
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurol. , 11 August 2020
Sec. Pediatric Neurology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00639
This article is part of the Research Topic Tuberous Sclerosis Complex - Diagnosis and Management View all 16 articles
 Nicola Specchio1,2*
Nicola Specchio1,2* Nicola Pietrafusa1
Nicola Pietrafusa1 Marina Trivisano1
Marina Trivisano1 Romina Moavero3,4
Romina Moavero3,4 Luca De Palma1
Luca De Palma1 Alessandro Ferretti1Federico Vigevano2,4
Alessandro Ferretti1Federico Vigevano2,4 Paolo Curatolo3
Paolo Curatolo3Introduction: Individuals with Tuberous Sclerosis Complex (TSC) are at increased risk of developing both epilepsy and autism spectrum disorder (ASD), but the relationship between these conditions is little understood. We reviewed published reports to elucidate the relationship between ASD, epilepsy, and TSC, and to define the genetic and neurological risk factors.
Methods: Articles (January 2004–May 2019) were identified via PubMed, EMBASE, and CENTRAL databases. Article inclusion required report on individuals with TSC-associated ASD and epilepsy with prevalence, odds ratio, or rate report on the comorbidity of ASD in epileptic patients due to TSC.
Results: A total of 841 abstracts were identified in the original search. Thirty-six articles were included, which identified study populations, ASD measures used, and study confounders as bias factors. This review included 2,666 TSC patients, with a mean age of 15.9 years (range 1.94–30.3 years). The percentage of TSC patients with epilepsy and autism was 33.7%. Patients with TSC and autism showed more frequent seizures and earlier epilepsy onset than TSC patients without autism. ASD and intractable epilepsy were both predicted by a higher number of areas with dysplastic features revealed in brain MR scans. ASD, the onset of seizures in children <2 years of age, and >3 tubers have all been associated with an increased risk of refractory epilepsy in TSC patients. However, the direction of the relationship is not clear because a history of epilepsy, or infantile spasms in patients with TSC is also associated with an increased likelihood of ASD. Overall, 73.2% of patients carried TSC2 genetic variant and, among patients with TSC and autism, the percentage of TSC2 individuals was 85.6%.
Conclusions: The complex interrelationship between TSC, autism, and epilepsy, coupled with limited knowledge on the neurobiological basis for the interrelationship, limits overall understanding and opportunities for management. The results of this review highlight the need for early identification and management to optimize favorable outcomes in the most vulnerable individuals with TSC. Regardless of whether studies are considered individually or collectively, interpretation is made difficult due to the differences between the studies, most notably between methods and diagnostic criteria used to assess intellectual ability.
Tuberous Sclerosis Complex (TSC) is a rare genetic multisystem disorder characterized by hamartoma formation in several organs and systems (1, 2), with an estimated birth incidence of 1 in 5,800 (3). TSC is caused by mutation in either TSC1 (chromosome 9q34) or TSC2 (16p13.3) gene, encoding for hamartin and tuberin, respectively (4). These two proteins, along with TBC1D7, form a heterotrimeric complex regulating the activity of mTOR complex 1 (mTORC1), which is a key regulator of cell metabolism and proliferation. mTORC1 dysregulation is the main reason for aberrant growth and differentiation underlying the formation of TSC-related lesions, either in the brain or other organs (1).
Neurologic and developmental issues such as epilepsy, autism spectrum disorder (ASD), and developmental delay (DD), are major sources of morbidity in people of all ages with TSC and typically present in infancy or early childhood (1). Epilepsy is estimated to occur in about 80% of TSC patients, typically within the first 3 years of life, and considered to be a result of the genetic mutation leading to an imbalance between excitation and inhibition of gamma-amino-butyric acid (GABA) receptors. Dysregulation of the neurotransmission of GABA has also been proposed as a neurobiological link between epilepsy and ASD in TSC patients (5).
ASD is an early onset, lifelong, neurobiological disorder characterized by impairments in communication and social interaction along with the presence of restricted and repetitive patterns of behavior, interests or activities, and is prevalent in 1.85% of children aged 8 years (6–8). In the last 5 years, some longitudinal studies have explored the early emerging symptoms and prompt intervention in infants with high familial risk of ASD (9, 10). In contrast, very few studies have addressed this topic in ASD associated with specific syndromes or genetic conditions (11–13).
