 Susanna Asseyer
Susanna Asseyer Graham Cooper
Graham Cooper Friedemann Paul
Friedemann Paul
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 21 August 2020
Sec. Neuroepidemiology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00778
This article is part of the Research Topic Epidemiology of Atypical Demyelinating Diseases View all 15 articles
Neuromyelitis optica spectrum disorders (NMOSDs) and myelin oligodendrocyte glycoprotein-antibody-associated disease (MOGAD) are autoimmune inflammatory disorders of the central nervous system (CNS). Pain is highly prevalent and debilitating in NMOSD and MOGAD with a severe impact on quality of life, and there is a critical need for further studies to successfully treat and manage pain in these rare disorders. In NMOSD, pain has a prevalence of over 80%, and pain syndromes include neuropathic, nociceptive, and mixed pain, which can emerge in acute relapse or become chronic during the disease course. The impact of pain in MOGAD has only recently received increased attention, with an estimated prevalence of over 70%. These patients typically experience not only severe headache, retrobulbar pain, and/or pain on eye movement in optic neuritis but also neuropathic and nociceptive pain. Given the high relevance of pain in MOGAD and NMOSD, this article provides a systematic review of the current literature pertaining to pain in both disorders, focusing on the etiology of their respective pain syndromes and their pathophysiological background. Acknowledging the challenge and complexity of diagnosing pain, we also provide a mechanism-based classification of NMOSD- and MOGAD-related pain syndromes and summarize current treatment strategies.
In 1894, Eugène Devic (1858–1930) and his doctoral student Fernand Gault (1873–1936) reported a historical case on a patient with optic neuritis (ON) and myelitis and proposed the name “neuro-myélite optique” for this syndrome. The patient, a 45-year-old woman, was admitted for suspected “neurasthenia,” suffering from disturbed sleep, gastrointestinal symptoms, neuromuscular asthenia, palpitations, and, especially, headache: “The pain occurs in attacks, both during the day and night. Pain attacks may be long or short, affecting one side of the face and the head, sometimes the right side, mostly the left, but the highest intensity is always at the occipital region: the neck and eyeballs. The pain is sometimes so strong that it causes the patient to cry.”
One month after admission, the patient suddenly developed acute complete paraparesis and visual loss. It is currently a matter of debate whether the patient suffered from a neuromyelitis optica spectrum disorder (NMOSD) or a myelin oligodendrocyte glycoprotein-antibody-associated disease (MOGAD) (1). Terrible, agonizing, and unbearable pain can arise as an acute or chronic symptom in both pathologies (2–4) (Table 1).
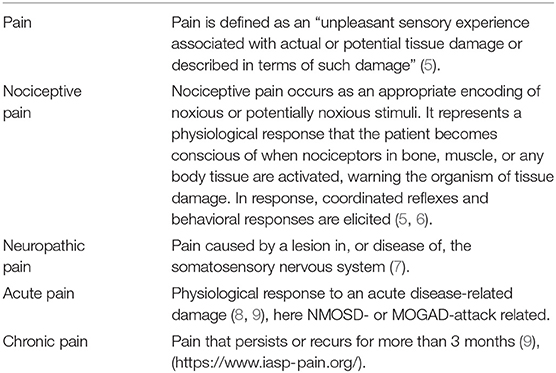
Table 1. Characteristics of different pain types.
Neuromyelitis optica spectrum disorders (NMOSDs) are rare and, in most cases, relapsing inflammatory diseases of the central nervous system (CNS) (10). In the majority of cases, NMOSDs are associated with serum immunoglobulin G (IgG) autoantibodies (Abs) targeting the astrocyte aquaporin-4 (AQP4) water channel (11, 12). Patients typically suffer from recurrent attacks of severe optic neuritis and/or myelitis (13, 14) and, less frequently, brainstem or brain involvement (15, 16), leading to a diverse range of symptoms, of which severe pain is one of the most frequent and disabling (2, 17–26). Chronic pain occurs in NMOSD with an estimated prevalence between 72 and 86% (2, 18, 27, 28). Over 50% of NMOSD (82% APQ4-Ab positive) patients recalled an increase in pain intensity as the first indicator of a relapse (26) and 25% of patients with NMOSD (82% AQP4-Ab positive) reported pain as their worst symptom, despite also experiencing severe weakness and bladder or bowel dysfunction (26). Neuropathic pain is the most common type of chronic pain with a prevalence of up to over 80% (2, 26), and painful tonic spasms occur with a prevalence of 25–40% (29–32).
MOGAD is another inflammatory autoimmune condition of the CNS, defined by IgG antibodies against conformationally intact myelin oligodendrocyte glycoprotein (MOG) localized on the surface of the myelin sheaths (13, 33, 34). Although there is some phenotypic overlap with AQP4-Ab-positive NMOSD, most researchers consider MOGAD to be a distinct disease entity (35–37). Affected patients may develop any combination of acute disseminated encephalomyelitis, transverse myelitis (long or short), optic neuritis (ON, typically anterior, often bilateral), brainstem pathology often affecting cerebellar peduncles, cranial nerve involvement, and, less frequently, brainstem encephalitis, encephalitis mimicking small vessel CNS vasculitis, and cortical disease with seizures (33, 38–44). Pain is also becoming increasingly recognized as a common and debilitating symptom in MOGAD. However, data in pain in MOGAD are scarce and have to be verified in larger studies: mild chronic pain has a reported prevalence of 86% (2), and severe acute pain in the context of attacks has a prevalence of 70% (38). Furthermore, in addition to the typical retrobulbar pain and/or pain on eye movement, severe and sometimes migraine-like headache can precede visual loss in MOG-Ab-related ON (45, 46), the most-common clinical feature at onset and subsequent relapse (33, 37, 38, 47, 48).
Pain is a very common feature of both diseases and has a higher prevalence and severity compared to multiple sclerosis (MS), where estimates of pain prevalence are ~50% (18, 27, 49). It also has a severe impact on the quality of life of affected patients (2, 18, 26, 27), interfering with physical, emotional, and cognitive aspects of well-being (2, 27, 50), as well as activities of daily life in NMOSD (60–83% AQP4-Ab positive) and MOGAD (2, 18, 26, 27). The higher the pain intensity, the worse the physical and emotional quality of life (2, 51).
The alleviation of pain through careful management and treatment should lead to significant improvement in the quality of life of patients with NMOSD and MOGAD. However, successfully controlling pain is highly challenging in these disorders (2, 26–28), and there is relatively little published literature on therapeutic intervention or treatment of pain as a primary outcome in these patient groups. In order to highlight this and facilitate future research in this critical area, we conduct a systematic review of the current literature on different pain syndromes in NMOSD and MOGAD. Based on this, we propose a mechanism-based classification of NMOSD- and MOGAD-related pain and additionally evaluate current treatment strategies.
We performed a search of PubMed (last updated on June 09, 2020), combining neuromyelitis optica or neuromyelitis optica spectrum disorders AND pain, as well as myelin oligodendrocyte glycoprotein AND pain. Additional searches were performed combining neuromyelitis optica and myelin oligodendrocyte glycoprotein, respectively, AND headache or dysesthesia or dystonia or Lhermitte's sign or neuralgia or spasms or spasticity. This search was limited to English language publications and yielded a total of ~200 articles including case reports, original clinical studies, and reviews, which were reviewed by title and abstract for potential relevance to this topic. When the title and abstract did not clearly indicate the degree of relevance to the topic, the article itself was reviewed. Bibliographies of topic-relevant articles were also examined to discover additional references not identified in the primary search. Finally, the authors' personal knowledge of the literature as well as congress contributions to ECTRIMS 2019 were used to supplement the above references.
As the impact of pain in patients with AQP4-Ab-positive and Ab-negative NMOSD is similar, we document both disease types together and report the percentage of AQP4-positive NMOSD patients whenever available. We note that some MOG-Ab-positive patients may have been included in former NMOSD studies. However, the percentage of MOG-Ab-positive patients within groups of Ab-negative NMOSD patients should be low.
We identified 18 studies evaluating pain in NMOSD (n = 17) and MOGAD (n = 2, one overlapping with NMOSD) (Table 2).
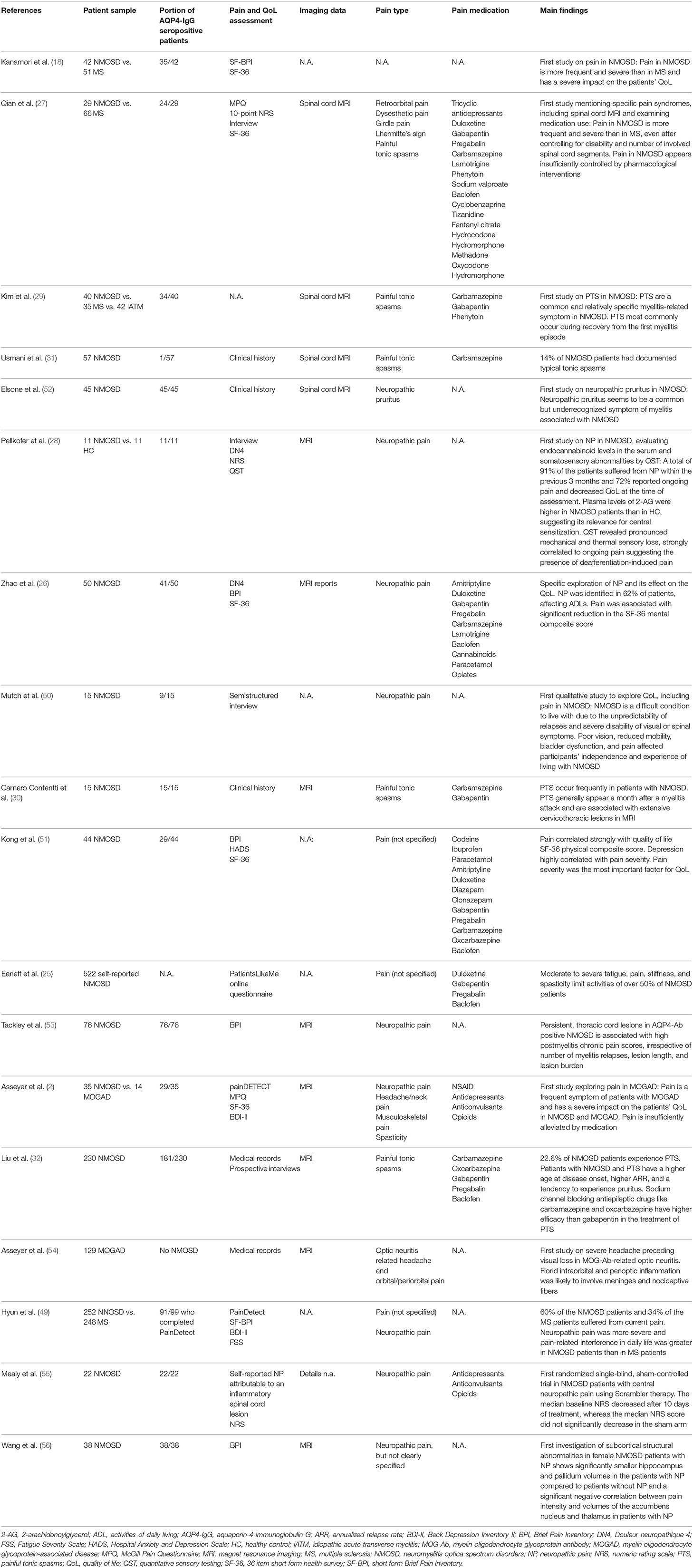
Table 2. Original publications on pain in neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein-antibody-associated disease (MOGAD) (listed in chronological order).
The studies focused on pain without diagnostic specification (18, 25, 51), neuropathic pain (26, 28, 49, 50, 53, 55, 56), one study on neuropathic pruritus (52), painful tonic spasms (29–32), ON-related headache (54), and a description of diverse pain types (2, 27). One randomized single blind sham-controlled trial studied the effect of Scrambler therapy in NMOSD patients with central neuropathic pain (55). All other studies (n = 17) were descriptive and non-interventional. Two reviews on pain in NMOSD are available, one focusing on potential mechanisms underlying the pathogenesis of pain in NMOSD and another focusing on the impact of neuropathic pain medication on patients' quality of life (3, 57). Moreover, we included 12 case reports describing pain as part of the patients' symptom complex (4, 58–68). We additionally reviewed studies (n = 131) in NMOSD that included pain but where it was not the primary outcome. Where available, we provide the information on the percentage of AQP4-Ab-positive patients of the respective NMOSD cohort. Our review is the first to provide an overview of (1) disease-associated lesion locations in relation to different pain syndromes, (2) different types of NMOSD- and MOGAD-related pain, (3) possibilities to classify acute and chronic pain in NMOSD and MOGAD, and (4) the impact of the currently available immunotherapy on pain.
