 Allison R. Peterson
Allison R. Peterson Devin K. Binder
Devin K. Binder- Division of Biomedical Sciences, Center for Glial-Neuronal Interactions, School of Medicine, University of California, Riverside, Riverside, CA, United States
Astrocytes regulate and respond to extracellular glutamate levels in the central nervous system (CNS) via the Na+-dependent glutamate transporters glutamate transporter-1 (GLT-1) and glutamate aspartate transporter (GLAST) and the metabotropic glutamate receptors (mGluR) 3 and mGluR5. Both impaired astrocytic glutamate clearance and changes in metabotropic glutamate signaling could contribute to the development of epilepsy. Dysregulation of astrocytic glutamate transporters, GLT-1 and GLAST, is a common finding across patients and preclinical seizure models. Astrocytic metabotropic glutamate receptors, particularly mGluR5, have been shown to be dysregulated in both humans and animal models of temporal lobe epilepsy (TLE). In this review, we synthesize the available evidence regarding astrocytic glutamate homeostasis and astrocytic mGluRs in the development of epilepsy. Modulation of astrocyte glutamate uptake and/or mGluR activation could lead to novel glial therapeutics for epilepsy.
Introduction
Epilepsy is a common neurological disorder and is characterized by the occurrence of unprovoked seizures. Epilepsy is a major public health problem, affecting more than 65 million people worldwide (1). Healthcare cost estimates associated with epilepsy in the United States range from $9.6 to $12 billion per year (2). TLE is the most common form of epilepsy with focal seizures. TLE is also frequently associated with refractory epilepsy. Approximately 1/4 of patients with TLE develop refractory epilepsies that are pharmaco-resistant to currently available antiepileptic drugs (AEDs) (3).
AEDs work primarily by targeting neurons through modulation of ion channels, enhancement of inhibitory neurotransmission or attenuation of excitatory neurotransmission (4). Most AEDs target channels on neurons to exert their antiepileptic effects. Newer generation AEDs still primarily target neurons but through novel mechanisms and unique binding sites [e.g., AMPA-R, CMRP2, SV2A, or inhibition of carbonic anhydrase activity (5)]. Modulation of neurotransmission can consequently lead to dose-dependent “neurotoxic” adverse effects which are common undesired effects associated with AED usage. Adverse cognitive and behavioral effects of AEDs have been shown to lead to AED discontinuation in up to one-third of patients (6). Therefore, new non-neuronal targets that could potentially have fewer side effects should be considered and further investigated.
Neuronal hyperexcitability is a major contributor to epilepsy but increased evidence suggests that changes in astrocytes can contribute to the development of epilepsy (7–13). Astrocytes are involved in ionic homeostasis, regulation of extracellular space volume and clearance of neurotransmitters. Astrocytes are a critical component of the tripartite synapse, where they are involved in the active control of neuronal activity and synaptic neurotransmission. Astrocytes regulate extracellular glutamate levels via Na+-dependent glutamate transporters, glutamate transporter-1 (GLT-1) and glutamate aspartate transporter (GLAST). GLT-1 is responsible for ~90% of glutamate uptake in the adult dorsal forebrain and is crucial for the maintenance of low extracellular glutamate to permit efficient synaptic transmission (14). The human homologs of GLAST and GLT-1 are EAAT1 and EAAT2, respectively. In this review we will be referring to these transporters in pre-clinical and clinical studies by GLAST and GLT-1. Aside from perisynaptic glutamate uptake, astrocytes can also sense extracellular glutamate to more readily adapt to their microenvironments through metabotropic glutamate receptors mGluR3 and mGluR5. These G-protein coupled receptors can differentially modulate the expression of glutamate transporters and glutamate release therefore indirectly regulating synaptic activity. This review will provide an overview of what we currently understand regarding the regulation of astrocytic glutamate transporters and receptors in the development of epilepsy. Targeting glutamate uptake and/or glutamate receptor activation through astrocytes could lead to novel treatment options for patients with refractory epilepsies.
