
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 05 August 2020
Sec. Neurotrauma
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00749
This article is part of the Research Topic On the Basis of Sex: Impact on Traumatic Brain Injury View all 14 articles
 Gokul Krishna1,2
Gokul Krishna1,2 Caitlin Bromberg1,2
Caitlin Bromberg1,2 Emily Charlotte Connell1,2,3
Emily Charlotte Connell1,2,3 Erum Mian1,2Chengcheng Hu4
Erum Mian1,2Chengcheng Hu4 Jonathan Lifshitz1,2,5
Jonathan Lifshitz1,2,5 P. David Adelson1,2
P. David Adelson1,2 Theresa Currier Thomas1,2,5*
Theresa Currier Thomas1,2,5*Women approximate one-third of the annual 2.8 million people in the United States who sustain traumatic brain injury (TBI). Several clinical reports support or refute that menstrual cycle-dependent fluctuations in sex hormones are associated with severity of persisting post-TBI symptoms. Previously, we reported late-onset sensory hypersensitivity to whisker stimulation that corresponded with changes in glutamate neurotransmission at 1-month following diffuse TBI in male rats. Here, we incorporated intact age-matched naturally cycling females into the experimental design while monitoring daily estrous cycle. We hypothesized that sex would not influence late-onset sensory hypersensitivity and associated in vivo amperometric extracellular recordings of glutamate neurotransmission within the behaviorally relevant thalamocortical circuit. At 28 days following midline fluid percussion injury (FPI) or sham surgery, young adult Sprague-Dawley rats were tested for hypersensitivity to whisker stimulation using the whisker nuisance task (WNT). As predicted, both male and female rats showed significantly increased sensory hypersensitivity to whisker stimulation after FPI, with females having an overall decrease in whisker nuisance scores (sex effect), but no injury and sex interaction. In males, FPI increased potassium chloride (KCl)-evoked glutamate overflow in primary somatosensory barrel cortex (S1BF) and ventral posteromedial nucleus of the thalamus (VPM), while in females the FPI effect was discernible only within the VPM. Similar to our previous report, we found the glutamate clearance parameters were not influenced by FPI, while a sex-specific effect was evident with female rats showing a lower uptake rate constant both in S1BF and VPM and longer clearance time (in S1BF) in comparison to male rats. Fluctuations in estrous cycle were evident among brain-injured females with longer diestrus (low circulating hormone) phase of the cycle over 28 days post-TBI. Together, these findings add to growing evidence indicating both similarities and differences between sexes in a chronic response to TBI. A better understanding of the influence of gonadal hormones on behavior, neurotransmission, secondary injury and repair processes after TBI is needed both clinically and translationally, with potential impact on acute treatment, rehabilitation, and symptom management.
- Diffuse traumatic brain injury (TBI)-induces sensory hypersensitivity in males and females.
- TBI increased evoked glutamate overflow in S1BF and VPM of males and VPM of females.
- Females displayed sex-specific changes in glutamate in comparison to males.
- TBI alters estrous cyclicity with prolonged low hormonal diestrus over 28 days post-injury.
The global burden of traumatic brain injury (TBI) is estimated at 69 million cases each year (1). Mild TBI makes up 75% of all reported TBIs, commonly associated with falls, assault, sports, and military activities (2). After mild TBI, patients are at higher risk for developing long-lasting neurological morbidities that have detrimental effects on functional ability (3). Persisting post-TBI symptoms include headaches, cognitive disabilities (4), sensory deficits (5), sexual dysfunction (6), mood and anxiety disorders (7, 8), and increases the risk for second brain injuries (9). Specifically, symptoms associated with cognition (memory, executive function, and processing speed), emotional processing (mood and social), and sensory perception (vision and auditory) (10) can hamper recovery. Reports of complaints associated with post-TBI symptoms vary widely in mild TBI literature (11, 12). In recent publications from the UPFRONT studies, 85–90% of mild TBI patients had complaints at 2-weeks post-injury (13, 14). When evaluating the 10% of patients that did not have complaints at 2-weeks post-injury, over 50% developed complaints over the following 12 months (14). Overall, more complaints were in mild TBI patients with a psychiatric history with more complaints in females compared to males. The De Koning study highlights the prevalence for late-onset symptoms, for which few preclinical models exist.
One-third of TBI patients each year in the United States are women, where reports support or refute whether sex differences influence severity and duration of chronic post-TBI symptoms, with a greater amount of data supporting sex differences (15–18). In particular, a largely overlooked population are victims of domestic violence who are predominantly women and children (19). Reports indicate that women of reproductive age are more likely than men to report severe persisting post-TBI symptoms (5, 20) and data suggest that they sustain significantly higher TBI rates (21). The number of women patients receiving health care from Veterans Affairs medical centers has also substantially increased over the past years (22). Moreover, clinical observations suggest many female TBI survivors experience menstrual problems such as amenorrhea, irregular cycles, infertility, and postpartum complications (23–25). Among the few known physiological factors, fluctuating sex hormone levels have been indicated in the disparity of chronic symptoms associated with persisting post-TBI symptoms in females, with injury during the luteal phase of the menstrual cycle associated with worse outcomes (26). Clinically and translationally, TBI-induced chronic ovarian hormonal deficiencies have been shown to contribute to behavioral deficits (27, 28). Further, TBI prolongs the low circulating ovarian hormone phase that is associated with cognitive and sensorimotor deficits in rodents (29). Since brain injury is characterized by a wide heterogeneity in pathophysiological mechanisms, evaluating for variability across the sexes may be crucial for patient stratification and treatment as well as translationally relevant study designs.
Mild TBI initiates shearing forces leading to diffuse axonal injury (DAI), synaptic deafferentation, vascular permeability, and inflammation that progress toward dysfunction of neural circuitry that can be replicated, in part, in experimental models of TBI using midline fluid percussion injury (FPI) (30). DAI initiates a long-term process that elicits both degenerative and regenerative (dendritic and synaptic sprouting) responses (31). Abnormal pathophysiology drives maladaptive compensation in neurotransmitter systems of long-range projections relevant to post-TBI symptoms [reviewed in (32)]. The somatosensory thalamocortical projections of the whisker barrel circuit (WBC) in rodents are essential components for sensory processing in rats (33). Within this circuitry, glutamatergic input from the principal trigeminal nucleus (PrV) projects to the contralateral ventral posteromedial nucleus of the thalamus (VPM) that extends long range glutamatergic projections to the primary somatosensory cortex (S1BF). Midline FPI causes pathological alterations along the whisker circuit that are characterized by changes in glutamate neurotransmission, axotomy, dendritic atrophy, circuit reorganization, and gliosis that parallel the development of late-onset sensory hypersensitivity in male rats after TBI (34–38), similar to agitation presented in human TBI (39). At 1-week post-injury, rats exposed to FPI did not exhibit sensitivity to whisker stimulation (40), similar to clinical reports in the De Koning study, where symptoms developed over time (14).
One elegant approach to studying the circuit function is taking advantage of biosensor technology to assess neurotransmission responsible for the development of chronic morbidities after TBI (41). Our previous work in male brain-injured animals demonstrated hypersensitive glutamate signaling associated with the severity of late-onset hypersensitivity to whisker stimulation as a consequence of pre-synaptic glutamate release (40). Here, we replicate previous studies in males with the inclusion of systematic estrous cycle assessment in naturally cycling female rats to determine if sex plays an intrinsic role in TBI-induced sensory hypersensitivity associated with changes in glutamate neurotransmission.
1,3 phenylenediamine (mPD 99%, cat. no. 78450, Acros Organics, NJ), bovine serum albumin (BSA, cat. no. A3059), glutaraldehyde, L-ascorbic acid (≥99%, cat. no. A5960), L-glutamic acid (≥99%, HPLC grade, cat. no. G1626) and L-Glutamate oxidase from Streptomyces sp. with rated activity of ≥10 U mg−1 (Lowry's method) (cat. no. G59211UN) were purchased from Millipore Sigma. Phosphate-buffered saline (PBS, pH 7.4) was composed of sodium phosphate dibasic (Na2HPO4, cat. no. BP332), sodium phosphate (NaHPO HO, cat. no. BP330), and sodium chloride (NaCl, cat. no. S271). Ultrapure water, generated using a Millipore Milli-Q Water Purification System, was used for preparation of all solutions used in this work.
Young adult male and naturally cycling female 3–4 month old Sprague-Dawley rats (330–350 g and 210–230 g, respectively, at the time of surgery; Envigo, Indianapolis, IN) were same-sex pair housed (2 animals/cage) and allowed 1-week of acclimation to a normal 12 h light/dark cycle with access to food (Teklad 2918, Envigo, Indianapolis, IN) and water ad libitum. All study protocols were approved by the Institutional Animal Care and Use Committee, University of Arizona College of Medicine—Phoenix (Protocol No. 18-384) and were conducted in adherence to guidelines established by the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals. Based on our previous reports in males (34, 40), group sizes of 14 rats are sufficient to detect a three-point change in whisker nuisance scores between groups with 90% power (2-tailed; μ1 = 3.8, μ2 = 6.8, σ = 2.5; significance level P = 0.05).
Following acclimation to the vivarium for 1 week, all rats were habituated to human handling for 5 days prior to surgery. Cages of rats were randomized into either injured (midline FPI) or sham surgery groups. We followed our standard midline FPI surgery protocol as previously described (42). In brief, rat surgeries were performed under isoflurane anesthesia (5% induction and 2.5% maintenance vaporized in 100% O2 with flow rate of 0.8 L/min via nosecone) while being secured on a stereotaxic frame (Kopf Instruments, Tujunga, CA) and absent toe pinch response. The body temperature was maintained at 37°C throughout the surgical period using a thermostatic heating pad. Ophthalmic ointment was applied to the eyes to prevent drying (07-888-2572, Patterson Veterinary, CO). The scalp was shaved and cleaned with alternating betadine and ethanol scrubs. A 4.8 mm diameter circular craniectomy was centered on the sagittal suture midway between bregma and lambda using a trephine, ensuring that the underlying dura, and superior sagittal sinus were not damaged. An injury hub, using the female portion of a 20-gauge Luer-Lock needle hub (cut and beveled), was placed directly above and in-line with the craniectomy site. A stainless-steel anchoring screw was then placed in the right frontal bone using a hand-drill. The injury hub was affixed over the craniectomy using cyanoacrylate gel and dental cement (Hygenic Corp., Akron, OH). After the dental cement hardened, the hub was filled with 0.9% sterile saline. The incision was then partially sutured closed on both the anterior and posterior edges with 4.0 Ethilon suture (Med-Vet International, Mettawa, IL) topical lidocaine and antibiotic ointment were applied. Rats were returned to a warmed holding cage and monitored until ambulatory (~60–90 min).