Inflammatory attacks in the CNS occur in both NMOSD and MOGAD and can lead to acute pain via the release of pronociceptive brain-derived neurotropic factor (BDNF), cytokines and chemokines [interleukin (IL)-1ß, IL-6, IL-17, and tumor necrosis factor (TNF)] (3, 69–71). Cytokine release enhances glutamatergic signaling, the main pronociceptive neurotransmitter in the spinal dorsal horn (3).
Under healthy conditions, AQP4 is coexpressed with the excitatory amino acid transporter 2, which enables glutamate uptake by astrocytes. Loss of AQP4 in AQP4-Ab-positive NMOSD may lead to an excessive accumulation of glutamate in the extracellular space. In the context of neuroinflammation and dysregulation of sensory neurons, persistent excessive BDNF, and glutamate concentrations affect vulnerable inhibitory alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and gamma-aminobutyric acid (GABA) neurons, respectively (72, 73). The resulting imbalance between excitation and inhibition can then facilitate the development of chronic pain (3, 74, 75). In addition, astrocytes release endocannabinoid 2-arachidonoylglycerol (2-AG), which strongly enhances GABAergic inhibition. Loss of astrocytes in NMOSD leads to 2-AG reduction, likely leading to nociceptive pain and hyperalgesia (28).
Structural cerebral alterations may also affect chronic pain perception in NMOSD. Recently, a study on subcortical abnormalities in female NMOSD patients showed smaller hippocampus and pallidum volumes in patients with neuropathic pain compared to patients without neuropathic pain, as well as a negative correlation between pain intensity and volumes of the accumbens nucleus and thalamus (56). A study on pain-related morphological abnormalities in AQP4-Ab-positive NMOSD described an association of the ventral posterior nucleus (VPN) volume with several measures of pain intensity (76). Both studies suggest that subcortical structures are substantially involved in cognitive, emotional, and modulatory pain processing in AQP4-Ab-positive NMOSD (56, 76).
While AQP4-Abs target astrocytes, MOG-Abs bind to myelin-forming oligodendrocytes. Therefore, inflammation in MOGAD primarily causes demyelination with a loss of the microtubule cytoskeleton of oligodendrocytes (13, 77–79). Under healthy conditions, the neuropeptide nerve growth factor (NGF) has a high affinity to bind MOG. Moreover, NGF is part of the nociceptive system: It binds tropomyosin receptor kinase A (TrkA). TrkA is expressed on unmyelinated nociceptive axons of the spinal cord and regulates synaptic strength and plasticity of sensory neurons. Thus, the loss of MOG by antibody-mediated destruction in MOGAD may cause abundant NGF concentrations in the CNS, leading to aberrant sprouting of unmyelinated nociceptive fibers in the posterolateral tract of the spinal cord and hence nociceptive pain (80).
Spinal cord lesions in NMOSD are typically extensive and occur predominantly in the cervical and thoracic spinal cord (17, 81–83). As AQP4 is mainly expressed in the gray matter, lesions concentrate around the central canal, and the adjacent gray matter in the dorsal and ventral horns, as well as in the dorsal root entry zone (84). Ascendant and descendent white matter tracts, including the spinothalamic tract (STT) (52, 85, 86), are affected by severe lesions (87). Tackley et al. report a significant relationship between persistent thoracic myelitis lesions and the severity of neuropathic pain. The presence of cervical lesions, in contrast, were predictive of lower pain scores (53).
In the brainstem, the dorsal medulla oblongata and area postrema have the highest distribution of AQP4 (74, 88). It has been shown that 27% of NMOSD patients with cervical longitudinally extensive transverse myelitis (LETM) showed lesions involving the brainstem (89). Such a distribution could include trigeminal nucleus or periaqueductal gray (PAG) pathology, causing headaches in affected patients (74). The PAG is considered to be a migraine generator and a modulator of headache in NMOSD. Moreover, the hypothalamospinal tract, localized in the dorsolateral medulla, activates the hypothalamus, and the trigeminovascular system. Both regions are considered to be involved in the pathogenesis of headache (90, 91).
Dorsal lesions of the medulla oblongata lead to substance P release, a transmitter that can cause and maintain nociceptive activation of the trigeminal tract nucleus (92). Besides headache, neuropathic pain was also reported more frequently in NMOSD patients with medulla oblongata lesions (85.7% AQP4-Ab positive) than in patients without such lesions (31.8 vs. 11.1% and 65.9 vs. 29.4%) (93). Increased neuropathic pain frequency could be explained by the severe and extensive spinal cord involvement associated with the medulla oblongata (93).
Moreover, AQP4-Ab-positive NMOSD has a predilection to affect the optic nerve (94–96). Astrocytes surrounding the optic nerve express high levels of AQP4, but the unmyelinated optic nerve head also expresses AQP4. Moreover, a high density of retinal astrocytic Müller cells, expressing AQP4, are located in the parafoveal area (97–101).
For a further and more detailed pathophysiological background of possible mechanisms explaining pain in NMOSD, we refer to a review by Bradl et al. (3).
Spinal cord lesions in MOG-Ab-positive myelitis are not always longitudinal and extensive but can still cause sensory symptoms like pain and dysesthesia (38). The axial lesion extension may be crucial for the risk of pain. Depending on the level of the lesion, aberrant nerve fiber sprouting could lead to occipital neuralgia or to more distal neuropathic pain syndromes. Moreover, it has been shown that central neuropathic pain can be induced by oligodendrocyte death and axonal pathology in the spinothalamic tract (102).
The brainstem is a critical region in the pathophysiology of headache. Brainstem lesions are present in up to one-third of patients suffering from MOGAD and could promote the risk for migraine and trigeminal neuralgia (103, 104).
MOG is highly expressed by oligodendrocytes myelinating the optic nerve (105) and is consequently a predominant target in MOG-Ab-related ON. ON-related pain is particularly severe in MOGAD and can present as a migraine-like headache (54). In these cases, severe edema may lead to irritation of the meningeal nerve sheath, which surrounds the optic nerve and contains nociceptive fibers of trigeminal origin (106–108). The trigeminal nerve provides sensory innervation to the ocular and periocular area, and its recurrent branches innervate the intracranial dura, venous sinuses, and cerebral vessels, likely leading to headache (109, 110).
Pain can occur during acute attacks and be an indicator of current damage, or it can become a chronic syndrome over the course of the disease. The main pain syndromes in NMOSD and MOGAD comprise ON-related pain, headache, neuropathic pain, and musculoskeletal pain including spasticity, painful tonic spasms, and back pain. We discuss these symptoms in the context of NMOSD and MOGAD below, highlighting any differences between the two diseases where information is available.
Optic neuritis is an inflammation of the optic nerve characterized by severe visual loss or blindness associated with ocular pain (111) and occurs in the context of many inflammatory diseases (112–116). ON-related eye pain and pain on eye movement is more common in MOGAD, with reports ranging from 65 to 86% (46, 117, 118), compared to AQP4-Ab-positive ON (28.6–50%) (46, 117) and idiopathic Ab-negative ON (10–46%) (117, 119).
AQP4-Ab-positive ON is typically accompanied by retrobulbar pain often worsened by eye movement (2, 27, 46).
MOGAD-related ON pain seems to be particularly severe, sometimes accompanied by migraine-like headaches that precede the visual deficit (54, 120).
Headache is an unspecific but common symptom in NMOSD (2, 74) and has also been described in MOGAD, here mainly associated with optic neuritis (2, 38, 54). It can occur as a first symptom or persist during the disease course (2, 38, 74). NMOSD-related headache can occur as a cervicogenic-like headache (2, 58, 74), neck pain (60, 68), paroxysmal hemicrania (62), or in the context of meningoencephalitis (74, 121). It is typically a mixed pain condition with neuropathic and nociceptive components (74).
Cervicogenic-like headache is caused by a lesion in or disorder of the cervical spine or soft tissues of the neck. While a few cases presenting with cervicogenic-like headache following myelitis have been mentioned in NMOSD and MOGAD (2, 58, 74), only a single case report has described it in detail: The patient had a left occipital headache spreading to the posterior neck associated with numbness and aching. Response to occipital nerve block was slight, and the headache progressed. MRI revealed an extensive myelitis from the medulla oblongata to the C5 level, a bilateral ocular or prechiasmatic lesion, and suspicious bilateral upper brainstem lesion. Symptoms and MRI pathology improved with steroid treatment (58).
Note that we suggest avoiding the diagnosis of cervicogenic headache in NMOSD and MOGAD in favor of the term cervicogenic-like headache or headache attributed to non-infectious inflammatory diseases (106). Classical cervicogenic headache, in contrast, is caused by a disorder of the cervical spine and its component bony disk and/or soft tissue elements (106).
Paroxysmal hemicrania is characterized by severe unilateral pain attacks, affecting orbital, supraorbital, and/or temporal regions. The attacks are mostly associated with autonomic features (ipsilateral conjunctival injection, lacrimation, nasal congestion, rhinorrhea, forehead and facial sweating, miosis, ptosis, and/or eyelid edema) (106). We are aware of one case report, describing a patient presenting with paroxysmal hemicrania as first symptom of an AQP4-Ab-positive NMOSD. MRI revealed a lesion extending from the lower medulla oblongata to the cervical cord (C4), possibly involving the spinal nucleus of the trigeminal nerve (62). As in primary paroxysmal hemicrania, indomethacin has been effective in the case of AQP4-Ab-positive NMOSD-related paroxysmal hemicrania (62), but evidence is limited. No reports of paroxysmal hemicrania in MOGAD were identified.
Meningoencephalitis-like pathology with fever, severe headache, and pleocytosis in the cerebrospinal fluid (CSF) has been reported in both disease complexes, NMOSD and MOGAD (74, 121), most likely due to meningeal inflammation (122, 123).
Neuropathic pain is particularly severe (2, 53) and patients typically characterize neuropathic pain as agonizing, shooting, and distressing (57). Neuropathic pain occurs more frequently in NMOSD (83% AQP4-Ab positive) than in MOGAD (80 vs. 40%) (2, 27). It can occur as an early myelitis-related symptom or develop during the disease course (3, 50, 53). Medication is currently not sufficient to control neuropathic pain (2, 27), particularly in patients with AQP4-Ab-positive NMOSD (51). A higher dosage of pain medication was not associated with being free of pain but rather with greater cognitive dysfunction and fatigue (27).
Neuropathic pain can be permanent or intermittent like Lhermitte's sign (27, 81, 124) and is localized either on the extremities or on the trunk, the latter often defined as a girdle sensation (18, 26, 28, 124, 125).
Lhermitte's sign is often painful and occurs in 35–60% of AQP4-Ab-positive NMOSD patients (27, 81, 124). It is defined as a brief, electric-shock-like sensation that runs from the back of the head down the spine, provoked by inclining the neck forward (124). It has been proposed that Lhermitte's sign occurs because demyelinated sensory fibers are hyperexcitable to percussion or elongation (124).
The girdle sensation describes an often burning sensation on the skin, localized with an extension of three or four dermatomes between T3 and T11 (124). It has been reported in 45.8–69% of NMOSD (83% AQP4-Ab positive) patients and can sometimes be misdiagnosed as acute abdomen (27, 124). Schöberl et al. describe an AQP4-Ab-positive NMOSD patient presenting with typical area postrema syndrome who developed an unusual painful segmental erythema resulting from a dorsolateral spinal cord lesion at C6/7 level. A dysregulated A-beta-fiber-evoked vasodilation has been discussed as a possible underlying pathophysiological mechanism (126). Pelvic pain has been reported to occur as an unusual presentation of AQP4-Ab-positive NMOSD, following a lesion of the conus medullaris (61).
Brainstem pathology can also cause neuropathic pain syndromes like trigeminal (2, 16, 74, 127) and occipital neuralgia (2, 128) in NMOSD and MOGAD. Trigeminal neuralgia is defined by pain in the area of the trigeminal innervation (usually V3 and/or V3 division). It is typically characterized by paroxysmal, sudden attacks of short severe stimulus-triggered and electric-like pain episodes (74). Interestingly, NMOSD patients with trigeminal neuralgia rarely show MRI pathology affecting the trigeminal root entry zone (129). It has been discussed whether or not a dual mechanism including pontine plaques and consecutive neurovascular compression may contribute to the pathophysiology (74). Neuropathic pruritus has also been described following brainstem and spinal cord lesions (52). Pruritus is defined as “an unpleasant cutaneous sensation provoking the desire to scratch.” Neuropathic pruritus is caused by affected pruritogenic neurons in the absence of a pruritogenic substance (52). Neuropathic pruritus associated with myelitis has been observed in 27.3% of ACQP4-Ab-positive NMOSD patients, either as a first symptom or a few days after the onset of other myelitis-related symptoms. It has a sudden onset of high intensity with a duration from seconds to minutes, associated with superficial sensory deficits and/or pain. It can occur on the trunk, the extremities, or the occipital region of the head (52). An inflammation-related demyelination involving second-order itch neurons in the dorsal horn of the spinal cord has been discussed as an underlying pathophysiological mechanism. The role of brainstem lesions affecting the spinal nucleus of the trigeminal nerve or periaqueductal pathways has also been discussed (52, 130).