Dysregulation of Glutamate Uptake in Epileptogenesis
GLT-1 and GLAST are the primary transporters responsible for glutamate clearance in the central nervous system (CNS) following excitatory neuronal transmission. It is crucial to maintain low levels of basal extracellular glutamate in the brain to permit efficient and localized synaptic transmission. Evidence of increased glutamate levels have been observed in patients suffering from TLE and in preclinical seizure models (15–17). The vast majority of GLT-1 is astrocytic with synaptic localization, with ~5–10% of expression in neurons (18, 19). Mice that globally lack GLT-1 develop lethal spontaneous seizures, while transgenic mice that overexpress GLT-1 have a higher seizure threshold than wild-type mice, suggesting that GLT-1 plays an important role in preventing seizures and protection against glutamate toxicity (20, 21). In multiple preclinical studies, GLT-1 protein levels have been shown to be downregulated during the development of epilepsy (Figure 1). Perisynaptic GLT-1 at the plasma membrane in astrocytes is significantly reduced around CA3-CA1 synapses during the latent period following systemic kainate-induced status epilepticus (SE) (22). Hippocampal GLT-1 total protein levels have been found to be downregulated following intrahippocampal kainate-induced SE (11). Crude synaptosomal GLT-1 levels, which include components of the tripartite synapse, are also reduced nearly 80% 1 week following intrahippocampal kainate induced-SE in the hippocampus early in the epileptogenic process (13). These data suggest that the pool of transporters available for glutamate uptake at excitatory synapses is substantially reduced in epileptogenesis. The kainic acid (KA) model of TLE is characterized by a period of SE, that serves as the initial insult, followed by a latency period where the mice are seizure-free followed by the occurrence of spontaneous recurrent seizures (23–25). Downregulation in GLT-1 protein levels observed in these studies interestingly coincides with the approximate onset of spontaneous seizures, demonstrating that glutamate transporter dysregulation could contribute to the development of epilepsy (13, 22, 26–28). Interestingly, GLT-1 protein levels were found to be upregulated in in a spontaneously epileptic rat, a double mutant (zi/zi, tm, tm), compared to control Wistar rats (29). GLT-1 protein levels have also been shown to be disrupted in patients with TLE (8, 9). GLT-1 levels have been found to be decreased in the hippocampus of TLE patients with hippocampal sclerosis (HS) in most (8, 9) but not all (30) studies. In patients with decreased GLT-1, severe neuronal cell loss was observed suggesting that loss of glutamate transporters could exacerbate neurotoxicity in epilepsy (8, 9).
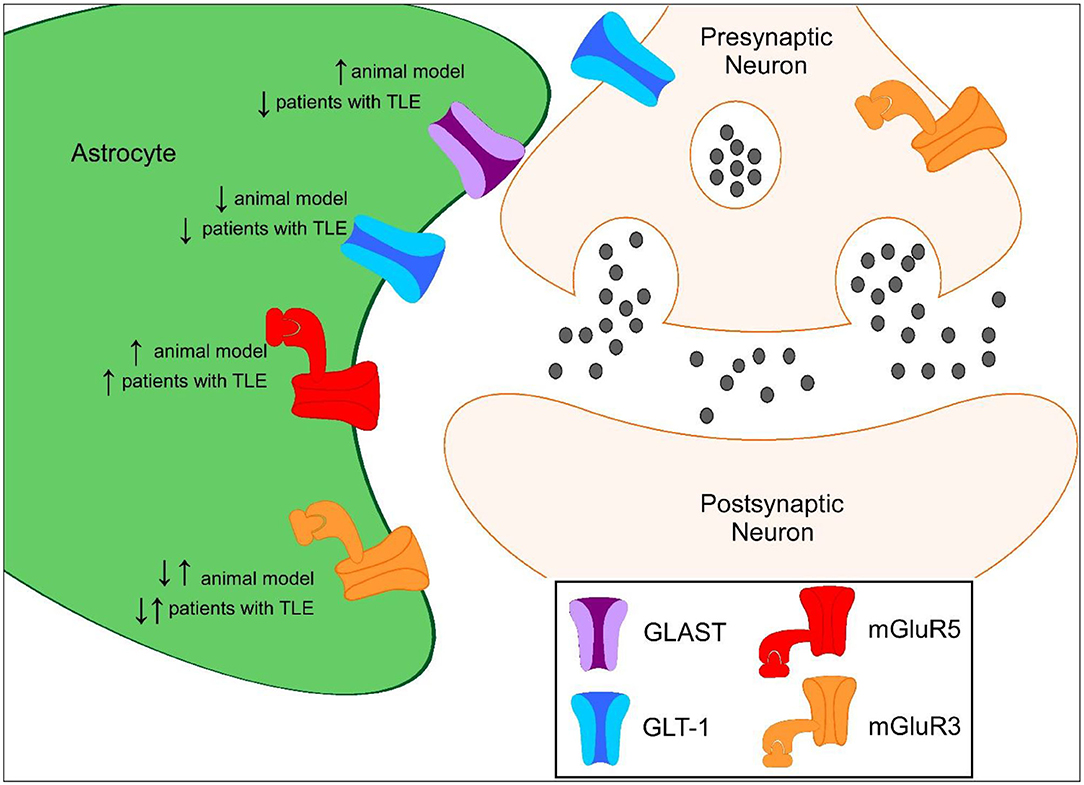
Figure 1. Proposed protein expression of glutamate transporters and mGluRs in the hippocampus during epileptogenesis compared to controls. ↑ represents increased protein expression observed compared to control. ↓ represents decreased protein expression observed compared to control. ↓↑ represents decreased and increased in protein expression observed compared to control.
