 Samantha E. Hoffman1
Samantha E. Hoffman1 Sally A. Al Abdulmohsen1
Sally A. Al Abdulmohsen1 Saksham Gupta1
Saksham Gupta1 Blake M. Hauser1David M. Meredith2Ian F. Dunn3
Blake M. Hauser1David M. Meredith2Ian F. Dunn3 Wenya Linda Bi1*
Wenya Linda Bi1*- 1Center for Skull Base and Pituitary Surgery, Harvard Medical School, Brigham and Women's Hospital, Boston, MA, United States
- 2Department of Pathology, Harvard Medical School, Brigham and Women's Hospital, Boston, MA, United States
- 3Department of Neurosurgery, University of Oklahoma College of Medicine, Oklahoma City, OK, United States
Chordomas are rare tumors that are notoriously refractory to chemotherapy and radiotherapy when radical surgical resection is not achieved or upon recurrence after maximally aggressive treatment. The study of chordomas has been complicated by small patient cohorts and few available model systems due to the rarity of these tumors. Emerging next-generation sequencing technologies have broadened understanding of this disease by implicating novel pathways for possible targeted therapy. Mutations in cell-cycle regulation and chromatin remodeling genes have been identified in chordomas, but their significance remains unknown. Investigation of the immune microenvironment of these tumors suggests that checkpoint protein expression may influence prognosis, and adjuvant immunotherapy may improve patient outcome. Finally, growing evidence supports aberrant growth factor signaling as potential pathogenic mechanisms in chordoma. In this review, we characterize the impact on treatment opportunities offered by the genomic and immunologic landscape of this tumor.
Introduction
Chordomas are locally aggressive tumors arising from fetal notochord remnants with an average annual incidence of 0.088 per 100,000 in the United States (1). These tumors frequently occur at the skull base, followed by sacrum and vertebral bodies, with increasing incidence with age (1). Gross total resection followed by adjuvant radiation offers long-term disease control in many patients (2, 3). However, encasement of vital neurovascular structures and concealed nests of tumor may hinder complete eradication, leading to repeated recurrences following subtotal resection (4, 5). They are also prone to seeding and late metastasis (6). Once recurrent, chordomas are extremely refractory to cure. Chemotherapy has been used for recurrent and metastatic disease, without any consistent efficacy thus far (7, 8).
The difficulties of managing aggressive and refractory chordoma have motivated study of the biological underpinnings of this disease. Genome-scale sequencing approaches and a resurgent interest in tumor immunology have been pivotal in expanding our understanding of chordoma. In this review, we discuss recent advances in the understanding of chordoma biology with a focus on its immune and extracellular microenvironment.
Histopathologic Characteristics
Although the notochord is derived from the ectoderm, chordoma shows both epithelial and mesenchymal differentiation. Their spectrum of inter- and intratumoral histological patterns render their level of differentiation and clinical behavior challenging to predict (9). The World Health Organization classification of chordoma subtypes includes conventional chordoma (Figure 1), chondroid chordoma (Figure 2), and dedifferentiated chordomas (Figure 3). Conventional chordomas show three neoplastic cell types in a myxoid stroma: (1) intermediate-sized physaliphorous cells are the main proliferating cell type and appear as distinctive vacuolated cells in cords and lobules, (2) large cells with prominent vesicular nuclei, and (3) small cells with pyknotic nuclei. Dedifferentiated chordomas are associated with poorer prognosis compared to conventional and chondroid subtypes, while chondroid chordoma is associated with worse survival than classic chordoma (10). Importantly, chondroid chordoma should be distinguished from chondrosarcoma based on brachyury immunopositivity, as chondrosarcoma has a significantly more indolent growth rate than any chordoma, and therefore may merit different treatment considerations than chordoma itself (11).
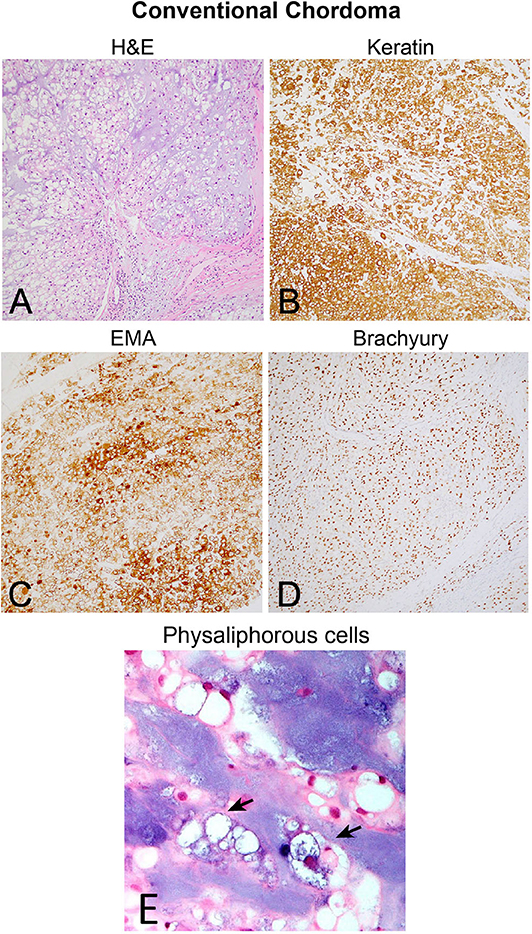
Figure 1. (A) Conventional chordoma showing classic histologic features of large, epithelioid tumor cells with eosinophilic cytoplasm containing multiple vacuoles arranged in nests and cords in an overall lobular growth pattern. Tumor cells are reliably positive for (B) keratins and (C) EMA. S100 staining may be seen in select cases but is not ubiquitous (not shown). (D) Nuclear positivity for brachyury is highly specific and observed in the vast majority of cases. (E) Physaliphorous cells (arrows) display abundant, “bubbly” cytoplasm with small, round nuclei on higher magnification. Large and small cells can be seen in the background of blue, myxoid stroma.
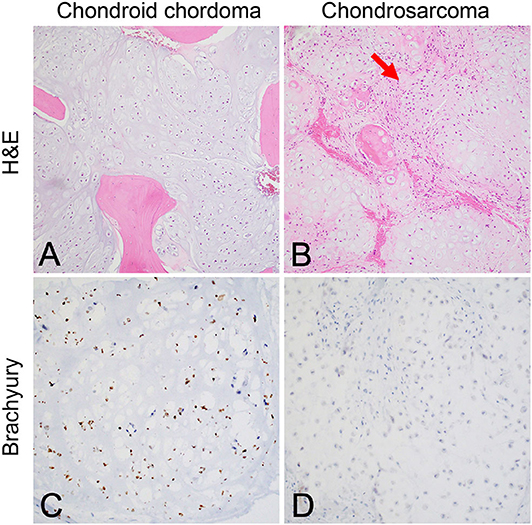
Figure 2. (A) Chondroid chordoma characteristically contains regions forming hyaline cartilage with tumor cells distributed individually in lacunar spaces that greatly resemble (B) low-grade chondrosarcoma, which classically feature hypercellular hyaline cartilage lobules with a permeative growth pattern. Chondrosarcomas of the skull base in particular have variably prominent myxoid areas (arrow) that resemble chondroid chordoma. (C) Brachyury nuclear positivity confirms the diagnosis of chordoma, while being (D) consistently negative in chondrosarcoma.