Very few studies have focused on neuropathic pain in MOGAD. Lhermitte's sign (38, 45), band-like girdle sensations (131), trigeminal and occipital neuralgia, and neuropathic extremity pain (2, 38) have been mentioned but have so far not been studied in detail. Myelitis in MOGAD may have a better tendency to recover (83) and therefore cause less severe central neuropathic pain syndromes than in NMOSD.
Some cases of possible peripheral nervous system (PNS) involvement in NMOSD have been published. Painful, flaccid paralysis (63), lumbosacral myelitis (132), clinical and electrophysiological second motor neuron involvement (133), and peripheral neuropathy (134, 135) have been described, and radicular pain has been reported to occur in up to 33% (81, 136). Recently, a few cases with PNS involvement in MOGAD have been described. Cranial nerve involvement, brachial neuritis, multifocal neuropathy, migratory paresthesia, myeloradicular symptoms, recurrent limb paresthesia, and pain have been mentioned (41, 64, 137, 138). As described above, the inflammatory process in the CNS could trigger an immune cascade targeting myelin-specific antigens in the nerve roots. Alternatively, low quantities of MOG may be expressed in the human peripheral myelin and the Schwann cells, as previously described in rodents and primates (64, 138, 139). However, current data are too scarce for pathophysiological conclusions. At present, we can only infer that PNS involvement should not prevent clinicians from investigating the presence of MOG- and AQP4-IgG Abs.
Spasticity is defined as “disordered sensorimotor control resulting from an upper motor neuron lesion, presenting as intermittent or sustained involuntary activation of muscles.” At the patient level, it can be defined as an “unusual tightening of muscles that feels like leg stiffness, jumping of legs, a repetitive bouncing of the foot, muscle cramping in the legs or arms, legs going out tight and straight or drawing up” (140). More than 50% of NMOSD patients are reported to suffer from moderate to severe spasticity (25), but very little is known about spasticity in MOGAD (1, 38).
Painful tonic spasms are defined as paroxysmal, recurrent muscle spasms in one or more limbs and/or the trunk, lasting seconds to minutes, accompanied by intense pain and dystonia (29, 30, 65). Several case reports and small series describe PTS in NMOSD (18, 29, 29–31, 65–67, 136, 141–144), but no reports were identified mentioning PTS in MOGAD. Abboud et al. reported that all patients with tonic spasms had associated neuropathic pain (145). PTS and pain occur more frequently in NMOSD than in MS (18, 29), and PTS-associated myelitis in AQP4-Ab-positive NMOSD has been described with a specificity of 98.7% compared to MS (143). Kim et al. showed that transverse myelitis at disease onset, but not optic neuritis, was predictive of future occurrence of PTS. PTS develop mainly during recovery from the first myelitis attack within a mean of 48 days without occurrence of new MRI lesions (3, 29, 30). A spinal cord syndrome with paroxysmal tonic spasms may be particularly suggestive for NMOSD (29, 81). PTS may occur following the loss of inhibitory motor neurons in the central gray matter of the spinal cord (142). Abnormal demyelination can cause ephaptic transmission between the tracts causing spasms (65). As nerve damage does not affect somatosensory pathways, PTS are not considered to be of neuropathic origin (146).
Like headache, back pain is an unspecific syndrome but occurs frequently in NMOSD and MOGAD (1, 38, 131, 147, 148). It can emerge in the context of myelitis following radiculitis as described above but is often a mixed syndrome including central and peripheral neuropathic as well as nociceptive pain components. Malposition and axial instability following paresis or spasticity, reduced mobility with wheelchair dependence, or long-term corticoid therapy leading to osteoporosis are important secondary aspects to consider in these disorders and can enhance pain, especially back pain (5).
Up to 45% of patients with NMOSD and ~10% of patients with MOGAD suffer from autoimmune comorbidities (13), including connective tissue disease, dermatomyositis, rheumatoid arthritis, Sjoegren's syndrome, systemic lupus erythematodes, vasculitis, and myasthenia gravis (2, 51, 148–155), which can themselves be associated with pain (156). A careful diagnostic workup is necessary to detect potentially overlapping pathologies.
Women are more often affected by autoimmune diseases than men, with a female/male ratio of up to 10:1 in NMOSD and, depending on the geographic region, between 1.1:1 and 3:1 in MOGAD (13, 157). However, no sex differences have been found concerning pain prevalence or intensity (26). Mixed results have been found regarding the correlation between pain intensity and age (18, 26). Severe overall disability, measured by the expanded disability status scale (EDSS), has been identified as a risk factor for more severe pain (27) and increasing disability scores correlated with pain intensity in NMOSD (83 and 66% AQP4-Ab positive) (27, 51). Moreover, an association of depression, fatigue, and NMOSD (66–83% AQP4-Ab positive) as well as MOGAD has been shown in several studies (2, 19, 27, 51, 158). Depression and pain are known to interact, and one cannot be certain whether depression enhances pain, occurs in response to pain, or both (159).
The International Association for the Study of Pain (IASP) defines pain as an “unpleasant sensory experience associated with actual or potential tissue damage or described in terms of such damage” (https://www.iasp-pain.org/). We propose a classification for pain in NMOSD and MOGAD (Table 3), which is similar to a previously provided MS-related pain classification (146). Our aim is to present a structure providing
1) the time course of pain development, to distinguish
a. pain as a warning signal of acute damage
b. pain as a self-sustaining chronic syndrome
2) the underlying pathophysiological mechanisms, to distinguish
a. ON-related pain
b. headache
c. neuropathic pain
i. intermittent (episodic), e.g., trigeminal and occipital neuralgia, Lhermitte's sign
ii. permanent (continuous), e.g., pain in the extremities
d. spasticity and painful tonic spasms
e. mixed pain, e.g., back pain
f. comorbidity-related pain
3) a reference for specific treatment strategies
4) a framework to generate future research hypotheses.
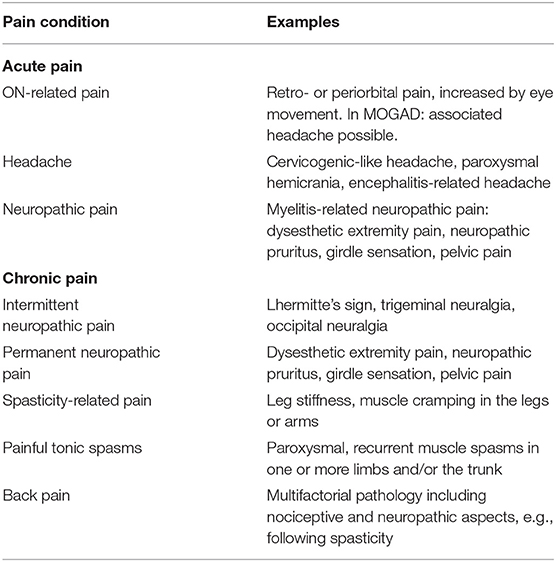
Table 3. Classification of pain in neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein-antibody-associated disease (MOGAD).
Of note, acute and chronic pain syndromes can overlap. For efficacious treatment, a detailed medical history is necessary.
Despite the use of multiple medications, pain is currently not sufficiently managed in NMOSD or MOGAD (2, 26–28), and there is relatively little published literature on therapeutic intervention or treatment of pain as a primary outcome in these patient groups. Three studies on immunosuppressive treatment in AQP4-Ab-positive NMOSD have shown promising results when examining pain as a secondary outcome: two in patients treated with the humanized monoclonal IL-6 antibody tocilizumab (125, 160, 161) and one in patients treated with low-dose mycophenolate mofetil (MMF) (162). One study on the positive effect of Scrambler therapy for the treatment of neuropathic pain in NMOSD was identified (55). No studies were found investigating pain treatment in MOGAD. We provide an overview of current strategies for relapse-related treatments and effects of immunosuppressive treatment focusing on acute and chronic pain, respectively. We additionally give a general overview on the management of chronic neuropathic pain, spasticity-related pain, and painful tonic spasms, although these are not specific to NMOSD or MOGAD.
Attack-related treatment aims to reduce pain by reducing the destruction of the CNS. In NMOSD, as well as in MOGAD, acute attacks are usually treated with 1,000 mg intravenous methylprednisolone (IVMP) for 3–5 days (163). Prompt treatment initiation should also be considered in patients who present with pain as their only symptom, in order to avoid rapid progression and attack-related disability (148). Of note, attack-related disability can cause the development of secondary pain, e.g., paresis- and malposition-related pain, reflecting attack-independent disease progression. Rapid corticoid therapy showed prompt recovery from pain in NMOSD (120), and Jarius et al. showed nearly complete recovery in 50% of IVMP-treated MOG-Ab-related attacks (38). In cases of poor outcome, IVMP therapy can be increased to 2,000 mg/day. Such a high-dose IVMP therapy, however, seems to be less effective than plasma exchange or immunoadsorption (13, 164–166). Especially in isolated myelitis, it has been shown that clinical response to immediate plasma exchange (PLEX) was better compared to high-dose steroid therapy (166). This could be of relevance in the treatment of patients presenting with neuropathic pain.
Bradl et al. suggest a multidrug treatment at an early disease stage to limit the previously discussed complex interactions of proinflammatory and pronociceptive molecules in order to avoid pain instauration. They propose an approach similar to the treatment for traumatic brain injury, involving minocycline, peroxisome proliferator-activated receptor agonists, cell cycle inhibitors, statins, and progesterone (3). However, currently, there are no data on possible preventive effects on pain development in NMOSD or MOGAD in this regard.
Immunosuppressive therapy is essential to reduce disease activity and to avoid relapses in NMOSD and MOGAD, again with the aim to reduce the risk of future CNS damage. Up to now, although recommendations for treatment of NMOSD are available, these are not based on a high level of evidence (163, 167, 168). It is strongly recommended that patients suffering from AQP4-ab-positive NMOSD should receive immunotherapy after the first attack. Currently used preventative treatments in NMOSD include prednisone, azathioprine, rituximab, MMF, intravenous immunoglobulins (IVIGs), eculizumab, and methotrexate (163, 168, 169). Data on the efficacy of IVIG, however, are scarce (13). Of note, in Canada, the USA, and Europe, Eculizumab is currently the only approved therapy for the treatment of NMOSD, and all other medications are used off-label and empirically. In clinical trials, the positive effects on relapse rates of inebelizumab and satralizumab NMOSD have been described (13, 160, 169–173). Satralizumab has shown no benefit on pain intensity in two phase III studies (171, 174), and no data on pain are available for eculizumab and inebilizumab (169, 170, 173). As mentioned above, tocilizumab and MMF in contrast have shown positive effects on pain in AQP4-Ab-positive NMOSD. Still, evidence has to be proven in prospective studies focusing on pain as a primary outcome.
Tocilizumab is an antibody against IL-6, a major cytokine involved in NMOSD pathophysiology (175). It has been shown that NMO-IgG binding to AQP4 on astrocytes selectively induces internalization of AQP4 and production of IL-6 (70), which is thought to enhance the survival time of plasmablasts, which generate anti-AQP4 antibodies (71). IL-6 is a pronociceptive cytokine, which plays an important role in the development of neuropathic pain (176). Treatment with tocilizumab leads to reduced immunological activity, as well as neuropathic pain reduction (59, 125, 160), and should therefore be considered in patients at risk for neuropathic pain.
MMF is an immunosuppressant inhibiting the inosine monophosphate dehydrogenase. Consequently, the synthesis of guanosine nucleotide is reduced, which leads to an inhibition of B- and T-lymphocyte proliferation. MMF can be administered in both NMOSD and MOGAD, in the latter preferably in combination with steroids (13, 162). MMF reduces immunological activity and has a positive effect on pain intensity in AQP4-Ab-positive NMOSD patients (162). Unfortunately, the type of pain was not defined in this study.
Of note, pain can occur as a side effect of some immunosuppressive therapy. Eculizumab, inebilizumab, MMF, and rituximab can lead to headache, MMF can cause abdominal pain (13, 120, 170), and inebilizumab can cause back pain, extremity pain, and chest pain (173).
