors in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Neuroscience. (2008) 156:203–15. doi: 10.1016/j.neuroscience.2008.06.073
124. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. (2009) 32:149–84. doi: 10.1146/annurev.neuro.051508.135600
125. Mo Z, Zecevic N. Human fetal radial glia cells generate oligodendrocytes in vitro. Glia. (2009) 57:490–8. doi: 10.1002/glia.20775
126. Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. (2005) 6:777–88. doi: 10.1038/nrm1739
127. Way SW, McKenna J III, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. (2009) 18:1252–65. doi: 10.1093/hmg/ddp025
128. Magri L, Cominelli M, Cambiaghi M, Cursi M, Leocani L, Minicucci F, et al. Timing of mTOR activation affects tuberous sclerosis complex neuropathology in mouse models. Dis Model Mech. (2013) 6:1185–97. doi: 10.1242/dmm.012096
129. Mietzsch U, McKenna J III, Reith RM, Way SW, Gambello MJ. Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J Comp Neurol. (2013) 521:3817–31. doi: 10.1002/cne.23380
130. Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci USA. (2009) 106:7840–5. doi: 10.1073/pnas.0901854106
131. Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. (2017) 358:1318–23. doi: 10.1126/science.aap8809
132. Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell. (2019) 176:743–56 e717. doi: 10.1016/j.cell.2019.01.017
133. Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. (2020) 578:142–8. doi: 10.1038/s41586-020-1962-0
134. Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. (2008) 29:541–51. doi: 10.1016/j.molcel.2007.12.023
135. Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. (2009) 29:5926–37. doi: 10.1523/JNEUROSCI.0778-09.2009
136. Urbach H, Scheffler B, Heinrichsmeier T, von Oertzen J, Kral T, Wellmer J, et al. Focal cortical dysplasia of Taylor's balloon cell type: a clinicopathological entity with characteristic neuroimaging and histopathological features, and favorable postsurgical outcome. Epilepsia. (2002) 43:33–40. doi: 10.1046/j.1528-1157.2002.38201.x
137. Baumer FM, Song JW, Mitchell PD, Pienaar R, Sahin M, Grant PE, et al. Longitudinal changes in diffusion properties in white matter pathways of children with tuberous sclerosis complex. Pediatr Neurol. (2015) 52:615–23. doi: 10.1016/j.pediatrneurol.2015.02.004
138. Scholl T, Muhlebner A, Ricken G, Gruber V, Fabing A, Samueli S, et al. Impaired oligodendroglial turnover is associated with myelin pathology in focal cortical dysplasia and tuberous sclerosis complex. Brain Pathol. (2017) 27:770–80. doi: 10.1111/bpa.12452
139. Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. (2010) 119:37–53. doi: 10.1007/s00401-009-0601-5
140. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q
141. Simao G, Raybaud C, Chuang S, Go C, Snead OC, Widjaja E. Diffusion tensor imaging of commissural and projection white matter in tuberous sclerosis complex and correlation with tuber load. Am J Neuroradiol. (2010) 31:1273–7. doi: 10.3174/ajnr.A2033
142. Krishnan ML, Commowick O, Jeste SS, Weisenfeld N, Hans A, Gregas MC, et al. Diffusion features of white matter in tuberous sclerosis with tractography. Pediatr Neurol. (2011) 42:101–6. doi: 10.1016/j.pediatrneurol.2009.08.001
143. Akins EJ, Moore ML, Tang S, Willingham MC, Tooze JA, Dubey P. In situ vaccination combined with androgen ablation and regulatory T-cell depletion reduces castration-resistant tumor burden in prostate-specific pten knockout mice. Cancer Res. (2010) 70:3473–82. doi: 10.1158/0008-5472.CAN-09-2490
144. Prohl AK, Scherrer B, Tomas-Fernandez X, Davis PE, Filip-Dhima R, Prabhu SP, et al. Early white matter development is abnormal in tuberous sclerosis complex patients who develop autism spectrum disorder. J Neurodev Disord. (2019) 11:36. doi: 10.1186/s11689-019-9293-x