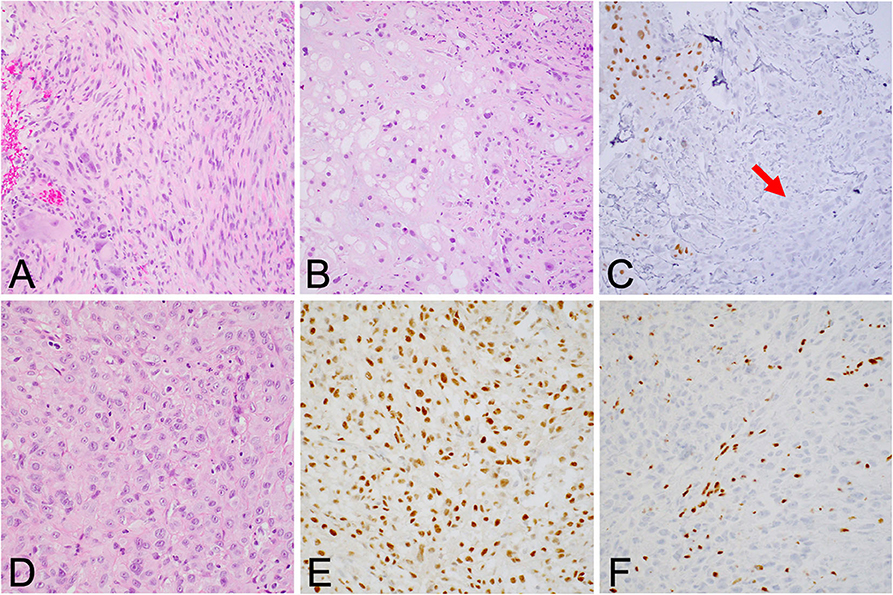
Figure 3. (A) Dedifferentiated chordoma contains regions of high-grade sarcoma, frequently juxtaposed with regions of conventional chordoma (B). (C) Brachyury positivity is often negative in the dedifferentiated component (arrow). (D) Poorly differentiated chordoma exhibits higher cellularity than conventional chordoma and occasionally shows rhabdoid morphology. (E) Brachyury is reliably positive, and (F) SMARCB1 (INI-1) loss in tumor cells is characteristic of this entity.
Genomic Alterations
Some de novo chordomas harbor chromosomal changes, which are associated with poorer prognoses (12, 13). The most common genomic alterations include large copy number losses in chromosomes 1p, 3, 4, 9, 10, 13, and 14; chromosomal deletions are more common than gains (14). Wide-scale deletions in 1p and 9p specifically have been associated with worse prognosis even after resection and radiation (15). Recurrence is not associated with increased copy number loss or gain compared to primary tumors (16).
Brachyury
Brachyury is a protein whose expression is the diagnostic hallmark of chordoma, regardless of subtype (17). Encoded by the TBXT or T gene on chromosome 6q, brachyury binds palindromic T sites to exert DNA-regulatory effects. Germline duplications of T are associated with familial chordomas, while somatic tandem T duplications have been identified in sporadic chordoma (18). Chordomas with increased brachyury expression are associated with poorer prognosis (18).
A member of the T-box protein family, brachyury is a transcription factor that regulates notochord cell fate determination during development (17). Brachyury is also a mediator of the epithelial-to-mesenchymal transition by downregulating E-cadherin expression, explaining both the heterogeneous histology and the metastatic tendency of chordomas (17).
The ubiquitous brachyury expression in chordoma makes it an attractive therapeutic target; however, its nuclear localization has barred access to targeted inhibitors, setting the stage for immunotherapeutic approaches (19). Targeting brachyury with a recombinant yeast vaccine has been found to activate human T cells in vitro and enhance immune response in phase I trials; brachyury vaccination is now being investigated in a phase II trial as well (Table 1) (20).
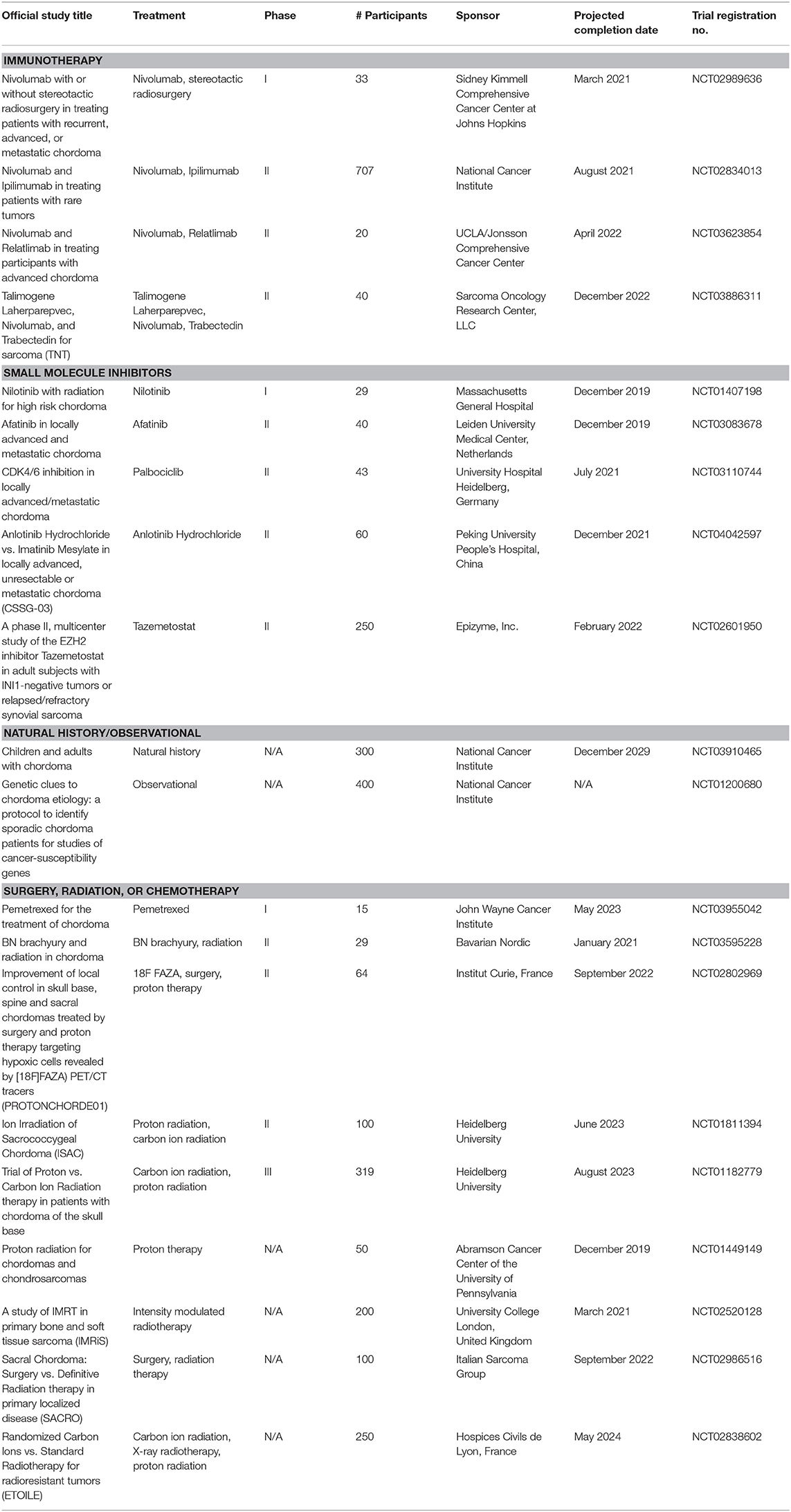
Table 1. Active trials for surgical, radiation, and medical therapies for chordoma (extrapolated from ClinicalTrials.gov; December 30, 2019).