GLAST has also shown to be dysregulated in the epileptic brain. GLAST is found in the dorsal forebrain postnatally and homogenously distributed among astrocytic soma and endfeet compared to its counterpart GLT-1 (18, 31). GLAST-deficient mice have significantly longer seizure duration compared to wild-type mice suggesting that GLAST also plays a role in seizure susceptibility (32). In a preclinical model of TLE, synaptosomal GLAST protein levels were elevated in the epileptic hippocampus while overall total protein levels were unchanged at chronic time points (13). GLAST protein levels were found to be significantly lower in a spontaneously epileptic rat, a double mutant (zi/zi, tm, tm), compared to control Wistar rats (29). In TLE patients with HS, GLAST protein levels have been shown to be downregulated while GLAST protein levels are unchanged in TLE patients without HS (8). Astrocytic glutamate synthetase is responsible for the rapid conversion of intracellular glutamate to glutamine and is a prerequisite for efficient glutamate clearance from the extracellular space. Loss of glutamine synthetase has also been observed in patients with TLE which could have an impact on glutamate transporter clearance (30). These findings suggest that glutamate transporter dysregulation could contribute to increased extracellular glutamate and ictogenesis in the epileptic brain.
Regulation of Astrocytic Glutamate Receptors in Epileptogenesis
Metabotropic glutamate receptors (mGluRs) are G-protein coupled receptors (GPCRs) important in synaptic neuromodulation. These receptors can be divided into three separate families: Group I, Group II, and Group III, based on their structure and downstream function (33). Metabotropic glutamate receptors found on astrocytes can influence astrocytic functions in physiology and disease. Astrocytes dominantly express mGluR3 and mGluR5 receptors and differential regulation of these receptors has been observed in epilepsy (10, 34, 35). Astrocytic mGluR5 signaling plays an important role in astrocytic motility, ensheathment and glutamate transport in the developing brain (36, 37). Expression of astrocytic mGluR5 is typically limited to the first few weeks of brain development (37–39). Activation of Group I mGluR5 receptors, coupled to Gαq proteins, has been shown to acutely alter GLT-1 activity by increasing glutamate clearance in astrocytes (40) but chronic stimulation can lead to a reduction in astrocytic GLT-1 and GLAST levels resulting in reduced glutamate transport (35).
Differential expression of astrocytic mGluRs have been reported in patients and preclinical models of epilepsy. mGluR5 levels have been shown to be overexpressed in murine seizure models. mGluR5 expression in reactive astrocytes is persistently upregulated following electrically induced SE in a kindling model and in TLE (34). Selective positive modulation of mGluR5 in the Theiler's murine encephalomyelitis virus (TMEV)-induced model of epilepsy attenuates seizures (41). Additionally, selectively knocking out astrocytic mGluR5 signaling during epilepsy slows glutamate clearance through glutamate transporters, suggesting that mGluR5 plays an important function in regulating these transporters in epileptogenesis (42).
Multiple studies have shown that mGluR5 levels are also increased in patients with TLE (10, 43). mGluR5 expression levels in patients have been associated with seizure frequency. Lower mGluR5 expression was found to be negatively correlated with seizure frequency and epilepsy duration in patients with TLE (non-HS) (10). Conditional knockout of mGluR5 signaling from astrocytes slowed glutamate clearance in epileptogenesis (42). These data support the hypothesis that mGluR5 upregulation could act as a compensatory mechanism to counterbalance the hyperexcitability observed in epilepsy.
It is important to note that activation of mGluR5 has also been shown to lead to increased excitability. For example, stimulation of group I mGluRs, including mGluR5, elicits ictal-like events in hippocampal slices (44). Following SE, mGluR5 activation has also been shown to enhance astrocytic calcium signals during the latency period of epileptogenesis in the pilocarpine model of TLE (45). Moreover, increases in astrocytic calcium transients can lead to release of gliotransmitters, including glutamate, and activation of NMDA receptors (46). In one study, intrahippocampal perfusion of the mGluR group 1 agonist, DHPG (R,S-3,5-dihydroxyphenylglycine), induced seizures while infusion of the mGluR5 receptor antagonist, MPEP (2-methyl-6-(phenylethynyl)-pyridin), attenuated pilocarpine-induced seizures (Table 1) (52).