After cranial surgery, rats were allowed to recover for ~2 h in the recovery chamber during which they were observed for ambulatory movement and any signs of ill health. After ensuring successful surgery, rats were re-anesthetized using 5% isoflurane in 100% oxygen for 3 min. The injury hub was filled with 0.9% sterile saline and attached to the fluid percussion device (Custom Design and Fabrication, Virginia Commonwealth University, Richmond, VA). At the return of the toe pinch withdrawal reflex, a moderate fluid pulse [(in atm): Males, 1.9–2.1 and Females, 1.8–2.0] was administered by releasing the pendulum (16.5 degrees for males and 16 degrees for females) onto the fluid-filled cylinder. Immediately after administration of the injury, the hub was removed en bloc, rats placed on the heated recovery chamber, and monitored for presence of apnea time, fencing response and return of righting reflex. The rats were then re-anesthetized to inspect for signs of hematoma and herniation at the site of injury. The surgical wound was cleaned, stapled closed, and topical lidocaine and antibiotic ointment were applied at the surgical site. Inclusion criteria required that injured rats have a righting reflex time ranging from 5 to 9 min and display a fencing response (43). After regaining the righting reflex, rats were placed in a holding cage for ~1 h until regaining normal ambulatory behavior before being returned to standard vivarium conditions. Sham animals underwent identical procedures without dropping the pendulum to induce the injury. Adequate post-operative care was administered for 3–5 days after surgery where all the animals were assessed for body weight changes, physical examination, suture site, and pain or distress using a standardized protocol. Rats that lost more than 20% body weight or scored poorly during this post-operative time were euthanized (<10%). All rats included in this study had (i) same-sex cagemate, (ii) same-injury cagemate, (iii) a righting reflex between 5 and 9 min (FPI only), and (iv) a visible fencing response (FPI only).
Estrous cycle was tracked using daily vaginal lavage with sterile saline beginning on the day of handling (7 days prior to surgery) and ending at 28 days post-injury (DPI) to determine the estrous cyclicity (Figure 1). Cycle tracking was done by an investigator blinded to injury status. Three hundred microliter of sterile saline was gently pipetted into and out of the vagina and smeared onto a glass microscope slide. Estrous stage was immediately determined under the microscope (Nikon, 77007) fitted with a 20× objective and classified according to previously reported methods (44). Briefly, the metestrus was characterized by the appearance of small leukocytes with equally round nucleated epithelial cells, whereas, the diestrus had fewer leukocytes along with cornified epithelia. The smears from proestrus rats had predominantly round nucleated cells. In the estrus phase, non-nucleated cornified epithelial cells were predominant. Estrous cycles were tracked daily, between 07:00 a.m. and 12:00 p.m. to minimize variability due to diurnal variations. Male rats were habituated to handling daily. Given that on a daily basis female rats normally fall in one of the different stages of the estrous cycle, all female rats underwent either sham or midline FPI (as described above) regardless of their estrous phase.
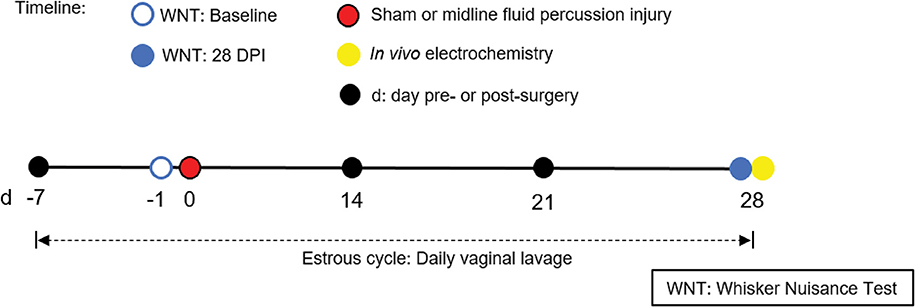
Figure 1. Timeline representing experimental design. Following acclimatization, young adult male and naturally cycling intact female Sprague-Dawley rats were habituated to handling. In females, estrous cycle was tracked daily, from 7 days pre-injury to 28 days post-injury. At day -1 (day before surgery), sham or midline FPI rats were tested with a baseline whisker nuisance task (WNT), where rats scoring >3 were excluded from the experiment. Then, cages of age-matched male and female rats were randomized to either sham surgery or midline fluid percussion injury (FPI). The WNT was performed again on day 28 post-injury. Rats were anesthetized 30 min following the final behavioral testing for in vivo amperometric recordings. Immediately following recordings, animals were decapitated and brains were cryosectioned to confirm microelectrode array (MEA) placement.
Behavioral response to whisker stimulation was assessed by whisker nuisance task (WNT), as described previously (34) at baseline (on the day before surgery) and 28 days post-injury by an investigator blinded to injury status. The test identifies sensory hypersensitivity when whiskers are manually stimulated in an open field (45). Briefly, each rat was individually placed in the middle of an open arena (16.5 × 38.1 × 60 cm, 7,362-cm2) and permitted free exploration of the chamber during which their whiskers of both mystacial pads were manually stimulated with a wooden applicator stick for 15 min with three consecutive 5-min periods and less than ≤ 60 s non-stimulation break between each period. For each 5 min period, the behavioral responses including movement, stance and body position, breathing, whisker position, whisking response, evading stimulation, response to stick presentation and grooming were assigned subjective behavioral scores on 0–2 point non-parametric scales. White noise of 70 dB was present at the testing times to mask external noise. Data collection: The primary method for data collection of behavioral phenotypes was evaluation and scoring using the published whisker nuisance task scoring sheet (34). A score of (0) indicated the rat showed no response or “absent” indicating normal response; (1) denoted the rat showed a minor degree of expression or “present”; and (2) the rat showed major degree of expression or “profound” exemplified with above behaviors indicating abnormal responses. The total scores were averaged across three 5-min stimulation bouts. The maximum whisker nuisance score was 16, with higher scores indicating multiple abnormal behavioral responses. From their homecages, rats were placed individually into another transport cage and moved to the separate behavioral testing room located within the same Laboratory Animal Care Facility and then returned to their home cages at the end of testing. The experimenter made minimal movements and no noise when collecting the behavioral data and used a timer to determine the testing period. To avoid the possibility of different experience or stressor condition, all rats spent brief, consistent time outside their homecages, during transport and in the behavioral testing suite to ensure they underwent similar experiences. Testing was conducted at the same time of day by the same observer, who was blinded to injury status. Animals were excluded if they had high (>3) basal whisker nuisance scores tested on the day before surgery.
Ceramic-based microelectrode arrays (MEAs) encompassing four platinum (Pt) recording surfaces (15 μm × 333 μm) aligned in a dual, paired configuration were prepared to measure glutamate for in vivo anesthetized recordings (S2 configuration; Quanteon, Nicholasville, KY). MEAs were fabricated for sensitive and selective measurements of glutamate as previously described (42, 46). The MEAs use glutamate oxidase (GluOx) to catalyze the oxidative deamination of glutamate leading to formation of hydrogen peroxide (H2O2) as a by-product. Electro-oxidation (at a potential of +0.7 V) of enzymatically generated H2O2 at the surface of the electrode generated the current output which was recorded and converted into glutamate concentration via an in vitro calibration factor. Further, interference from other electroactive neurotransmitters was excluded from the amperometric recordings by application of mPD coating to the electrode sites (40).
Briefly, a solution of GluOx, bovine serum albumin (BSA), and glutaraldehyde (GA) was immobilized by chemical cross-linking onto two of the Pt electrode recording sites, enabling these sites to selectively detect glutamate levels with low limits of detection (42). The two sentinel channels were coated with only BSA and glutaraldehyde, recording everything except for glutamate (47). Enzyme immobilization was accomplished by chemical cross-linking using a solution of GluOx (400 U/ml), BSA (6 mg/ml), and GA (0.075%). A needle attached to a 10 μL Hamilton syringe tip was used to coat MEAs under a stereomicroscope with a ~1 μL drop of the solution. The recording channels were carefully coated 3–4 times (allowing drying between each coat) with the solution droplet. The sentinel channels were coated with a solution that did not contain GluOx (BSA-GA). MEAs were cured for at least 72 h prior to use. One day prior to recordings, all four Pt recording sites on the MEAs were electroplated with a size exclusion layer of mPD. Representative schematic of MEA coating can be found in Figure 3A (left).
Electrochemical preparation of the MEAs was performed using a multichannel Potentiostat (model VMP3). In vitro and in vivo measurements were conducted using a multichannel FAST16 mk-IV system (Fast Analytical Sensor Technology Mark IV, Quanteon, LLC, Nicholasville, KY) with reference electrodes consisting of a glass enclosed Ag/AgCl wire.
On the day of amperometric recording, each MEA was calibrated in vitro to determine slope (nA/μM; sensitivity to glutamate), limit of detection (μM; lowest amount of glutamate reliably recorded), and selectivity (ratio of glutamate to ascorbic acid). For calibration, a constant potential (+0.7 V) was applied to the MEA against an Ag/AgCl reference in 40 ml of stirred 0.05 M PBS (pH 7.1–7.4; 37°C) in a beaker. After the current detected by the MEAs equilibrated to baseline (~20 min), 500 μL of ascorbic acid was added to the beaker to assess a readily oxidizable potential interferent that is in high concentration within the brain. This was followed by three subsequent additions of 40 μL of L-glutamate (20 μM) to confirm selectivity for glutamate and provide the slope of change in current as a function of changes in glutamate concentration. Last, 40 μL of H2O2 (8.8 μM) was also added to the beaker solution to test the sensitivity of the MEAs to the reporter molecule, peroxide. The final concentration consisted of (in μM): 250 ascorbic acid, 20, 40, and 60 glutamate, and 8.8 H2O2. In the present study, the average slope was 4.4 pA/μM, LOD 2.62 μM and selectivity 50.7–1. Figure 3A (right) depicts a representative MEA calibration.
For recordings in anesthetized rats, a glass micropipette was attached to the MEA for the local application of solutions. A single-barrel glass micropipette (1.0 × 0.58 mm2, 6″ A-M Systems, Inc., Sequim, WA) was pulled using a Kopf Pipette Puller (David Kopf Instruments, Tujunga, CA). Using a microscope with an eyepiece reticle, the pulled micropipette was bumped against a glass rod to have an inner diameter of 7–13 μm (10.5 μm ± 0.2). Clay was used to place the tip of the micropipette equidistant between the four Pt recording sites. The assembly was secured using Sticky Wax (Kerr Manufacturing Co.) and while the wax was still soft the micropipette was adjusted such that its tip was within 65 ± 6 μm from the surface of MEA. The assembly was allowed to cure for ~10–15 min and rechecked for distance before use in experiments.
Immediately after behavioral assessment, sham and FPI rats were anesthetized (1.5 g/kg urethane, i.p.). Following cessation of a toe pinch withdrawal reflex, each rat was then placed in a stereotaxic frame (David Kopf Instruments) with terminal ear bars. Body temperature was maintained at 37°C with Deltaphase® isothermal pads (Braintree Scientific, Inc., Braintree, MA). A midline incision was made, and the skin, fascia, and temporal muscles were reflected to expose the skull. A bilateral craniectomy exposed the stereotaxic coordinates for the S1BF and VPM. Dura was then removed prior to the implantation of the MEA. Brain tissue was kept moist through the application of saline soaked cotton balls and gauze. Finally, using blunt dissection, a 200 μM diameter Ag/AgCl reference electrode was placed in a subcutaneous pocket site remote from the recording areas.