TSC is one of the major syndromes associated with ASD. The prevalence of ASD in TSC ranges from 26 to 45%, depending on the sample, ASD criteria, and the testing methodologies employed (14, 15). Some autistic features are present in about half of patients with TSC. A number of factors have been identified as being associated with ASD in TSC, including brain lesion load, prominent lesion type, the size and location of the tubers, cyst-like tubers, TSC2 mutation, early onset and refractory seizures, and the presence and severity of cognitive impairment (1, 16). Prompt cessation of early seizures can, in at least some cases, improve neuropsychiatric outcome (17, 18).
To our knowledge, no review has yet examined the relationship between ASD and epilepsy in patients with TSC. We performed a review of the literature to assess the prevalence and risk factors for ASD in patients with TSC and epilepsy, and to investigate the relationship and comorbidity between these conditions. The main aims of this review were: to identify the frequency of both ASD and epilepsy within the TSC population, and to elucidate the relationship between ASD and epilepsy in individuals with TSC.
The results of the present review were reported according to the preferred reporting items for reviews and meta-analyses (PRISMA) and adheres to a structured review protocol (19).
Two authors (NP and NS) performed a search of PubMed, EMBASE, and CENTRAL databases using the following search strategy: “autism” OR “autistic” OR “asperger” OR “autism spectrum disorder” OR “pervasive” OR “pervasive developmental disorder” OR “PDD” OR “ASD” AND “epilepsy” OR “seizure” OR “epileptic” OR “convulsion” AND “tuberous sclerosis complex” OR “tuberous sclerosis” OR “TSC.”
Studies were initially included if they:
1) Involved individuals with ASD and epilepsy symptomatic of TSC.
2) Reported prevalence, odds ratio, or numerical report of the comorbidity of ASD in patients with epilepsy due to TSC.
3) Were written in English.
4) Were based on human research.
5) Were published within 15 years of the search date (January 2004–May 2019), which was considered a sufficient period to capture publications with the most reliable and appropriate diagnostic and management procedures.
Two authors independently screened all titles and abstracts of studies identified by the initial search. The full text of an article was obtained when either reviewer thought that it might fulfill the inclusion criteria. Upon uncertainty for inclusion of a publication, an additional author was consulted (LDP).
Full articles were reviewed for relevance and articles were excluded if they did not include data relating to the prevalence of epilepsy/seizures in the TSC population. Articles also had to contain a reported or calculable prevalence for ASD in the text (if not provided in the abstract).
Based on the Quality in Prognosis Strategy (QUIPS) tool, the most commonly found risk factors for bias in the studies reviewed included study participation, ASD measure, and study confounders. Many [14] of the reviewed articles included participants drawn from one clinic or hospital (18, 20–32); others [5] had a specific age range (12, 13, 33–35) or a particular subset of the TSC population (18, 35–47, 49, 50). Only 18 of the included articles reported the diagnostic criteria for ASD (Table 1). Large variations were noted in the measures and criteria used to define ASD and many of the articles relied on reports of ASD by parents and caregivers. Comparisons between various studies were subject to a number of potential confounders, including a failure to report seizure onset, type, and frequency for epilepsy, antiseizure medication (ASM), genetic susceptibility, or other relevant baseline measures. Only articles that unequivocally reported the above-mentioned information were included in Tables 2–4. From Table 2, eight articles were excluded because of no mention of onset, type, or frequency of epileptic seizures; 16 articles were excluded from Table 3 because of no mention of number of patients with epilepsy, TSC and autism; 14 articles were excluded from Table 4 because of no mention of genetic mutation in TSC1 and TSC2. In this review we have used the terminology “infantile spasms” for infants with ES (with or without hypsarrhythmia), who may or may not have had cognitive regression. This operational definition was chosen because it was not always possible to determine whether the infants had hypsarrhythmia or cognitive regression. In the tables and figures, however, the term “epileptic spasms” has been used because this refers to that specific type of seizure.