145. Gaesser JM, Fyffe-Maricich SL. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp Neurol. (2016) 283:501–11. doi: 10.1016/j.expneurol.2016.03.008
146. Ishii A, Furusho M, Macklin W, Bansal R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia. (2019) 67:1277–95. doi: 10.1002/glia.23602
147. Musah AS, Brown TL, Jeffries MA, Shang Q, Hashimoto H, Evangelou AV, et al. Mechanistic target of rapamycin regulates the oligodendrocyte cytoskeleton during myelination. J Neurosci. (2020) 40:2993–3007. doi: 10.1523/JNEUROSCI.1434-18.2020
148. Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, et al. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. (2009) 29:6367–78. doi: 10.1523/JNEUROSCI.0234-09.2009
149. Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, Macklin WB. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J Neurosci. (2014) 34:4466–80. doi: 10.1523/JNEUROSCI.4314-13.2014
150. Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. (2014) 34:4453–65. doi: 10.1523/JNEUROSCI.4311-13.2014
151. Lebrun-Julien F, Bachmann L, Norrmen C, Trotzmuller M, Kofeler H, Ruegg MA, et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci. (2014) 34:8432–48. doi: 10.1523/JNEUROSCI.1105-14.2014
152. Carson RP, Kelm ND, West KL, Does MD, Fu C, Weaver G, et al. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann Clin Transl Neurol. (2015) 2:1041–54. doi: 10.1002/acn3.254
153. Grier MD, West KL, Kelm ND, Fu C, Does MD, Parker B, et al. Loss of mTORC2 signaling in oligodendrocyte precursor cells delays myelination. PLoS ONE. (2017) 12:e0188417. doi: 10.1371/journal.pone.0188417
154. Nadadhur AG, Alsaqati M, Gasparotto L, Cornelissen-Steijger P, van Hugte E, Dooves S, et al. Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem Cell Rep. (2019) 12:42–56. doi: 10.1016/j.stemcr.2018.11.019
155. Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. (2007) 27:5546–58. doi: 10.1523/JNEUROSCI.5540-06.2007
156. Ercan E, Han JM, Di Nardo A, Winden K, Han MJ, Hoyo L, et al. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J Exp Med. (2017) 214:681–97. doi: 10.1084/jem.20160446
157. Figlia G, Gerber D, Suter U. Myelination and mTOR. Glia. (2018) 66:693–707. doi: 10.1002/glia.23273
158. Chari DM. Remyelination in multiple sclerosis. Int Rev Neurobiol. (2007) 79:589–620. doi: 10.1016/S0074-7742(07)79026-8
159. Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kim HJ, Padmanabhan K, et al. A regenerative approach to the treatment of multiple sclerosis. Nature. (2015) 502:327–32. doi: 10.1038/nature12647.A
160. Tillema JM, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology. (2012) 78:526–31. doi: 10.1212/WNL.0b013e318247ca8d
161. Peters JM, Prohl A, Kapur K, Nath A, Scherrer B, Clancy S, et al. Longitudinal effects of everolimus on white matter diffusion in tuberous sclerosis complex. Pediatr Neurol. (2019) 90:24–30. doi: 10.1016/j.pediatrneurol.2018.10.005
162. Bedner P, Jabs R, Steinhauser C. Properties of human astrocytes and NG2 glia. Glia. (2020) 68:756–67. doi: 10.1002/glia.23725
163. Steinhauser C, Jabs R, Kettenmann H. Properties of GABA and glutamate responses in identified glial cells of the mouse hippocampal slice. Hippocampus. (1994) 4:19–35. doi: 10.1002/hipo.450040105
164. Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. (2010) 63:2–10. doi: 10.1016/j.brainresrev.2009.12.001
165. Degen J, Dublin P, Zhang J, Dobrowolski R, Jokwitz M, Karram K, et al. Dual reporter approaches for identification of Cre efficacy and astrocyte heterogeneity. FASEB J. (2012) 26:4576–83. doi: 10.1096/fj.12-207183
166. Moshrefi-Ravasdjani B, Dublin P, Seifert G, Jennissen K, Steinhauser C, Kafitz KW, et al. Changes in the proliferative capacity of NG2 cell subpopulations during postnatal development of the mouse hippocampus. Brain Struct Funct. (2017) 222:831–47. doi: 10.1007/s00429-016-1249-2
167. Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. (2003) 24:476–88. doi: 10.1016/s1044-7431(03)00210-0
168. Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. (2008) 28:10434–42. doi: 10.1523/JNEUROSCI.2831-08.2008
169. Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. (2008) 135:145–57. doi: 10.1242/dev.004895
170. Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. (2011) 138:745–53. doi: 10.1242/dev.047951
171. Levine JM, Reynolds R. Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Exp Neurol. (1999) 160:333–47. doi: 10.1006/exnr.1999.7224
172. Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. (2000) 405:187–91. doi: 10.1038/35012083
173. Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. (2004) 7:24–32. doi: 10.1038/nn1162
174. Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. (2009) 10:9–22. doi: 10.1038/nrn2495
175. Bergles DE, Jabs R, Steinhauser C. Neuron-glia synapses in the brain. Brain Res Rev. (2010) 63:130–7. doi: 10.1016/j.brainresrev.2009.12.003
176. Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. (2002) 19:447–58. doi: 10.1006/mcne.2001.1080
177. Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. (1996) 16:2659–70.
178. Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci. (2015) 18:674–82. doi: 10.1038/nn.3990
179. Sakry D, Neitz A, Singh J, Frischknecht R, Marongiu D, Biname F, et al. Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. (2014) 12:e1001993. doi: 10.1371/journal.pbio.1001993
180. Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. (2015) 138:1208–22. doi: 10.1093/brain/awv067
181. Nakano M, Tamura Y, Yamato M, Kume S, Eguchi A, Takata K, et al. NG2 glial cells regulate neuroimmunological responses to maintain neuronal function and survival. Sci Rep. (2017) 7:42041. doi: 10.1038/srep42041
182. Zhang SZ, Wang QQ, Yang QQ, Gu HY, Yin YQ, Li YD, et al. NG2 glia regulate brain innate immunity via TGF-beta2/TGFBR2 axis. BMC Med. (2019) 17:204. doi: 10.1186/s12916-019-1439-x
183. Nayak T, Trotter J, Sakry D. The intracellular cleavage product of the NG2 proteoglycan modulates translation and cell-cycle kinetics via effects on mTORC1/FMRP signaling. Front Cell Neurosci. (2018) 12:231. doi: 10.3389/fncel.2018.00231
184. González-Fernández E, Jeong H-K, Fukaya M, Kim H, Khawaja RR, Srivastava IN, et al. PTEN negatively regulates the cell lineage progression from NG2 + glial progenitor to oligodendrocyte via mTOR-independent signaling. eLife 7:e32021. doi: 10.7554/eLife.32021.001
185. McLane LE, Bourne JN, Evangelou AV, Khandker L, Macklin WB, Wood TL. Loss of tuberous sclerosis complex1 in adult oligodendrocyte progenitor cells enhances axon remyelination and increases myelin thickness after a focal demyelination. J Neurosci. (2017) 37:7534–46. doi: 10.1523/JNEUROSCI.3454-16.2017
186. Takahashi K, Naito M, Takeya M. Development and heterogeneity of macrophages and their related cells through their differentiation pathways. Pathol Int. (1996) 46:473–85. doi: 10.1111/j.1440-1827.1996.tb03641.x
187. Lichanska AM, Hume DA. Origins and functions of phagocytes in the embryo. Exp Hematol. (2000) 28:601–11. doi: 10.1016/s0301-472x(00)00157-0
188. Monier A, Adle-Biassette H, Delezoide AL, Evrard P, Gressens P, Verney C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol. (2007) 66:372–82. doi: 10.1097/nen.0b013e3180517b46
189. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
190. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
191. Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. (2001) 238:274–88. doi: 10.1006/dbio.2001.0393
192. Swinnen N, Smolders S, Avila A, Notelaers K, Paesen R, Ameloot M, et al. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia. (2013) 61:150–63. doi: 10.1002/glia.22421
193. Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. (2015) 7:a020537. doi: 10.1101/cshperspect.a020537
194. Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
195. Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. (2013) 33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013
196. Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. (2013) 61:24–36. doi: 10.1002/glia.22389
197. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
198. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
199. Schlegelmilch T, Henke K, Peri F. Microglia in the developing brain: from immunity to behaviour. Curr Opin Neurobiol. (2011) 21:5–10. doi: 10.1016/j.conb.2010.08.004
200. Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. (2015) 131:65–86. doi: 10.1016/j.pneurobio.2015.05.003
201. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.13139
202. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. (2016) 53:1181–94. doi: 10.1007/s12035-014-9070-5
203. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. (2012) 13:1118–28. doi: 10.1038/ni.2419
204. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. (2013) 16:1896–905. doi: 10.1038/nn.3554
205. Cherry JD, Olschowka JA, O'Banion MK. Are “resting” microglia more “m2”? Front Immunol. (2014) 5:594. doi: 10.3389/fimmu.2014.00594
206. Greter M, Lelios I, Croxford AL. Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol. (2015) 6:249. doi: 10.3389/fimmu.2015.00249
207. Roseti C, Fucile S, Lauro C, Martinello K, Bertollini C, Esposito V, et al. Fractalkine/CX3CL1 modulates GABAA currents in human temporal lobe epilepsy. Epilepsia. (2013) 54:1834–44. doi: 10.1111/epi.12354
208. Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. (2014) 15:43–53. doi: 10.1038/nrn3617
209. Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, et al. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun. (2014) 2:71. doi: 10.1186/2051-5960-2-71
210. Boer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E. Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol. (2006) 173:188–95. doi: 10.1016/j.jneuroim.2006.01.002
211. Choi J, Nordli DRJr, Alden TD, DiPatri AJr, et al. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J Neuroinflamm. (2009) 6:38. doi: 10.1186/1742-2094-6-38
212. Wirenfeldt M, Clare R, Tung S, Bottini A, Mathern GW, Vinters HV. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen's encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol Dis. (2009) 34:432–40. doi: 10.1016/j.nbd.2009.02.015
213. Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia. (2010) 51:1763–73. doi: 10.1111/j.1528-1167.2010.02547.x
214. Aronica E, Gorter JA, Redeker S, Ramkema M, Spliet WG, van Rijen PC, et al. Distribution, characterization and clinical significance of microglia in glioneuronal tumours from patients with chronic intractable epilepsy. Neuropathol Appl Neurobiol. (2005) 31:280–91. doi: 10.1111/j.1365-2990.2004.00636.x
215. Sun FJ, Zhang CQ, Chen X, Wei YJ, Li S, Liu SY, et al. Downregulation of CD47 and CD200 in patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. J Neuroinflamm. (2016) 13:85. doi: 10.1186/s12974-016-0546-2
216. Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘on’ and ‘off’ signals control microglia. Trends Neurosci. (2007) 30:596–602. doi: 10.1016/j.tins.2007.08.007
217. Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. (2018) 22:2080–93. doi: 10.1016/j.celrep.2018.02.004
218. Zhang B, Zou J, Han L, Beeler B, Friedman JL, Griffin E, et al. The specificity and role of microglia in epileptogenesis in mouse models of tuberous sclerosis complex. Epilepsia. (2018) 59:1796–806. doi: 10.1111/epi.14526
219. Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. (2012) 75:1022–34. doi: 10.1016/j.neuron.2012.08.002