Amperometric recordings performed here were made similar to published methods (40, 42, 46). Solutions of either (in mM): KCl (120), NaCl (29), CaCl2 (2.5) in ddH2O, pH (7.2–7.5) or L-glutamate (in μM) [L-glutamate (100) in 0.9% sterile saline, pH 7.2–7.6] were filtered through a 0.20 μm sterile syringe filter (Sarstedt AG & Co., Numbrecht, Germany) and loaded into the affixed single-barrel glass micropipette using a 4-inch, 30- gauge stainless steel needle with a beveled tip (Popper and Son, Inc., NY). The open end of the single-barrel glass micropipette was connected to a Picospritzer III (Parker-Hannifin Corp., Mayfield Heights, OH). Solutions were locally applied from the glass micropipette with settings to dispense nanoliter (nL) quantities over a 1 s time period using the necessary pressure of nitrogen (inert) gas. The volume ejected was monitored using a stereomicroscope (Meiji Techno, San Jose, CA) fitted with a calibrated reticle. In vivo recordings were performed at an applied potential of +0.7 V compared to the Ag/AgCl reference electrode. All data were recorded at a frequency of 40 Hz, amplified by the headstage (2 pA/mV) without signal processing or filtering of the data. Glutamate and KCl-evoked measures were recorded in both hemispheres in a randomized and balanced experimental design to mitigate possible hemispheric variations or effect of anesthesia duration by investigators blinded to the injury status. The amperometric recordings were collected from multiple independent cohorts on consecutive days that contained both male and female rats from sham and FPI groups.
Using the Dual Precise Small Animal Stereotaxic Frame (Kopf, Model 962), the MEA assembly was slowly vertically lowered at 0.3 mm steps from the dorsal site. The MEA-micropipette assembly was lowered through the S1BF for males [from bregma (in mm): anteroposterior, ±2.3; mediolateral, ±5.0; dorsoventral, −1.1 to −2.1)] and females [from bregma (in mm): anteroposterior, ±2.2; mediolateral, ±5.0; dorsoventral, −0.8 to −2.0)]. The MEA-micropipette assembly was lowered through VPM for males [from bregma (in mm): anteroposterior, ±3.5; mediolateral, ±2.68; dorsoventral, −4.3 to −6.2)] and females [from bregma (in mm): anteroposterior, ±2.3; mediolateral, ±2.68; dorsoventral, −4.0 to −5.8)] (48).
Once the electrochemical signal had reached baseline, 120 mM KCl was locally applied to produce an evoked glutamate overflow. Additional ejections of KCl were completed at 2-min intervals. Criteria for analysis required that the peak represent the maximum amount of glutamate overflow within the surrounding neuronal tissue, this is confirmed by smaller peak amplitudes from consecutive KCl ejections. Primary outcome measure was peak amplitude (μM) taken as the absolute height of the recorded peak.
Once the baseline was reached and maintained for at least 10 min, 100 μM glutamate was locally applied into the extracellular space. Exogenous glutamate was released at 30 s intervals. In analysis, up to three peaks (with <10% variability) were selected based on a predetermined amplitude range of 8 to 18 μM to maintain similar Michaelis-Menten clearance kinetics. The parameters for the three peaks were then averaged to create a single representative value per recorded region per rat. Outcome measures analyzed included the uptake rate constant (k−1) measured as the first order decay rate of the glutamate signal (sec−1) and T80 duration (seconds) calculated as the time taken for 80% of the maximum amplitude of glutamate to clear the extracellular space. The uptake rate can be calculated using the uptake rate constant (k−1) multiplied by the peak's maximum amplitude, which normalizes for small variations (data not shown). We are presenting k−1 data that represent a similar outcome as the uptake rate due to amplitude matching. For a diagrammatic representation of these calculations, see Figure 2.
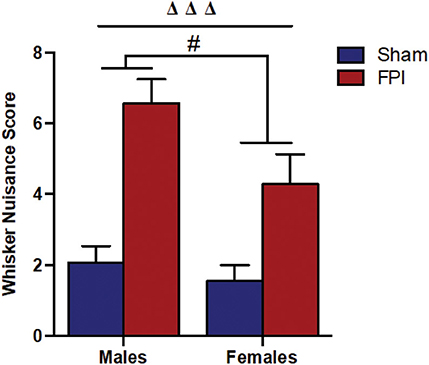
Figure 2. Comparison of whisker nuisance scores in WNT among male (Left) and female (Right) rats, subjected to either sham or midline fluid percussion injury (FPI). Data were collected during three 5-min time bins across a 15-min testing session in an open box. Two-way ANOVA [(injury: sham vs. FPI) and (sex: male vs. female)] where adjusted WNS (WNS + 1) was log-transformed to remove skewness. Sham: male, N = 12 rats; FPI: male, N = 17 rats; sham: female, N = 16 rats and FPI: female, N = 16 rats. Data expressed as mean ± SEM on original scale. ΔΔΔP < 0.001 compared to sham animals (overall FPI effect) and #P < 0.05 compared to males (overall sex effect).
Immediately following in vivo anesthetized recordings, rats were decapitated, brains post-fixed with 4% paraformaldehyde in PBS, and cryoprotected in serial solutions of 15 and 30% sucrose in Tris-buffered saline. Later, brains were sectioned at 40 μm to confirm MEA placement (see Figure S1).
Whisker nuisance score (WNS) is from 0 to 16. Electrochemical data were organized using Microsoft Excel (version 16.39). All outcome measures, unless otherwise stated, were averaged from multiple depths to create a single representative value for each rat prior to statistical analysis. Distribution of WNS and all electrochemical outcomes were evaluated for variability, and logarithmic transformation was applied to remove skewness for statistical methods. For log-transformation of the WNS, we adjusted the score by adding 1 to each reading since the lowest score was 0. Two-way ANOVA was used to compare each outcome measure between male and female rats and between FPI and sham groups. When there was evidence of an interaction between these two sets of factors, the FPI effect was reported separately for males and females; when there was no evidence of an interaction, FPI effect was reported for male and female rats combined, and sex difference was reported for FPI and sham rats combined. Linear mixed effects model was used to analyze data with multiple measurements from the same animal (e.g., electrochemical measurements at multiple depths). Spearman's rank correlation was employed for correlation analysis between WNS and evoked glutamate overflow or uptake rate constant (k−1) to assess whether the severity of hypersensitivity to the WNT was correlated with glutamate neurotransmission. This was performed separately for FPI male and FPI female rats. The estrous cycle for females was tracked daily for the entire duration of the study. Estrous cycle data are represented as percentage days spent in each phase over 28 days post-injury analyzed by two-way ANOVA followed by Fischer's comparisons. Significant changes in the time spent in each phase were a posteriori analyzed as a function of time (binned weekly) to determine whether the test was picking up acute or chronic changes. Graph Pad Prism (version 8.0) or R (version 3.5.3) were used to create graphs and perform statistical analyses. All tests were two-sided and P-levels < 0.05 were considered to be statistically significant for all tests except indicated. Data were graphically represented as mean ± standard error of the mean (SEM), regardless of any transformation performed in the statistical tests.
Injury characteristics including apnea, fencing response and righting reflex times were monitored in all rats immediately after injury as indicators of TBI severity. Apnea times were determined from injury to the return of spontaneous breathing. Righting reflex time was noted as the time of injury until return of an upright position. Baseline body weight for males was ~34% higher in comparison to female rats at the time of surgery. Due to differences in body weights, females received a marginally lower (6%) FPI impact with atmospheric pressure ranging from 1.9 to 2.1 for males and 1.8 to 2.0 for females. The weighted pendulum arm was adjusted for female sex to produce a righting reflex time of 5–9 min and survival rates of matched male rats based on previous publications (49, 50) and procedures previously established in our research program indicating less injury force in females. The average righting reflex times, similar among the brain-injured male and female rats, were 389.3 ± 17.08 and 411.0 ± 25.46 s, respectively (see Table S1).
It is understood that TBI impairs sensory processing contributing to behavioral morbidity (51–53). Previously, we reported late-onset post-TBI sensory hypersensitivity to whisker stimulation in male rats (40). The WNT serves as a useful test to measure late-onset sensory hypersensitivity associated with impaired sensory processing after brain injury (34). Two-way ANOVA showed that there was no evidence of interaction between injury and sex (P = 0.46), indicating that male and female rats shared a similar injury effect. In the absence of interaction effects, median adjusted WNS (WNS + 1) showed FPI rats had higher (127%) scores compared to sham rats [95% confidence interval (CI): 66.4–210%, P < 0.0001] and the overall scores were lower (31.8%) in females compared to males (sex effect) (95% CI: 7.0–50.0%, P = 0.019; Figure 2).
Vaginal cytology was used to track the four stages of the estrous cycle in all females. Table 1 presents a summary of the effects of sham and FPI on percentage of days female rats spent in each phase of estrous out of the 28 days post-injury (28 DPI). The two-way ANOVA revealed a significant injury × phase interaction (P = 0.012). The follow-up comparisons indicated that FPI females spent significantly more time in diestrus (P = 0.02) and less time in the estrus (P = 0.002). However, no significant differences were observed after FPI on number of days in proestrus and metestrus. An a posteriori assessment of diestrus and estrus over weeks post-injury in female rats was carried out to identify if the 28-day assessment was influenced by FPI (see Figures S2A, S2B). In the case of diestrus, a two-way ANOVA (week post-injury × injury; on log-transformed measurements) revealed a significant effect of injury (P = 0.008) with increased number of days in diestrus among FPI females, but not as a function of weeks (P = 0.452). There was a reduction in number of days in estrus as a function of weeks that approached significance as a function of injury (P = 0.053), but not as a function of weeks (P = 0.338). Thus, changes in the estrous cycle after FPI were not skewed by earlier time points. The key point is that FPI-induced chronic changes to the estrous cycle, similar to clinical observations (24) and capable of influencing long-term changes in circulating gonadal hormones.
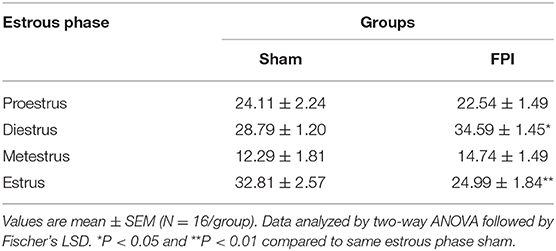
Table 1. Percentage of days female rats spent in each phase of estrous over the 28 DPI for sham or midline fluid percussion injury (FPI).
Isotonic KCl solution (120 mM) was locally applied to depolarize the synaptic microenvironment to assess glutamate stores. As shown in Figure 3B, two-way ANOVA (on log-transformed measurements) showed marginally significant evidence of interaction between injury and sex (P = 0.091), so FPI effects were reported separately for males and females. The follow-up comparisons indicated that FPI significantly enhanced evoked glutamate overflow in S1BF among male rats (median 82.6% higher, 95% CI 6.8–212%, P = 0.034), whereas FPI had no effect in female rats (P = 0.84). While depth profile (dorsal-ventral axis) of the S1BF showed increased glutamate overflow in FPI males (median 132% higher, 95% CI 20–347%; P = 0.012) in comparison to sham males (Figure 3C), no such effect was observable in females (FPI females vs. sham females) (Figure 3D). In VPM, the two-way ANOVA revealed a significant main effect of injury (P = 0.005), such that FPI significantly increased evoked glutamate overflow in male and female rats when compared to sham (see Figure 3E).