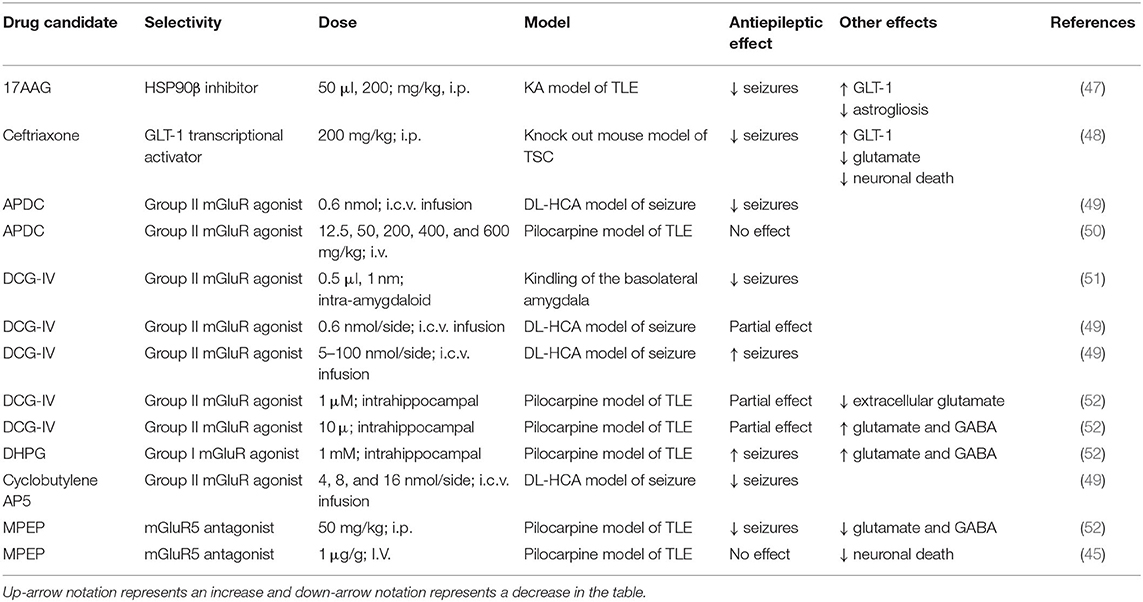
Table 1. Positive and negative outcomes of glutamate transporter modulation and mGluR agonists/antagonist in preclinical seizure models.
Hyperexcitability associated with mGluR5 activation has also been observed in other neurological diseases. For example, preclinical data suggest that in Fragile X Syndrome, a genetic form of autism, the absence of fragile X mental retardation protein (FMRP) leads to overstimulation of the mGluR5 pathway enhancing glutamatergic signaling contributing to phenotypes observed in this disease (53–55). Interestingly, treatment of Fmr1 knockout mice with negative modulators of mGluR5 ameliorates phenotypes (56, 57). These studies indicate that although acute activation of mGluR5 can decrease excitability, chronic stimulation, which could occur in a diseased state, can be detrimental.
Activation of Group II mGluR3 receptors, which are coupled to Gαi proteins in astrocytes, may have neuroprotective functions including increasing the capacity for glutamate clearance in the CNS through upregulation of glutamate transporters (35, 58). mGluR3 receptor activation has been shown to upregulate GLT-1 and GLAST protein levels promoting increased glutamate uptake in astrocytes (35, 58). mGluR3 receptors are also found in the presynaptic terminals of glutamatergic neurons (59). mGluR3 receptor agonists have also been shown to protect neurons from excitotoxicity and astrocytes from nitric oxide-induced death (60). Astrocyte-specific mGluR3 expression is markedly increased at early and chronic time points following SE in CA3 and hilar region following electrically induced SE in a kindling model and in TLE (34). A reduction in astrocyte-specific mGluR3 was observed in the molecular layer and stratum lacunosum moleculare of the hippocampus at chronic time points (34). mGluR2/3 expression was also found to be markedly decreased both acute and chronic time points following pilocarpine-induced SE (50).
Whether mGluR3 expression levels are upregulated or downregulated in patients with TLE is controversial. One study found mGluR2/3 expression is downregulated (50) while a separate study showed that mGluR2/3 is upregulated in the hippocampi of TLE patients (61). Whether this discrepancy is due to study design, severity or stage of epilepsy, region-specific effects, or technical differences remains to be determined. Future studies could further examine the use of selective negative modulators of mGluR5 or positive modulators of mGluR3 as an alternative therapeutic approach to treat epilepsy.
Astrocytic Glutamate Uptake and Targeting of Glutamate Receptors as Therapies for Refractory Epilepsies
Non-neuronal targets, including glial cells, are an attractive alternative approach to treat patients whose seizures are not well-controlled with currently available AEDs. Increasing astrocytic glutamate uptake capacity by upregulation of glutamate transporters has been shown to have neuroprotective and anti-epileptic effects. Seizures were significantly reduced and astrogliosis was attenuated when mice were administered an HSP90β inhibitor to increase GLT-1 expression in a mouse model of TLE (47). Ceftriaxone, a β-lactam antibiotic, has also been shown to upregulate GLT-1 protein expression and reduce seizures in multiple preclinical studies (48, 62, 63). Treatment with ceftriaxone has shown negative adverse side effects including impairment in synaptic plasticity and memory recognition (64, 65). Ceftriaxone affects many pathways in the CNS, therefore, it is currently not well-understood if these adverse effects are a result of GLT-1 activation. Nevertheless, selectively targeting aberrant astrocytes could reduce adverse side effects. Intraspinal delivery of AAV8-Gfa2-GLT1 has been used to selectively increase GLT-1 protein expression under the truncated glial fibrillary acidic protein promotor in a model of spinal cord injury showing promising results (66). Gene therapy could potentially be used to target subpopulations of astrocytes by selecting genes known to be overexpressed in the epileptic brain. For example, adenosine kinase is strikingly upregulated in reactive astrocytes after kainic acid-induced SE and its promotor could be used to selectively target this cell population (67).
