 Suzanne Lesage1,2,3,4*
Suzanne Lesage1,2,3,4* Marion Houot5,6
Marion Houot5,6 Graziella Mangone1,2,3,4,6
Graziella Mangone1,2,3,4,6 Christelle Tesson1,2,3,4Hélène Bertrand1,2,3,4Sylvie Forlani1,2,3,4
Christelle Tesson1,2,3,4Hélène Bertrand1,2,3,4Sylvie Forlani1,2,3,4 Mathieu Anheim7,8,9
Mathieu Anheim7,8,9 Christine Brefel-Courbon10,11Emmanuel Broussolle12,13,14
Christine Brefel-Courbon10,11Emmanuel Broussolle12,13,14 Stéphane Thobois12,13,14
Stéphane Thobois12,13,14 Philippe Damier15
Philippe Damier15 Franck Durif16
Franck Durif16 Emmanuel Roze1,2,3,4,17François Tison18David Grabli1,2,3,4,6Fabienne Ory-Magne19,20
Emmanuel Roze1,2,3,4,17François Tison18David Grabli1,2,3,4,6Fabienne Ory-Magne19,20 Bertrand Degos21,22François Viallet23,24Florence Cormier-Dequaire1,2,3,4,6Anne-Marie Ouvrard-Hernandez25
Bertrand Degos21,22François Viallet23,24Florence Cormier-Dequaire1,2,3,4,6Anne-Marie Ouvrard-Hernandez25 Marie Vidailhet1,2,3,4,17
Marie Vidailhet1,2,3,4,17 Ebba Lohmann26Andrew Singleton27
Ebba Lohmann26Andrew Singleton27 Jean-Christophe Corvol1,2,3,4,6Alexis Brice1,2,3,4 †
Jean-Christophe Corvol1,2,3,4,6Alexis Brice1,2,3,4 †- 1Sorbonne Université, Unité Mixte de Recherche (UMR) 1127, Paris, France
- 2Unité de Recherche U1127 à l'Institut National de la Santé et de la Recherche Médicale (INSERM), Paris, France
- 3Unité de Recherche Unité Mixte de Recherche (UMR) 7225 au Centre National de la Recherche Scientifique (CNRS), Paris, France
- 4Institut du Cerveau (ICM), Paris, France
- 5Institut de la Mémoire et de la Maladie d'Alzheimer (IM2A), Centre d'Excellence sur les Maladies Neurodégénératives (CoEN), Assistance Publique – Hôpitaux de Paris (AP-HP), Département de Neurologie, Groupe Hospitalier Pitié-Salpêtrière, Université Paris 6, Paris, France
- 6Centre d'Investigation Clinique Pitié Neurosciences CIC-1422, Paris, France
- 7Département de Neurologie aux Hôpitaux Universitaires de Strasbourg, Strasbourg, France
- 8Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France
- 9Fédération de Médecine Translationnelle de Strasbourg (FMTS), Université de Strasbourg, Strasbourg, France
- 10Service de Pharmacologie Clinique, Faculté de Médecine, Hôpital Universitaire, Toulouse, France
- 11Service de Neurologie B8, Hôpital Pierre Paul Riquet, Hôpital Universitaire, Toulouse, France
- 12Université de Lyon, Institut des Sciences Cognitives Marc-Jeannerod, Unité Mixte de Recherche (UMR) 5229, Centre National de la Recherche Scientifique (CNRS), Bron, France
- 13Hospices Civils de Lyon, Hôpital Neurologique Pierre-Wertheimer, Département de Neurologie C, Bron, France
- 14Université de Lyon, Faculté de Médecine Lyon-Sud Charles-Mérieux, Oullins, France
- 15Centre Hospitalier Universitaire de Nantes, Centre d'Investigation Clinique, Nantes, France
- 16Département de Neurologie A, Centre Hospitalier Universitaire de Clermont-Ferrand, Clermont-Ferrand, France
- 17Département de Neurologie, Groupe Hospitalier Pitié-Salpêtrière, Paris, France
- 18Institut des Maladies Neurodégénératives, Centre Hospitalier Universitaire et Université de Bordeaux, Bordeaux, France
- 19Centre de Neuroimagerie de Toulouse, Université de Toulouse - Institut National de la Santé et de la Recherche Médicale (INSERM) - Université de Toulouse, Toulouse, France
- 20Centre des Neurosciences, Hôpital Universitaire de Toulouse, Toulouse, France
- 21Unité de Neurologie, Hôpital Universitaire Avicenne, Hôpitaux Universitaires de Paris-Seine Saint Denis, Assistance Publique – Hôpitaux de Paris (AP-HP), Sorbonne Paris Nord, Bobigny, France
- 22Equipe Dynamique et Physiopathologie des Réseaux Neuronaux, Centre pour la Recherche Interdisciplinaire en Biologie, Collège de France, Centre National de la Recherche Scientifique (CNRS) Unité Mixte de Recherche (UMR) 7241, Institut National de la Santé et de la Recherche Médicale (INSERM) U1050, Labex MemoLife, Paris, France
- 23Département de Neurologie, Centre Hospitalier Intercommunal d'Aix-Pertuis, Aix-en-Provence, France
- 24Laboratoire Parole et Langage, Unité Mixte de Recherche (UMR) 7309, Centre National de la Recherche Scientifique (CNRS) et Université d'Aix-Marseille, Aix-en-Provence, France
- 25Centre Hospitalier Universitaire de Grenoble, Service de Neurologie, Grenoble, France
- 26Department of Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany
- 27Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, United States
LRRK2, SNCA, and VPS35 are unequivocally associated with autosomal dominant Parkinson's disease (PD). We evaluated the prevalence of LRRK2, SNCA, and VPS35 mutations and associated clinical features in a large French multi-center cohort of PD patients. Demographic and clinical data were collected for 1,805 index cases (592 with autosomal dominant inheritance and 1,213 isolated cases) since 1990. All probands were screened with TaqMan assays for LRRK2 Gly2019Ser. In the absence of this mutation, the coding sequences of the three genes were analyzed by Sanger sequencing and/or next-generation sequencing. The data for the three genes were analyzed according to age at onset, family history, ethnic origin and clinical features. We identified 160 index cases (8.9%) with known pathogenic variants: 138 with pathogenic LRRK2 variants (7.6%), including 136 with the Gly2019Ser mutation, 19 with SNCA point mutations or genomic rearrangements (1.1%), and three with the VPS35 Asp620Asn mutation (0.16%). Mutation frequencies were higher in familial than isolated cases, consistent with autosomal dominant inheritance (12.0 vs. 7.3%; OR 1.7, 95% CI [1.2–2.4], p = 0.001). PD patients with LRRK2 variants were more likely to have higher rates of late-onset PD (>50 years; OR 1.5, 95% CI [1.0–2.1], p = 0.03), whereas those with SNCA mutations tended to have earlier age at onset disease (≤ 50 years, p = 0.06). The clinical features of LRRK2 carriers and those without any pathogenic variants in known PD-associated genes were similar. The likelihood of detecting disease-causing mutations was higher in cases compatible with autosomal dominant inheritance.