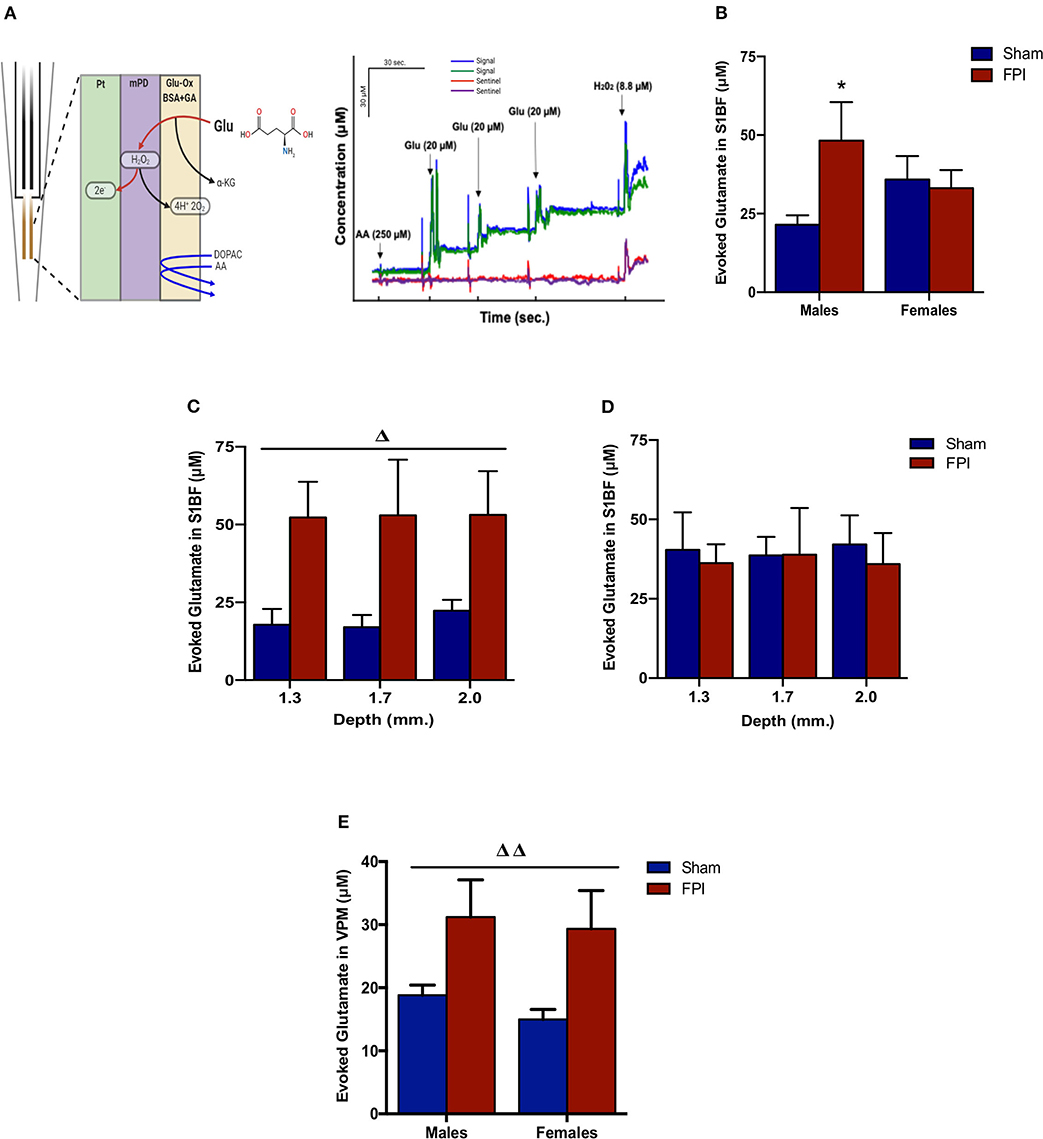
Figure 3. Evoked glutamate overflow in brain-injured circuitry among male and female rats subjected to either sham or midline fluid percussion injury (FPI). (A) Representative schematic of MEA coatings (left) and in vitro calibration (right). The ceramic-based MEA is equipped with four separate platinum (Pt) recording sites, where coatings are applied to the front sites (green and blue) to make the MEA selective to glutamate detection. In 3A (right), arrows represent aliquots of solution of either 250 μM ascorbic acid (AA), 20 μM glutamate (Glu), or 8.8 μM H2O2. (B) KCl-evoked glutamate overflow analyzed at 28 days post-injury by enzyme-based MEAs coupled with amperometry in the barrel fields of S1BF of male and female rats subjected to either sham or midline fluid percussion injury (FPI). (C) The depth profile of evoked glutamate overflow in the S1BF of male rats. Linear mixed effects model was applied to log-transformed measurements. (D) The depth profile of evoked glutamate overflow in the S1BF of female rats. (E) Evoked glutamate overflow in the VPM of male and female rats. Two-way ANOVA (injury and sex) applied to the log-transformed measurements to remove skewness. Data expressed as mean ± SEM on original scale. ΔP < 0.05 and ΔΔP < 0.01 compared to sham rats (overall injury effect) and *P < 0.05 compared to sex-matched sham. Figure 3A (left) created with BioRender.com.
Exogenous glutamate was locally applied to the extracellular space of the VPM and S1BF to evaluate glutamate clearance kinetics. Glutamate transporters regulate extracellular neurotransmitter levels based on Michaelis-Menten kinetics (54), so peaks were amplitude matched for analysis of glutamate clearance parameters. A sex-specific influence was observed with glutamate clearance within the thalamocortical circuit (representative signals in Figure 4A). For the glutamate uptake rate constant within the S1BF, there was no significant interaction between injury (sham vs. FPI) (P = 0.36), indicating that the uptake rate constant in males and females was not influenced by FPI. In contrast, as shown in Figure 4B, a significant sex effect was evident (P = 0.002), with females showing a lower glutamate uptake rate constant. As depicted in Figure 4C, while there was no evidence of a significant interaction between injury and sex for the T80 (P = 0.43), the T80 varied as a function of sex (P = 0.037), such that glutamate took longer to clear from the extracellular space in females in comparison to their male counterparts. However, there was no evidence of an FPI effect on T80 (P = 0.68). Likewise, the glutamate uptake rate constant in the VPM was also influenced by sex, with females having a lower uptake rate constant in comparison to males (P = 0.002; see Figure 5A), that was not altered by FPI (P = 0.36). Finally, there was neither FPI (P = 0.85) nor sex (P = 0.34) effect on the T80 (Figure 5B). These results are indicative of sex-dependent alterations in glutamate clearance as a function of sex, not injury. For additional validation, results of all electrochemical parameters were tested for rank order correlation with the corresponding WNS. While the increase in evoked overflow after TBI replicated previous experiments, the correlation between evoked glutamate overflow and severity of WNS was only significant (P = 0.01) for sham males in the S1BF. Further, no significant relationships were found in FPI animals (see Table S2).
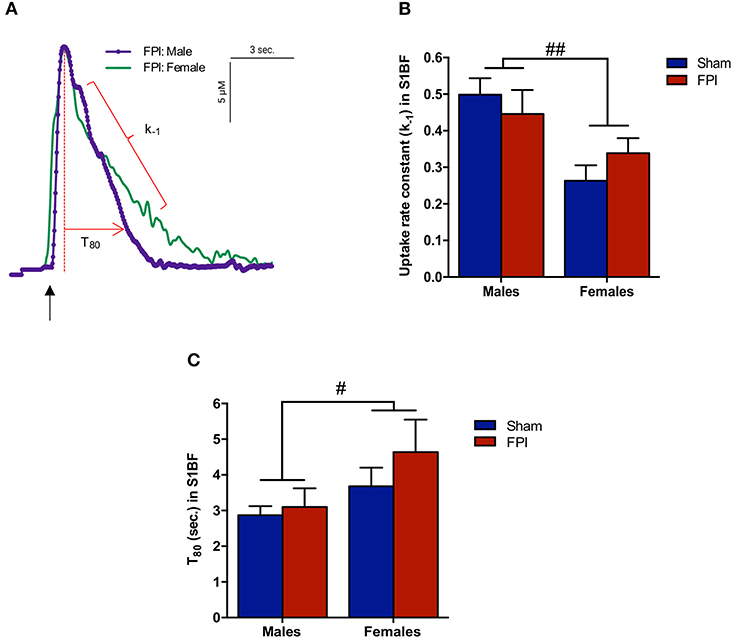
Figure 4. Extracellular glutamate clearance parameters among male and female rats subjected to either sham or midline fluid percussion injury (FPI). (A) Representative signals of extracellular glutamate clearance following local applications of glutamate through a micropipette in the S1BF (blue; males and green; females). An arrow represents application of 100 μM glutamate. The amplitude (μM) is calculated as the peak amplitude of the transient. Uptake rate constant (k−1) is calculated as the linear fit of the first order decay of the glutamate signal (s−1). T80 (sec.) is the time for the signal to decay 80% from the peak amplitude. (B) Extracellular glutamate uptake rate constant (k−1) in the S1BF of male and female rats subjected to either sham or midline fluid percussion injury (FPI). (C) T80 clearance time in S1BF of male and female rats at 28 days post-injury. Two-way ANOVA applied to the log-transformed measurements to remove skewness. Data represented as mean ± SEM. #P < 0.05 and ##P < 0.01 compared to male rats.
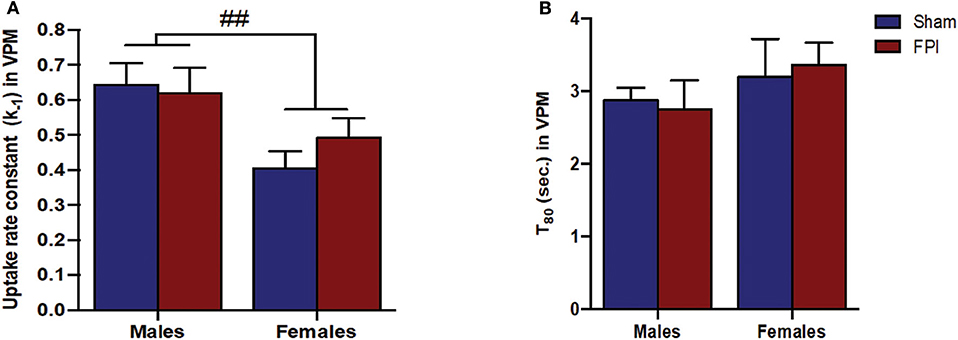
Figure 5. (A) The glutamate uptake rate constant (k−1) in the VPM of male and female rats subjected to either sham or midline fluid percussion injury (FPI). Data were analyzed by two-way ANOVA. (B) T80 clearance time (sec.) of glutamate in the VPM of male and female rats subjected to either sham or midline FPI. Two-way ANOVA applied to the log-transformed measurements to remove skewness. Data expressed as mean ± SEM. ##P < 0.01 compared to male rats.