The mGluR5 receptor antagonist MPEP reduced seizures when administered i.p. in the pilocarpine seizure model (52). In contrast, another study also using the pilocarpine seizure model found that MPEP suppressed neuronal death but did not result in a change in synaptic activity, suggesting that astrocytes could have neurotoxic roles in epilepsy through increased gliotransmission (45). Future studies should further examine mGluR5 antagonists as potential adjunctive therapies to decrease the severe neuronal loss observed in TLE patients with HS. The mGluR2/3 agonist, APDC, was shown to reduce seizure in the DL-homocysteic acid (DL-HCA) seizure model (49). In the pilocarpine seizure model, APDC did not reduce seizures nor neuronal death (50). These studies indicate that selection of agonist/antagonists can have differential outcomes. Two Group II mGluR agonists targeting mGluR2/3, cyclobutylene AP5 and DCG-IV, have both demonstrated positive effects on seizure control in the DL-homocysteic acid (DL-HCA) seizure model and kindling model of TLE (49, 51). Interestingly, at higher doses DL-HCA has been shown to have pro-epileptic effects (49, 52). Thus, activation of Group II mGluRs may be another promising avenue for alternative therapies for treating epilepsy.
Conclusion
Astrocytes play a critical role in the development and progression of epilepsy (7, 8, 30, 68–76). Astrocytic glutamate uptake is dysregulated in both preclinical models and in patients with TLE leading to increases in basal glutamate levels, and activation and signaling of astrocytic metabotropic glutamate receptors, mGluR3 and mGluR5, is also altered in animal models and patients with TLE. It is not clear yet whether targeting glutamate transporters and receptors would be more effective as a novel antiepileptic (controlling seizures in pharmacoresistant epilepsies) or antiepileptogenic (disease-modifying prevention of development of epilepsy after epileptogenic insults) strategy. Future studies should distinguish antiepileptic vs. antiepileptogenic effects, for example of GLT1 upregulation in appropriate animal models. Targeting of altered “epileptic” glutamate metabolism and signaling in astrocytes has the potential of efficacy with fewer side effects compared to traditional suppression of glutamatergic neurotransmission in neurons. This could lead to novel approaches to antiepileptic, antiepileptogenic, and/or neuroprotective therapies.
Author Contributions
AP did an exhaustive literature search, generated a complete draft of the review, and prepared the table and figure. DB also reviewed the literature, provided detailed comments, and edits to the review and the table and figure. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Clossen BL, Reddy DS. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1519–38. doi: 10.1016/j.bbadis.2017.02.003
2. Lekoubou A, Bishu KG, Ovbiagele B. Nationwide trends in medical expenditures among adults with epilepsy, 2003–2014. J Neurol Sci. (2018) 384:113–20. doi: 10.1016/j.jns.2017.11.025
3. Kwan P, Schachter SC, Brodie MJ. Drug-resistant. Epilepsy. (2011) 365:919–26. doi: 10.1056/NEJMra1004418
4. Stafstrom CE. Mechanisms of action of antiepileptic drugs: the search for synergy. Curr Opin Neurol. (2010) 23:157–63. doi: 10.1097/WCO.0b013e32833735b5
5. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. (2007) 4:18–61. doi: 10.1016/j.nurt.2006.11.010
6. Bootsma HP, Ricker L, Hekster YA, Hulsman J, Lambrechts D, Majoie M, et al. The impact of side effects on long-term retention in three new antiepileptic drugs. Seizure. (2009) 18:327–31. doi: 10.1016/j.seizure.2008.11.006
7. Tessler S, Danbolt NC, Faull RL, Storm-Mathisen J, Emson PC. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. (1999) 88:1083–91. doi: 10.1016/S0306-4522(98)00301-7
8. Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. (2002) 125:32–43. doi: 10.1093/brain/awf001
9. Sarac S, Afzal S, Broholm H, Madsen FF, Ploug T, Laursen H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS. (2009) 117:291–301. doi: 10.1111/j.1600-0463.2009.02443.x
10. Kandratavicius L, Rosa-Neto P, Monteiro MR, Guiot MC, Assirati JA Jr, Carlotti CG Jr, et al. Distinct increased metabotropic glutamate receptor type 5 (mGluR5) in temporal lobe epilepsy with and without hippocampal sclerosis. Hippocampus. (2013) 23:1212–30. doi: 10.1002/hipo.22160