Introduction
Heterozygous sequence variants of LRRK2 or VPS35, and mutations or genomic rearrangements in SNCA cause monogenic Parkinson's disease (PD) with autosomal dominant (AD) inheritance [reviewed in (1)]. Only seven of the hundred or so variants of LRRK2 reported to date (Asn1437His, Arg1441Gly/Cys/His, Tyr1699Cys, Gly2019Ser, and Ile2020Thr), which appear to be clustered in functionally important regions highly conserved throughout evolution, have been demonstrated to be pathogenic on the basis of co-segregation with the disease and absence or rarity in specific control populations (2). The most common of these mutations, LRRK2 Gly2019Ser, has a reported frequency of 0% to above 40%, depending on the population considered (3), mostly due to a common founder effect (4). SNCA mutations are the second most common cause of autosomal dominant inherited PD; genomic duplications have been detected in ~1–2% of families with AD PD. Other SNCA mutations, such as whole-locus triplications and a few missense mutations (Ala53Thr/Glu/Val, Glu46Lys, Ala30Pro, and Gly51Asp), are extremely rare [reviewed in (1)]. Finally, VPS35 was the first PD-causing gene to be identified by next-generation sequencing (NGS) in large multi-incident families (5, 6). Subsequent studies in multiple ethnic groups, including a large multi-center study, indicated that Asp620Asn was the only pathogenic variant, with a relative frequency ranging from 0.1 to 1% in familial PD, depending on population background (7). As the phenotype of AD PD closely resembles that of idiopathic PD, we assumed that rare variants of genes causing AD PD might also contribute to the etiology of isolated PD in the French population.
In this study, we aimed at determining the relative frequencies of known mutations of three genes, LRRK2, SNCA, and VPS35, in a large cohort of familial and isolated cases. The high prevalence of the LRRK2 Gly2019Ser mutation provided us with a unique opportunity to compare in details clinical characteristics between carriers of this mutation and patients with no known mutations of PD-associated genes.
Materials and Methods
Patients
In total, 673 PD patients from 592 families and 1,213 isolated cases without known consanguinity were recruited from 1990 onwards, through the French Parkinson Disease Genetics Network (the PDG group, Supplementary Material). All participants underwent a detailed medical and family history, and a family tree were drawn. Familial cases compatible with AD inheritance, and referred to here as AD PD cases, were defined as AD cases with at least one other affected relative in a different generation, identified by an examination of secondary cases (n = 146) or on the basis of family history (n = 446). PD was diagnosed according to the clinical diagnostic criteria of the UK Parkinson Disease Society Brain Bank (PDSBB) (8). Comprehensive standardized interviews and neurological examinations were performed by movement disorder experts. Motor and non-motor symptoms were assessed by evaluating Unified Parkinson Disease Rating Scale (UPDRS) scores, Hoehn and Yahr staging, autonomic dysfunction, sleep, cognitive [Mini Mental State Examination (MMSE)], neuropsychological and behavioral scores. Early onset was defined as the onset of symptoms before the age of 51 years.
Informed consent was obtained from all participants, and genetic studies were approved by local ethics committees.
Most index cases were of European ancestry (n = 1,530; 84.8%), mostly French (n = 1,202; 78.6%); the others were North African (n = 221; 12.2%) or of other origins, including Asian, Sub-Saharan African or “mixed” origins (n = 42; 2.3%); or of unknown origin (n = 12; 0.7%). This study included 226 index cases from families with PD compatible with AD inheritance reported elsewhere (9, 10).
Methods
Genomic DNA was obtained from peripheral blood lymphocyte or saliva samples (OrageneTM DNA Self-Collection Kit, DNA Genotek), by standard protocols. Patients with variants of the GBA risk factor (n = 153), or with variants of genes for which the causal role in AD PD was uncertain, such as GIGYF2 (n = 6), EIF4G1 (n = 2), and c9ORF72 (n = 4), were not included in this study. In addition, we excluded 25 (23 with bi-allelic PRKN mutations and two with bi-allelic PINK1 mutations) of the 1,089 PD index cases who have been screened for autosomal recessive (AR)-PD associated genes [814 by gene panel/exome sequencing (see below) and 275 by direct sequencing of the two most frequent AR PD genes, PRKN and PINK1].
All index cases were genotyped in duplicate for LRRK2 Gly2019ser, by the TaqMan allelic discrimination Assay-By-Design method, in accordance with the manufacturer's instructions, with 8 ng of DNA mixed with the TaqMan Genotyping Master Mix (Thermo Fisher Scientific Inc.) and custom-produced TaqMan SNP genotyping assays [C_63498123_10 (rs34637584), Thermo Fisher Scientific Inc.] on an Applied Biosystems PRISM 7000 sequence detection system (Thermo Fisher Scientific Inc.) or LightCycler® 480 machine (Roche, Life Technologies SAS). All patients found not to carry LRRK2 Gly2019ser were then screened for pathogenic variants of the coding sequences of LRRK2, SNCA, and VPS35, by Sanger sequencing (n = 855), targeted sequencing of a customized next-generation sequencing (NGS) gene panel containing the 22 most prevalent PD-associated genes (n = 404; Supplementary Table 1), or available whole-exome sequencing (n = 410), as previously described (11, 12). We considered known pathogenic mutations of the three genes.
Sanger sequencing was used to confirm variants and co-segregation analyses were performed, where possible. SNCA rearrangements were detected by semi-quantitative multiplex PCR (13) or by the SALSA multiplex ligation-dependent probe amplification method (MLPA, MRC Holland, Amsterdam, the Netherlands; http://www.mlpa.com), according to the manufacturer's instructions.
Statistical Analysis
Demographic and clinical characteristics are expressed as means and standard deviations for continuous variables and as counts and percentages for qualitative variables, separately for each group (i.e., with and without mutation). These characteristics were compared between patients with the LRRK2 Gly2019ser mutation (LRRK2+) and those with no mutations of genes known to be associated with PD (genetically undefined PD), in Welch's t-tests for continuous variables and Fisher's exact tests for qualitative variables.