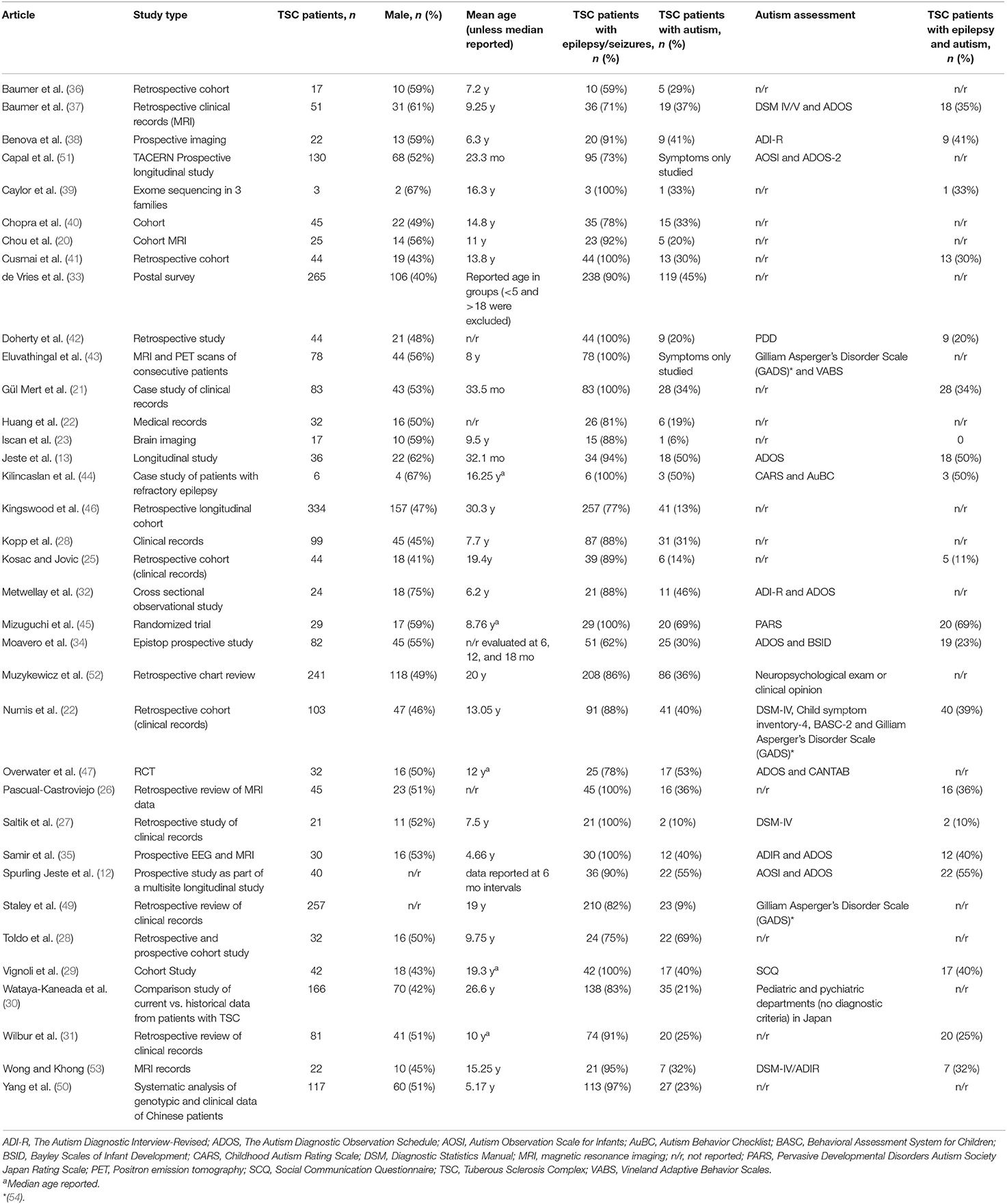
Table 1. Demographic information and prevalence rates of autism and epilepsy/seizures in Tuberous Sclerosis Complex patients reported within each of the articles included in this review.