Cell Cycle Regulation (CDKN2A, PTEN)
Upregulation of other T-box genes in malignancy is thought to repress cell-cycle regulators like Cyclin-dependent kinase inhibitor 2A (CDKN2A). The CDKN2A gene encodes two tumor suppressor proteins that inhibit progression through the cell cycle; mutations in this gene therefore result in an uncontrolled rate of cell proliferation and differentiation via activation of cyclin-dependent kinases 4 and 6 (CDK4/6). Similarly, the phosphate and tensin homolog (PTEN) gene also encodes for a tumor suppressor protein and regulates cell growth (Figure 5).
Genomic analysis of chordoma samples reveals widespread cell cycle dysregulation (14, 21, 22). Both homozygous and heterozygous copy number losses of CDKN2A have been identified in chordomas (14, 18, 21, 22). Of note, tumors with copy number losses in CDKN2A and PTEN also harbored point mutations in those genes, though no activating mutations were identified (21).
The success of CDK4/6 inhibitors in other malignancies has motivated their application to chordomas. Treatment of a CDKN2A-deleted chordoma cell line in vitro with palbociclib, for example, resulted in significant growth inhibition (23). Preserved CDK4/6 expression may serve as a potential biomarker for future screening.
Many chordomas feature PTEN loss, which may drive oncogenesis both through loss of tumor suppression and an increase in PD-L1 expression (7). To date, no studies have examined existing molecular therapies for chordomas with PTEN loss. Instead, PTEN upregulation has been investigated as a sensitizing factor to improve response to PDGFR and mTOR pathway inhibitors in these tumors (24, 25).
Chromatin Remodeling
The pathogenesis of chordoma may involve DNA-level dysregulation that promotes oncogene expression or tumor suppressor silencing. A cardinal regulator of chromatin organization and subsequent regulation of gene expression is the Switch/Sucrose Non-Fermentable (SWI/SNF) protein complex. Mutations of SWI/SNF genes result in a loss of chromatin regulation, facilitating neoplastic development via uncontrolled DNA replication and subsequent cell proliferation.
The SMARCB1 gene is a SWI/SNF component that is thought to serve a tumor suppressor role by regulating the histone methylation activity of the transcription factor EZH2 (26). SMARCB1 loss is the defining marker of poorly differentiated chordomas (PDCs), a highly aggressive subtype distinct from dedifferentiated chordomas that occur most frequently in children and at the skull base. Specifically, PDCs display a loss of nuclear SMARCB1 expression and homozygous deletions of SMARCB1, implicating SWI/SNF aberrations in their rapidly progressive course (21, 27–29).
In other chordoma subtypes where SMARCB1 was downregulated but not lost, upregulation of microRNAs associated with transforming growth factor β signaling (TGF- β) was observed (30). TGF-β signaling may drive chordoma progression and survival through both augmented bone formation (31) and anti-inflammatory effects (32).
Mutations in three other SWI/SNF members—PBRM1, SETD2, ARID1A—have been identified as potential drivers in chordomas (18, 29); however, the relevance of these mutations to chordoma etiology remains to be elucidated.
Immune Microenvironment
The immune microenvironment offers a therapeutic window for chordomas since there are no reliable molecular markers to predict clinical outcome and drug response in these tumors (Table 2) (45). The tumor microenvironment is often more genetically stable than tumor cells, which can rapidly mutate and acquire drug resistance. A lack of successful treatment options and sparse genetic drivers has driven exploration of immunomodulation strategies such as vaccination with tumor-specific neoantigens, chimeric antigen receptor T-cell (CAR-T cell) engineering, and blocking immune checkpoints.
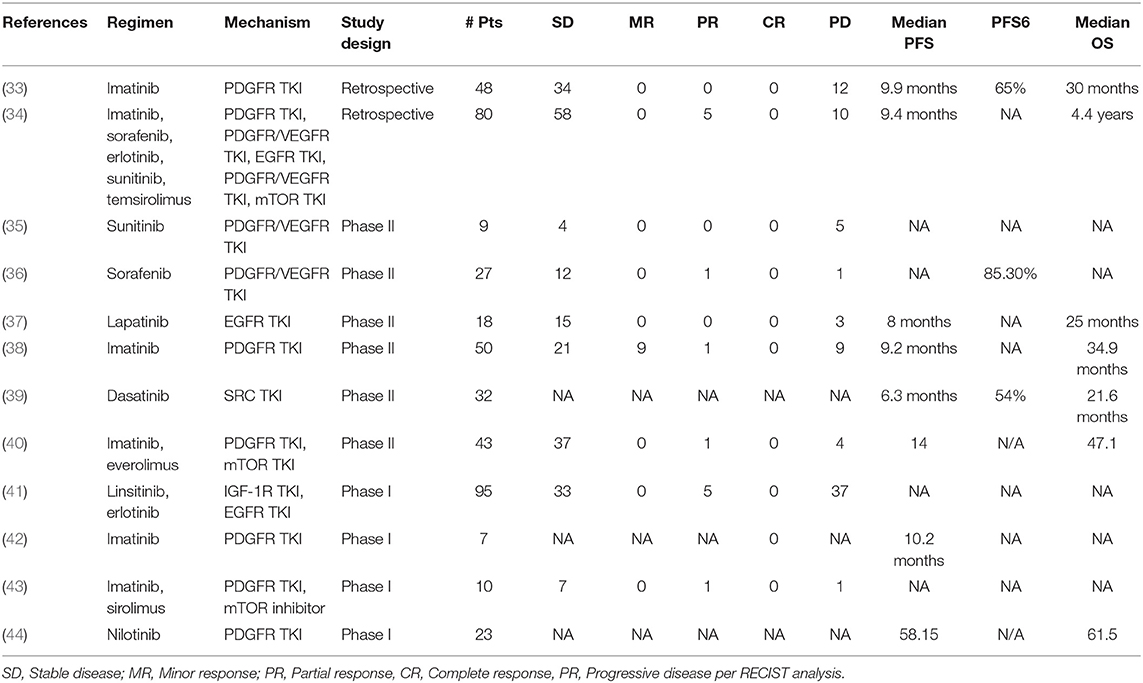
Table 2. Completed clinical trials for targeted therapies in chordoma.
Though chordomas display an elevated immune infiltration, the immune effectors are likely generating an anti-inflammatory tumor microenvironment (46). For instance, infiltrating macrophages in chordomas are predominantly M2 (anti-inflammatory) macrophages; elevated CD47 expression on chordoma cells also downregulates pro-inflammatory macrophage activity (47).