11. Hubbard JA, Szu JI, Yonan JM, Binder DK. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol. (2016) 283:85–96. doi: 10.1016/j.expneurol.2016.05.003
12. Munoz-Ballester C, Berthier A, Viana R, Sanz P. Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim Biophys Acta. (2016) 1862:1074–83. doi: 10.1016/j.bbadis.2016.03.008
13. Peterson A, Binder RDK. Regulation of Synaptosomal GLT-1 and GLAST during Epileptogenesis. Neuroscience. (2019) 411:185–201. doi: 10.1016/j.neuroscience.2019.05.048
14. Danbolt NC. Glutamate uptake. Prog Neurobiol. (2001) 65:1–105. doi: 10.1016/S0301-0082(00)00067-8
15. During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. (1993) 341:1607–10. doi: 10.1016/0140-6736(93)90754-5
16. Cavus I, Romanyshyn JC, Kennard JT, Farooque P, Williamson A, Eid T, et al. Elevated basal glutamate and unchanged glutamine and GABA in refractory epilepsy: microdialysis study of 79 patients at the yale epilepsy surgery program. Ann Neurol. (2016) 80:35–45. doi: 10.1002/ana.24673
17. Kanamori K. Faster flux of neurotransmitter glutamate during seizure—evidence from 13C-enrichment of extracellular glutamate in kainate rat model. PLoS ONE. (2017) 12:e0174845. doi: 10.1371/journal.pone.0174845
18. Schreiner AE, Durry S, Aida T, Stock MC, Ruther U, Tanaka K, et al. Laminar and subcellular heterogeneity of GLAST and GLT-1 immunoreactivity in the developing postnatal mouse hippocampus. J Comp Neurol. (2014) 522:204–24. doi: 10.1002/cne.23450
19. Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, et al. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. (2015) 35:5187–201. doi: 10.1523/JNEUROSCI.4255-14.2015
20. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. (1997) 276:1699–702. doi: 10.1126/science.276.5319.1699
21. Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis. (2012) 47:145–54. doi: 10.1016/j.nbd.2012.03.032
22. Clarkson C, Smeal RM, Hasenoehrl MG, White JA, Rubio ME, Wilcox KS. Ultrastructural and functional changes at the tripartite synapse during epileptogenesis in a model of temporal lobe epilepsy. Exp Neurol. (2020) 326:113196. doi: 10.1016/j.expneurol.2020.113196
23. Bouilleret V, Ridoux V, Depaulis A, Marescaux C, Nehlig A, Le Gal La Salle G. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience. (1999) 89:717–29. doi: 10.1016/S0306-4522(98)00401-1
24. Riban V, Bouilleret V, Pham-Le BT, Fritschy JM, Marescaux C, Depaulis A. Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience. (2002) 112:101–11. doi: 10.1016/S0306-4522(02)00064-7
25. Levesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. (2013) 37:2887–99. doi: 10.1016/j.neubiorev.2013.10.011
26. Raedt R, Van Dycke A, Van Melkebeke D, De Smedt T, Claeys P, Wyckhuys T, et al. Seizures in the intrahippocampal kainic acid epilepsy model: characterization using long-term video-EEG monitoring in the rat. Acta Neurol Scand. (2009) 119:293–303. doi: 10.1111/j.1600-0404.2008.01108.x
27. Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. (2009) 29:2103–12. doi: 10.1523/JNEUROSCI.0980-08.2009
28. Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol. (2012) 235:246–55. doi: 10.1016/j.expneurol.2012.02.002
29. Guo F, Sun F, Yu JL, Wang QH, Tu DY, Mao XY, et al. Abnormal expressions of glutamate transporters and metabotropic glutamate receptor 1 in the spontaneously epileptic rat hippocampus. Brain Res Bull. (2010) 81:510–6. doi: 10.1016/j.brainresbull.2009.10.008
30. Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. (2004) 363:28–37. doi: 10.1016/S0140-6736(03)15166-5
31. Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. (1997) 17:8363–75. doi: 10.1523/JNEUROSCI.17-21-08363.1997
32. Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. (1999) 845:92–6. doi: 10.1016/S0006-8993(99)01945-9
33. Spampinato SF, Copani A, Nicoletti F, Sortino MA, Caraci F. Metabotropic glutamate receptors in glial cells: a new potential target for neuroprotection? Front Mol Neurosci. (2018) 11:414. doi: 10.3389/fnmol.2018.00414
34. Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. (2000) 12:2333–44. doi: 10.1046/j.1460-9568.2000.00131.x
35. Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. (2003) 17:2106–18. doi: 10.1046/j.1460-9568.2003.02657.x