It has to be kept in mind that NMOSD and MOGAD are distinct nosologic entities regarding their underlying pathogenesis (36). In MOGAD, long-term immunotherapy is often considered and recommended only after a second attack in light of the presumably high proportion of monophasic cases and the overall good recovery. Empirical data suggest oral steroids as mainstay of treatment, and slow tapering is crucial to avoid recurrence of disease activity (33, 177, 178). In contrast to NMOSD, the efficacy of rituximab in MOGAD is controversial. Two recent studies showed that up to 45% of the patients under rituximab treatment still relapsed, despite an effective biological effect of rituximab. Consequently, memory B-cell depletion seems to be unable to prevent relapses in a subset of patients suffering from MOGAD (179, 180). Currently, a long-term treatment with intravenous immunoglobulins, or in some cases with methotrexate, may be preferred (13, 38). Like in NMOSD, treatment of MOGAD with classical MS drugs should be avoided, as they can worsen the disease course (181). Up to now, no treatment guidelines with high grade evidence are available for the treatment of MOGAD, and all medications are used off-label and empirically. Of note, none of the immunotherapies have been studied with regard to a potential effect on pain in MOGAD.
Symptomatic therapies aim to treat pain. Of note, the efficacy of the following treatment strategies have not been specifically demonstrated in NMOSD or MOGAD-related pain.
Based on the pathophysiological course of neuropathic pain development and the mechanisms of action, Bradl et al. suggest inducing pharmacological inhibition of glutamatergic signal transduction early in the disease course, e.g., by N-methyl-D-aspartate (NMDA)-receptor blockade with low-dose ketamine or memantine. In patients with established lesions and reduced antinociceptive inhibition in advanced disease stages, Bradl et al. propose medication with GABA agonists, e.g., baclofen, and monoamine reuptake inhibitors (3). However, evidence on its effects is limited, and none of these agents are routinely used clinically (3, 182).
Regarding the current state of pain research, multidisciplinary care in combination with tricyclic antidepressants (TCAs), serotonin norepinephrine reuptake inhibitors (SNRIs), gabapentanoids, and tramadol are the most effective options to treat central neuropathic pain (7, 183–185). Depending on the type of medication, a 3–8-week trial is recommended to evaluate its effect. If no significant pain relief can be achieved, the dosage should be adjusted if the medication is tolerated by the patient. In a second step, alternative medication, combination therapy, or evaluation for neurostimulation may be considered (182, 186).
For the effect of medical neuropathic pain treatment on the patients' self-reported quality of life, we recommend the review by Mealy et al. (57).
Tricyclic antidepressants like nortriptyline and amitriptyline show pain-relieving effects by inhibiting serotonin and noradrenaline reuptake (5, 182, 183, 187–189). Nortriptyline and amitriptyline should be started with a daily dose of 10–25 mg per os (p.o.) and increased to a maximal daily dose of 150 mg. Side effects comprise falls, cardiac arrhythmias, orthostatic dysregulation, urinary retention, and dry mouth, and occur especially in elderly people (182, 184, 187, 190, 191).
Serotonin and norepinephrine reuptake inhibitors (SNRIs) like duloxetine and venlafaxine enhance monoamine neurotransmission in the descending inhibitory spinal pathways, resulting in decreased sensation of pain (183–185, 187–189). SNRIs showed positive effects on neuropathic pain in MS but without a corresponding positive effect on the patients' quality of life. Duloxetine should be started with a daily dose of 30 mg p.o. and increased to a maximal daily dose of 60 mg. Venlafaxine should be prescribed with an initial daily dose of 37.5 mg p.o. and escalated to a maximal daily dose of 200 mg. Side effects include mainly renal and liver pathology (7, 57, 182–185, 187, 191).
Gabapentanoids are anticonvulsant drugs, including gabapentin and pregabalin. These drugs inhibit neurotransmitter release in the dorsal horn of the spinal cord by blocking presynaptic alpha-2-delta calcium channels, leading to pain relief. Gabapentin has been shown to effectively decrease pain intensity and improve quality of life of patients suffering from neuropathic pain after spinal cord injury. Gabapentin dosage should also be increased slowly, starting with a daily dose up to 600 mg p.o., and escalating to a maximum daily dose of 3,600 mg. Pregabalin should be initiated with a daily dose of 150 mg p.o. and escalated to a maximal daily dose of 600 mg. Effective pain release by gabapentanoids should be evaluated after a 4–6-week period with 2 weeks at the maximum tolerated dose. Side effects include mainly renal pathology (57, 182–184, 187–190, 192, 193).
For the treatment of trigeminal neuralgia, carbamazepine is considered to be a first-line therapy (184). Carbamazepine can be induced with a daily dose of 200–400 mg. Slowly increasing the dosage by 50 mg/day can be continued up to 600–1,200 mg/day. Especially in elderly people, the tolerance of dosages above 600 mg/day is often poor with important motor and sedative side effects. Apart from the treatment of trigeminal neuralgia, carbamazepine is considered a third-line therapy for neuropathic pain (5, 194, 195).
Medication of first-line treatment should be trialed over an average time period of 4–6 weeks. If sufficient pain relief is not achieved, progression to the next medication or next line of treatment should occur (182, 184, 187, 189, 191).
Most guidelines consider tramadol as a second-line therapy (182, 189–191, 196). However, for acute neuropathic pain and intermittent exacerbations of neuropathic pain, it is considered first-line medication (182, 189, 191). Tramadol primarily acts as a weak μ-opioid agonist and inhibits serotonin and norepinephrine reuptake. One study on neuropathic pain after spinal cord injury showed a positive effect of tramadol, in addition to stable regimen (57).
Tramadol should be started with a daily dose of 50 mg p.o. and escalated to a maximal daily dose of 400 mg. Side effects comprise seizure disorder and renal impairment, notably in the elderly (182).
Combination therapy is common in the treatment of neuropathic pain. The patient should be closely observed due to an increased risk for side effects (182, 187).
Cannabinoids have shown a positive impact on pain, sometimes additionally improving quality of life (5, 57). Cannabinoids bind to the presynaptic cannabinoid receptor, reducing calcium influx from voltage-gated calcium channels, and hyperpolarization. Consequently, cellular excitability decreases. However, cannabinoids are currently only licensed in Canada, Israel, and New Zealand for the treatment of neuropathic pain and the safety profile remains a matter of debate (5, 57).
For patients who do not tolerate first- or second-line therapy or do not benefit from adequate pain relief, medication with serotonin-specific reuptake inhibitors (SSRIs), anticonvulsants such as lamotrigine, carbamazepine, topiramate, sodium valproate, and NMDA antagonists, as well as tapentadol, can be considered in a specialized setting. Tapentadol is a newer weak μ-opioid agonist, and strong norepinephrine reuptake inhibitor that does not affect serotonin reuptake. Due to its increased potency compared to tramadol, it is currently considered third- or fourth-line treatment. Evidence grades of third-line treatments are currently relatively low (182, 184, 187–189, 191).
Neuromodulation, including intracranial stimulation, spinal cord stimulation, high-frequency and burst spinal cord stimulation, and dorsal root ganglion stimulation, is considered to be fourth-line treatment before starting medication with long-term opioids (55, 182). As mentioned above, one phase II study has shown a positive effect of Scrambler therapy for the treatment of neuropathic pain in 22 AQP4-positive NMOSD patients (55). Scrambler therapy is non-invasive technology with Food and Drug Administration (FDA) 510(k) approval for acute, chronic, and postoperative pain. Scrambler is a transcutaneous electric nerve stimulation (TENS) technique that stimulates ascending peripheral C-fibers. It aims to modify nociceptive pain by reorganizing maladaptive signaling pathways in the sensory cortex (197). The trial showed pain reduction from a median baseline numeric rating scale (NRS) pain score of 5.0–1.5 after 10 days of treatment. The median NRS score did not significantly decrease in the sham arm (55). Currently, the lack of clear guidelines regarding the frequency and stimulation amplitude necessary to achieve sufficient pain reduction currently limits the use of TENS (57, 198, 199). A phase III study would be necessary to prove the effect of Scrambler therapy on pain, reduction in analgesic medication, and QoL in a larger NMOSD cohort (57, 198, 199).
Low-dose opioid medication to treat permanent neuropathic pain is currently considered as fourth- and fifth-line treatment, if appropriate conservative pharmacological and interventional management (neurostimulation) has failed (182). Opioids bind to an opioid receptor, inhibit adenylyl-cyclase, lead to neuronal hyperpolarization, and decrease neuronal excitability. However, opioids are considered to have a limited efficacy on neuropathic pain, and safety concerns require strict monitoring (7). Combination therapy of gabapentin and opioids provided better neuropathic pain relief than gabapentin or opioids alone but was associated with increased levels of adverse events (182).
Baclofen has shown a positive effect on myelitis-related neuropathic pain in MS patients after intrathecal administration (5–1,200 μg/day). However, baclofen is currently not licensed for the treatment of neuropathic pain but rather indicated for medical treatment of spasticity (5, 146). Some patients may benefit from its positive overlapping effects.
Spasticity can cause discomfort and stiffness and lead to pain, e.g., back pain (194). Management should be patient focused and target function rather than aiming to reduce the degree of spasticity. Effectively reduced spasticity can accentuate profound underlying weakness, which contributes to the disability and potential complications of malposition. To avoid complications like pain, early treatment of spasticity should emphasize self-management strategies, education, and physiotherapy (200).
Oral pharmacological agents most commonly used to treat spasticity are baclofen, tizanidine, benzodiazepines, dantrolene, and gabapentin (3, 200). If oral medication does not reach the sufficient effect, antispastic agents such as botulinum toxin, intrathecal baclofen, phenol, and cannabinoids can be administered (200, 201). A positive effect on both spasticity and pain has been shown for baclofen, gabapentin, botulinum toxin, and cannabinoids (194, 202).
Baclofen is a derivate of ɤ-aminobutyric acid (GABA), which can cross the blood–brain barrier to a limited extent. GABA is a major inhibiting CNS transmitter of impulse transmission, and baclofen is thought have an antispastic effect through the inhibition of reflex neurological transmissions in the spinal cord. Baclofen should be administered starting with a daily dose of three times 5 mg p.o. and increased to a maximal daily dose of 80–100 mg. Common side effects include drowsiness, weakness, paresthesia, and dry mouth (194).
As oral baclofen crosses the blood–brain barrier only to a small extent, the administration of baclofen directly to the site of antispastic action into the spinal canal improves efficacy and reduces potential side effects. A programmable infusion pump allows a continuous supply of the drug. Dosage has to be titrated over time. Long-term dosage used in MS-related spasticity ranged from 21 to 648 μg/day (194).
The effect of botulinum toxin (botox, dysport) is to inhibit acetylcholine release at the neuromuscular junction. Despite permanent blockade, the clinical effect of botulinum toxin injections is reversible because of nerve sprouting and muscle reinnervation (200). The total dosage of botox should be ≤200 units and the dosage at one site ≤50 units. Dysport should be started with a total dosage of 500 units per patient. Depending on the clinical response, the dosage of dysport can range from 250 to 1,000 units (200).
Gabapentin is increasingly used as first-line treatment for spasticity, most particularly since it is licensed for neuropathic pain. Its mode of action, administration, and side effects are described in the section of first-line neuropathic pain treatment.
The medical use of cannabinoids remains controversial. The two most studied cannabinoids in cannabis are tetrahydrocannabinol (THC) and cannabidiol (CBD). THC is the most psychoactive substance and CBD is the major non-psychotropic substance in cannabis. Two cannabinoid receptors, CB1 and CB2, have been identified. CB1 receptors are located in the CNS and on peripheral nerves. CB2 receptors are found on the cells of the immune system. Evidence for successful treatment of both spasticity and pain in MS is available for nabiximols (trade mark: sativex oral spray), oral cannabis extract (OCE) (trade mark: cannador), and synthetic THC (trade mark: dronabinol, nabilone). OCE and THC, however, show only patient-reported spasticity reduction but were not found to be effective to reduce objective measures of spasticity (201, 202).
Nabiximols is a natural cannabis extract with a 1:1 ratio of THC and CBD activating CB1 and CB2 receptors. Nabiximols is available as oromucosal spray with 2.7 mg of THC and 2.5 mg of CBD per actuation (202). Nabiximols has also shown good efficacy for painful tonic spasms (202).
Cannador is a natural cannabis extract with 2.5 mg of THC and 1.25 mg of CBD per capsule and is currently only available in a research setting in Europe. Dronabinol and nabilone are currently not licensed for the treatment of spasticity and pain (202).