Cytokine signaling generated by the immune microenvironment may augment the malignant behavior of these tumors. Tumor necrosis factor alpha (TNF-α) is an inflammatory cytokine produced by tumor-associated macrophages that can promote tumor growth and progression. Leukemia inhibitory factor (LIF) is a cytokine in the interleukin-6 family whose functions include regulating proliferation of cancer stem cells, like those often found in chordoma (48). Expression of either TNF-α or LIF in chordoma cell lines promoted cell migration, invasive capabilities, and anchorage-independent growth, demonstrating their pro-tumoral and metastatic potential (49). Exposure of chordomas to TNF-α upregulates not only its own gene expression but also LIF expression, and vice versa; both cytokines are correlated with increased tumor size, implicating these factors in chordoma growth (49, 50).
Immune cell signaling also induces alterations in gene expression in chordoma cells. Epithelial marker expression was downregulated while mesenchymal marker expression was upregulated following exposure to TNF-α, implicating TNF-α in the chordoma epithelial-mesenchymal transition (49). Differential gene expression analysis post-TNF-α exposure demonstrated upregulation of the phosphoinositide 3-kinase (PI3K)/Akt, Ras, and Ras-related protein 1 (Rap1) signaling pathways, implicated in progression of chordomas and other malignancies, respectively (49, 51). Pro-angiogenic and anti-apoptotic pathways were also upregulated with longer TNF-α exposure, which may contribute to the survival and metastatic potential of chordomas (49).
The chordoma microenvironment can impact treatment efficacy by limiting the penetration of pro-inflammatory effectors and altering the expression of putative therapeutic targets. LIF has been shown to inhibit CD8+ T-cell entry while increasing tumor-associated macrophage and regulatory T-cell entry into other tumors, decreasing the efficacy of immune checkpoint blockade (48). The abundance of extracellular matrix associated with chordomas may similarly contribute to lymphocyte distribution. Elevated stroma-tumor ratio was associated with decreased infiltration of effector T-cells and an elevated density of regulatory T cells within the tumor (52). TNF-α-exposed chordoma cells displayed increased programmed death-ligand 1 (PD-L1) but downregulated T expression; brachyury-targeted treatments may be impacted as a result (49, 50). Finally, resistance to cytotoxic chemotherapies also increases in these chordoma cells exposed to LIR and TNF-α (49, 50).
Immune Checkpoint
Immune activation is physiologically downregulated by “immune checkpoints” pathways. Regulatory T cells express immune checkpoint ligands that bind to receptors on activated cytotoxic T-cells, inducing T-cell exhaustion. Immune checkpoint proteins normally inhibit autoimmune responses, but cancer cells can also upregulate these ligands to decrease inflammatory response to the tumor. Pharmacologic blockade of these immune checkpoints, known as checkpoint inhibitor drugs, have proven efficacious immunotherapies in several types of cancer, motivating their trial in chordoma (53).
PD-1/PD-L1
Current immunotherapy trials of chordoma have focused on blockade of PD-L1, the immune checkpoint regulator responsible for suppressing regulatory T cell apoptosis (Figure 4). Whether or not chordomas harbor PD-L1 natively remains in contention, but chordoma cell lines uniformly upregulated PD-L1 expression after interferon-γ treatment (46). PD-L1 expression was also found on tumor-infiltrating macrophages and lymphocytes at the tumor-stroma interface (7). Chordomas with negative PD-L1 expression tended to have more PD-L1 positive tumor-infiltrating lymphocytes (TIL), and the prevalence of these cells correlated with metastatic potential (7).
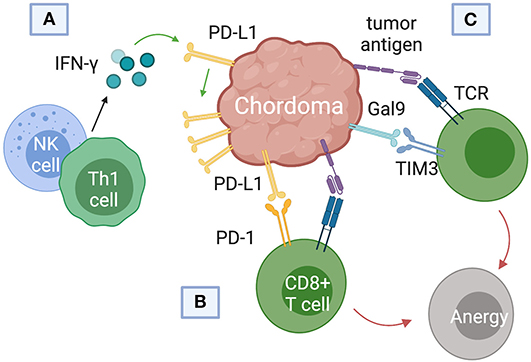
Figure 4. Immune checkpoint pathways implicated in chordoma pathogenesis and survival. (A) Interferon-gamma released from natural killer (NK) cells and helper CD4+ T (Th1) cells was found to upregulate programmed death ligand (PD-L1) expression in chordomas. (B) PD-L1 binds the programmed cell death protein 1 (PD-1) on cytotoxic T cells, inducing T cell anergy. (C) Chordomas also induce immunosuppression via the Gal9 immune-checkpoint protein binding to TIM3 on cytotoxic T cells.
The putative correlation between PD-L1 expression and malignancy augmented interest in checkpoint blockade in chordomas. Avelumab, an anti-PD-L1 monoclonal antibody, enhanced the immune-mediated killing of chordoma cells in vitro; upregulation of PD-L1 on the chordoma cells further increased their sensitivity to lysis (54). Cancer stem cells, which are resistant to radiation and chemotherapy, were also eliminated at higher rates by avelumab, suggesting that chordoma progression and recurrence may rely on immunologic escape mechanisms (54).
PD-L1 expression in chordomas may be further regulated by microRNAs, small non-coding RNA molecules responsible for post-transcriptional regulation of gene expression. Comparison of chordoma tissue with fetal notochords reveals significant microRNA dysregulation that may precede and augment the tumorigenic effects of PD-L1 upregulation (55). MiR-574-3p, for example, is a microRNA whose expression is negatively correlated with PD-L1 expression (56). Chordomas with low miR-574-3p and high PD-L1 expression were associated with higher muscle invasion, more tumor necrosis, and poorer patient outcomes (55). Elucidating the mechanism underlying this anticorrelation could augment checkpoint blockade implementation in these tumors.
TIM3/Gal9
T-cell immunoglobulin and mucin-domain 3 (TIM3) has only recently emerged as a target of interest in chordoma immunotherapy and cancer therapy in general (57–59). First identified on cells of the adaptive immune system, TIM3 is also constitutively expressed on innate immune cells (59–61). TIM3 promotes tumor survival by binding to galectin-9 (Gal9) on tumor cells and inducing T cell exhaustion (Figure 4C). (61). Combining anti-TIM3 and anti-PD-1 regimens may better preclude immunotherapy failure compared to monotherapy, invigorating research into the TIM3/Gal9 pathway as a novel pharmaceutical target (62).
Only one study has investigated TIM3/Gal9 in the setting of chordoma, but its results are consistent with corresponding studies in solid and hematological malignancies (63). In a retrospective analysis of 93 skull base chordomas, the only parameter that independently predicted local recurrence-free survival on both univariate and multivariate analysis was the density of TIM3+ lymphocytes (63). Patients with low TIM3+ TILs in their tumors also had better overall survival compared to patients with high TIM3+ TIL density (63). A putative regulator of the Gal9 native to the chordoma microenvironment has been identified: microRNA 455-5p (miR-455-5p) (63). Tumors that are Gal9+/miR-455-5p-high are associated with longer overall survival than Gal9+/miR-455-5p-low tumors, suggesting an intrinsic downregulation mechanism that could be exploited for future immunotherapy design (63).