36. Cai Z, Schools GP, Kimelberg HK. Metabotropic glutamate receptors in acutely isolated hippocampal astrocytes: developmental changes of mGluR5 mRNA and functional expression. Glia. (2000) 29:70–80. doi: 10.1002/(SICI)1098-1136(20000101)29:1<70::AID-GLIA7>3.0.CO;2-V
37. Bernardinelli Y, Randall J, Janett E, Nikonenko I, Konig S, Jones EV, et al. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol. (2014) 24:1679–88. doi: 10.1016/j.cub.2014.06.025
38. Devaraju P, Sun MY, Myers TL, Lauderdale K, Fiacco TA. Astrocytic group I mGluR-dependent potentiation of astrocytic glutamate and potassium uptake. J Neurophysiol. (2013) 109:2404–14. doi: 10.1152/jn.00517.2012
39. Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. (2013) 339:197–200. doi: 10.1126/science.1226740
40. Vermeiren C, Najimi M, Vanhoutte N, Tilleux S, de Hemptinne I, Maloteaux JM, et al. Acute up-regulation of glutamate uptake mediated by mGluR5a in reactive astrocytes. J Neurochem. (2005) 94:405–16. doi: 10.1111/j.1471-4159.2005.03216.x
41. Hanak TJ, Libbey JE, Doty DJ, Sim JT, DePaula-Silva AB, Fujinami RS. Positive modulation of mGluR5 attenuates seizures and reduces TNF-alpha(+) macrophages and microglia in the brain in a murine model of virus-induced temporal lobe epilepsy. Exp Neurol. (2019) 311:194–204. doi: 10.1016/j.expneurol.2018.10.006
42. Umpierre AD, West PJ, White JA, Wilcox KS. Conditional knock-out of mGluR5 from astrocytes during epilepsy development impairs high-frequency glutamate uptake. J Neurosci. (2019) 39:727–42. doi: 10.1523/JNEUROSCI.1148-18.2018
43. Notenboom RG, Hampson DR, Jansen GH, van Rijen PC, van Veelen CW, van Nieuwenhuizen O, et al. Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain. (2006) 129:96–107. doi: 10.1093/brain/awh673
44. Bianchi R, Wong RKS, Merlin LR. Glutamate Receptors in Epilepsy: Group I mGluR-Mediated Epileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper's Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda, MD: National Center for Biotechnology Information (US) (2012).
45. Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, et al. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. (2007) 27:10674–84. doi: 10.1523/JNEUROSCI.2001-07.2007
46. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. (2016) 19:182–9. doi: 10.1038/nn.4201
47. Sha L, Wang X, Li J, Shi X, Shen Y. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J Exp Med. (2016) 214:547–63. doi: 10.1084/jem.20160667
48. Zeng LH, Bero AW, Zhang B, Holtzman DM, Wong M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol Dis. (2010) 37:764–71. doi: 10.1016/j.nbd.2009.12.020
49. Folbergrova J, Haugvicova R, Mares P. Attenuation of seizures induced by homocysteic acid in immature rats by metabotropic glutamate group II and group III receptor agonists. Brain Res. (2001) 908:120–9. doi: 10.1016/S0006-8993(01)02620-8
50. Tang FR, Chia SC, Chen PM, Gao H, Lee WL, Yeo TS, et al. Metabotropic glutamate receptor 2/3 in the hippocampus of patients with mesial temporal lobe epilepsy, and of rats and mice after pilocarpine-induced status epilepticus. Epilepsy Res. (2004) 59:167–80. doi: 10.1016/j.eplepsyres.2004.04.002
51. Attwell PJ, Singh Kent N, Jane DE, Croucher MJ, Bradford HF. Anticonvulsant and glutamate release-inhibiting properties of the highly potent metabotropic glutamate receptor agonist (2S,2'R, 3'R)-2-(2',3'-dicarboxycyclopropyl)glycine (DCG-IV). Brain Res. (1998) 805:138–43. doi: 10.1016/S0006-8993(98)00698-2
52. Smolders I, Lindekens H, Clinckers R, Meurs A, O'Neill MJ, Lodge D, et al. In vivo modulation of extracellular hippocampal glutamate and GABA levels and limbic seizures by group I and II metabotropic glutamate receptor ligands. J Neurochem. (2004) 88:1068–77. doi: 10.1046/j.1471-4159.2003.02251.x
53. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. (2004) 27:370–7. doi: 10.1016/j.tins.2004.04.009
54. Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. (2008) 586:1503–8. doi: 10.1113/jphysiol.2008.150722
55. Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. (2012) 15:431–40. doi: 10.1038/nn.3033
56. Pop AS, Gomez-Mancilla B, Neri G, Willemsen R, Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology. (2014) 231:1217–26. doi: 10.1007/s00213-013-3330-3