Poor long-term outcomes after TBI are frequently associated with the number and severity of chronic post-TBI symptoms, with clinical data indicating that sex could influence such outcomes (55–57). Yet, studies focusing on post-traumatic circuit disruption and repair in naturally cycling females have received little attention. Our present findings support that differential manifestations of post-traumatic symptoms can be, in part, related to sex. We found TBI-induced sex-dependent changes in glutamate signaling, in terms of release and uptake profiles, associated with sensory hypersensitivity to whisker stimulation. Although the precise factors responsible for sex-dependent changes were not addressed in these experiments, they may be related, in part, to sex hormones. Additional studies are warranted to further investigate the role of specific hormones and receptor mediated events to draw more conclusive evidence in their control of neurotransmission and response to repair and compensation over time following diffuse brain injury. These results shed light on sex-dependent influences on glutamate neurotransmission and associated late-onset sensory hypersensitivity with and without TBI.
Axons that survive the diffuse insult (adjacent to damaged axons) can impair circuit function creating maladaptive neuronal circuitry and activation, axonal damage, altered signaling cascades, and synaptic dysfunction (35, 58, 59). Impaired circuit function in the whisker barrel disrupts somatosensory processing through the whiskers of rats, a primary sensory modality, similar to vision in humans, and is implicated in the agitated response displayed during whisker stimulation (52, 60). In agreement with previous findings (34, 40), we found that TBI induced sensory hypersensitivity to whisker stimulation in both sexes, however, females displayed an overall lower magnitude of sensory hypersensitivity than males. The whisker barrel circuit is structurally well-organized with somatotopic arrangement that, while being a simple circuit, mediates complex behaviors (61). Our research and others demonstrate that late-onset sensory hypersensitivity after FPI parallels neuropathology, neuroinflammation, gliosis, circuit reorganization, changes in electrophysiological activity, and neurotransmission indicating a number of ongoing sequelae of events in the relays of the whisker barrel circuit leading up to the development of this aberrant behavioral phenotype (36, 38, 62–65). The sensory hypersensitivity to stimulation is indicative of a sensory gating defect after TBI that manifests as a defensive and apprehensive phenotype to tactile stimuli (66). Moreover, the apprehensive behaviors observed here after experimental TBI are similar to the hypersensitivity to visual stimuli, agitation, irritability, and hyperarousal reported in male and female TBI patients (39, 60, 67). Our results lend further credence to the whisker barrel circuit representing a useful somatosensory model to elucidate circuit disruption and repair that causes abnormalities in sensory processing after TBI (68), however, further pathological assessment in the whisker barrel circuit of female rats may highlight important sex differences.
TBI has been reported to cause hypothalamic-pituitary-gonadal (HPG) axis dysfunction and endocrinopathy that can impact the menstrual cycle in women (69–71). In these experiments, we evaluated the estrous cycle for 28 days after injury or sham surgery. Our data indicate that FPI females spend more time in the diestrus phase (low estrogen and high progesterone levels) and less time in the estrus phase over 28 days post-injury. When breaking this down to a week-by-week a posteriori analysis, the differences are a function of injury and not time, indicating that by 1 month post-injury normal cycling had not resumed (Figure S2). Previous TBI reports in clinical and rodent studies have also showed disruption of normal hormonal cyclicity (25, 29). Prolonged diestrus indicates longer secretion of progesterone with low levels of luteinizing hormone (LH) (72) that can interfere with ovulation. Additional clinical observations in women have reported abnormal menstrual pattern after TBI associated with amenorrhea (23) linked to variations in cortisol levels (73). Daily assessment of estrous cycle in these subjects was the optimal approach to link hormonal profiles of the cycle phases at any given time due to the repeated stress associated with blood draws that can downregulate neurotropic factors, including BDNF, that could mediate circuit repair after injury [reviewed by (74)]. Future studies directly evaluating the role of ovarian hormones in neurotransmission and neuroplasticity after TBI are necessary to further explain the sex-differences detected in these studies. Also, more detailed studies are required to test the influence of circulating hormones at the time of injury and at time of behavioral testing.
Age-matched naturally cycling females were used in these experiments to complement the history of FPI experiments using male Sprague-Dawley rats (300–350 g; 10–12 weeks old) with several studies of comparable behavioral, pathological, physiological, and molecular data available in males (40, 50, 75). At 10–12 weeks of age, rats are sexually mature, but have not reached social maturity, with literature estimating the translational relevance of this age to be late-adolescence/young adult (76, 77). In juvenile and aging TBI research, women and female rodents would not be actively cycling, thereby having lower circulating gonadal hormones levels, where post-menopausal women have been indicated to respond similar to males (78). However, an epidemiological study of TBI in pediatric patients indicates that endocrinopathies peak within 2 years of the initial TBI, were more prevalent in females, and were predominantly reported as precocious sexual development (79). In the geriatric population, non-survivors of TBI were more likely to be males (80). Together, these data indicate that sex-differences may be prevalent in all age groups after TBI, where severity and type of morbidities (and mortality) may change based on the sex and age at injury, warranting further investigation and inclusion of females cohorts across all age groups in translational studies. In fact, impaired circuit function can also contribute to gonadal hormone deficiency, recognized as an important consequence of TBI-induced hypothalamic-pituitary-gonadal (HPG) dysfunction and has translational implications for therapeutic strategies.
Females have several factors that can influence outcome measures, including (but not limited too) circulating gonadal hormones, thickness of skull, size of brain, smaller mean axonal diameter (similar to clinical reports), muscle mass, and metabolic processes (81–83). The presence of estrogen in females has been shown to be neuroprotective in most of the animal models of neurodegenerative diseases and genetic mechanisms that control for the sex differences may also influence the pathophysiology. For these experiments, in order to maintain similar righting reflex times (our primary inclusion factor) between males and females, the force of the injury was decreased, with the potential that the pathology is decreased. Another option is to hold the injury force constant with the potential to induce greater pathology. With the paucity of data available in sex differences, these sex-related factors should be considered in the context of the questions asked and approach toward the answers. At this time, inclusion of detailed methodology and transparency is necessary as we evaluate for specific mechanisms responsible for increasing reports of sex differences accumulating in the TBI literature.
Several studies have documented the sequelae of events associated with glutamate dysregulation after mild TBI [reviewed by (84)]. Our previous report indicated that TBI-induced hypersensitivity to whisker stimulation is correlated with alterations of thalamocortical glutamate activity (40) suggesting that the neural correlates of hypersensitivity to sensory stimuli can be associated with evoked neurotransmitter release. In males, we found evoked glutamate overflow was elevated in S1BF and VPM 1 month after TBI, similar to our previous reports (40), which parallels with damage and repair of corticothalamic projections over time post-injury (85). Further, our results showed that in females, elevated evoked glutamate overflow was restricted to the VPM, with no change in the S1BF. Extensive investigations on estrogen after ischemia reveal both neuroprotective and neurotoxic influences over neuroinflammation, apoptosis, growth factor regulation, vascular modulation, and excitotoxicity that could mediate differential outcomes in diffuse TBI [reviewed in (86, 87)]. More studies are needed to understand the role of sex hormones in influencing evoked-glutamate overflow.
Previous studies indicate that elevated evoked glutamate overflow after TBI in males was mediated presynaptically, and not due to changes in glutamate transporters at 28 DPI (40). We have previously reported changes in VPM neuron morphology over time, where there is an acute and subacute loss of processes followed by return of the number of processes by 28 DPI in male rats, that could contribute to changes in evoked glutamate overflow (38). There are several other mechanisms that could contribute to the loss of presynaptic homeostasis at 1 month post-injury that require further investigation. Elevated evoked-glutamate overflow in the VPM could be due to influence on the components of the neurotransmitter vesicle release machinery to increase glutamate release from the releasable pools of synaptic vesicles. Diffuse TBI has been shown to enhance synapse-specific complexin levels that function as a vesicle fusion clamp to regulate neurotransmitter release to enhance neuronal excitability (88). Hyperexcitability of dendrites has also been linked to altered expression of channel function and deafferentation (89, 90) suggesting that the sensory hypersensitivity and increased evoked glutamate overflow observed in the present study could arise from excessive circuit hyperexcitability after TBI. Further, cellular signaling pathways could also be chronically influenced, affecting glutamate release through synaptic changes in the glutamate receptors and/or voltage-gated calcium channels (84, 91). Moreover, presynaptic NMDA-type glutamate receptors (alpha amino- 3-hydroxy-5-methyl-4-isoxazole-propionic acid, AMPA) are also involved in neuroexcitatory activity that is initially activated by post-injury increases in glutamate that chronically augment neurotransmitter release in a feedback fashion (92, 93). Other factors that could play a role, but have not been investigated are; (i) anterograde microglial reactions to axonal injury (94) to release glutamate as a result of oxidative stress (95) or (ii) the dense white matter connectivity in the thalamus with glutamatergic bidirectional inputs from the S1BF and PrV that make the thalamus twice as vulnerable to DAI (96, 97). Primary inhibition to the whisker barrel circuit comes through the thalamic reticular nucleus, where controlled cortical impact injury has been indicated in the triggering of neuroinflammation and delayed reactive astrogliosis associated with the development of sleep disruption, indicating that GABAergic inhibition could be impaired to the whisker barrel circuit as well (98). These previous reports indicate a number of feasible approaches to further investigate the role of inhibitory: excitatory balance in the development of late-onset sensory hypersensitivity from whisker stimulation on the level of cells and circuits as well as a function of sex.
The lack of a TBI effect on glutamate clearance is consistent with our previous report (40). In addition, we presently observed a robust sex-difference, with female rats displaying lower glutamate k−1 in S1BF and VPM with increased T80 in S1BF, indicative of slower glutamate clearance in the female brain. However, T80 only increased in the S1BF, and not the VPM, indicating that while the rate of clearance is slower in females, the time glutamate spends in the extracellular space is the same in the VPM, supporting a change in how glutamate is being cleared from the extracellular space. The synaptic level and clearance of glutamate is primarily regulated by astrocytic sodium dependent excitatory amino acid transporters (EAATs) involving GLT1 and GLAST expressed by glia (99). We have previously reported that expression of glutamate transporters is not changed at 28 DPI in males within the whisker barrel circuit (40). An ex vivo radioactive uptake study reports that the female estrous cycle influences glutamate clearance (100). Although our data indicated TBI-induced changes in estrous cyclicity, there was no TBI effect on glutamate clearance, a differential effect involving in vivo assessment or possibly that the changes in estrous cycle were not robust enough to impact glutamate clearance. Given that glutamate homeostasis is tightly regulated under normal physiological conditions, the present results provide evidence that glutamate homeostasis may be differentially regulated between males and females. Sex differences have also been measured in glutamate receptor-mediated regulation of dopamine in rats, further supporting sexual dimorphism in the regulation of neurotransmission (101). There are also reports of estrogen receptor mediated inhibition of glutamate uptake activity (102, 103), where 17β-estradiol can increase protein levels of GLAST and GLT-1 and enhance glutamate clearance function [reviewed by (104)], which could normally play a role, in part, in how homeostasis is achieved. Another factor that warrants consideration is that the astrocytes express all estrogen receptor subtypes which provides multiple mechanistic pathways by which estrogen could mediate glutamate homeostasis, including upregulation of neuroprotective growth factors (105). In males, brain-injury induced astrocyte activation (indicated by GFAP staining) has been observed at 7 and 28 DPI in the VPM (38), indicating an active role for astrocytes in ongoing circuit reorganization and behavioral morbidity after TBI. Nevertheless, given that estrogen influences glutamate homeostasis, further exploration into its role in recovery following neurological insults is required. Ionic glutamate receptor perturbations have been associated with TBI, where sex differences in NMDA receptors on astrocytes can mediate regulation of glutamate neurotransmission and have a sexually dimorphic influence on behavioral and hormonal responses (106–108). Along these lines, we posit that sex-specific differences support the need for future studies assessing glutamate neurotransmission to power for females independently, or depending on outcome measures, power for increased variability in cohorts combining male and female animals.