We used generalized linear models (GLMs) to compare clinical features between patients with the LRRK2 Gly2019ser mutation and those with genetically undefined PD, with adjustment for sex, age at onset (AAO), and disease duration. Disease duration was not included in models of clinical features at onset. We used GLMs with identity links and normal distributions for continuous clinical features, and GLMs with logit links and Bernoulli distributions for binary clinical features. Fisher type II tests were performed to test each effect, and effect size was estimated with Cohen's f2. We corrected for multiple testing by the Benjamini-Hochberg method. Residual normality and heteroskedasticity were checked visually. Influencers and outliers were checked by calculating hat values and Cook distances. Only patients with no missing data for the covariables included in the models, such as AAO, sex, and disease duration, were retained for analysis. Statistical analyses were performed with R 3.6.1.
Results
Demographic and Clinical Data
Table 1 shows the baseline demographic and clinical characteristics of the 1,805 PD index cases included in the study. Men (n = 1,106, 61.3%) and early AAO cases (mean 47.4 [SD 12.5]; range 9–86 years; Table 1) were overrepresented, particularly among isolated cases (mean 46.1 [SD 12.5]; range: 9–79 years) as opposed to familial cases (mean 50.0 [SD 12.1]; range 10–86 years).
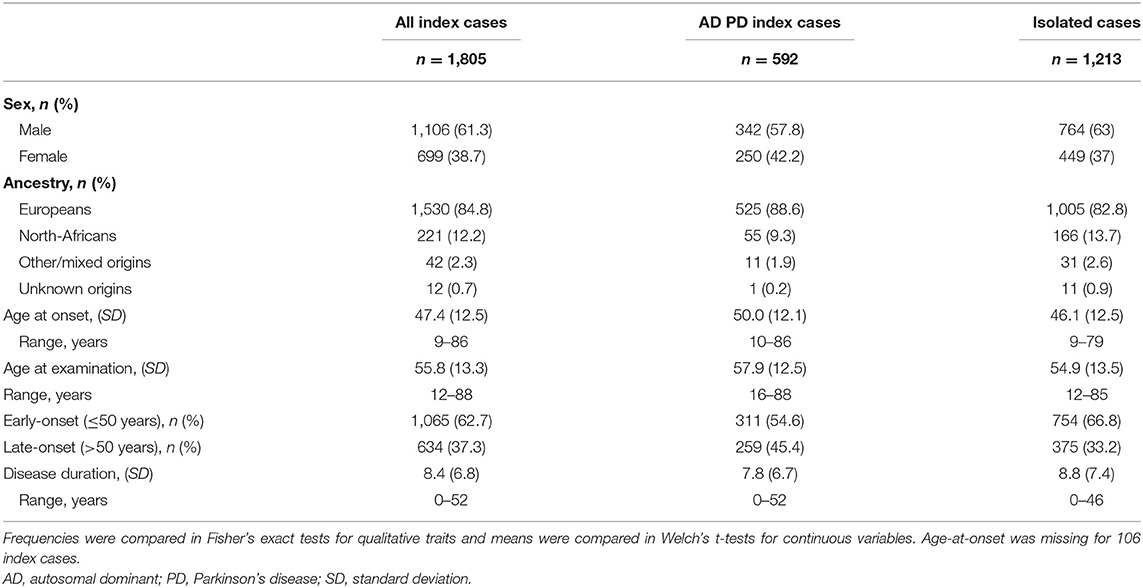
Table 1. Baseline demographics for our study population.
Summary of the Mutations Identified
We identified seven different known mutations of LRRK2, SNCA, and VPS35 in 160 of the 1,805 PD index cases (8.9%, 95% confidence interval (CI): [7.6–10.2]). With the exception of rare cases with the homozygous LRRK2 Gly2019Ser mutation (n = 6), all the known mutations and gene multiplications identified were heterozygous. We found that 136 of the 138 LRRK2 mutation carriers (7.5% of index cases, 95% CI [6.4–8.9]) carried the Gly2019Ser mutation; 13 of these patients had already been reported elsewhere (9). Two index cases carried the rare LRRK2 Arg1441His mutation (9). SNCA mutations were found in 19 index cases (1.1%, 95% CI [0.63–1.6]), including 11 families already reported (13–15): one with whole-gene triplications, 14 with duplications, one with the Gly51Asp mutation, and three with the Ala53Thr mutation. The three PD index cases with the only VPS35 mutation identified, Asp620Asn (0.17%) were described in a previous report (10).
The LRRK2 Gly2019Ser mutation was more common in PD index cases of North-African origin (100/221, 45.2%; 95% CI [38.7–51.8]) than in Europeans (36/1,530, 2.4%; 95% CI [1.7–3.2]) (Fisher's exact test: odds ratio (OR) = 34.3, 95% CI [22.0–53.8], p < 0.0001; Table 2). By contrast, SNCA point mutations and locus triplications/duplications were found mostly in PD index cases of European ancestry, accounting for 1.2% (18/1,530) of such patients, whereas only one case of North-African ancestry (0.45%, 1/221) carried an SNCA duplication. VPS35 mutations were identified exclusively in patients of European origin, accounting for 0.20% (3/1,530) of these individuals.