In addition to physiotherapy, most frequent medications used to treat PTS are sodium-channel-blocking antiepileptic agents such as carbamazepine, oxcarbazepine, gabapentin, clonazepam, and phenytoin sodium, as well as benzodiazepines, barbiturates, baclofen, and cannabinoids (3, 5, 31, 202, 203). It has been reported that topiramate at a daily dose of 400 mg can lead to the alleviation of PTS in AQP4-Ab-positive NMOSD (67) and one AQP4-Ab-positive NMOSD case with a favorable response to levetiracetam has been described (142). The highest efficacy for NMOSD-related PTS has been reported for carbamazepine, oxcarbazepine, and gabapentin (29, 32), with carbamazepine and oxcarbazepine outperforming gabapentin (32). These recommendations refer to a daily dose of 600–1,200 mg of oxcarbazepine and 100 mg three times a day of carbamazepine compared to 300 or 600 mg three times a day of gabapentin (32). Carbamazepine and oxcarbazepine act as voltage-gated sodium channel blockers and decrease neuronal excitability (32). Considering the emergence of important side effects of carbamazepine, oxcarbazepine has been recommended as a first-line treatment, preferably in combination with antispastic medication or antidepressants such as baclofen, pregabalin, or duloxetine (32).
Side effects of carbamazepine comprise ataxia, dizziness, somnolence, leukopenia, Steven–Johnson syndrome, and hyponatremia (204). In MS, carbamazepine can lead to a reversible exacerbation of neurological symptoms (205). Oxcarbazepine is better tolerated and safer than carbamazepine, especially with respect to CNS secondary side effects (ataxia, somnolence, and dizziness) and interaction with other medications (206). Side effects are often resolved after the titration period or with dosage adjustment. Frequently reported adverse effects include dizziness, headache, nausea, somnolence, fatigue, vomiting, back pain, diarrhea, tremor, skin rash, and blurred vision (206).
Pain is more than just an unpleasant physical sensation. It can comprise emotional, social, and spiritual suffering. Therefore, treatment strategies should not only directly target pain relief. Besides psychotherapy or behavioral therapy, exercise programs for physical reconditioning, relaxation techniques, and patient education should be considered to target functional, affective, social, and spiritual consequences affecting the patients' quality of life (182, 207, 208). Currently, pain syndromes in NMOSD and MOGAD are insufficiently controlled by medication, and multidrug therapy has been associated with worse fatigue and depression (2, 27, 209). Therefore, future studies should explore the efficacy of a multimodal and multidisciplinary approach of pain management (27).
Pain is a very frequent symptom in NMOSD and MOGAD and has a prevalence of over 80% with a severe impact on the quality of life of affected patients. Pain syndromes differ between NMOSD and MOGAD and can be an indicator for the respective disease type. Acute pain syndromes like retro-orbital pain, headache, or dysesthetic pain can be indicative for a first disease-related attack or a relapse of MOGAD-related optic neuritis, or NMOSD-related myelitis, brainstem, or cerebral affection. Chronic pain syndromes occur during the disease course and comprise primarily neuropathic pain and painful tonic spasms but also spasticity-related pain, back pain, and treatment-associated pain like osteoporosis. Acute ON-related pain seems to be particularly severe in MOGAD, while chronic neuropathic pain is more severe in NMOSD. Symptomatic treatment is currently insufficient to reduce pain intensity and improve the patients' quality of life. However, disease preventative immunosuppressive agents like tocilizumab and mycophenolate mofetil have shown a positive effect on pain reduction and should be further investigated. Patient care and future research should concentrate on a multidisciplinary approach of pain management, focusing on the respective pain type.
All authors contributed equally to the submitted work, analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript.
SA received a travel grant from Celgene and speaker honoraria from Roche and Bayer. GC has received speaker honoraria from Merck Serono and Bayer unrelated to this review. FP serves on the scientific advisory board for Novartis; received speaker honoraria and travel funding from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Merck Serono, Alexion, Chugai, MedImmune, and Shire; is an academic editor for PLoS ONE; is an associate editor for Neurology® Neuroimmunology and Neuroinflammation; consulted for SanofiGenzyme, Biogen Idec, MedImmune, Shire, and Alexion; and received research support from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Alexion, Merck Serono, German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, Arthur Arnstein Founda-tion Berlin, Guthy Jackson Charitable Foundation, and National Multiple Sclerosis of the USA.
1. Jarius S, Wildemann B. Devic's index case: a critical reappraisal – AQP4-IgG-mediated neuromyelitis optica spectrum disorder, or rather MOG encephalomyelitis? J Neurol Sci. (2019) 407:116396. doi: 10.1016/j.jns.2019.07.014
2. Asseyer S, Schmidt F, Chien C, Scheel M, Ruprecht K, Bellmann-Strobl J, et al. Pain in AQP4-IgG-positive and MOG-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. (2018) 4:1–12. doi: 10.1177/2055217318796684
3. Bradl M, Kanamori Y, Nakashima I, Misu T, Fujihara K, Lassmann H, et al. Pain in neuromyelitis optica - prevalence, pathogenesis and therapy. Nat Rev Neurol. (2014) 10:529–36. doi: 10.1038/nrneurol.2014.129
4. Gault F. De la neuromyélite optique aiguë. (Thèse). Faculté de Médecine et de Pharmacie. Lyon: Alexandre Rey (1894).
5. Solaro C, Trabucco E, Uccelli MM. Pain and multiple sclerosis: pathophysiology and treatment. Curr Neurol Neurosci Rep. (2012) 13:1–9. doi: 10.1007/s11910-012-0320-5
6. Treede R-D, Jensen TS, Campbell JN, Dostrocsky JO, Griffin JW, Hansson P, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. (2008) 70:1630–5. doi: 10.1212/01.wnl.0000282763.29778.59
7. Finnerup NB, Haroutounian S, Kamerman P, Baron R, Bennett DLH, Bouhassira D, et al. Neuropathic pain: an updated grading system for research and clinical practice. Pain. (2016) 157:1599–606. doi: 10.1097/j.pain.0000000000000492
9. Aziz Q, Barke A, Bennett MI, Benoliel R, Cohen M, Evers S, et al. A classification of chronic pain for ICD-11. Pain. (2015) 156:1003–7. doi: 10.1097/j.pain.0000000000000160
10. Cook LJ, Rose JW, Alvey JS, Jolley AM, Kuhn R, Marron B, et al. Collaborative international research in clinical and longitudinal experience study in NMOSD. Neurol Neuroimmunol NeuroInflamm. (2019) 6:e583. doi: 10.1212/NXI.0000000000000583
11. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
12. Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, et al. Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. (2007) 4:669–74. doi: 10.1371/journal.pmed.0040133
13. Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol. (2018) 9:888. doi: 10.3389/fneur.2018.00888
14. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176:149–64. doi: 10.1111/cei.12271
15. Sato DK, Lana-Peixoto MA, Fujihara K, de Seze J. Clinical spectrum and treatment of neuromyelitis optica spectrum disorders: evolution and current status. Brain Pathol. (2013) 23:647–60. doi: 10.1111/bpa.12087
16. Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, et al. Brainstem manifestations in neuromyelitis optica: A multicenter study of 258 patients. Mult Scler J. (2014) 20:843–7. doi: 10.1177/1352458513507822
17. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. (2012) 9:1–17. doi: 10.1186/1742-2094-9-14
18. Kanamori Y, Nakashima I, Takai Y, Nishiyama S, Kuroda H, Takahashi T, et al. Pain in neuromyelitis optica and its effect on quality of life a cross-sctional study. Neurology. (2011) 77:652–8. doi: 10.1212/WNL.0b013e318229e694
19. Chavarro VS, Mealy MA, Simpson A, Lacheta A, Pache F, Ruprecht K, et al. Insufficient treatment of severe depression in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e286. doi: 10.1212/NXI.0000000000000286
20. Oertel FC, Schließeit J, Brandt AU, Paul F. Cognitive impairment in neuromyelitis optica spectrum disorders: a review of clinical and neuroradiological features. Front Neurol. (2019) 10:608. doi: 10.3389/fneur.2019.00608
21. Song Y, Pan L, Fu Y, Sun N, Li YJ, Cai H, et al. Sleep abnormality in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol NeuroInflamm. (2015) 2:e94. doi: 10.1212/NXI.0000000000000094
22. Penner I, Paul F. Fatigue as a symptom or comorbidity of neurological diseases. Nat Publ Gr. (2017) 13:662–75. doi: 10.1038/nrneurol.2017.117
23. Beekman J, Keisler A, Pedraza O, Haramura M, Gianella-Borradori A, Katz E, et al. Neuromyelitis optica spectrum disorder: patient experience and quality of life. Neurol Neuroimmunol NeuroInflamm. (2019) 6:580. doi: 10.1212/NXI.0000000000000580
24. D'Souza M, Papadopoulou A, Levy M, Jacob A, Yeaman MR, Kümpfel T, et al. Diagnostic procedures in suspected attacks in patients with neuromyelitis optica spectrum disorders: results of an international survey. Mult Scler Relat Disord. (2020) 41:102027. doi: 10.1016/j.msard.2020.102027
25. Eaneff S, Wang V, Hanger M, Levy M, Mealy MA, Brandt AU, et al. Patient perspectives on neuromyelitis optica spectrum disorders: data from the patients like me online community. Mult Scler Relat Disord. (2017) 17:116–22. doi: 10.1016/j.msard.2017.07.014
26. Zhao S, Mutch K, Elsone L, Nurmikko T, Jacob A. Neuropathic pain in neuromyelitis optica affects activities of daily living and quality of life. Mult Scler. (2014) 20:1658–61. doi: 10.1177/1352458514522103
27. Qian P, Lancia S, Alvarez E, Klawiter EC, Cross AH, Naismith RT. Association of neuromyelitis optica with severe and intractable pain. Arch Neurol. (2012) 69:1482–7. doi: 10.1001/archneurol.2012.768
28. Pellkofer HL, Havla J, Hauer D, Schelling G, Azad SC, Kuempfel T, et al. The major brain endocannabinoid 2-AG controls neuropathic pain and mechanical hyperalgesia in patients with neuromyelitis optica. PLoS ONE. (2013) 8:e71500. doi: 10.1371/journal.pone.0071500
29. Kim SM, Go MJ, Sung JJ, Park KS, Lee KW. Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol. (2012) 69:1026–31. doi: 10.1001/archneurol.2012.112
30. Carnero Contentti E, Leguizamon F, Hryb JP, Celso J, Pace JL, Ferrari J, et al. Neuromyelitis optica: association with paroxysmal painful tonic spasms. Neurologia. (2015) 31:511–5. doi: 10.1016/j.nrleng.2014.12.015
31. Usmani N. Association between paroxysmal tonic spasms and neuromyelitis optica. Arch Neurol. (2012) 69:121–4. doi: 10.1001/archneurol.2011.832
32. Liu J, Zhang Q, Lian Z, Chen H, Shi Z, Feng H, et al. Painful tonic spasm in neuromyelitis optica spectrum disorders: prevalence, clinical implications and treatment options. Mult Scler Relat Disord. (2017) 17:99–102. doi: 10.1016/j.msard.2017.07.004
33. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease : a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
34. Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol. (2018) 9:217. doi: 10.3389/fneur.2018.00217
35. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, Seze J De, et al. MOG encephalomyelitis : international recommendations on diagnosis and antibody testing. J Neuroinflam. (2018) 15:1–10. doi: 10.1186/s12974-018-1144-2
36. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol NeuroInflamm. (2015) 2:e62. doi: 10.1212/NXI.0000000000000062
37. Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas MC. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol NeuroInflamm. (2015) 2:e131. doi: 10.1212/NXI.0000000000000131
38. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm. (2016) 13:1–45. doi: 10.1186/s12974-016-0718-0
39. Biotti D, Bonneville F, Tournaire E, Ayrignac X, Dallière CC, Mahieu L, et al. Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol. (2017) 264:2173–5. doi: 10.1007/s00415-017-8615-8
40. Papeix C, Moreau T, Biotti D, Pelletier J, Audoin B, Ruiz A, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults. Neurology. (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
41. Cobo-Calvo A, Ayrignac X, Kerschen P, Horellou P, Cotton F, Labauge P, et al. Cranial nerve involvement in patients with MOG antibody-associated disease. Neurol Neuroimmunol NeuroInflamm. (2019) 6:e543. doi: 10.1212/NXI.0000000000000543
42. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol NeuroInflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
43. Matesanz S, Kotch C, Perrone C, Waanders AJ, Hill B, Narula S. Expanding the MOG phenotype: brainstem encephalitis with punctate and curvilinear enhancement. Neurol Neuroimmunol neuroinflamm. (2019) 6:e619. doi: 10.1212/NXI.0000000000000619