In these experiments, we tested the hypothesis that sex would not influence sensory hypersensitivity and associated in vivo amperometric extracellular recordings of glutamate neurotransmission within the behaviorally relevant thalamocortical circuit. Based on the findings from this study, we reject the hypothesis. Our results indicate that sensory hypersensitivity to whisker stimulation is present in both male and female rats at 1 month post-injury, however, overall scores were lower in females compared to males. Similar to previous results, evoked overflow of glutamate was elevated in the S1BF and VPM of males, yet this only occurred in the VPM of females. Also, similar to previous results, glutamate clearance was not impacted by injury at 28 DPI, however, there is a robust sex-difference indicating glutamate clearance in females is slower than in males. In addition, injured females had prolonged diestrus over the duration of 1 month post-injury in comparison to sham females (Figure 6), supporting clinical reports that TBI has a long-term impact on menstrual cycle. These results highlight the need to consider the effects of female hormonal status in TBI studies on the development of functional morbidity.
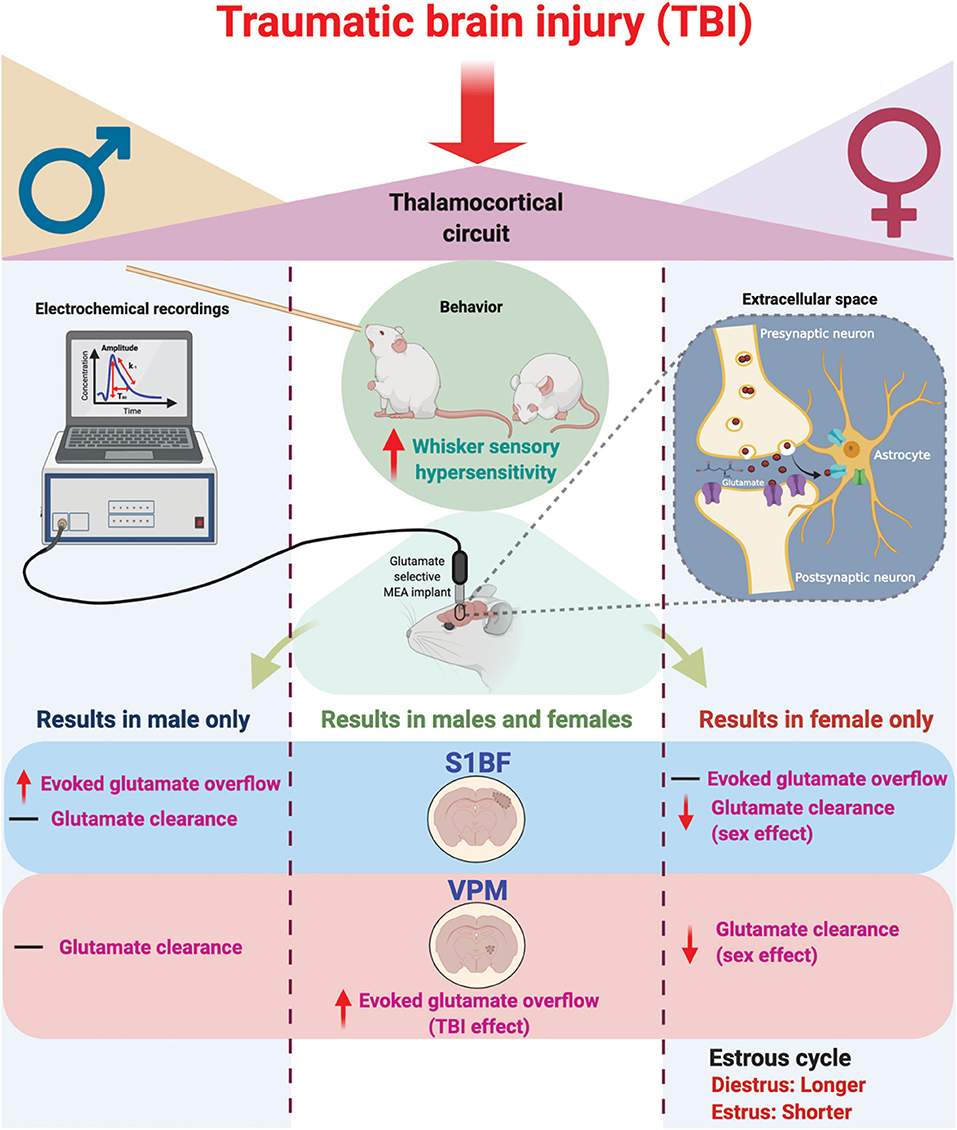
Figure 6. Schematic summary of chronic traumatic brain injury (TBI)-induced circuit disruption with sex-dependent late-onset sensory hypersensitivity and altered glutamate neurotransmission in whisker thalamocortical relays. In these experiments, we assessed the impact of diffuse TBI on changes in glutamate neurotransmission in the thalamocortical system that underlies the manifestation of late-onset sensory hypersensitivity to whisker stimulation in male (Left) and female (Right) rats. At 28 days post-injury (DPI), behavioral morbidity was detected as a function of injury in both males and females (Center). Glutamate selective microelectrode arrays (MEAs) were used for electrochemical recordings within the extracellular space of primary somatosensory barrel field of the cortex (S1BF) and ventral posteromedial nucleus of the thalamus (VPM) relays of the whisker barrel circuit of anesthetized rats. In the S1BF, increased evoked glutamate overflow was observed only in male rats (Left). In the VPM, there was an overall TBI effect on the depolarization-evoked overflow of glutamate in both sexes (Center). Further, both in the S1BF and VPM, female rats had overall slower glutamate clearance parameters in comparison to males (Right), with no injury effects detected. TBI females spent a significantly greater percentage of time in diestrus compared to shams, supported by an a posteriori analysis over weeks post-injury confirming increased time in diestrus. These data indicate irregular estrous cycles chronically after TBI. In summary, TBI induces sex-dependent post-TBI changes in somatosensory circuit function and long-term disruption of the estrous cycle (↑, increase; ↓, decrease; –, no change). Figure created with BioRender.com.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This animal study was reviewed and approved by Institutional Animal Care and Use Committee Protocol (18–384) at the University of Arizona College of Medicine-Phoenix.
GK performed estrous cycle monitoring, amperometric analysis, analyzed the data, wrote the manuscript, and prepared the figures. CB performed estrous cycle monitoring, amperometric analysis and behavioral experiments. EC and EM assisted with amperometric experiments. CH performed statistical analyses and assisted with writing the results. PA and JL provided feedback on the manuscript. TT designed the research, assisted with experiments, analyzed the data, wrote the manuscript, and approved the final version. All authors approved the final version of the paper.
This work was supported by the National Institute of Neurological Disorders and Stroke/National Institutes of Health (Grant No. R01NS100793) and Phoenix Children's Hospital Mission Support to TT. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Phoenix VA Health Care System.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Phoenix VA Health Care System.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Heather Bimonte-Nelson for providing the estrous cycle tracking protocol, Mr. Andrew Condon and Ms. Tala Curry for assistance with the amperometric experiments and Dr. Rachel K. Rowe for optimization of female surgeries. The authors acknowledge the assistance of Ms. Carol A. Haussler for proof reading the article and the Phoenix Children's Hospital Leadership Circle for purchasing the FASTmk-IV.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00749/full#supplementary-material
TBI, traumatic brain injury; S1BF, primary somatosensory (barrel) cortex; VPM, ventral posteromedial nucleus of the thalamus; MEA, microelectrode array; FPI, fluid percussion injury.
1. Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. (2018) 130:1080–97. doi: 10.3171/2017.10.JNS17352
2. NCIPC. Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Atlanta GA Centers for Disease Control and Prevention (2003).
3. Fleminger S, Ponsford J. Long term outcome after traumatic brain injury. BMJ. (2005) 331:1419–20. doi: 10.1136/bmj.331.7530.1419
4. Whitnall L, McMillan TM, Murray GD, Teasdale GM. Disability in young people and adults after head injury: 5–7 year follow up of a prospective cohort study. J Neurol Neurosurg Psychiatry. (2006) 77:640–5. doi: 10.1136/jnnp.2005.078246
5. Farace E, Alves WM. Do women fare worse: a metaanalysis of gender differences in traumatic brain injury outcome. J Neurosurg. (2000) 93:539–45. doi: 10.3171/jns.2000.93.4.0539
6. Sander AM, Maestas KL, Pappadis MR, Sherer M, Hammond FM, Hanks R, et al. Sexual functioning 1 year after traumatic brain injury: findings from a prospective traumatic brain injury model systems collaborative study. Arch Phys Med Rehabil. (2012) 93:1331–7. doi: 10.1016/j.apmr.2012.03.037
7. Bay E, Sikorskii A, Saint-Arnault D. Sex differences in depressive symptoms and their correlates after mild-to-moderate traumatic brain injury. J Head Trauma Rehabil. (2008) 23:351. doi: 10.1097/01.HTR.0000336876.99972.cd
8. Colantonio A, Harris JE, Ratcliff G, Chase S, Ellis K. Gender differences in self reported long term outcomes following moderate to severe traumatic brain injury. BMC Neurol. (2010) 10:102. doi: 10.1186/1471-2377-10-102
9. Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. (2007) 161:125–41. doi: 10.1016/S0079-6123(06)61009-1
10. Mayer AR, Hanlon FM, Dodd AB, Ling JM, Klimaj SD, Meier TB. A functional magnetic resonance imaging study of cognitive control and neurosensory deficits in mild traumatic brain injury. Hum Brain Mapp. (2015) 36:4394–406. doi: 10.1002/hbm.22930
11. Ryan LM, Warden DL. Post concussion syndrome. Int Rev Psychiatry. (2003) 15:310–6. doi: 10.1080/09540260310001606692
12. Waljas M, Iverson GL, Lange RT, Hakulinen U, Dastidar P, Huhtala H, et al. A prospective biopsychosocial study of the persistent post-concussion symptoms following mild traumatic brain injury. J Neurotrauma. (2015) 32:534–47. doi: 10.1089/neu.2014.3339
13. Scheenen ME, Spikman JM, de Koning ME, van der Horn HJ, Roks G, Hageman G, et al. Patients “at risk” of suffering from persistent complaints after mild traumatic brain injury: The role of coping, mood disorders, and post-traumatic stress. J Neurotrauma. (2017) 34:31–7. doi: 10.1089/neu.2015.4381
14. De Koning ME, Scheenen ME, Van Der Horn HJ, Spikman JM, Van Der Naalt J. From ‘miserable minority’ to the ‘fortunate few’: the other end of the mild traumatic brain injury spectrum. Brain Inj. (2018) 32:540–3. doi: 10.1080/02699052.2018.1431844
15. Tsushima WT, Lum M, Geling O. Sex differences in the long-term neuropsychological outcome of mild traumatic brain injury. Brain Inj. (2009) 23:809–14. doi: 10.1080/02699050903200530
16. Bazarian JJ, Blyth B, Mookerjee S, He H, McDermott MP. Sex differences in outcome after mild traumatic brain injury. J Neurotrauma. (2010) 27:527–39. doi: 10.1089/neu.2009.1068
17. Brooks BL, Mrazik M, Barlow KM, McKay CD, Meeuwisse WH, Emery CA. Absence of differences between male and female adolescents with prior sport concussion. J Head Trauma Rehabil. (2014) 29:257–64. doi: 10.1097/HTR.0000000000000016
18. Li SH, Graham BM. Why are women so vulnerable to anxiety, trauma-related and stress-related disorders? The potential role of sex hormones. Lancet Psychiatry. (2017) 4:73–82. doi: 10.1016/S2215-0366(16)30358-3
19. Valera E, Kucyi A. Brain injury in women experiencing intimate partner-violence: neural mechanistic evidence of an “invisible” trauma. Brain Imaging Behav. (2017) 11:1664–77. doi: 10.1007/s11682-016-9643-1
20. Chiang Colvin A, Mullen J, Lovell MR, Vereeke West R, Collins MW, Groh M. The role of concussion history and gender in recovery from soccer-related concussion. Am J Sports Med. (2009) 37:1699–704. doi: 10.1177/0363546509332497
21. Covassin T, Swanik CB, Sachs ML. Sex differences and the incidence of concussions among collegiate athletes. J Athl Train. (2003) 38:238–44.