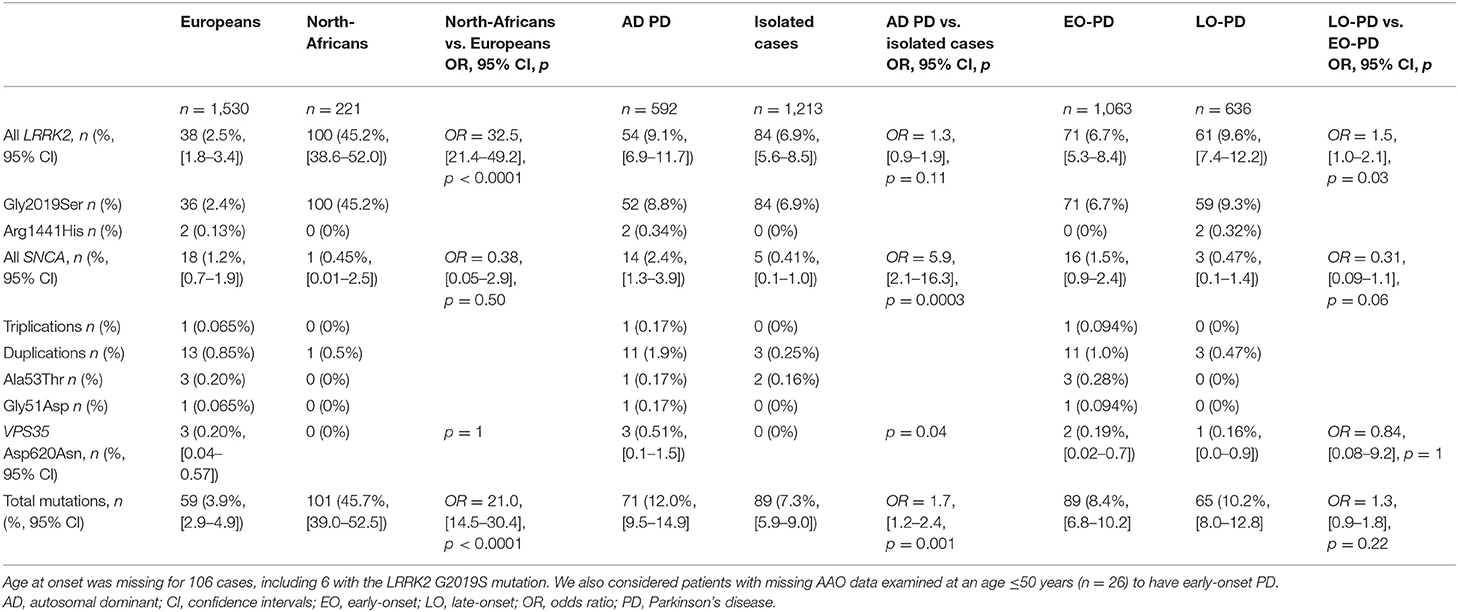
Table 2. Overall frequencies of LRRK2, SNCA, and VPS35 mutations according to index case ethnicity, family history of Parkinson's disease and age at onset.
Overall, mutations were more frequently identified in familial (71/592, 12.0%; 95% CI [9.5–14.9]) than in isolated cases (89/1,213, 7.3%; 95% CI [5.9–9.0], Fisher's exact test: OR 1.7, 95% CI [1.2–2.4], p = 0.001), particularly for SNCA (2.4%, 14/592 vs. 0.41%, 5/1,213, Fisher's exact test: OR 5.9, 95% CI [2.1–16.3], p = 0.0003; Table 2). An analysis of PD cases according to age at onset (≤ 50 years vs. >50 years) showed that LRRK2 mutations were more frequent among late-onset PD cases (61/636, 9.6%; 95% CI [7.4–12.2] vs. 71/1063, 6.7%; 95% CI [5.3–8.4], Fisher's exact test: OR 1.5, 95% CI [1.0–2.1], p = 0.03; Table 2). By contrast, SNCA mutation carriers tended to have an earlier AAO (16/1063, 1.5% vs. 3/636, 0.5%, p = 0.06).
Clinical Characteristics of Mutation Carriers and Comparison of LRRK2 Gly2019Ser Mutation Carriers (LRRK2+) With Individuals With No Known PD Mutations (Genetically Undefined PD)
Co-segregation analyses identified 193 PD patients as mutation carriers: 151 with LRRK2 Gly2019Ser and five with Arg1441His mutations, 29 with SNCA (see below) and eight with VPS35 Asp620Asn mutations.
SNCA
The clinical characteristics of the 29 PD patients carrying either SNCA rearrangements [triplications (n = 2) and duplications (n = 21)] or missense mutations [Ala53Thr (n = 3) and Gly51Asp (n = 3)] are shown in Table 3. All but three of the families concerned originated from France. The remaining three families, originating from Italy, Turkey and Morocco, all had SNCA duplications. Within this cohort, SNCA duplications were the most frequent mutation identified (14/19, 73.7%), followed by the Ala53Thr mutation (3/19, 18.8%). Disease onset occurred earliest in patients with the Ala53Thr mutation (mean 34.7 [SD 7.6], range 26–40 years).
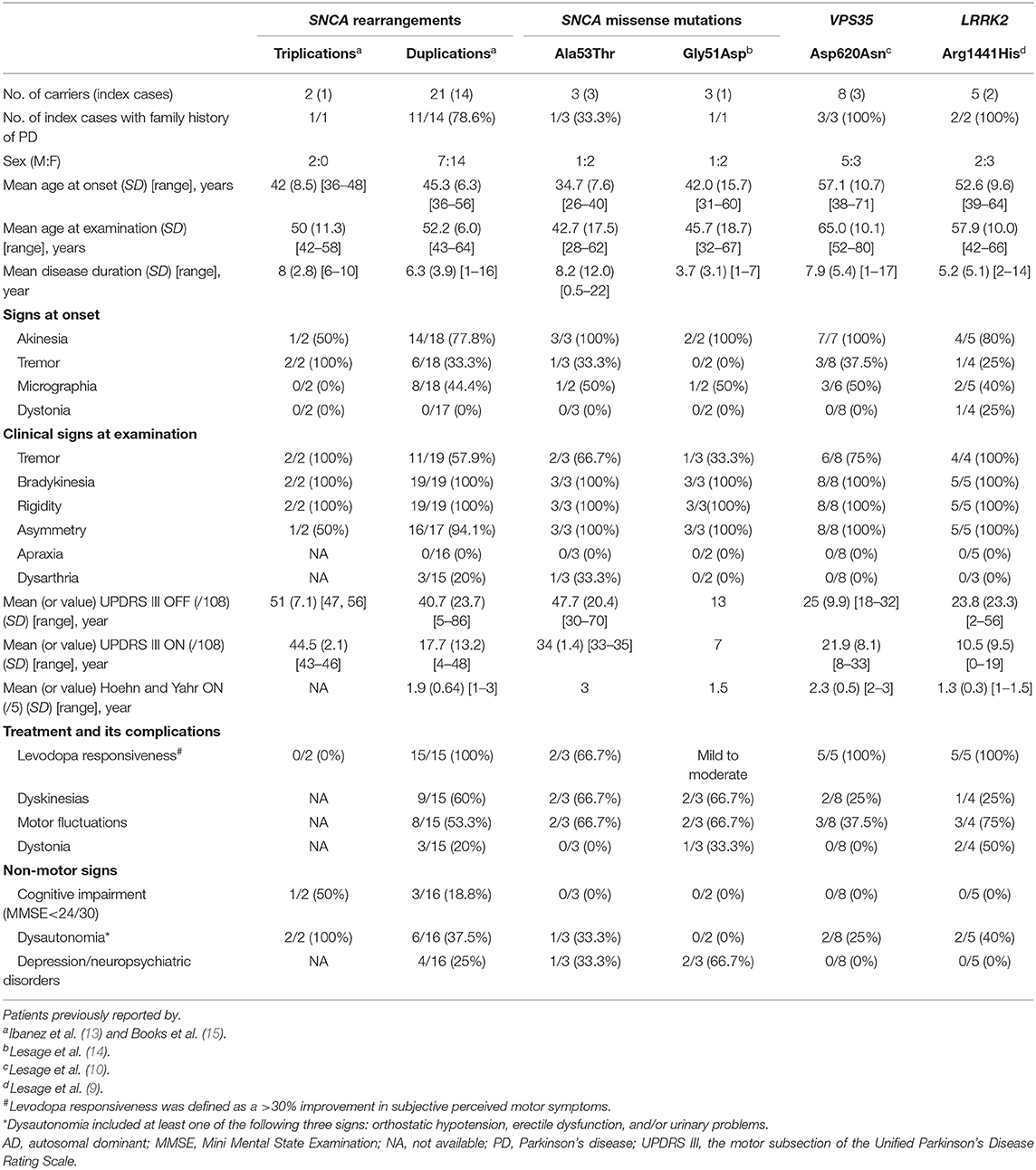
Table 3. Summary of the clinical data for patients carrying SNCA, VPS35, and LRRK2 Arg1441His mutations identified in this study.