44. Patterson K, Iglesias E, Nasrallah M, González-Álvarez V, Sunõl M, Anton J, et al. Anti-MOG encephalitis mimicking small vessel CNS vasculitis. Neurol Neuroimmunol NeuroInflamm. (2019) 6:e538. doi: 10.1212/NXI.0000000000000538
45. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders : a multicenter study of 50 patients. Part 3 : Brainstem involvement - frequency, presentation and outcome. J Neuroinflamm. (2016) 13:281. doi: 10.1186/s12974-016-0719-z
46. Zhou L, Huang Y, Li H, Fan J, Zhangbao J, Yu H, et al. MOG-antibody associated demyelinating disease of the CNS : a clinical and pathological study in Chinese Han patients. J Neuroimmunol. (2017) 305:19–28. doi: 10.1016/j.jneuroim.2017.01.007
47. Narayan R, Simpson A, Fritsche K, Salama S, Pardo S, Mealy M, et al. MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. (2018) 25:66–72. doi: 10.1016/j.msard.2018.07.025
48. Cobo-Calvo A, Vukusic S, Marignier R. Clinical spectrum of central nervous system myelin oligodendrocyte glycoprotein autoimmunity in adults. Curr Opin Neurol. (2019) 32:459–66. doi: 10.1097/WCO.0000000000000681
49. Hyun J-W, Jang H, Yu J, Park NY, Kim S-H, Huh S-Y, et al. Comparison of neuropathic pain in neuromyelitis optica and multiple sclerosis. Korean Neurol Assoc. (2020) 16:124–30. doi: 10.3988/jcn.2020.16.1.124
50. Mutch K, Methley A, Moore P, Jacob A. Life on hold: the experience of living with neuromyelitis optica. Disabil Rehabil. (2014) 36:1100–7. doi: 10.3109/09638288.2013.833301
51. Kong Y, Okoruwa H, Revis J, Tackley G, Leite MI, Lee M, et al. Pain in patients with transverse myelitis and its relationship to aquaporin 4 antibody status. J Neurol Sci. (2016) 368:84–8. doi: 10.1016/j.jns.2016.06.041
52. Elsone L, Townsend T, Mutch K, Das K, Boggild M, Nurmikko T, et al. Neuropathic pruritus (itch) in neuromyelitis optica. Mult Scler J. (2012) 19:475–9. doi: 10.1177/1352458512457720
53. Tackley G, Vecchio D, Hamid S, Jurynczyk M, Kong Y, Gore R, et al. Chronic neuropathic pain severity is determined by lesion level in aquaporin 4-antibody-positive myelitis. J Neurol Neurosurg Psychiatry. (2017) 88:165–9. doi: 10.1136/jnnp-2016-314991
54. Asseyer S, Hamblin J, Messina S, Mariano R, Siebert N, Everett R, et al. Prodromal headache in MOG-antibody positive optic neuritis. Mult Scler Relat Disord. (2020) 40:101965. doi: 10.1016/j.msard.2020.101965
55. Mealy MA, Kozachik SL, Cook LJ, Totonis L, Salazar RA, Allen JK, et al. Scrambler therapy improves pain in neuromyelitis optica. Neurology. (2020) 94:e1900–7. doi: 10.1212/WNL.0000000000009370
56. Wang T, Lian Z, Wu X, Kong Y, Zhou H, Wei M. Subcortical structural abnormalities in female neuromyelitis optica patients with neuropathic pain. Mult Scler Relat Disord. (2020) 37:101432. doi: 10.1016/j.msard.2019.101432
57. Mealy MA, Kozachik SL, Levy M. Review of treatment for central spinal neuropathic pain and its effect on quality of life : implications for neuromyelitis optica spectrum disorder. Pain Manag Nurs. (2019) 20:580–91. doi: 10.1016/j.pmn.2019.03.003
58. Choi S Il, Lee YJ, Kim DW, Yang JY. A case of neuromyelitis optica misdiagnosed as cervicogenic headache. Korean J Pain. (2014) 27:77–80. doi: 10.3344/kjp.2014.27.1.77
59. Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. (2013) 23:827–31. doi: 10.3109/s10165-012-0715-9
60. Horbinski C, Pollack IF, Wiley C, Murdoch G. A 10-year old girl with neck pain. Brain Pathol. (2010) 20:519–22. doi: 10.1111/j.1750-3639.2009.00365.x
61. Loschner AL, Snyder JE. Pelvic pain as an unusual first presentation of a demyelinating disease. J Gen Intern Med. (2008) 23:1917–20. doi: 10.1007/s11606-008-0767-x
62. Mathew T, Nadimpally USUS, Sarma GRKRK, Nadig R. Trigeminal autonomic cephalalgia as a presenting feature of neuromyelitis optica: “a rare combination of two uncommon disorders.” Mult Scler Relat Disord. (2016) 6:73–4. doi: 10.1016/j.msard.2016.01.006
63. Grüter T, Ayzenberg I, Gahlen A, Kneiphof J, Gold R, Kleiter I. Flaccid paralysis in neuromyelitis optica: an atypical presentation with possible involvement of the peripheral nervous system. Mult Scler Relat Disord. (2018) 25:83–6. doi: 10.1016/j.msard.2018.07.032
64. Sundaram S, Nair SS, Jaganmohan D, Unnikrishnan G, Nair M. Relapsing lumbosacral myeloradiculitis: an unusual presentation of MOG antibody disease. Mult Scler J. (2019) 26:509–11. doi: 10.1177/1352458519840747
65. Roman-Filip C, Ungureanu A, Cernuşcă-Mitaru M. Painful tonic spasms and brainstem involvement in a patient with neuromyelitis optica spectrum disorder. Neurol Neurochir Pol. (2016) 50:55–8. doi: 10.1016/j.pjnns.2015.10.010
66. El Otmani H, Dany F, El Moutawakil B, Abdoh-Rafai M, Slassi I. Intractable hiccup and vomiting, neuropathic pruritus and tonic spasms in a case of neuromyelitis optica spectrum disorder. Acta Neurol Belg. (2015) 115:797–9. doi: 10.1007/s13760-014-0418-4
67. Iida S, Nakamura M, Wate R, Kaneko S, Kusaka H. Successful treatment of paroxysmal tonic spasms with topiramate in a patient with neuromyelitis optica. Mult Scler Relat Disord. (2015) 4:457–9. doi: 10.1016/j.msard.2015.07.011
68. Lovera L, Jay WM, Biller J. Horner syndrome in a case of neuromyelitis optica. Neuro Ophthalmol. (2014) 38:78–81. doi: 10.3109/01658107.2013.856027
69. Uzawa A, Masahiro M, Kuwabara S. Cytokines and chemokines in neuromyelitis optica: pathogenetic and therapeutic implications. Brain Pathol. (2014) 24:67–73. doi: 10.1111/bpa.12097
70. Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, et al. Effects of neuromyelitis optica - IgG at the blood-brain barrier in vitro. Neurol Neuroimmunol NeuroInflamm. (2017) 4:e311. doi: 10.1212/NXI.0000000000000311
71. Melamed E, Levy M, Waters PJ, Sato DK, Bennett JL, John GR, et al. Update on biomarkers in neuromyelitis optica. Neurol Neuroimmunol NeuroInflamm. (2015) 2:e134. doi: 10.1212/NXI.0000000000000134
72. Pezet S, McMahon SB. Neutrophins: mediators and modulators of pain. Annu Rev Neurosci. (2006) 29:507–38. doi: 10.1146/annurev.neuro.29.051605.112929
73. Lewin GR, Nykjaer A. Pro-neurotrophins, sortilin, and nociception. Eur J Neurosci. (2014) 39:363–74. doi: 10.1111/ejn.12466
74. Masters-Israilov A, Robbins MS. Headache in neuromyelitis optica. Curr Pain Headache Rep. (2017) 21:1–8. doi: 10.1007/s11916-017-0620-1
75. Dawes JM, Vincent A. Autoantibodies and pain. Curr Opin. (2016) 10:137–42. doi: 10.1097/SPC.0000000000000211
76. Asseyer S, Kuchling J, Gaetano L, Siebert N, Chien C, Scheel M, et al. Associations Between Neuropathic Pain and Thalamic Ventral Posterior Nucleus Volume In Neuromyelitis Optica Spectrum Disorders. ECTRIMS Online Library (2019). p. 279087; P. 727.
77. Hacohen Y, Palace J. Time to separate MOG-Ab-associated disease from AQP4-Ab-positive neuromyelitis optica spectrum disorder. Neurology. (2018) 90:947–8. doi: 10.1212/WNL.0000000000005619
78. Kawachi I, Lassmann H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2017) 88:137–45. doi: 10.1136/jnnp-2016-313300
79. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, et al. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. (2008) 4:202–14. doi: 10.1038/ncpneuro0764
80. von Büdingen HC, Mei F, Greenfield A, Jahn S, Shen YAA, Reid HH, et al. The myelin oligodendrocyte glycoprotein directly binds nerve growth factor to modulate central axon circuitry. J Cell Biol. (2015) 210:891–8. doi: 10.1083/jcb.201504106
81. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. (1999) 53:1107–14. doi: 10.1212/WNL.53.5.1107
82. Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. (2015) 84:1165–73. doi: 10.1212/WNL.0000000000001367
83. Chien C, Scheel M, Schmitz-hübsch T, Borisow N, Ruprecht K, Bellmann-strobl J, et al. Spinal cord lesions and atrophy in NMOSD with AQP4-IgG and MOG-IgG associated autoimmunity. Mult Scler J. (2018) 25:1926–36. doi: 10.1177/1352458518815596
84. Pekcevik Y, Mitchell CH, Mealy MA, Orman G, Lee IH, Scott D, et al. Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler J. (2016) 22:302–11. doi: 10.1177/1352458515591069
85. Hayashida S, Masaki K, Yonekawa T, Suzuki SO, Hiwatashi A. Early and extensive spinal white matter involvement in neuromyelitis optica. Brain Pathol. (2017) 27:249–65. doi: 10.1111/bpa.12386
86. Krampla W, Jecel J, Fertl E. Spinal cord lesions in patients with neuromyelitis optica : a retrospective long-term MRI follow-up study. Eur Soc Radiol. (2009) 19:2535–43. doi: 10.1007/s00330-009-1425-3
87. Nakamura M, Miyazawa I, Fujihara K, Nakashima I, Misu T, Watanabe S, et al. Preferential spinal central gray matter involvement in neuromyelitis optica: An MRI study. J Neurol. (2008) 255:163–70. doi: 10.1007/s00415-008-0545-z
88. Asgari N, Khorooshi R, Lillevang ST, Owens T. Complement-dependent pathogenicity of brain-specific antibodies in cerebrospinal fluid. J Neuroimmunol. (2013) 254:76–82. doi: 10.1016/j.jneuroim.2012.09.010
89. Asgari N, Skejoe HPB, Lillevang ST, Steenstrup T, Stenager E, Kyvik KO. Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol. (2013) 13:33. doi: 10.1186/1471-2377-13-33
90. Doi H, Matsushita T, Isobe N, Ishizu T, Ohyagi Y, Kira JI. Frequency of chronic headaches in japanese patients with multiple sclerosis: with special reference to opticospinal and common forms of multiple sclerosis. Headache. (2009) 49:1513–20. doi: 10.1111/j.1526-4610.2009.01427.x
91. Leone M, Bussone G, Besta NC. Pathophysiology of trigeminal autonomic cephalalgias. Lancet Neurol. (2009) 8:755–64. doi: 10.1016/S1474-4422(09)70133-4
92. Messlinger K, Ebersberger A, Schaible H-G. Release of immunoreactive substance P in the brain stem upon stimulation of the cranial dura mater with low pH - inhibition by the serotonin (5-HT1) recetpr agonist CP 93,129. Br J Pharmacol. (1998) 125:1726–32. doi: 10.1038/sj.bjp.0702247
93. Wang Y, Zhang L, Zhang B, Dai Y, Kang Z, Lu C, et al. Comparative clinical characteristics of neuromyelitis optica spectrum disorders with and without medulla oblongata lesions. J Neurol. (2014) 261:954–62. doi: 10.1007/s00415-014-7298-7
94. Oertel FC, Zimmermann H, Paul F, Brandt AU. Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J. (2018) 9:21–33. doi: 10.1007/s13167-017-0123-5
95. Finke C, Zimmermann H, Pache F, Oertel FC, Chavarro VS, Kramarenko Y, et al. Association of visual impairment in neuromyelitis optica spectrum disorder with visual network reorganization. JAMA Neurol. (2018) 75:296–303. doi: 10.1001/jamaneurol.2017.3890
96. Bennett JL, de Seze J, Lana-Peixoto M, Palace J, Waldman A, Banwell B, et al. Neuromyelitis optica and multiple sclerosis: seeing differences through optical coherence tomography. Mult Scler J. (2015) 21:678–88. doi: 10.1177/1352458514567216
97. Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, et al. Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol NeuroInflamm. (2017) 4:e334. doi: 10.1212/NXI.0000000000000334
98. Bradl M, Reindl M, Lassmann H. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol. (2018) 31:325–33. doi: 10.1097/WCO.0000000000000551
99. Matiello M, Schaefer-Klein J, Sun D, Weinshenker B. Aquaporin 4 expression and tissue susceptibility to neuromyelitis optica. JAMA Neurol. (2013) 70:1118–25. doi: 10.1001/jamaneurol.2013.3124
100. Graber DJ, Levy M, Kerr D, Wade WF. Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflamm. (2008) 5:1–21. doi: 10.1186/1742-2094-5-22
101. Yamamura T, Nakashima I. Foveal thinning in neuromyelitis optica a sign of retinal astrocytopathy? Neurol Neuroimmunol NeuroInflamm. (2017) 4:e347. doi: 10.1212/NXI.0000000000000347
102. Gritsch S, Lu J, Thilemann S, Wo S, Bruttger J, Karram K, et al. Oligodendrocyte ablation triggers central pain independently of innate or adaptive immune responses in mice. Nat Commun. (2014) 5:1–17. doi: 10.1038/ncomms6472
103. Kister I, Caminero AB, Herbert J, Lipton RB. Tension-type headache and migraine in multiple sclerosis. Curr Pain Headache Rep. (2010) 14:441–8. doi: 10.1007/s11916-010-0143-5
104. Russo A, Silvestro M, Tedeschi G, Tessitore A. Physiopathology of migraine: what have we learned from functional imaging? Curr Neurol Neurosci Rep. (2017) 17:2–11. doi: 10.1007/s11910-017-0803-5
105. Solly SK, Thomas J-L, Monge M, Demerens C, Lubetzki C, Gardier MV, et al. Myelid/Oligodendrocyte glycoprotein (MOG) Expression is associated with myelin deposition. Glia. (1996) 18:39–48. doi: 10.1002/(SICI)1098-1136(199609)18:1<39::AID-GLIA4>3.0.CO;2-Z
106. Vincent M, Wang S. The international classification of headache disorders, 3rd edition. Cephalgia. (2018) 38:1–211. doi: 10.1177/0333102417738202
107. Marzoli SB, Criscuoli A. Pain in optic neuropathies. Neurol Sci. (2018) 39:25–31. doi: 10.1007/s10072-018-3334-1
108. Evers S. Facial pain : overlapping syndromes. Cephalgia. (2017) 37:705–13. doi: 10.1177/0333102417703761
109. Marzoli SB, Criscuoli A. Headaches attributed to visual disturbances. Neurol Sci. (2015) 36:85–8. doi: 10.1007/s10072-015-2167-4
110. Agostini E, Frigerio R, Protti A. Controversies in optic neuritis pain diagnosis. J Mec Theor Appl. (1987) 6:145–64.