22. Friedman SA, Phibbs CS, Schmitt SK, Hayes PM, Herrera L, Frayne SM. New women veterans in the VHA: a longitudinal profile. Womens Health Issues. (2011) 21:S103–11. doi: 10.1016/j.whi.2011.04.025
23. Ripley DL, Harrison-Felix C, Sendroy-Terrill M, Cusick CP, Dannels-McClure A, Morey C. The impact of female reproductive function on outcomes after traumatic brain injury. Arch Phys Med Rehabil. (2008) 89:1090–6. doi: 10.1016/j.apmr.2007.10.038
24. Colantonio A, Mar W, Escobar M, Yoshida K, Velikonja D, Rizoli S, et al. Women's health outcomes after traumatic brain injury. J Womens Health. (2010) 19:1109–16. doi: 10.1089/jwh.2009.1740
25. Snook ML, Henry LC, Sanfilippo JS, Zeleznik AJ, Kontos AP. Association of concussion with abnormal menstrual patterns in adolescent and young women. JAMA Pediatr. (2017) 171:879–86. doi: 10.1001/jamapediatrics.2017.1140
26. Wunderle MK, Hoeger KM, Wasserman ME, Bazarian JJ. Menstrual phase as predictor of outcome after mild traumatic brain injury in women. J Head Trauma Rehabil. (2014) 29:E1. doi: 10.1097/HTR.0000000000000006
27. Bavisetty S, Bavisetty S, McArthur DL, Dusick JR, Wang C, Cohan P, et al. Chronic hypopituitarism after traumatic brain injury: risk assessment and relationship to outcome. Neurosurgery. (2008) 62:1080–94. doi: 10.1227/01.neu.0000325870.60129.6a
28. Russell AL, Richardson MR, Bauman BM, Hernandez IM, Saperstein S, Handa RJ, et al. Differential responses of the HPA axis to mild blast traumatic brain injury in male and female mice. Endocrinology. (2018) 159:2363–75. doi: 10.1210/en.2018-00203
29. Fortress AM, Avcu P, Wagner AK, Dixon CE, Pang KC. Experimental traumatic brain injury results in estrous cycle disruption, neurobehavioral deficits, and impaired GSK3β/β-catenin signaling in female rats. Exp Neurol. (2019) 315:42–51. doi: 10.1016/j.expneurol.2019.01.017
30. Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. (2007) 161:43–59. doi: 10.1016/S0079-6123(06)61004-2
31. Emery DL, Royo NC, Fischer I, Saatman KE, McIntosh TK. Plasticity following injury to the adult central nervous system: is recapitulation of a developmental state worth promoting? J Neurotrauma. (2003) 20:1271–92. doi: 10.1089/089771503322686085
32. Krishna G, Beitchman JA, Bromberg CE, Currier Thomas T. Approaches to monitor circuit disruption after traumatic brain injury: frontiers in preclinical research. Int J Mol Sci. (2020) 21:588. doi: 10.3390/ijms21020588
33. Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Rev. (1998) 26:113–35. doi: 10.1016/S0165-0173(97)00061-1
34. McNamara KC, Lisembee AM, Lifshitz J. The whisker nuisance task identifies a late-onset, persistent sensory sensitivity in diffuse brain-injured rats. J Neurotrauma. (2010) 27:695–706. doi: 10.1089/neu.2009.1237
35. Hall KD, Lifshitz J. Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain Res. (2010) 1323:161–73. doi: 10.1016/j.brainres.2010.01.067
36. Lifshitz J, Lisembee AM. Neurodegeneration in the somatosensory cortex after experimental diffuse brain injury. Brain Struct Funct. (2012) 217:49–61. doi: 10.1007/s00429-011-0323-z
37. Miremami JD, Talauliker PM, Harrison JL, Lifshitz J. Neuropathology in sensory, but not motor, brainstem nuclei of the rat whisker circuit after diffuse brain injury. Somatosens Mot Res. (2014) 31:127–35. doi: 10.3109/08990220.2014.897602
38. Thomas TC, Ogle SB, Rumney BM, May HG, Adelson PD, Lifshitz J. Does time heal all wounds? Experimental diffuse traumatic brain injury results in persisting histopathology in the thalamus. Behav Brain Res. (2018) 340:137–46. doi: 10.1016/j.bbr.2016.12.038
39. Singh R, Venkateshwara G, Nair KP, Khan M, Saad R. Agitation after traumatic brain injury and predictors of outcome. Brain Inj. (2014) 28:336–40. doi: 10.3109/02699052.2013.873142
40. Thomas TC, Hinzman JM, Gerhardt GA, Lifshitz J. Hypersensitive glutamate signaling correlates with the development of late-onset behavioral morbidity in diffuse brain-injured circuitry. J Neurotrauma. (2012) 29:187–200. doi: 10.1089/neu.2011.2091
41. Hinzman JM, Thomas TC, Burmeister JJ, Quintero JE, Huettl P, Pomerleau F, et al. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. J Neurotrauma. (2010) 27:889–99. doi: 10.1089/neu.2009.1238
42. Beitchman JA, Griffiths DR, Hur Y, Ogle SB, Bromberg CE, Morrison HW, et al. Experimental traumatic brain injury induces chronic glutamatergic dysfunction in amygdala circuitry known to regulate anxiety-like behavior. Front Neurosci. (2020) 13:1434. doi: 10.3389/fnins.2019.01434
43. Hosseini AH, Lifshitz J. Brain injury forces of moderate magnitude elicit the fencing response. Med Sci Sports Exerc. (2009) 41:1687–97. doi: 10.1249/MSS.0b013e31819fcd1b
44. Kippin TE, Fuchs RA, Mehta RH, Case JM, Parker MP, Bimonte-Nelson HA, et al. Potentiation of cocaine-primed reinstatement of drug seeking in female rats during estrus. Psychopharmacology. (2005) 182:245–52. doi: 10.1007/s00213-005-0071-y
45. Balasco L, Chelini G, Bozzi Y, Provenzano G. Whisker Nuisance Test: a valuable tool to assess tactile hypersensitivity in mice. Bio Protoc. (2019) 9:e3331. doi: 10.21769/BioProtoc.3331
46. Thomas TC, Beitchman JA, Pomerleau F, Noel T, Jungsuwadee P, Butterfield DA, et al. Acute treatment with doxorubicin affects glutamate neurotransmission in the mouse frontal cortex and hippocampus. Brain Res. (2017) 1672:10–7. doi: 10.1016/j.brainres.2017.07.003
47. Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal Chem. (2001) 73:1037–42. doi: 10.1021/ac0010429
48. Paxinos G, Watson C. The Rat Brain In Stereotaxic Coordinates. New York, NY: Academic Press (2007).
49. Eakin K, Rowe RK, Lifshitz J. Frontiers in neuroengineering: modeling fluid percussion injury: relevance to human traumatic brain injury. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: CRC Press/Taylor and Francis (2015) 259–72. doi: 10.1201/b18126-23
50. Rowe RK, Griffiths DR, Lifshitz J. Midline (central) fluid percussion injury model of traumatic brain injury. In: Kobeissy F, Dixon CE, Hayes RL, Mondello S, editors. Methods in Molecular Biology. New York, NY: Humana Press (2016). p. 211–30. doi: 10.1007/978-1-4939-3816-2_13
51. Galvin J, Froude EH, Imms C. Sensory processing abilities of children who have sustained traumatic brain injuries. Am J Occup Ther. (2009) 63:701–9. doi: 10.5014/ajot.63.6.701
52. Alwis DS, Yan EB, Morganti-Kossmann MC, Rajan R. Sensory cortex underpinnings of traumatic brain injury deficits. PLoS ONE. (2012) 7:e52169. doi: 10.1371/journal.pone.0052169
53. Alwis DS, Johnstone V, Yan E, Rajan R. Diffuse traumatic brain injury and the sensory brain. Clin Exp Pharmacol Physiol. (2013) 40:473–83. doi: 10.1111/1440-1681.12100
54. Nicholson C. Interaction between diffusion and Michaelis-Menten uptake of dopamine after iontophoresis in striatum. Biophys J. (1995) 68:1699–715. doi: 10.1016/S0006-3495(95)80348-6
55. McAllister TW. Neuropsychiatry sequelae of head injuries. Psychiatr Clin. (1992) 15:395–413. doi: 10.1016/S0193-953X(18)30245-4
56. Lafrenaye AD, Krahe TE, Povlishock JT. Moderately elevated intracranial pressure after diffuse traumatic brain injury is associated with exacerbated neuronal pathology and behavioral morbidity in the rat. J Cereb Blood Flow Metab. (2014) 34:1628–36. doi: 10.1038/jcbfm.2014.122
57. Nillni YI, Pineles SL, Patton SC, Rouse MH, Sawyer AT, Rasmusson AM. Menstrual cycle effects on psychological symptoms in women with PTSD. J Trauma Stress. (2015) 28:1–7. doi: 10.1002/jts.21984
58. Povlishock JT, Stone JR. Traumatic axonal injury. In: Miller LP, Hayes RL, Newcomb JK, editors. Head Trauma: Basic, Preclinical and Clinical Directions. New York, NY: Wiley (2001). p. 281–302.