Growth Factor Signaling
Growth factor signaling pathways are key modulators of cell growth, proliferation, and survival in normal biology. Aberrant and amplified cascades also play a significant role in tumor pathogenesis. Targeted inhibition of known oncogenic growth factors has driven chordoma clinical trials, and genomic analysis of these tumors has provided several new candidate pathways whose roles remain fully unexplored.
PDGF and EGF Pathways
Amplification of platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) signaling has been shown to increase tumor hyperplasia and survival (Figure 5). Both PDGF and EGF bind to a receptor tyrosine kinase (PDGFR and EGFR), resulting in activation of downstream signaling cascades that regulate proliferation and differentiation. Tyrosine kinase inhibitors (TKIs) can selectively block PDGFR and EGFR phosphorylation and have revolutionized cancer management.
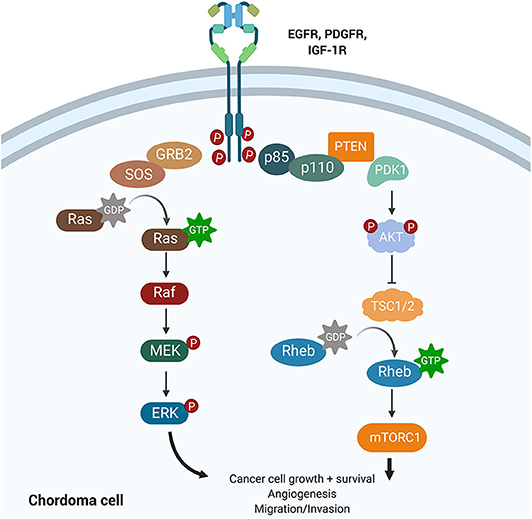
Figure 5. Growth factor signaling pathways implicated in chordoma pathogenesis. The PI3K/AKT and the RAS pathways lie downstream of EGFR, PDGFR, and IGF-1R and have been implicated in cancer promotion and progression. Upregulation of PI3K/AKT was associated with high brachyury expression in chordomas and may also regulate tuberous sclerosis comorbidity with chordomas. IGF-1R is a novel target whose activation was associated with poorer prognosis and increased tumor volume in chordomas.
There is significant burgeoning interest in applying TKIs to chordomas (Tables 1, 2). Numerous clinical trials to date have investigated the use of PDGFR and EGFR TKIs as either monotherapies or in combination with other small molecule inhibitors (Table 1). Complete response was not achieved by RECIST analysis in any of the trials or retrospective analyses, with the majority of patients experiencing either stable or progressive disease.
PI3K/Akt/mTOR Pathway
The phosphoinositide 3-kinase (PI3K)/Akt signaling pathway regulates cell cycle progression, cellular proliferation, survival, and cell growth (Figure 5). Activating mutations of this pathway, downstream of PDGF and EGF signaling, are some of the most commonly identified driver mutations in malignancy. For example, mutations in mTOR and the tumor suppressors TSC1 and TSC2, components of the PI3K/Akt cascade, have been associated with the development of tuberous sclerosis complex syndrome (TSC) in children. The dual diagnosis of TSC and chordoma in several children motivated the study of the PI3K/Akt pathway in chordomas (64). TSC-associated chordomas emerge significantly earlier than other pediatric chordomas, and sacral chordomas are associated with a significantly better prognosis when concurrent with TSC (64).
Protein analysis reveals a possible mechanism underlying the TSC-chordoma association. Positive expression of phosphorylated AKT and TSC2 identified across chordoma samples indicates loss of tumor suppression stemming from activation of the PI3K pathway (65). Phosphorylated p70S6kinase expression (p-p70S6K), which lies downstream of TSC2 activation, was also identified; together, these data support the use of mTOR inhibitors to treat chordoma (65).
Analysis of chordoma cell lines not associated with TSC also revealed a correlation between PI3K/AKT pathway upregulation and brachyury expression (66). Cells from skull-base chordomas with high brachyury expression had significant upregulation of PI3K/AKT pathway genes compared to low-brachyury tumors (66). Treatment with PI3K/AKT pathway inhibitors resulted in decreased brachyury expression that corresponded with impaired cell growth (66). These results confirmed that PI3K/AKT pathway activation augmented brachyury expression (66). Importantly, mTOR, TSC2, EGFR, and PDGFR were not among the upregulated genes identified in the high-brachyury tumors (66).
Few clinical trials investigating PI3K/AKT/mTOR inhibition in chordomas exist to date; current trials implement mTOR inhibitors secondary to other small-molecule inhibitors (43). A small number of published case reports found neither rapamycin or everolimus monotherapy to be efficacious (34, 67), despite prior suggestion of rapamycin-responsiveness in patient-derived cell lines (68). A randomized investigation of these therapies may further clarify their efficacy in chordoma treatment.
IGF-1 Pathway
Chordoma's osseous localization has prompted investigation of bone growth regulation as a possible pathogenic pathway. Interest in insulin-like growth factor 1 (IGF-1), one such growth mediator, in chordomas has been spurred by two features of the IGF family. First, IGF-1 effects strong mitogenic activity in bone, which, if dysregulated, could accelerate chordoma development. The IGF-1/IGF-1R axis is also easily targetable by existing small molecular inhibitors.
Emerging literature supports a relationship between IGF-1/IGF-1R expression and prognosis in chordomas. The majority of chordoma samples studied were positive for both IGF-1 and IGF-1R; the level of IGF-1R staining correlated with tumor volume (69). Phosphorylated-IGF-1R expression was also associated with decreased median progression-free survival (70). Activation of IGF-1R signaling may therefore contribute to chordoma growth or progression, presenting a potential new biomarker and targetable pathway (69, 70). Given that the AKT/mTOR pathway lies downstream of IGF-1/IGF-1R signaling (71) and the success of combined mTOR/IGF-1R inhibition in sarcomas (72), future clinical trials could benefit from looking at a similar combined approach to chordoma management. Clinical trials of IGF-1R inhibitors in chordoma have thus far comprised phase I studies of combined anti-IGF-1R/EGFR inhibition and suggest some efficacy to this approach (41, 73).
Extracellular Matrix
Chordomas produce abundant extracellular matrix. High molecular weight melanoma-associated antigen expression is detected in 62% of chordomas, making them an attractive matrix target (74). Cathepsin K is a cysteine protease that is thought to play a role in osteoclast-mediated bone resorption. Its high expression in the invasion fronts of chordoma, compared to chorda dorsalis and chondrosarcoma controls, incriminates Cathepsin K in chordoma's infiltrative growth (75). Morphogens, signaling molecules that govern embryological tissue development in the process of morphogenesis, along with extracellular signals that regulate embryonic notochord development may also play key roles in establishing a microenvironment that promotes chordoma pathogenesis (76).