Patients carrying the Ala53Thr mutation had an extra-pyramidal parkinsonian syndrome, but with heterogeneity between patients with the same mutation: Patient 1172-001, with both SNCA Ala53Thr and a heterozygous PRKN Thr240Met variant, had atypical PD, with a poor response to levodopa, early motor fluctuations and cerebellar signs. He rapidly developed impulse control disorders. This patient currently displays no cognitive decline. He had a bilateral subthalamic deep brain stimulation (STN-DBS). He received clozapine treatment for delusions with a beneficial effect. Patient 1219-001 presented a parkinsonian syndrome that responded well to levodopa, but developed severe dysarthria. She underwent unilateral internal globus pallidus (GPi)-DBS. This patient presented no major cognitive and behavioral signs other than an alteration of executive functions. A third PD patient, 196-016 presented early-onset (26 years) typical PD that responded well to levodopa. Detailed clinical data are provided in Supplementary Table 2.
Both SNCA locus triplications and Gly51Asp mutation were associated with early-onset atypical parkinsonism (mean AAO: 42.0 years [SD 8.5], range: 36–48 years and mean AAO: 42.0 [SD 15.7], range: 31–60 years, respectively). The patients with SNCA triplications were characterized by severe cognitive impairment in one of two carriers, dysautonomia, a poor response to levodopa in both patients. The three Gly51Asp mutation carriers had shorter disease duration (mean: 3.7 years [SD 3.1], range: 1–7 years), a mild-to-moderate response to levodopa, frequent psychiatric symptoms but no dementia or autonomic dysfunction (Supplementary Table 2). By contrast, AAO was highest in patients with SNCA duplications (mean AAO: 45.3 years [SD 6.3], range: 36–56 years), consistent with typical PD and a good response to levodopa (100%), with more than 50% of these patients reporting levodopa-induced motor complications. Non-motor symptoms, including depression/psychosis and dysautonomia, were present in about one third of the reported cases, but cognitive decline was less frequent (18.8%, 3/16). Detailed clinical characteristics for PD patients with SNCA multiplications are provided in Supplementary Table 3.
VPS35
Five of the eight PD patients carrying VPS35 Asp620Asn mutations have been described before (10). The three newly genotyped patients were relatives of patient 838–006 (Supplementary Table 4). Briefly, patients carrying VPS35 mutations had features similar to those with idiopathic PD, with a mean AAO of ~57 years (range: 38–71 years, Table 3): all patients presented the classical triad, with akinesia as the predominant symptom at onset (100%), but a much lower frequency of tremor as an initial symptom (37.5%), a good response to levodopa (100%), with <37% of those treated developing dyskinesias and motor fluctuations, and a low rate of dysautonomia (2/8, 25%), with no cognitive or neuropsychiatric symptoms or atypical signs.
LRRK2
We compared the clinical features of the LRRK2 G2019S+ PD patient (LRRK2+) group with those of PD patients with no mutations of known PD-associated genes, excluding subjects with missing data for the covariables included in the models, such as sex, AAO, and disease duration: 135/151 LRRK2+ and 1,552/1,693 PD patients without mutations were included in the final analysis (Table 4). The proportion of men was greater in the genetically undefined PD patient group than in the LRRK2+ group (61.1 vs. 51.9%, p = 0.04). The mean AAO of the LRRK2 Gly2019Ser carriers was 4 years higher than that of non-carriers (p < 0.001). The Gly2019Ser carriers were more likely to be of North-African ancestry (p < 0.001) and to report a family history of PD (p < 0.02). They had a higher UPDRS III score during the “OFF” state and a higher Hoehn and Yahr score during the “ON” state than non-carriers, but these results were no longer significant after adjustment for AAO and disease duration. The frequencies of signs at onset and at examination, the degree of response to treatment, motor complications and non-motor signs, including cognitive impairment and autonomic dysfunction were similar in both groups.