111. Bruscolini A, Sacchetti M, La M, Gharbiya M, Ralli M, Lambiase A, et al. Autoimmunity reviews diagnosis and management of neuromyelitis optica spectrum disorders - an update. Autoimmun Rev. (2018) 17:195–200. doi: 10.1016/j.autrev.2018.01.001
112. Galetta SL, Villoslada P, Levin N, Shindler K, Ishikawa H, Parr E, et al. Acute optic neuritis: unmet clinical needs and model for new therapies. Neurol Neuroimmunol NeuroInflamm. (2015) 2:e135. doi: 10.1212/NXI.0000000000000135
113. Petzold A, Wattjes MP, Costello F, Flores-Rivera J, Fraser CL, Fujihara K, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol. (2014) 10:447–58. doi: 10.1038/nrneurol.2014.108
114. Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol NeuroInflamm. (2019) 6:1–7. doi: 10.1212/NXI.0000000000000572
115. Kidd DP, Burton BJ, Graham EM, Plant GT. Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e270. doi: 10.1212/NXI.0000000000000270
116. Soelberg K, Jarius S, Skejoe HPB, Engberg H, Mehlsen JJ, Nilsson AC, et al. A population-based prospective study of optic neuritis. Mult Scler. (2017) 23:1893–901. doi: 10.1177/1352458517734070
117. Ishikawa H, Kezuka T, Shikishima K, Yamagami A. Epidemiologic and clinical characteristics of optic neuritis in Japan. Ophthalmology. (2019) 126:1385–98. doi: 10.1016/j.ophtha.2019.04.042
118. Chen JJ, Flanagan EP, Jitprapaikulsan J, López- ASS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody–positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. (2018) 195:8–15. doi: 10.1016/j.ajo.2018.07.020
119. Nakajima H, Motomura M, Tanaka K, Fujikawa A, Nakata R, Maeda Y, et al. Antibodies to myelin oligodendrocyte glycoprotein in idiopathic optic neuritis. BMJ Open. (2015) 5:e007766. doi: 10.1136/bmjopen-2015-007766
120. Zabad RK, Stewart R, Healey KM. Pattern recognition of the multiple sclerosis syndrome. Brain Sci. (2017) 7:1–51. doi: 10.3390/brainsci7100138
121. Zhou L, ZhangBao J, Li H, Li X, Huang Y, Wang M, et al. Cerebral cortical encephalitis followed by recurrent NS demyelination in a patient with concomitant anti-MOG and anti-NMDA receptor antibodies. Mult Scler Relat Disord. (2017) 18:90–2. doi: 10.1016/j.msard.2017.09.023
122. Gebhardt M, Kropp P, Hoffmann F, Zettl UK. Headache at the time of first symptom manifestation of multiple sclerosis: a prospective, longitudinal study. EurNeurol. (2018) 80:115–20. doi: 10.1159/000494092
123. Wang J, Wang K, Chen X-W, Wang J-W, Zhang K, XU M-W, et al. Meningoencephalitis as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler J. (2012) 19:639–43. doi: 10.1177/1352458512459785
124. Muto M, Mori M, Sato Y, Uzawa A, Masuda S, Uchida T, et al. Current symptomatology in multiple sclerosis and neuromyelitis optica. Eur J Neurol. (2015) 22:299–304. doi: 10.1111/ene.12566
125. Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. (2014) 82:1302–6. doi: 10.1212/WNL.0000000000000317
126. Schöberl F, Csanadi E, Eren O, Dieterich M, Kümpfel T. NMOSD triggered by yellow fever vaccination – an unusual clinical presentation with segmental painful erythema. Mult Scler Relat Disord. (2017) 11:43–4. doi: 10.1016/j.msard.2016.11.009
127. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol NeuroInflamm. (2015) 2:e110. doi: 10.1212/NXI.0000000000000110
128. Hayashi Y, Koumura A, Yamada M, Kimura A, Shibata T, Inuzuka T. Acute-onset severe occipital neuralgia associated with high cervical lesion in patients with neuromyelitis optica spectrum disorder. Headache. (2017) 57:1145–51. doi: 10.1111/head.13126
129. Sugiyama A, Mori M, Masuda H, Uchida T, Muto M, Uzawa A, et al. Trigeminal root entry zone involvement in neuromyelitis optica and multiple sclerosis. J Neurol Sci. (2015) 355:147–9. doi: 10.1016/j.jns.2015.06.004
130. Lee S, Lee H, Baek S. Paroxysmal pruritus as the first relapsing symptom of neuromyelitis optica. Neurol Asia. (2010) 15:185–7.
131. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
132. Takai Y, Misu T, Nakashima I, Takahashi T, Itoyama Y, Fujihara K, et al. Two cases of lumbosacral myeloradiculitis with anti-aquaporin-4 antibody. Neurology. (2012) 79:1826–9. doi: 10.1212/WNL.0b013e3182703ff7
133. Laemmer AB, Maihöfer C, Gölitz P, Schwab S, Lee DH, Linker RA, et al. Possible second motor neuron damage in neuromyelitis optica. Clin Neurophysiol. (2014) 125:859–61. doi: 10.1016/j.clinph.2013.08.023
134. Warabi Y, Yamazaki M, Shimizu T, Nagao M. Abnormal nerve conduction study findings indicating the existence of peripheral neuropathy in multiple sclerosis and neuromyelitis optica. Biomed Res Int. (2013) 2013:847670. doi: 10.1155/2013/847670
135. Kim S, Park J, Kwon BS, Park J, Lee HJ, Choi J. Radiculopathy in neuromyelitis optica. how does anti-AQP4 Ab involve PNS? Mult Scler Relat Disord. (2017) 18:77–81. doi: 10.1016/j.msard.2017.09.006
136. Zhang W, Jiao Y, Cui L, Liu L, Zhang L, Jiao J. Etiological, clinical, and radiological features of longitudinally extensive myelopathy in Chinese patients. J Clin Neurosci. (2016) 32:61–6. doi: 10.1016/j.jocn.2015.12.048
137. Ramanathan S, Davies A, Monif M, Shuey N, Walt A, Van Der Hardy T, et al. Peripheral Nervous System Involvement in Mog Antibody-Associated Demyelination. ECTRIMS Online Library (2019). p. 279523; 245.
138. Do Campo RV, Stephens A, Marin Collazo IV, Rubin DI. MOG antibodies in combined central and peripheral demyelination syndromes. Neurol Neuroimmunol NeuroInflamm. (2018) 5:e503. doi: 10.1212/NXI.0000000000000503
139. Pagany M, Jagodic M, Schubart A, Pham-dinh D, Bachelin C, Baron A, et al. Myelin oligodendrocyte glycoprotein is expressed in the peripheral nervous system of rodents and primates. Neurosci Lett. (2003) 350:165–8. doi: 10.1016/S0304-3940(03)00899-1
140. Pozzilli C. Advances in the management of multiple sclerosis spasticity: experiences from recent studies and everyday clinical practice. Expert Rev Neurother. (2013) 13:49–54. doi: 10.1586/14737175.2013.865877
141. Li Y, Jiang B, Chen B, Zhao M, Zhou C. Neuromyelitis optica spectrum disorders with multiple brainstem manifestations : a case report. Neurol Sci. (2016) 37:309–13. doi: 10.1007/s10072-015-2196-z
142. Rodríguez-Quiroga SA, Abaroa L, Arakaki T, Garretto NS, Villa AM. Commentary on neuromyelitis optica associated with painful paroxysmal dystonia: case report and literature review. Acta Neurol Belg. (2014) 115:523–4. doi: 10.1007/s13760-014-0389-5
143. Kim SM, Waters P, Woodhall M, Kim JY, Kim JE, Yang JW, et al. Utility of aquaporin-4 antibody assay in patients with neuromyelitis optica spectrum disorders. Mult Scler J. (2013) 19:1060–7. doi: 10.1177/1352458512472748
144. Li X, Wang YQ. Membranous nephropathy with Neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. (2017) 15:49–51. doi: 10.1016/j.msard.2017.05.007
145. Abboud H, Fernandez HH, Mealy MA, Levy M. Spinal movement disorders in neuromyelitis optica: an under-recognized phenomenon. Mov Disord. (2016) 3:596–602. doi: 10.1002/mdc3.12321
146. O'Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. (2008) 137:96–111. doi: 10.1016/j.pain.2007.08.024
147. Ciccarelli O, Thomas DL, De Vita E, Wheeler-kingshott CAM, Kachramanoglou C, Kapoor R, et al. Low myo-inositol indicating astrocytic damage in a case series of neuromyelitis optica. Ann Neurol. (2013) 74:301–5. doi: 10.1002/ana.23909
148. Pittock SJ, Lucchinetti CF, Clinic M, Clinic M. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci. (2017) 1366:20–39. doi: 10.1111/nyas.12794
149. Jarius S, Jacobi C, De Seze J, Zephir H, Paul F, Franciotta D, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler J. (2011) 17:1067–73. doi: 10.1177/1352458511403958
150. Shahmohammadi S, Doosti R, Shahmohammadi A, Mohammadianinejad SE, Sahraian MA, Azimi AR, et al. Autoimmune diseases associated with neuromyelitis optica spectrum disorders: a literature review. Mult Scler Relat Disord. (2018) 27:350–63. doi: 10.1016/j.msard.2018.11.008
151. Jarius S, Paul F, Franciotta D, De Seze J, Münch C, Salvetti M, et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler J. (2012) 18:1135–43. doi: 10.1177/1352458511431728
152. Gkaniatsou T, Papadopoulou A, Paul F, Brandt AU, Oertel FC. Frequency of autoimmune disorders and autoantibodies in European patients with neuromyelitis optica spectrum disorders. Acta Neurol Belg. (2019) 120:223–22. doi: 10.1007/s13760-019-01176-6
153. Jobling K, Ledingham D, Ng W-F, Guadagno J. Positive anti-MOG antibodies in a patient with sjögren's syndrome and transverse myelitis. Eur J Rheumatol. (2019) 6:100–2. doi: 10.5152/eurjrheum.2018.18041
154. Mader S, Jeganathan V, Arinuma Y, Fujieda Y, Dujmovic I, Drulovic J, et al. Understanding the antibody repertoire in neuropsychiatric systemic lupus erythematosus and neuromyelitis optica spectrum disorder: do they share common targets? Arthritis Rheumatol. (2018) 70:277–86. doi: 10.1002/art.40356
155. Pröbstel AK, Thanei M, Erni B, Lecourt AC, Branco L, André R, et al. Association of antibodies against myelin and neuronal antigens with neuroinflammation in systemic lupus erythematosus. Rheumatology. (2019) 58:908–13. doi: 10.1093/rheumatology/key282
156. Mifflin KA, Kerr BJ. Pain in autoimmune disorders. J Neurosci Res. (2017) 95:1282–94. doi: 10.1002/jnr.23844
157. Gold SM, Willing A, Leypoldt F, Paul F, Friese MA. Sex differences in autoimmune disorders of the central nervous system. Semin Immunopathol. (2019) 41:177–88. doi: 10.1007/s00281-018-0723-8
158. Yeo T, dos Passos GR, Muhammed L, Everett R, Reeve S, Messina S, et al. Factors associated with fatigue in CNS inflammatory diseases with AQP4 and MOG antibodies. Ann Clin Transl Neurol. (2020) 7:375–83. doi: 10.1002/acn3.51008
159. Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: antecedent or consequence of chronic pain? A Rev Clin J Pain. (1997) 13:116–37. doi: 10.1097/00002508-199706000-00006
160. Ringelstein M, Ayzenberg I, Harmel J, Lauenstein A-S, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. (2015) 72:1–8. doi: 10.1001/jamaneurol.2015.0533
161. Duchow A, Paul F, Bellmann-Strobl J. Current and emerging biologics for the treatment of neuromyelitis optica spectrum disorders. Expert Opin Biol Ther. (2020). doi: 10.1080/14712598.2020.1749259. [Epub ahead of print].