Future Directions
Novel therapies for chordoma will need to demonstrate efficacy via clinical trials, but several disease and patient-intrinsic factors complicate the execution of these trials (77, 78). First, durability of long-term disease control with complete surgical removal of tumor and invaded bone followed by radiation remains salient for newly diagnosed chordomas. Clinical trials, therefore, center on recurrent and therapy-resistant chordomas for which effective control proves more challenging. The low incidence of these tumors, especially in the recurrent setting, leads to an even smaller population of eligible patients for accrual into diverse clinical trials, which may underpower the testing of any specific hypothesis. The increased average age of chordoma patients and associated comorbidities may further winnow this population following trial exclusion criteria. Lack of access to clinical trials and physician referral patterns may also impact patient enrollment in clinical trials (78). In general, chordoma remains understudied compared to more prevalent malignancies; efforts by patient advocacy groups like the Chordoma Foundation and research consortiums continue to raise awareness and funding for this rare disease.
Future clinical trials for chordoma will benefit from the adoption of innovative and integrative methods to overcome these obstacles (77, 78). Existing trials, such as the Bayesian design of the glioblastoma AGILE trial, may provide a model for efficient assessment of a large armament of therapy options in a rare disease cohort; optimal assignment of patients to clinical trials requires stratification based on clinical indication and prognostic biomarkers, which have yet to be fully elucidated in chordoma (79, 80).
Finally, increased understanding of the biological crosstalk within chordomas highlight a possible role for combination immunogenomic therapy moving forward. The tumor-intrinsic and microenvironmental heterogeneity of chordomas portends risk of emerging resistance to any rationally designed, molecularly targeted therapies, while therapies themselves have been shown to induce genomic alterations in multiple cancers. The promise of immunotherapies, either independently or synergistic with molecularly targeted therapies, presents a particularly promising adaptive strategy for future investigation.
In all, clinical and scientific investigations into chordoma hold promise for the identification of genetic, molecular, and immunomodulatory agents involved in the etiology of this malignancy. The results of these and future studies will continue to combat this rare malignancy.
Author Contributions
WB conceived of the paper. DM contributed data. SH, SA, SG, BH, and WB contributed to the initial drafting and data synthesis of the manuscript. All authors contributed to and reviewed the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge support from the following grants: National Institute of General Medical Sciences T32 GM007753 (BH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
1. Das P, Soni P, Jones J, Habboub G, Barnholtz-Sloan JS, Recinos PF, et al. Descriptive epidemiology of chordomas in the United States. J Neurooncol. (2020) 148:173–78. doi: 10.1007/s11060-020-03511-x
2. Catton C, O'Sullivan B, Bell R, Laperriere N, Cummings B, Fornasier V, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. (1996) 41:67–72. doi: 10.1016/S0167-8140(96)91805-8
3. Rassi MS, Hulou MM, Almefty K, Bi WL, Pravdenkova S, Dunn IF, et al. Pediatric clival chordoma: a curable disease that conforms to collins' law. Neurosurgery. (2018) 82:652–60. doi: 10.1093/neuros/nyx254
4. Hulen CA, Temple HT, Fox WP, Sama AA, Green BA, Eismont FJ. Oncologic and functional outcome following sacrectomy for sacral chordoma. J Bone Joint Surg Am. (2006) 88:1532–9. doi: 10.2106/00004623-200607000-00014
5. Kayani B, Hanna SA, Sewell MD, Saifuddin A, Molloy S, Briggs TWR. A review of the surgical management of sacral chordoma. Eur J Surg Oncol. (2014) 40:1412–20. doi: 10.1016/j.ejso.2014.04.008
6. Chambers PW, Schwinn CP. Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol. (1979) 72:765–76. doi: 10.1093/ajcp/72.5.765
7. Feng Z, Xiang-Lei L, Hai-Tao W, Zuo-Pei W, Bao-Li H, Hai-Feng Z, et al. Programmed cell death 1 expression in esophageal squamous cell carcinoma and association with clinical characteristics. Indian J Cancer. (2015) 52(Suppl. 3):E176–8. doi: 10.4103/0019-509X.186574
8. Stacchiotti S, Casali PG. Systemic therapy options for unresectable and metastatic chordomas. Curr Oncol Rep. (2011) 13:323–30. doi: 10.1007/s11912-011-0176-x
9. Volpe R, Mazabraud A. A clinicopathologic review of 25 cases of chordoma (a pleomorphic and metastasizing neoplasm). Am J Surg Pathol. (1983) 7:161–70. doi: 10.1097/00000478-198303000-00006
10. Wojno KJ, Hruban RH, Garin-Chesa P, Huvos AG. Chondroid chordomas and low-grade chondrosarcomas of the craniospinal axis. An immunohistochemical analysis of 17 cases. Am J Surg Pathol. (1992) 16:1144–52. doi: 10.1097/00000478-199212000-00002
11. Almefty K, Pravdenkova S, Colli BO, Al-Mefty O, Gokden, Chordoma M. chondrosarcoma: similar. but quite different, skull base tumors. Cancer. (2007) 110:2457–67. doi: 10.1002/cncr.23073
12. Almefty KK, Pravdenkova S, Sawyer J, Al-Mefty O. Impact of cytogenetic abnormalities on the management of skull base chordomas. J Neurosurg. (2009) 110:715–24. doi: 10.3171/2008.9.JNS08285
13. Sandberg AA. Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: chondrosarcoma and other cartilaginous neoplasms. Cancer Genet Cytogenet. (2003) 143:1–31. doi: 10.1016/S0165-4608(03)00002-5
14. Le LP, Nielsen GP, Rosenberg AE, Thomas D, Batten JM, Deshpande V, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS ONE. (2011) 6:e18846. doi: 10.1371/journal.pone.0018846
15. Zenosos GA, Fernandez-Miranda JC, Mukherjee D, Chang YF, Panayidou K, Snyderman CH, et al. Prospective validation of a molecular prognostication panel for clival chordoma. J Neurosurg. (2018) 130:1528–37. doi: 10.3171/2018.3.JNS172321
16. Scheil S, Brüderlein S, Liehr T, Starke H, Herms J, Schulte M, Moller P. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. (2001) 32:203–11. doi: 10.1002/gcc.1184
17. Nelson AC, Pillay N, Henderson S, Presneau N, Tirabosco R, Halai D, et al. An integrated functional genomics approach identifies the regulatory network directed by brachyury (T) in chordoma. J Pathol. (2012) 228:274–85. doi: 10.1002/path.4082
18. Tarpey PS, Behjati S, Young MD, Martincorena I, Alexandrov LB, Farndon SJ, et al. The driver landscape of sporadic chordoma. Nat Commun. (2017) 8:890. doi: 10.1038/s41467-017-01026-0