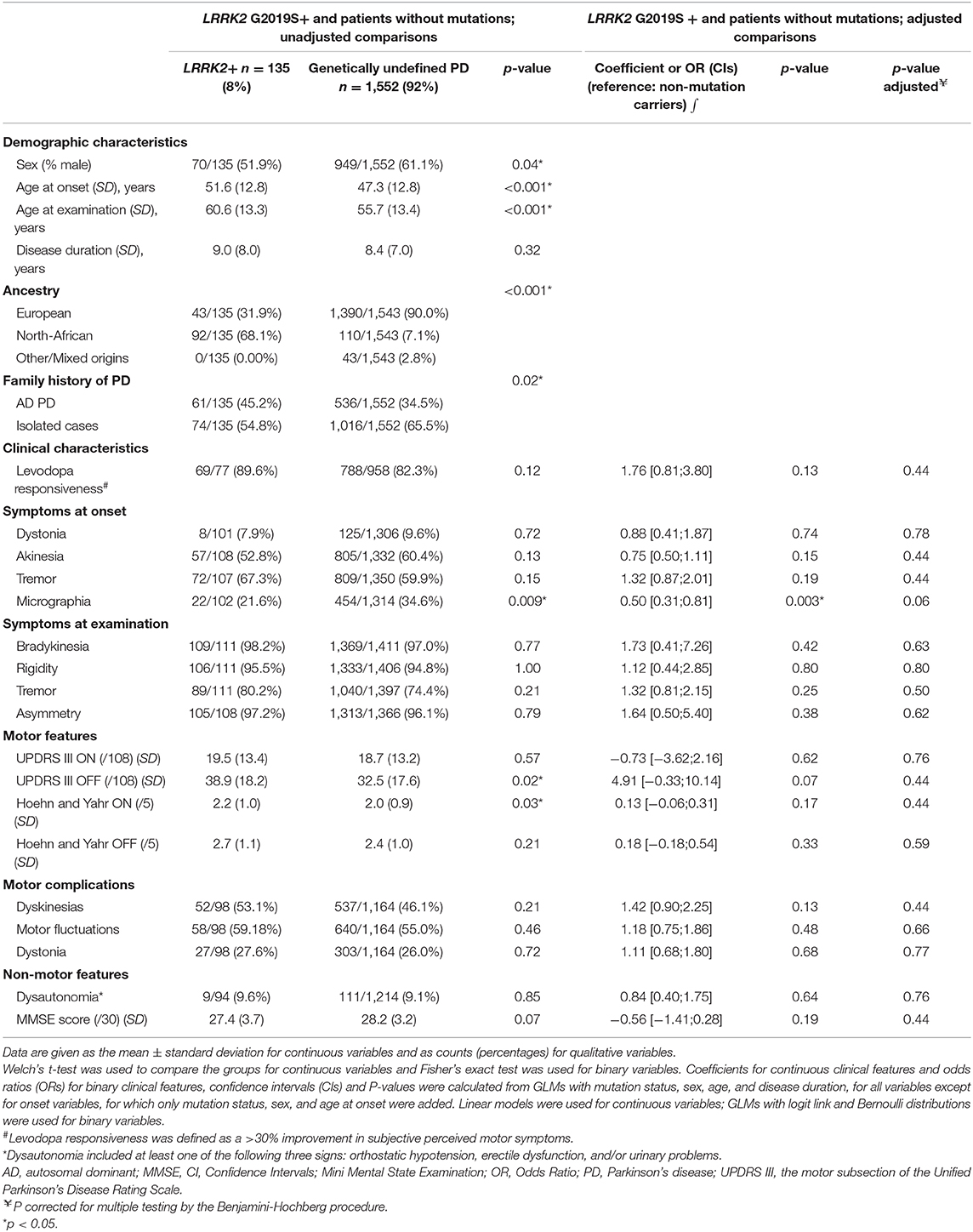
Table 4. Comparison of demographic and clinical characteristics of patients with Parkinson's disease by LRRK2 mutation status (LRRK2 carriers vs. patients without mutations in known Parkinson's disease-associated genes).
Clinical comparisons between heterozygous (n = 144) and homozygous (n = 7) Gly2019Ser mutation carriers revealed no significant differences in sex (men: 52.1 vs. 42.9%, OR 1.4, CI [0.31–6,7], p = 0.71), AAO (mean 51.4 [SD 12.1] vs. mean 53.7 [SD 11.2] years, p = 0.62), disease duration (mean 8.7 [SD 6.7] vs. 9.1 [SD 4.5] years, p = 0.87) or clinical presentation, but the number of homozygous carriers was small.
Unlike LRRK2 Gly2019Ser carriers, all patients with the Arg1441His mutation were French and all reported a family history of PD. They had a shorter disease duration (mean: 5.2 years [SD 5.1], range: 2–14 years vs. mean 9.0 years [SD 8.0], range: 0.5–63 years), were more likely to develop an akinetic-rigid motor phenotype (80 vs. 53%), had a slightly better response to levodopa (100 vs. 90%), and an absence of cognitive and neuropsychiatric symptoms, but a similar mean age at onset (52.6 years [SD 9.6], range: 39–64 years vs. 51.4 years [SD 12.1], range: 29–86 years).
Discussion
This is one of the largest national multi-center studies to investigate the frequency of variants of the three major genes unequivocally linked to AD PD—LRRK2, SNCA, and VPS35—and their associated phenotypes in a large cohort of >1,800 French and North African index PD cases. We report an overall mutation frequency of 8.9% across both populations, the LRRK2 G2019S mutation being the most frequently identified variant (7.5%), particularly in familial rather than isolated cases. However, the frequency of mutations differed considerably between populations. We confirm here that the LRRK2 Gly2019Ser mutation is the principal genetic cause of PD in our cases of North-African ancestry, reaching an overall frequency of 45% (100/221) and 62% (34/55) in familial cases. By contrast, this mutation was present at a much lower rate of 2.4% (36/1,530) in our native French PD cases. Our findings are consistent with those of previous multi-center studies (16). SNCA duplications were the second most common type of mutations, identified in 14 index cases (0.78%). SNCA duplication carriers were mostly of European ancestry, particularly French (93%), tended to be predominantly females, probably due to random or recruitment bias, and had a higher frequency of a family history of PD. We also identified three unrelated PD patients carrying the SNCA Ala53Thr mutation. Although generally rare, this mutation appears to be particularly common in the Italian and Greek populations, due to a founder effect (17, 18). Only four individuals without Greek or Italian ancestry have been reported to carry this mutation, in haplotypes different from those reported in Greek and Italian families (19–22). Additional haplotype analysis would determine the ancestral origin of our three French mutation carriers. Other rare known mutations were also identified in our study: VPS35 Asp620Asn and LRRK2 Arg1441His in three and two AD PD families, respectively. At least 25 LRRK2 Arg1441His carriers, including those described here, have been reported to date [(3); www.mdsgene.org]. Most were Caucasian, and all but one case reported a family history of the disease. This pathogenic variant was not found in more than 6,000 healthy controls tested (23) and is very rare in the Genome Aggregation Database (GnomAD) (1/31,298 alleles); it is therefore very likely to be pathogenic. Consistent with this conclusion, the Arg1441His variant affects the same amino-acid residue as two other recurrent PD-causing mutations (Arg1441Cys and Arg1441Gly). Finally, previous haplotype analyses did not support the hypothesis of a common founder for the Arg1441His variant, instead suggesting that there might be a mutational hotspot. Following initial reports of the existence of several VPS35 variants (5, 6), pathogenicity has been confirmed only for the Asp620Asn variant. Consistent with our findings, this recurrent mutation has been identified predominantly in families of Caucasian descent affected by AD PD. A meta-analysis of 21,824 PD patients from 15 case-control studies performed worldwide from 2011 to 2016 identified an overall mutation frequency of 0.12% (0.29% in familial cases and 0.023% in isolated cases) [reviewed in (24)], and an absence of this mutation from healthy controls and the GnomAD public database.