59. McGinn MJ, Povlishock JT. Pathophysiology of traumatic brain injury. Neurosurg Clin N Am. (2016) 27:397–407. doi: 10.1016/j.nec.2016.06.002
60. Brosseau-Lachaine O, Gagnon I, Forget R, Faubert J. Mild traumatic brain injury induces prolonged visual processing deficits in children. Brain Inj. (2008) 22:657–68. doi: 10.1080/02699050802203353
61. Adibi M. Whisker-mediated touch system in rodents: from neuron to behavior. Front Syst Neurosci. (2019) 13:40. doi: 10.3389/fnsys.2019.00040
62. Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. (2012) 225:65–75. doi: 10.1016/j.neuroscience.2012.08.058
63. Johnstone VP, Shultz SR, Yan EB, O'Brien TJ, Rajan R. The acute phase of mild traumatic brain injury is characterized by a distance-dependent neuronal hypoactivity. J Neurotrauma. (2014) 31:1881–95. doi: 10.1089/neu.2014.3343
64. Johnstone VP, Wright DK, Wong K, O'Brien TJ, Rajan R, Shultz SR. Experimental traumatic brain injury results in long-term recovery of functional responsiveness in sensory cortex but persisting structural changes and sensorimotor, cognitive, and emotional deficits. J Neurotrauma. (2015) 32:1333–46. doi: 10.1089/neu.2014.3785
65. Morrison H, Young K, Qureshi M, Rowe RK, Lifshitz J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci Rep. (2017) 7:1–12. doi: 10.1038/s41598-017-13581-z
66. Kumar S, Rao SL, Nair RG, Pillai S, Chandramouli BA, Subbakrishna DK. Sensory gating impairment in development of post-concussive symptoms in mild head injury. Psychiatry Clin Neurosci. (2005) 59:466–72. doi: 10.1111/j.1440-1819.2005.01400.x
67. Kadyan V, Mysiw WJ, Bogner JA, Corrigan JD, Fugate LP, Clinchot DM. Gender differences in agitation after traumatic brain injury. Am J Phys Med Rehabil. (2004) 83:747–52. doi: 10.1097/01.PHM.0000140790.30468.F4
68. Wu CS, Ballester Rosado CJ, Lu HC. What can we get from ‘barrels’: the rodent barrel cortex as a model for studying the establishment of neural circuits. Eur J Neurosci. (2011) 34:1663–76. doi: 10.1111/j.1460-9568.2011.07892.x
69. Giuliano S, Talarico S, Bruno L, Nicoletti FB, Ceccotti C, Belfiore A. Growth hormone deficiency and hypopituitarism in adults after complicated mild traumatic brain injury. Endocrine. (2017) 58:115–23. doi: 10.1007/s12020-016-1183-3
70. Holtzman B, Ackerman KE. Hypothalamic-pituitary-gonadal axis in women's sport: injuries, manipulations, and aberrations. Curr Opin Endocr Metab Res. (2019) 9:78–85. doi: 10.1016/j.coemr.2019.08.003
71. Ntali G, Tsagarakis S. Traumatic brain injury induced neuroendocrine changes: acute hormonal changes of anterior pituitary function. Pituitary. (2019) 22:283–95. doi: 10.1007/s11102-019-00944-0
72. Wagner AK, McCullough EH, Niyonkuru C, Ozawa H, Loucks TL, Dobos JA, et al. Acute serum hormone levels: characterization and prognosis after severe traumatic brain injury. J Neurotrauma. (2011) 28:871–88. doi: 10.1089/neu.2010.1586
73. Ranganathan P, Kumar RG, Davis K, McCullough EH, Berga SL, Wagner AK. Longitudinal sex and stress hormone profiles among reproductive age and post-menopausal women after severe TBI: a case series analysis. Brain Inj. (2016) 30:452–61. doi: 10.3109/02699052.2016.1144081
74. Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: stressor-and cytokine-induced alterations of neuroplasticity. Neuroscience. (2005) 135:659–78. doi: 10.1016/j.neuroscience.2005.03.051
75. Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, et al. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. (2005) 22:42–75. doi: 10.1089/neu.2005.22.42
76. Andreollo NA, Santos EF, Araujo MR, Lopes LR. Rat's age versus human's age: what is the relationship? Arq Bras Cir Dig. (2012) 25:49–51. doi: 10.1590/S0102-67202012000100011
77. Cyrenne DL, Brown GR. Ontogeny of sex differences in response to novel objects from adolescence to adulthood in lister-hopded rats. Dev Psychobiol. (2011) 52:670–6. doi: 10.1002/dev.20542
78. Davis DP, Douglas DJ, Smith W, Sise MJ, Vilke GM, Holbrook TL. Traumatic brain injury outcomes in pre-and post-menopausal females versus age-matched males. J Neurotrauma. (2006) 23:140–8. doi: 10.1089/neu.2006.23.140
79. Ortiz JB, Sukhina A, Balkan B, Harootunian G, Adelson PD, Lewis KS, et al. Epidemiology of pediatric traumatic brain injury and hypothalamic-pituitary disorders in Arizona. Front Neurol. (2020) 10:1410. doi: 10.3389/fneur.2019.01410
80. McIntyre A, Mehta S, Aubut J, Dijkers M, Teasell RW. Mortality among older adults after a traumatic brain injury: a meta-analysis. Brain Inj. (2013) 27:31–40. doi: 10.3109/02699052.2012.700086
81. Bishop KM, Wahlsten D. Sex and species differences in mouse and rat forebrain commissures depend on the method of adjusting for brain size. Brain Res. (1999) 815:358–66. doi: 10.1016/S0006-8993(98)01088-9
82. Claassen V (editor). Neglected factors in pharmacology and neuroscience research. In: Techniques in the Behavioral and Neural Sciences. Vol. 12. Amsterdam: Elsevier. (1994). p. 1–486.
83. Dollé JP, Jaye A, Anderson SA, Ahmadzadeh H, Shenoy VB, Smith DH. Newfound sex differences in axonal structure underlie differential outcomes from in vitro traumatic axonal injury. Exp Neurol. (2018) 300:121–34. doi: 10.1016/j.expneurol.2017.11.001
84. Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. (2015) 15:27. doi: 10.1007/s11910-015-0545-1
85. Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury–associated traumatic brain injury. Arch Phys Med Rehabil. (2001) 82:1461–71. doi: 10.1053/apmr.2001.25137
86. Schwarz JM, Liang S-L, Thompson SM, McCarthy MM. Estradiol induces hypothalamic dendritic spines by enhancing glutamate release: a mechanism for organizational sex differences. Neuron. (2008) 58:584–98. doi: 10.1016/j.neuron.2008.03.008
87. Strom JO, Theodorsson A, Theodorsson E. Mechanisms of estrogens' dose-dependent neuroprotective and neurodamaging effects in experimental models of cerebral ischemia. Int J Mol Sci. (2011) 12:1533–62. doi: 10.3390/ijms12031533
88. Yi JH, Hoover R, McIntosh TK, Hazell AS. Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. J Neurotrauma. (2006) 23:86–96. doi: 10.1089/neu.2006.23.86
89. Akasu T, Muraoka N, Hasuo H. Hyperexcitability of hippocampal CA1 neurons after fluid percussion injury of the rat cerebral cortex. Neurosci Lett. (2002) 329:305–8. doi: 10.1016/S0304-3940(02)00707-3
90. Cai X, Wei DS, Gallagher SE, Bagal A, Mei YA, Kao JP, et al. Hyperexcitability of distal dendrites in hippocampal pyramidal cells after chronic partial deafferentation. J Neurosci. (2007) 27:59–68. doi: 10.1523/JNEUROSCI.4502-06.2007
91. Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett. (2009) 459:52–6. doi: 10.1016/j.neulet.2009.04.064
92. Patel DR, Croucher MJ. Evidence for a role of presynaptic AMPA receptors in the control of neuronal glutamate release in the rat forebrain. Eur J Pharmacol. (1997) 332:143–51. doi: 10.1016/S0014-2999(97)01077-7
93. Reeves TM, Kao CQ, Phillips LL, Bullock MR, Povlishock JT. Presynaptic excitability changes following traumatic brain injury in the rat. J Neurosci Res. (2000) 60:370–9. doi: 10.1002/(SICI)1097-4547(20000501)60:3<370::AID-JNR12>3.0.CO;2-B
94. Banati RB. Visualising microglial activation in vivo. Glia. (2002) 40:206–17. doi: 10.1002/glia.10144
95. Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem.. (2007) 101:1205–13. doi: 10.1111/j.1471-4159.2007.04487.x
97. Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM, Sharp DJ. Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage. J Neuroinflammation. (2015) 12:224. doi: 10.1186/s12974-015-0445-y
98. Hazra A, Macolino C, Elliott MB, Chin J. Delayed thalamic astrocytosis and disrupted sleep–wake patterns in a preclinical model of traumatic brain injury. J Neurosci Res.. (2014) 92:1434–45. doi: 10.1002/jnr.23430
99. Gadea A, López-Colomé AM. Glial transporters for glutamate, glycine and GABA I. Glutamate transporters. J Neurosci Res. (2001) 63:453–60. doi: 10.1002/jnr.1039
100. Sajjad J, Felice VD, Golubeva AV, Cryan JF, O'Mahony SM. Sex-dependent activity of the spinal excitatory amino acid transporter: role of estrous cycle. Neuroscience. (2016) 333:311–9. doi: 10.1016/j.neuroscience.2016.07.036
101. Locklear MN, Cohen AB, Jone A, Kritzer MF. Sex differences distinguish intracortical glutamate receptor-mediated regulation of extracellular dopamine levels in the prefrontal cortex of adult rats. Cereb Cortex. (2016) 26:599–610. doi: 10.1093/cercor/bhu222
102. Sato K, Matsuki N, Ohno Y, Nakazawa K. Effects of 17β-estradiol and xenoestrogens on the neuronal survival in an organotypic hippocampal culture. Neuroendocrinology. (2002) 76:223–34. doi: 10.1159/000065948
103. Sato K, Matsuki N, Ohno Y, Nakazawa K. Estrogens inhibit L-glutamate uptake activity of astrocytes via membrane estrogen receptor α. J Neurochem. (2003) 86:1498–505. doi: 10.1046/j.1471-4159.2003.01953.x
104. Karki P, Smith K, Johnson J Jr, Lee E. Astrocyte-derived growth factors and estrogen neuroprotection: role of transforming growth factor-α in estrogen-induced upregulation of glutamate transporters in astrocytes. Mol Cell Endocrinol. (2014) 389:58–64. doi: 10.1016/j.mce.2014.01.010
105. Dhandapani KM, Brann DW. Estrogen-astrocyte interactions: implications for neuroprotection. BMC Neurosci. (2002) 3:6. doi: 10.1186/1471-2202-3-6
106. Ho D, Lo W. Sex differences in NMDA receptor mediated responses in rats. Brain Res.. (1993) 620:167–70. doi: 10.1016/0006-8993(93)90287-W
107. Schumann J, Alexandrovich GA, Biegon A, Yaka R. Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma. (2008) 25:945–57. doi: 10.1089/neu.2008.0521
Keywords: traumatic brain injury, sex difference, glutamate, behavior, neurotransmitters, whisker, microelectrode arrays, estrous
Citation: Krishna G, Bromberg C, Connell EC, Mian E, Hu C, Lifshitz J, Adelson PD and Thomas TC (2020) Traumatic Brain Injury-Induced Sex-Dependent Changes in Late-Onset Sensory Hypersensitivity and Glutamate Neurotransmission. Front. Neurol. 11:749. doi: 10.3389/fneur.2020.00749
Received: 19 April 2020; Accepted: 17 June 2020;
Published: 05 August 2020.
Edited by:
Peter John Crack, The University of Melbourne, AustraliaReviewed by:
Sandy R. Shultz, Monash University, AustraliaCopyright © 2020 Krishna, Bromberg, Connell, Mian, Hu, Lifshitz, Adelson and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theresa Currier Thomas, dGhlcmVzYXRob21hc0BlbWFpbC5hcml6b25hLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.