220. Zhuang X, Yu Y, Jiang Y, Zhao S, Wang Y, Su L, et al. Molecular hydrogen attenuates sepsis-induced neuroinflammation through regulation of microglia polarization through an mTOR-autophagy-dependent pathway. Int Immunopharmacol. (2020) 81:106287. doi: 10.1016/j.intimp.2020.106287
221. Brewster AL, Lugo JN, Patil VV, Lee WL, Qian Y, Vanegas F, et al. Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage. PLoS ONE. (2013) 8:e57808. doi: 10.1371/journal.pone.0057808
222. Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. (2014) 192:6009–19. doi: 10.4049/jimmunol.1303492
223. Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R, et al. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. (2016) 30:3388–99. doi: 10.1096/fj.201600495R
224. Chen L, Zhang Y, Li D, Zhang N, Liu R, Han B, et al. Everolimus (RAD001) ameliorates vascular cognitive impairment by regulating microglial function via the mTORC1 signaling pathway. J Neuroimmunol. (2016) 299:164–71. doi: 10.1016/j.jneuroim.2016.09.008
225. Guo JR, Wang H, Jin XJ, Jia DL, Zhou X, Tao Q. Effect and mechanism of inhibition of PI3K/Akt/mTOR signal pathway on chronic neuropathic pain and spinal microglia in a rat model of chronic constriction injury. Oncotarget. (2017) 8:52923–34. doi: 10.18632/oncotarget.17629
226. Yang MT, Lin YC, Ho WH, Liu CL, Lee WT. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J Neuroinflamm. (2017) 14:15. doi: 10.1186/s12974-017-0797-6
227. Dello Russo C, Lisi L, Tringali G, Navarra P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol. (2009) 78:1242–51. doi: 10.1016/j.bcp.2009.06.097
228. Gao C, Wang H, Wang T, Luo C, Wang Z, Zhang M, et al. Platelet regulates neuroinflammation and restores blood-brain barrier integrity in a mouse model of traumatic brain injury. J Neurochem. (2020) 154:190–204. doi: 10.1111/jnc.14983
229. Ye X, Zhu M, Che X, Wang H, Liang XJ, Wu C, et al. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J Neuroinflamm. (2020) 17:18. doi: 10.1186/s12974-019-1644-8
230. Abraham J, Fox PD, Condello C, Bartolini A, Koh S. Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiol Dis. (2012) 46:425–30. doi: 10.1016/j.nbd.2012.02.006
231. Han J, Zhu K, Zhang XM, Harris RA. Enforced microglial depletion and repopulation as a promising strategy for the treatment of neurological disorders. Glia. (2019) 67:217–31. doi: 10.1002/glia.23529
232. Zhang B, Zou J, Han L, Rensing N, Wong M. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia. (2016) 57:1317–25. doi: 10.1111/epi.13429
233. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
234. Jha MK, Jo M, Kim JH, Suk K. Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist. (2019) 25:227–40. doi: 10.1177/1073858418783959
235. Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. (2013) 36:174–84. doi: 10.1016/j.tins.2012.11.008
236. Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. (2016) 4:71. doi: 10.3389/fcell.2016.00071
237. Moore CS, Abdullah SL, Brown A, Arulpragasam A, Crocker SJ. How factors secreted from astrocytes impact myelin repair. J Neurosci Res. (2011) 89:13–21. doi: 10.1002/jnr.22482
238. Li J, Zhang L, Chu Y, Namaka M, Deng B, Kong J, et al. Astrocytes in oligodendrocyte lineage development and white matter pathology. Front Cell Neurosci. (2016) 10:119. doi: 10.3389/fncel.2016.00119
239. Bugiani M, van der Knaap MS. Childhood white matter disorders: much more than just diseases of myelin. Acta Neuropathol. (2017) 134:329–30. doi: 10.1007/s00401-017-1750-6
240. Bugiani M, Vuong C, Breur M, van der Knaap MS. Vanishing white matter: a leukodystrophy due to astrocytic dysfunction. Brain Pathol. (2018) 28:408–21. doi: 10.1111/bpa.12606
241. Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci. (2008) 35:101–16. doi: 10.1007/s12031-007-9027-5