The clinical features of our LRRK2 mutation carriers, whether heterozygous or homozygous, were indistinguishable from those of patients with no mutations in known PD-associated genes. These features overlapped those of typical, idiopathic PD. In our study, patients with the G2019S mutation had a mean AAO of ~52 years, a high proportion of patients with late AAO (>50 years), a good response to levodopa, a predominance of tremor as a first symptom of PD, about a quarter had cognitive impairment, about 10% had dysautonomia, but no other atypical signs after a mean disease duration of ~10 years. Although the clinical features of the LRRK2 Gly2019Ser carriers compared with patients with idiopathic PD in literature are conflicting [meta-analysis in (25)], even for the same ethnic PD population [i.e., of North-African origin; (26–31)], our data are consistent with those of 724 LRRK2 mutation carriers listed in the MDSGene database. Like LRRK2 mutation carriers, VPS35 Asp620Asn carriers had a phenotype very similar overall to that of idiopathic PD: absence of atypical signs, excellent levodopa response, normal cognition, and absence of neuropsychiatric features. However, the mean AAO of our patients appeared to be later (57 years) than that reported in a recent meta-analysis (51 years) (32), due to the presence of multiple affected relatives with a late onset of disease within the same family (see Supplementary Table 4). Lastly, SNCA mutation carriers had motor features similar to those of idiopathic PD, but an overall earlier AAO, a shorter disease course, a higher frequency of motor complications, a higher frequency of non-motor signs and symptoms (cognitive decline in 17%, autonomic dysfunction in 39%, and psychotic symptoms, and depression in 32%). Atypical signs have also been observed in rare PD patients carrying the SNCA Gly51Asp mutation (14). In this study, we also identified a rare known variant of SNCA, His50Gln, in the homozygous state. This variant has been described as a causal variant associated with late-onset PD, dementia, and dystonia (33, 34), but a revaluation in larger datasets of PD patients and controls, including the GnomAD database (23/282,808 alleles), provided no evidence of pathogenicity for this variant (35). However, interestingly, the 42 year-old female patient carrying the His50Gln variant in our cohort had clinical features similar to those observed in carriers of other types of SNCA mutation carriers. She presented an early AAO (32 years), an excellent response to levodopa, motor complications, akinetic-rigid parkinsonism, and dystonia, an absence of cognitive decline and neuropsychiatric symptoms, but the presence of autonomic dysfunction and atypical neurological signs, such as postural instability, REM sleep behavior disorder (RBD) and impulse control disorders (see Supplementary Table 2). However, in this study, the SNCA His50Gln was found using our customized gene panel and in absence of whole exome/genome sequencing to detect other possible pathogenic mutations, its pathogenicity remains inconclusive.
The principal strength of this national multi-center study is the large group of well-phenotyped and genotyped patients and family members recruited at the 16 different PDG centers, and the use of a standardized protocol, ensuring comparable, and consistent clinical data reporting and diagnoses at each center. This enabled us to refine the estimated prevalence of mutations in genes causing AD PD in France. We show that our population, although mixed, has a relatively high frequency of SNCA, LRRK2, and VPS35 mutations. However, the clinical data were cross-sectional, most patients were European or North African, and our populations were biased toward EO cases.
In most instances, the phenotypes of cases due to AD PD mutations are indistinguishable from those of cases without mutations, demonstrating the need for genetic analysis for their identification. Gene-specific disease-modifying therapies are currently being developed and tested. More generalized genetic testing is therefore required in PD patients, to identify those most likely to benefit from personalized care (36).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by INSERM, CCPPRB (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) du Groupe Hospitalier Pitié-Salpêtrière, Paris, France. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
SL conceived, designed and organized the study, wrote the first draft, reviewed, and critically revised the manuscript. J-CC and AB conceived the project, reviewed, and critically revised the manuscript. MH contributed to the statistical analysis and critically revised the manuscript. GM, CT, HB, SF, MA, CB-C, EB, ST, PD, FD, ER, FT, DG, FO-M, BD, FV, FC-D, A-MO-H, MV, EL, and AS contributed to the execution of the research project and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Fondation de France, France-Parkinson Association, la Fédération pour la Recherche sur le Cerveau (FRC) and the French program Investissements d'avenir (ANR-10-IAIHU-06). This work was also supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; Project No. ZO1 AG000949.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients and their families. We thank the DNA and Cell Bank of the ICM for sample preparation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00682/full#supplementary-material
Abbreviations
AAO, age at onset; AD, autosomal dominant; LRRK2, leucine-rich repeat kinase 2; MMSE, mini mental state examination; PD, Parkinson's disease; VPS35, vacuolar protein sorting 35.
References
1. Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson's disease. Rev Neurol. (2018) 174:628–43. doi: 10.1016/j.neurol.2018.08.004
2. Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case-control study. Lancet Neurol. (2011) 10:898–908. doi: 10.1016/S1474-4422(11)70175-2
3. Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. (2018) 33:1857–70. doi: 10.1002/mds.27527
4. Lesage S, Patin E, Condroyer C, Leutenegger AL, Lohmann E, Giladi N, et al. Parkinson's disease-related LRRK2 G2019S mutation results from independent mutational events in humans. Hum Mol Genet. (2010) 19:1998–2004. doi: 10.1093/hmg/ddq081
5. Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. (2011) 89:162–7. doi: 10.1016/j.ajhg.2011.06.001
6. Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. (2011) 89:168–75. doi: 10.1016/j.ajhg.2011.06.008
7. Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Bertram L, et al. A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. (2012) 49:721–6. doi: 10.1136/jmedgenet-2012-101155
8. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. (1992) 55:181–4. doi: 10.1136/jnnp.55.3.181