19. Palena C, Fernando RI, Hamilton DH. An immunotherapeutic intervention against tumor progression: targeting a driver of the epithelial-to-mesenchymal transition. Oncoimmunology. (2014) 3:e27220. doi: 10.4161/onci.27220
20. Heery CR, Singh BH, Rauckhorst M, Marté JL, Donahue RN, Grenga I, et al. Phase I trial of a yeast-based therapeutic cancer vaccine (GI-6301) targeting the transcription factor brachyury. Cancer Immunol Res. (2015) 3:1248–56. doi: 10.1158/2326-6066.CIR-15-0119
21. Choy E, MacConaill LE, Cote GM, Le LP, Shen JK, Nielson GP, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS ONE. (2014) 9:e101283. doi: 10.1371/journal.pone.0101283
22. Hallor KH, Staaf J, Jonsson G, Heidenblad M, von Steyern FV, Bauer HCF, et al. Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Br J Cancer. (2007) 98:434–42. doi: 10.1038/sj.bjc.6604130
23. von Witzleben A, Goerttler LT, Marienfeld R, Barth H, Lechel A, Mellert K, et al. Preclinical characterization of novel chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res. (2015) 75:3823–31. doi: 10.1158/0008-5472.CAN-14-3270
24. Zhao K, Li X, Chen X, Zhu Q, Yin F, Ruan Q, et al. Inhibition of miR-140-3p or miR-155-5p by antagomir treatment sensitize chordoma cells to chemotherapy drug treatment by increasing PTEN expression. Eur J Pharmacol. (2019) 854:298–306. doi: 10.1016/j.ejphar.2019.03.034
25. Lee DH, Zhang Y, Kassam AB, Park MJ, Gardner P, Prevedello, et al. Combined PDGFR and HDAC inhibition overcomes PTEN disruption in chordoma. PLoS ONE. (2015) 10:e0134426. doi: 10.1371/journal.pone.0134426
26. Shih AR, Chebib I, Deshpande V, Dickson BC, Iafrate AJ, Nielsen GP, et al. Molecular characteristics of poorly differentiated chordoma. Genes Chromosomes Cancer. (2019) 58:804–8. doi: 10.1002/gcc.22782
27. Owosho AA, Zhang L, Rosenblum MK, Antonescu CR. High sensitivity of FISH analysis in detecting homozygous SMARCB1 deletions in poorly differentiated chordoma: a clinicopathologic and molecular study of nine cases. Genes Chromosomes Cancer. (2018) 57:89–95. doi: 10.1002/gcc.22511
28. Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, et al. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. (2010) 120:745–53. doi: 10.1007/s00401-010-0767-x
29. Wang L, Zehir A, Nafa K, Zhou N, Berger MF, Casanova J, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosomes Cancer. (2016) 55:591–600. doi: 10.1002/gcc.22362
30. Malgulwar PB, Pathak P, Singh M, Kale SS, Suri V, Sarkar C, et al. Downregulation of SMARCB1/INI1 expression in pediatric chordomas correlates with upregulation of miR-671-5p and miR-193a-5p expressions. Brain Tumor Pathol. (2017) 34:155–9. doi: 10.1007/s10014-017-0295-7
31. Chen G, Deng C, Li Y-P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. (2012) 8:272–88. doi: 10.7150/ijbs.2929
33. Hindi N, Casali PG, Morosi C, Messina A, Palassini E, Pilotti S, et al. Imatinib in advanced chordoma: a retrospective case series analysis. Eur J cancer. (2015) 51:2609–14. doi: 10.1016/j.ejca.2015.07.038
34. Lebellec L, Chauffert B, Blay JY, Le Cesne A, Chevreau C, Bompas E, et al. Advanced chordoma treated by first-line molecular targeted therapies: Outcomes and prognostic factors. A retrospective study of the French sarcoma group (GSF/GETO) and the association des neuro-oncologues d'expression francaise (ANOCEF). Eur J Cancer. (2017) 79:119–28. doi: 10.1016/j.ejca.2017.03.037
35. George S, Merriam P, Maki RG, Van den Abbeele AD, Yap JT, Akhurst T, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J clin oncol. (2009) 27:3154–60. doi: 10.1200/JCO.2008.20.9890
36. Bompas E, Le Cesne A, Tresch-Bruneel E, Lebellec L, Laurence V, Collard O, et al. Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO). Ann oncol. (2015) 26:2168–73. doi: 10.1093/annonc/mdv300
37. Stacchiotti S, Tamborini E, Lo Vullo S, Bozzi F, Messina A, Morosi C, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann oncol. (2013) 24:1931–6. doi: 10.1093/annonc/mdt117
38. Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, et al. Phase II study of imatinib in advanced chordoma. J clin oncol. (2012) 30:914–20. doi: 10.1200/JCO.2011.35.3656
39. Schuetze SM, Bolejack V, Choy E, Ganjoo KN, Staddon AP, Chow WA, et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer. (2017) 123:90–7. doi: 10.1002/cncr.30379
40. Stacchiotti S, Morosi C, Lo Vullo S, Casale A, Palassini E, Frezza AM, et al. Imatinib and everolimus in patients with progressing advanced chordoma: A phase 2 clinical study. Cancer. (2018) 124:4056–63. doi: 10.1002/cncr.31685
41. Macaulay VM, Middleton MR, Eckhardt SG, Rudin CM, Juergens RA, Gedrich R, et al. Phase I dose-escalation study of linsitinib (OSI-906) and erlotinib in patients with advanced solid tumors. Clin Cancer Res. (2016) 22:2897–907. doi: 10.1158/1078-0432.CCR-15-2218
42. Adenis A, Ray-Coquard I, Italiano A, Chauzit E, Bui-Nguyen B, Blay JY, et al. A dose-escalating phase I of imatinib mesylate with fixed dose of metronomic cyclophosphamide in targeted solid tumours. Br J cancer. (2013) 109:2574–8. doi: 10.1038/bjc.2013.648
43. Stacchiotti S, Marrari A, Tamborini E, Palassini E, Virdis E, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol. (2009) 20:1886–94. doi: 10.1093/annonc/mdp210
44. Cote GM, Barysauskas CM, DeLaney TF, Schwab J, Raskin K, Lozano-Calderon S, et al. A Phase 1 Study of Nilotinib Plus Radiation in High-Risk Chordoma. Int J Radiat oncol biol phys. (2018) 102:1496–504. doi: 10.1016/j.ijrobp.2018.07.2013
45. Fritzsching B, Fellenberg J, Moskovszky L, Sapi Z, Krenacs T, Machado I, et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology. (2015) 4:e990800. doi: 10.4161/2162402X.2014.990800
46. Mathios D, Ruzevick J, Jackson CM, Xu H, Shah SR, Taube M, et al. PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J Neurooncol. (2015) 121:251–9. doi: 10.1007/s11060-014-1637-5
47. Dancsok AR, Gao D, Lee AF, Steigen SE, Blay YJ, Thomas DM, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology. (2020) 9:1747340. doi: 10.1080/2162402X.2020.1747340
48. Pascual-García M, Bonfill-Teixidor E, Planas-Rigol E, Rubio-Perez C, Iurlaro R, Arias A, et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy. Nat Commun. (2019) 10:2416. doi: 10.1038/s41467-019-10369-9
49. Gulluoglu S, Tuysuz EC, Sahin M, Yaltirik CK, Kuskucu A, Ozkan F, et al. The role of TNF-α in chordoma progression and inflammatory pathways. Cell Oncol. (2019) 42:663–77. doi: 10.1007/s13402-019-00454-y