57. Youssef EA, Berry-Kravis E, Czech C, Hagerman RJ, Hessl D, Wong CY, et al. Effect of the mGluR5-NAM basimglurant on behavior in adolescents and adults with fragile X syndrome in a randomized, double-blind, placebo-controlled trial: FragXis phase 2 results. Neuropsychopharmacology. (2018) 43:503–12. doi: 10.1038/npp.2017.177
58. Zhou F, Yao HH, Wu JY, Yang YJ, Ding JH, Zhang J, et al. Activation of Group II/III metabotropic glutamate receptors attenuates LPS-induced astroglial neurotoxicity via promoting glutamate uptake. J Neurosci Res. (2006) 84:268–77. doi: 10.1002/jnr.20897
59. Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. (1996) 71:949–76. doi: 10.1016/0306-4522(95)00533-1
60. Bruno V, Caraci F, Copani A, Matrisciano F, Nicoletti F, Battaglia G. The impact of metabotropic glutamate receptors into active neurodegenerative processes: a “dark side” in the development of new symptomatic treatments for neurologic and psychiatric disorders. Neuropharmacology. (2017) 115:180–92. doi: 10.1016/j.neuropharm.2016.04.044
61. Das A, Wallace GCt, Holmes C, McDowell ML, Smith JA, Marshall JD, et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience. (2012) 220:237–46. doi: 10.1016/j.neuroscience.2012.06.002
62. Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma. (2013) 30:1434–41. doi: 10.1089/neu.2012.2712
63. Hussein AM, Ghalwash M, Magdy K, Abulseoud OA. Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J Epilepsy Res. (2016) 6:8–15. doi: 10.14581/jer.16002
64. Omrani A, Melone M, Bellesi M, Safiulina V, Aida T, Tanaka K, et al. Up-regulation of GLT-1 severely impairs LTD at mossy fibre–CA3 synapses. J Physiol. (2009) 587:4575–88. doi: 10.1113/jphysiol.2009.177881
65. Matos-Ocasio F, Hernandez-Lopez A, Thompson KJ. Ceftriaxone, a GLT-1 transporter activator, disrupts hippocampal learning in rats. Pharmacol Biochem Behav. (2014) 122:118–21. doi: 10.1016/j.pbb.2014.03.011
66. Falnikar A, Hala TJ, Poulsen DJ, Lepore AC. GLT1 Overexpression reverses established neuropathic pain-related behavior and attenuates chronic dorsal horn neuron activation following cervical spinal cord injury. Glia. (2016) 64:396–406. doi: 10.1002/glia.22936
67. Fedele DE, Gouder N, Guttinger M, Gabernet L, Scheurer L, Rulicke T, et al. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain. (2005) 128:2383–95. doi: 10.1093/brain/awh555
68. Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. (1998) 32:286–303. doi: 10.1016/S0920-1211(98)00059-X
69. Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. (1999) 52:453–72. doi: 10.1212/WNL.52.3.453
70. Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. (2000) 12:2087–96. doi: 10.1046/j.1460-9568.2000.00104.x
71. Bordey A, Spencer DD. Distinct electrophysiological alterations in dentate gyrus versus CA1 glial cells from epileptic humans with temporal lobe sclerosis. Epilepsy Res. (2004) 59:107–22. doi: 10.1016/j.eplepsyres.2004.04.004
72. Hsu MS, Lee DJ, Binder DK. Potential role of the glial water channel aquaporin-4 in epilepsy. Neuron Glia Biol. (2007) 3:287–97. doi: 10.1017/S1740925X08000112
73. Takahashi DK, Vargas JR, Wilcox KS. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol Dis. (2010) 40:573–85. doi: 10.1016/j.nbd.2010.07.018
74. Hubbard JA, Binder DK. Astrocytes and Epilepsy. Amsterdam: Elsevier (2016). doi: 10.1016/B978-0-12-802401-0.00004-1
75. Binder DK. Astrocytes: stars of the sacred disease. Epilepsy Curr. (2018) 15:172–9. doi: 10.5698/1535-7597.18.3.172
Keywords: epilepsy, astrocytes, glutamate transporters, metabotropic glutamate receptors, GLT-1, GLAST, mGluR3, mGluR5
Citation: Peterson AR and Binder DK (2020) Astrocyte Glutamate Uptake and Signaling as Novel Targets for Antiepileptogenic Therapy. Front. Neurol. 11:1006. doi: 10.3389/fneur.2020.01006
Received: 27 May 2020; Accepted: 30 July 2020;
Published: 08 September 2020.
Edited by:
Kjell Heuser, Oslo University Hospital, NorwayReviewed by:
Peter Bedner, University Hospital Bonn, GermanyDetlev Boison, Rutgers, The State University of New Jersey, United States
Tore Eid, Yale University, United States
Copyright © 2020 Peterson and Binder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Devin K. Binder, ZGJpbmRlckB1Y3IuZWR1