9. Lesage S, Condroyer C, Lannuzel A, Lohmann E, Troiano A, Tison F, et al. Molecular analyses of the LRRK2 gene in European and North African autosomal dominant Parkinson's disease. J Med Genet. (2009) 46:458–64. doi: 10.1136/jmg.2008.062612
10. Lesage S, Condroyer C, Klebe S, Honore A, Tison F, Brefel-Courbon C, et al. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology. (2012) 78:1449–50. doi: 10.1212/WNL.0b013e318253d5f2
11. Bouhouche A, Tesson C, Regragui W, Rahmani M, Drouet V, Tibar H, et al. Mutation analysis of consanguineous Moroccan patients with Parkinson's disease combining microarray and gene panel. Front Neurol. (2017) 8:567. doi: 10.3389/fneur.2017.00567
12. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016/j.ajhg.2016.01.014
13. Ibanez P, Lesage S, Janin S, Lohmann E, Durif F, Destee A, et al. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. (2009) 66:102–8. doi: 10.1001/archneurol.2008.555
14. Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
15. Book A, Guella I, Candido T, Brice A, Hattori N, Jeon B, et al. A meta-analysis of α-synuclein multiplication in familial parkinsonism. Front Neurol. (2018) 9:1021. doi: 10.3389/fneur.2018.01021
16. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
17. Papadimitriou D, Antonelou R, Miligkos M, Maniati M, Papagiannakis N, Bostantjopoulou S, et al. motor and nonmotor features of carriers of the p.a53t alpha-synuclein mutation: a longitudinal study. Mov Disord. (2016) 31:1226–30. doi: 10.1002/mds.26615
18. Tambasco N, Nigro P, Romoli M, Prontera P, Simoni S, Calabresi P. A53T in a parkinsonian family: a clinical update of the SNCA phenotypes. J Neural Transm. (2016) 123:1301–7. doi: 10.1007/s00702-016-1578-6
19. Michell AW, Barker RA, Raha SK, Raha-Chowdhury R. A case of late onset sporadic Parkinson's disease with an A53T mutation in alpha-synuclein. J Neurol Neurosurg Psychiatry. (2005) 76:596–7. doi: 10.1136/jnnp.2004.046425
20. Ki CS, Stavrou EF, Davanos N, Lee WY, Chung EJ, Kim JY, et al. The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. Clin Genet. (2007) 71:471–3. doi: 10.1111/j.1399-0004.2007.00781.x
21. Puschmann A, Ross OA, Vilariño-Güell C, Lincoln SJ, Kachergus JM, Cobb SA, et al. A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat Disord. (2009) 15:627–32. doi: 10.1016/j.parkreldis.2009.06.007
22. Xiong WX, Sun YM, Guan RY, Luo SS, Chen C, An Y, et al. The heterozygous A53T mutation in the alpha-synuclein gene in a Chinese Han patient with Parkinson disease: case report and literature review. J Neurol. (2016) 263:1984–92. doi: 10.1007/s00415-016-8213-1
23. Lubbe SJ, Escott-Price V, Gibbs JR, Nalls MA, Bras J, Price TR, et al. Additional rare variant analysis in Parkinson's disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet. (2016) 25:5483–9. doi: 10.1093/hmg/ddw348
24. Gambardella S, Biagioni F, Ferese R, Busceti CL, Frati A, Novelli G, et al. vacuolar protein sorting genes in Parkinson's disease: a re-appraisal of mutations detection rate and neurobiology of disease. Front Neurosci. (2016) 10:532. doi: 10.3389/fnins.2016.00532
25. Shu L, Zhang Y, Pan H, Xu Q, Guo J, Tang B, et al. Clinical heterogeneity among LRRK2 variants in Parkinson's disease: a meta-analysis. Front Aging Neurosci. (2018) 10:283. doi: 10.3389/fnagi.2018.00283
26. Ishihara L, Gibson RA, Warren L, Amouri R, Lyons K, Wielinski C, et al. Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson's disease families. Mov Disord. (2007) 22:55–61. doi: 10.1002/mds.21180
27. Lesage S, Belarbi S, Troiano A, Condroyer C, Hecham N, Pollak P, et al. Is the common LRRK2 G2019S mutation related to dyskinesias in North African Parkinson disease? Neurology. (2008) 71:1550–2. doi: 10.1212/01.wnl.0000338460.89796.06
28. Nishioka K, Kefi M, Jasinska-Myga B, Wider C, Vilariño-Güell C, Ross OA, et al. (2010). A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson's disease. J Neurol Neurosurg Psychiatry. (2010) 81:391–5. doi: 10.1136/jnnp.2009.185231
29. Trinh J, Amouri R, Duda JE, Morley JF, Read M, Donald A, et al. Comparative study of Parkinson's disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol Aging. (2014) 35:1125–31. doi: 10.1016/j.neurobiolaging.2013.11.015
30. Nabli F, Ben Sassi S, Amouri R, Duda JE, Farrer MJ, Hentati F. Motor phenotype of LRRK2-associated Parkinson's disease: a Tunisian longitudinal study. Mov Disord. (2015) 30:253–8. doi: 10.1002/mds.26097
31. Ben Romdhan S, Farhat N, Nasri A, Lesage S, Hdiji O, Ben Djebara M, et al. LRRK2 G2019S Parkinson's disease with more benign phenotype than idiopathic. Acta Neurol Scand. (2018) 138:425–31. doi: 10.1111/ane.12996
32. Deutschländer A, Ross OA, Wszolek ZK. VPS35-related Parkinson disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington (2017). p. 1993–2020.
33. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. (2013) 28:811–3. doi: 10.1002/mds.25421
34. Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel α-synuclein missense mutation in Parkinson disease. Neurology. (2013) 80:1062–4. doi: 10.1212/WNL.0b013e31828727ba
35. Blauwendraat C, Kia DA, Pihlstrøm L, Gan-Or Z, Lesage S, Gibbs JR. Insufficient evidence for pathogenicity of SNCA His50Gln (H50Q) in Parkinson's disease. Neurobiol Aging. (2018) 64:159.e5–159.e8. doi: 10.1016/j.neurobiolaging.2017.12.012
Keywords: Parkinson's disease, LRRK2, G2019S, SNCA, VPS35, autosomal dominant inheritance, genotype-phenotype correlations
Citation: Lesage S, Houot M, Mangone G, Tesson C, Bertrand H, Forlani S, Anheim M, Brefel-Courbon C, Broussolle E, Thobois S, Damier P, Durif F, Roze E, Tison F, Grabli D, Ory-Magne F, Degos B, Viallet F, Cormier-Dequaire F, Ouvrard-Hernandez A-M, Vidailhet M, Lohmann E, Singleton A, Corvol J-C and Brice A (2020) Genetic and Phenotypic Basis of Autosomal Dominant Parkinson's Disease in a Large Multi-Center Cohort. Front. Neurol. 11:682. doi: 10.3389/fneur.2020.00682
Received: 16 April 2020; Accepted: 08 June 2020;
Published: 28 July 2020.
Edited by:
Chin-Hsien Lin, National Taiwan University Hospital, TaiwanReviewed by:
Manabu Funayama, Juntendo University, JapanMin-Yu Lan, Kaohsiung Chang Gung Memorial Hospital, Taiwan
Adeline Ng, National Neuroscience Institute (NNI), Singapore
Copyright © 2020 Lesage, Houot, Mangone, Tesson, Bertrand, Forlani, Anheim, Brefel-Courbon, Broussolle, Thobois, Damier, Durif, Roze, Tison, Grabli, Ory-Magne, Degos, Viallet, Cormier-Dequaire, Ouvrard-Hernandez, Vidailhet, Lohmann, Singleton, Corvol and Brice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suzanne Lesage, c3V6YW5uZS5sZXNhZ2VAdXBtYy5mcg==
†See Supplementary Information for a full list of members of the PDG group