162. Huang Q, Wang J, Zhou Y, Yang H, Wang Z, Yan Z. Low-dose mycophenolate mofetil for treatment of neuromyelitis optica spectrum disorders: a prospective multicenter study in south China. Front Immunol. (2018) 9:2066. doi: 10.3389/fimmu.2018.02066
163. Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the neuromyelitis optica study group (NEMOS). J Neurol. (2014) 261:1–16. doi: 10.1007/s00415-013-7169-7
164. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, et al. Apheresis therapies for NMOSD attacks a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol NeuroInflamm. (2018) 5:e504. doi: 10.1212/NXI.0000000000000504
165. Levy M. Plasmapheresis for acute attacks in neuromyelitis optica spectrum disorders. Neurol Neuroimmunol NeuroInflamm. (2018) 5:e504. doi: 10.1212/NXI.0000000000000510
166. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. (2016) 79:206–16. doi: 10.1002/ana.24554
167. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. (2010) 17:1019–32. doi: 10.1111/j.1468-1331.2010.03066.x
168. Kimbrough DJ, Fujihara K, Jacob A, Lana-Peixoto MA, Isabel Leite M, Levy M, et al. Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord. (2012) 1:180–7. doi: 10.1016/j.msard.2012.06.002
169. Terzi M, Totolyan N, Viswanathan S, Wang K, Pace A, Fujita KP, et al. Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. New Engl J Med Orig. (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
170. Pittock SJ, Lennon VA, Mckeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. (2013) 12:554–62. doi: 10.1016/S1474-4422(13)70076-0
171. Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. (2019) 381:2114–24. doi: 10.1056/NEJMoa1901747
172. Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs. (2018) 27:265–71. doi: 10.1080/13543784.2018.1443077
173. Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. (2019) 394:1352–63. doi: 10.1016/S0140-6736(19)31817-3
174. Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. (2020) 19:402–12. doi: 10.1016/S1474-4422(20)30078-8
175. Agasing AM, Wu Q, Khatri B, Borisow N, Ruprecht K, Brandt AU, et al. Transcriptomics and proteomics reveal a cooperation between interferon and T-helper 17 cells in neuromyelitis optica. Nat Commun. (2020) 11:1–13. doi: 10.1038/s41467-020-16625-7
176. Zhou Y, Liu Z, Liu Z, Chen S, Li M, Shahveranov A, et al. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflamm. (2016) 13:1–9. doi: 10.1186/s12974-016-0607-6
177. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
178. Hyun JW, Woodhall MR, Kim SH, Jeong IH, Kong B, Kim G, et al. Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry. (2017) 88:811–7. doi: 10.1136/jnnp-2017-315998
179. Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. (2020) 87:256–66. doi: 10.1002/ana.25648
180. Whittam DH, Cobo-calvo A, Lopez-chiriboga AS, Pardo S, Gornall M, Cicconi S, et al. Treatment of MOG-IgG-associated disorder with rituximab: an international study of 121 patients. Mult Scler Relat Disord. (2020) 44:102251. doi: 10.1016/j.msard.2020.102251
181. Spadaro M, Gerdes LA, Krumbholz M, Ertl-Wagner B, Thaler FS, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e257. doi: 10.1212/NXI.0000000000000257
182. Bates D, Schultheis BC, Hanes MC, Jolly SM, Chakravarthy K V, Deer TR, et al. A comprehensive algorithm for management of neuropathic pain. Pain Med. (2019) 20:2–12. doi: 10.1093/pm/pnz075
183. Attal N. Pharmacological treatments of neuropathic pain : the latest recommendations. Rev Neurol. (2018) 175:46–50. doi: 10.1016/j.neurol.2018.08.005
184. Macone A, Otis JAD. Neuropathic Pain. Semin Neurol. (2018) 38:644–53. doi: 10.1055/s-0038-1673679
185. Gierthmühlen J, Baron R. Neuropathic Pain. Semin Neurol. (2016) 36:462–8. doi: 10.1055/s-0036-1584950
186. Haanpää M, Attal N, Backonja M, Baron R, Bennett M, Bouhassira D, et al. NeuPSIG guidelines on neuropathic pain assessment. Pain. (2011) 152:14–27. doi: 10.1016/j.pain.2010.07.031
187. O'Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. (2009) 122(Suppl. 10):S22–32. doi: 10.1016/j.amjmed.2009.04.007
188. Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. (2015) 14:162–73. doi: 10.1016/S1474-4422(14)70251-0
189. Dworkin RH, Connor ABO, Audette J, Baron R, Gourlay GK, Haanpää ML, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. (2010) 85:S3–14. doi: 10.4065/mcp.2009.0649
190. Mu A, Weinberg E, Clarke H. Pharmacologic management of chronic neuropathic pain. Can Fam Physician. (2017) 63:844–52.
191. Hansson P, Jensen TS, Attal N, Cruccu G, Baron R, Haanpa M, et al. Efns guidelines efns guidelines on the pharmacological treatment of neuropathic pain : 2010 revision. Eur J Neurol. (2010) 17:1113–23. doi: 10.1111/j.1468-1331.2010.02999.x
192. Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci. (2017) 18:1–12. doi: 10.3390/ijms18112483
193. Attal N, Bouhassira D. Translational neuropathic pain research. (2019) 160(Suppl. 1):S23–8. doi: 10.1097/j.pain.0000000000001522
194. Beard S, Hunn A, Wight J. Treatments for spasticity and pain in multiple sclerosis. Health Technol Assess. (2003) 7:1–111. doi: 10.3310/hta7400
195. Stefano G Di, Maarbjerg S, Truini A. Trigeminal neuralgia secondary to multiple sclerosis: from the clinical picture to the treatment options. J Headache Pain. (2019) 20:1–10. doi: 10.1186/s10194-019-0969-0
196. Neuropathic pain in adults : pharmacological management in non-specialist settings. NICE Clin Guidel (2013). Available online at: nice.org.uk/guidance/cg173 (accessed June 9, 2020)
197. Majithia N, Smith TJ, Coyne PJ, Abdi S, Pachman DR, Lachance D, et al. Scrambler therapy for the management of chronic pain. Support Care Cancer. (2016) 24:2807–14. doi: 10.1007/s00520-016-3177-3
198. Volkers R, Giesen E, Heiden M, Van Der, Kerperien M, Lange S, Kurt E, et al. Invasive motor cortex stimulation influences intracerebral structures in patients with neuropathic pain: an activation likelihood estimation meta-analysis of imaging data. Neuromodulation. (2020) 23:436–43. doi: 10.1111/ner.13119
199. Lefaucheur JP, Aleman A, Baeken C, Benninger DH, Brunelin J, Di Lazzaro V, et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS): an update (2014–2018). Clin Neurophysiol. (2020) 131:474–528. doi: 10.1016/j.clinph.2020.02.003
200. Thompson AJ, Jarrett L, Lockley L, Marsden J, Stevenson VL. Clinical management of spasticity. J Neurol Neurosurg Psychiatry. (2005) 76:459–63. doi: 10.1136/jnnp.2004.035972
201. Whiting PF, Wolff RF, Deshpande S, Nisio M Di, Duffy S, Hernandez A V, et al. Cannabinoids for medical use a systematic review and meta-analysis. JAMA Neurol. (2015) 313:2456–73. doi: 10.1001/jama.2015.6358
202. Rice J, Cameron M, Cameron M. Cannabinoids for treatment of ms symptoms : state of the evidence. Curr Neurol Neurosci Rep. (2018) 18:1–10. doi: 10.1007/s11910-018-0859-x
203. Schmidt FR, Costa FHR, Silva FMLC, Maultasch H, Rosso AL, Nicaretta DH, et al. Paroxysmal dystonia and neuromyelitis optica. Arq Neuropsiquiatr. (2011) 70:271–3. doi: 10.1590/S0004-282X2012005000011
204. Sommer C, Richter H, Rogausch JP, Frettlöh J, Lungenhausen M, Maier C. A modified score to identify and discriminate neuropathic pain: a study on the german version of the neuropathic pain symptom inventory (NPSI). BMC Neurol. (2011) 11:104. doi: 10.1186/1471-2377-11-104
205. Brola W, Mitosek-Szewczyk K, Opara J. Symptomatology and pathogenesis of different types of pain in multiple sclerosis. Neurol Neurochir Pol. (2014) 48:272–9. doi: 10.1016/j.pjnns.2014.07.009
206. Zhou M, Chen N, He L, Yang M, Zhu C, Wu F. Oxcarbazepine for neuropathic pain. Cochrane Database Syst Rev. (2017) 12:1–54. doi: 10.1002/14651858.CD007963.pub3
207. Wojcikowski K, Vigar V, Oliver C. New concepts of chronic pain and the potential role of complementary therapies. Altern Ther Health Med. (2018) 26:18–31.
208. Gauthier K, Dulong C, Argáez C. Multidisciplinary treatment programs for patients with chronic non-malignant pain : a review of clinical effectiveness, cost- effectiveness, and guidelines. CADTH Rapid Response Rep. (2017) 1–46. Available from: https://www.cadth.ca/sites/default/files/pdf/htis/2019/RC1110MultidcpProgramChronic~Pain_Final.pdf
Keywords: aquaporin 4, headache, myelin oligodendrocyte glycoprotein-antibody-associated disease, neuromyelitis optica spectrum disorders, neuropathic pain, pain, painful tonic spasms
Citation: Asseyer S, Cooper G and Paul F (2020) Pain in NMOSD and MOGAD: A Systematic Literature Review of Pathophysiology, Symptoms, and Current Treatment Strategies. Front. Neurol. 11:778. doi: 10.3389/fneur.2020.00778
Received: 13 January 2020; Accepted: 24 June 2020;
Published: 21 August 2020.
Edited by:
Dalia L. Rotstein, University of Toronto, CanadaReviewed by:
Natalie Elizabeth Parks, Dalhousie University, CanadaCopyright © 2020 Asseyer, Cooper and Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedemann Paul, ZnJpZWRlbWFubi5wYXVsQGNoYXJpdGUuZGU=
†ORCID: Friedemann Paul orcid.org/0000-0002-6378-0070
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.