242. Tress O, Maglione M, May D, Pivneva T, Richter N, Seyfarth J, et al. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J Neurosci. (2012) 32:7499–518. doi: 10.1523/JNEUROSCI.0392-12.2012
243. Markoullis K, Sargiannidou I, Schiza N, Roncaroli F, Reynolds R, Kleopa KA. Oligodendrocyte gap junction loss and disconnection from reactive astrocytes in multiple sclerosis gray matter. J Neuropathol Exp Neurol. (2014) 73:865–79. doi: 10.1097/NEN.0000000000000106
244. Yoshioka A, Hardy M, Younkin DP, Grinspan JB, Stern JL, Pleasure D. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors mediate excitotoxicity in the oligodendroglial lineage. J Neurochem. (1995) 64:2442–8. doi: 10.1046/j.1471-4159.1995.64062442.x
245. McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. (1998) 4:291–7. doi: 10.1038/nm0398-291
246. Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. (2005) 438:1162–6. doi: 10.1038/nature04302
247. Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. (2006) 439:988–92. doi: 10.1038/nature04474
248. Brambilla R, Morton PD, Ashbaugh JJ, Karmally S, Lambertsen KL, Bethea JR. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia. (2014) 62:452–67. doi: 10.1002/glia.22616
249. Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, et al. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci. (2006) 26:4383–93. doi: 10.1523/JNEUROSCI.4531-05.2006
250. Pang Y, Fan LW, Tien LT, Dai X, Zheng B, Cai Z, et al. Differential roles of astrocyte and microglia in supporting oligodendrocyte development and myelination in vitro. Brain Behav. (2013) 3:503–14. doi: 10.1002/brb3.152
251. Pang Y, Cai Z, Rhodes PG. Effects of lipopolysaccharide on oligodendrocyte progenitor cells are mediated by astrocytes and microglia. J Neurosci Res. (2000) 62:510–20. doi: 10.1002/1097-4547(20001115)62:4<510::AID-JNR5>3.0.CO;2-F
252. French HM, Reid M, Mamontov P, Simmons RA, Grinspan JB. Oxidative stress disrupts oligodendrocyte maturation. J Neurosci Res. (2009) 87:3076–87. doi: 10.1002/jnr.22139
253. Pang Y, Campbell L, Zheng B, Fan L, Cai Z, Rhodes P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience. (2010) 166:464–75. doi: 10.1016/j.neuroscience.2009.12.040
254. Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. (2006) 54:795–804. doi: 10.1002/glia.20416
255. Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. (2012) 45:369–80. doi: 10.1016/j.nbd.2011.08.024
256. Rensing N, Han L, Wong M. Intermittent dosing of rapamycin maintains antiepileptogenic effects in a mouse model of tuberous sclerosis complex. Epilepsia. (2015) 56:1088–97. doi: 10.1111/epi.13031
257. Moavero R, Curatolo P. Long-term use of mTORC1 inhibitors in tuberous sclerosis complex associated neurological aspects. Expert Opin Orphan Drugs. (2020) 8:215–25. doi: 10.1080/21678707.2020.1789862
258. Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. (2010) 40:193–9. doi: 10.1016/j.nbd.2010.05.024
Keywords: tuberous sclerosis (TSC), mammalian target of rapamycin (mTOR), epilepsy, astrocyte, microglia, oligodendrocyte, glia, epileptogenesis
Citation: Zimmer TS, Broekaart DWM, Gruber V, van Vliet EA, Mühlebner A and Aronica E (2020) Tuberous Sclerosis Complex as Disease Model for Investigating mTOR-Related Gliopathy During Epileptogenesis. Front. Neurol. 11:1028. doi: 10.3389/fneur.2020.01028
Received: 07 June 2020; Accepted: 06 August 2020;
Published: 17 September 2020.
Edited by:
Christian Steinhaeuser, Universität Bonn, GermanyReviewed by:
Peter Bedner, University Hospital Bonn, GermanyCopyright © 2020 Zimmer, Broekaart, Gruber, van Vliet, Mühlebner and Aronica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Aronica, ZS5hcm9uaWNhQGFtc3RlcmRhbXVtYy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.