50. Gulluoglu S, Sahin M, Tuysuz EC, Yaltirik CK, Kuskucu A, Ozkan F, et al. Leukemia inhibitory factor promotes aggressiveness of chordoma. Oncol Res. (2017) 25:1177–88. doi: 10.3727/096504017X14874349473815
51. Xu G, Liu C, Liang T, Zhang Z, Jiang J, Chen J, et al. Gene expression profile and bioinformatics analysis revealed key molecular characteristics of chordoma—before and after TNF- a treatment. Medicine. (2020) 99:e18790. doi: 10.1097/MD.0000000000018790
52. Zou MX, Zheng BW, Liu FS, Wang XB, Hu JR, Huang W, et al. The relationship between tumor-stroma ratio, the immune microenvironment, and survival in patients with spinal chordoma. Neurosurgery. (2019) 85:E1095–110. doi: 10.1093/neuros/nyz333
53. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
54. Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, et al. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. (2016) 7:33498–511. doi: 10.18632/oncotarget.9256
55. Zou MX, Guo KM, Lv GH, Huang W, Li J, Wang XB, et al. Clinicopathologic implications of CD8+/Foxp3+ ratio and miR-574-3p/PD-L1 axis in spinal chordoma patients. Cancer Immunol Immunother. (2018) 67:209–24. doi: 10.1007/s00262-017-2080-1
56. Gulluoglu S, Tuysuz EC, Kuskucu A, Ture U, Atalay B, Sahin F, et al. The potential function of microRNA in chordomas. Gene. (2016) 585:76–83. doi: 10.1016/j.gene.2016.03.032
57. He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch, et al. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. (2018) 11:7005–9. doi: 10.2147/OTT.S170385
58. Romero D. PD-1 says goodbye, TIM-3 says hello. Nat Rev Clin Oncol. (2016) 13:202–3. doi: 10.1038/nrclinonc.2016.40
59. Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng, X X, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. (2003) 4:1093–101. doi: 10.1038/ni987
60. Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth J, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. (2013) 2:e23849. doi: 10.4161/onci.23849
61. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
62. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
63. Zhou J, Jiang Y, Zhang H, Chen L, Luo P, Li L, et al. Clinicopathological implications of TIM3+ tumor-infiltrating lymphocytes and the miR-455-5p/Galectin-9 axis in skull base chordoma patients. Cancer Immunol Immunother. (2019) 68:1157–69. doi: 10.1007/s00262-019-02349-1
64. McMaster ML, Goldstein AM, Parry DM. Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J Med Genet. (2011) 48:444–9. doi: 10.1136/jmg.2010.085092
65. Presneau N, Shalaby A, Idowu B, Gikas P, Cannon SR, Gout I, et al. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br J Cancer. (2009) 100:1406–14. doi: 10.1038/sj.bjc.6605019
66. Otani R, Mukasa A, Shin M, Omata M, Takayanagi S, Tanaka S, et al. Brachyury gene copy number gain and activation of the PI3K/Akt pathway: association with upregulation of oncogenic Brachyury expression in skull base chordoma. J Neurosurg. (2018) 128:1428–37. doi: 10.3171/2016.12.JNS161444
67. Chay WY, Teo M, Sittampalam K, Toh HC. Effective use of thalidomide in the treatment of recurrent metastatic chordoma. J Clin Oncol. (2011) 29:e477–80. doi: 10.1200/JCO.2010.34.2139
68. Ricci-Vitiani L, Runci D, D'Alessandris QG, Cenci T, Martini M, Bianchi F, et al. Chemotherapy of skull base chordoma tailored on responsiveness of patient-derived tumor cells to rapamycin. Neoplasia. (2013) 15:773–82. doi: 10.1593/neo.13150
69. Scheipl S, Froehlich EV, Leithner A, Beham A, Quehenberger F, Mokry, et al. Does insulin-like growth factor 1 receptor (IGF-1R) targeting provide new treatment options for chordomas? A retrospective clinical and immunohistochemical study. Histopathology. (2012) 60:999–1003. doi: 10.1111/j.1365-2559.2012.04186.x
70. Sommer J, Itani DM, Homlar KC, Keedy VL, Halpern JL, Holt GE, et al. Methylthioadenosine phosphorylase and activated insulin-like growth factor-1 receptor/insulin receptor: potential therapeutic targets in chordoma. J Pathol. (2010) 220:608–17. doi: 10.1002/path.2679
71. Kurmasheva RT, Dudkin L, Billups C, Debelenko LV, Morton CL, Houghton PJ. The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer res. (2009) 69:7662–71. doi: 10.1158/0008-5472.CAN-09-1693
72. May CD, Landers SM, Bolshakov S, Ma XY, Ingram DR, Kivlin CM, et al. Co-targeting PI3K, mTOR, and IGF1R with small molecule inhibitors for treating undifferentiated pleomorphic sarcoma. Cancer Biol Ther. (2017) 18:816–26. doi: 10.1080/15384047.2017.1373230
73. Aleksic T, Browning L, Woodward M, Phillips R, Page S, Henderson S, et al. Durable response of spinal chordoma to combined inhibition of IGF-1R and EGFR. Front Oncol. (2016) 6:98. doi: 10.3389/fonc.2016.00098
74. Migliorini D, Mach N, Aguiar D, Vernet R, Landis BN, Becker M, et al. First report of clinical responses to immunotherapy in 3 relapsing cases of chordoma after failure of standard therapies. Oncoimmunology. (2017) 6:e1338235. doi: 10.1080/2162402X.2017.1338235
75. Haeckel C, Krueger S, Kuester D, Ostertag H, Samii M, Buehling F, et al. Expression of cathepsin K in chordoma. Hum Pathol. (2000) 31:834–40. doi: 10.1053/hupa.2000.8448
76. Patel P, Brooks C, Seneviratne A, Hess DA, Seguin CA. Investigating microenvironmental regulation of human chordoma cell behaviour. PLoS ONE. (2014) 9:e115909. doi: 10.1371/journal.pone.0115909
77. Galanis E, Wu W, Cloughesy T, Lamborn K, Mann B, Wen PY, et al. Phase 2 trial design in neuro-oncology revisited: a report from the RANO group. Lancet Oncol. (2012) 13:e196–204. doi: 10.1016/S1470-2045(11)70406-5
78. Lee EQ, Chukwueke UN, Hervey-Jumper SL, de Groot JF, Leone JP, Armstrong TS, et al. Barriers to accrual and enrollment in brain tumor trials. Neuro Oncol. (2019) 21:1100–17. doi: 10.1093/neuonc/noz104
79. Alexander BM, Ba S, Berger MS, Berry DA, Cavenee WK, Chang SM, et al. Adaptive global innovative learning environment for glioblastoma: GBM AGILE. Clin Cancer Res. (2018) 24:737–43. doi: 10.1158/1078-0432.CCR-17-0764
Keywords: chordoma, genomics, immunology, targeted therapy, checkpoint inhibition
Citation: Hoffman SE, Al Abdulmohsen SA, Gupta S, Hauser BM, Meredith DM, Dunn IF and Bi WL (2020) Translational Windows in Chordoma: A Target Appraisal. Front. Neurol. 11:657. doi: 10.3389/fneur.2020.00657
Received: 03 March 2020; Accepted: 02 June 2020;
Published: 08 July 2020.
Edited by:
Abdulrazag Ajlan, King Saud University, Saudi ArabiaReviewed by:
Alireza Mansouri, Pennsylvania State University (PSU), United StatesChristian Senft, University Hospital Frankfurt, Germany
Copyright © 2020 Hoffman, Al Abdulmohsen, Gupta, Hauser, Meredith, Dunn and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenya Linda Bi, d2JpQGJ3aC5oYXJ2YXJkLmVkdQ==