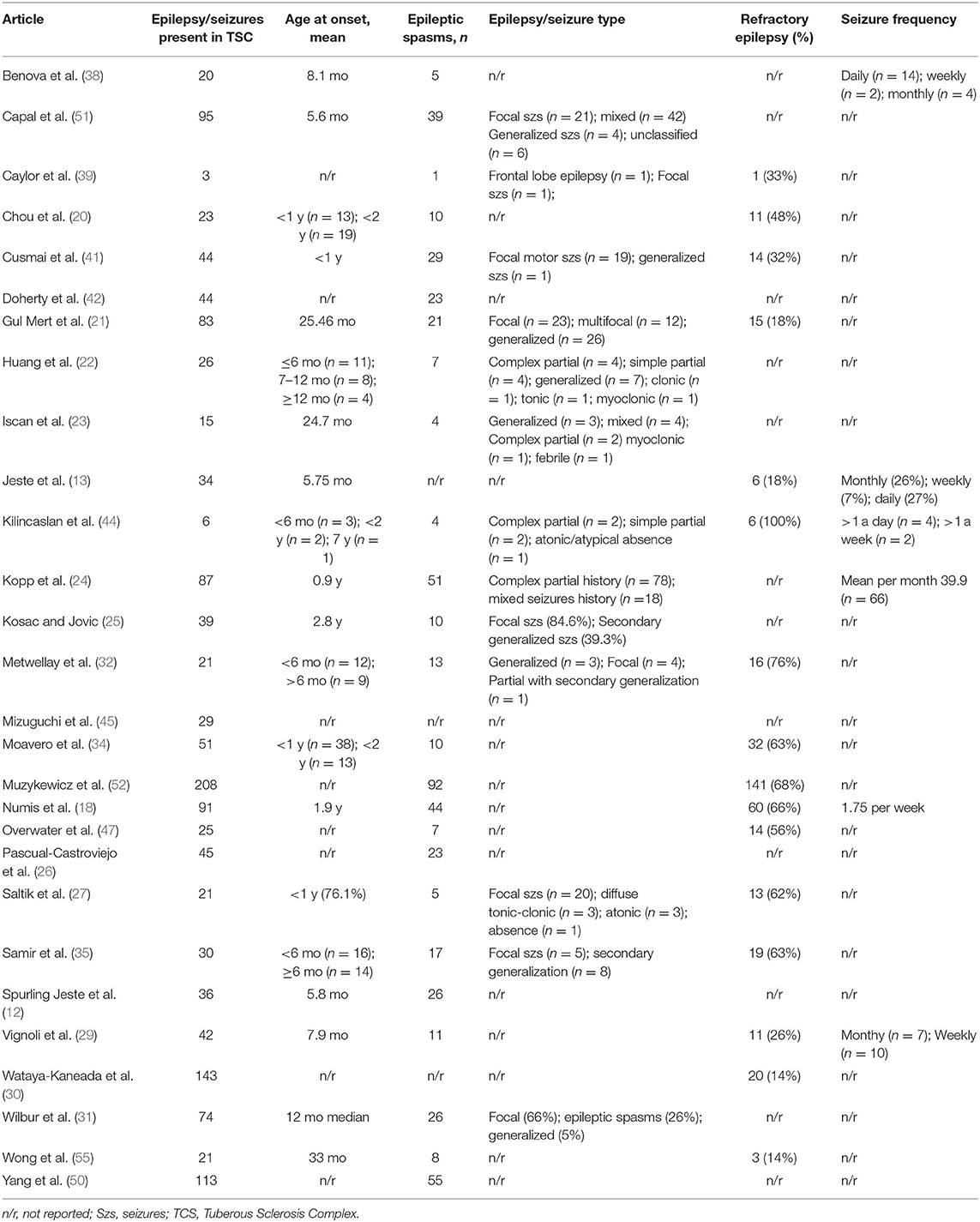
Table 2. Summary of history of epilepsy in patients with Tuberous Sclerosis Complex.
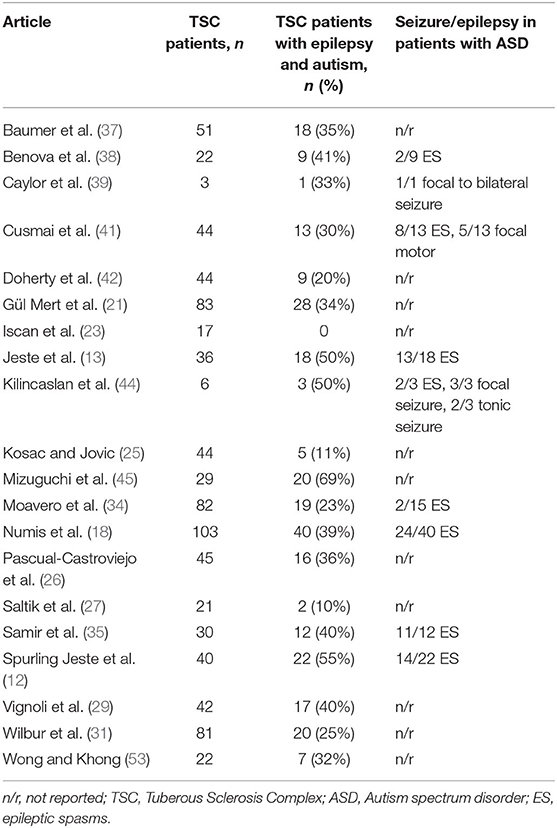
Table 3. Summary of family history of Tuberous Sclerosis Complex (TSC) and genetic mutations in patients with TSC.
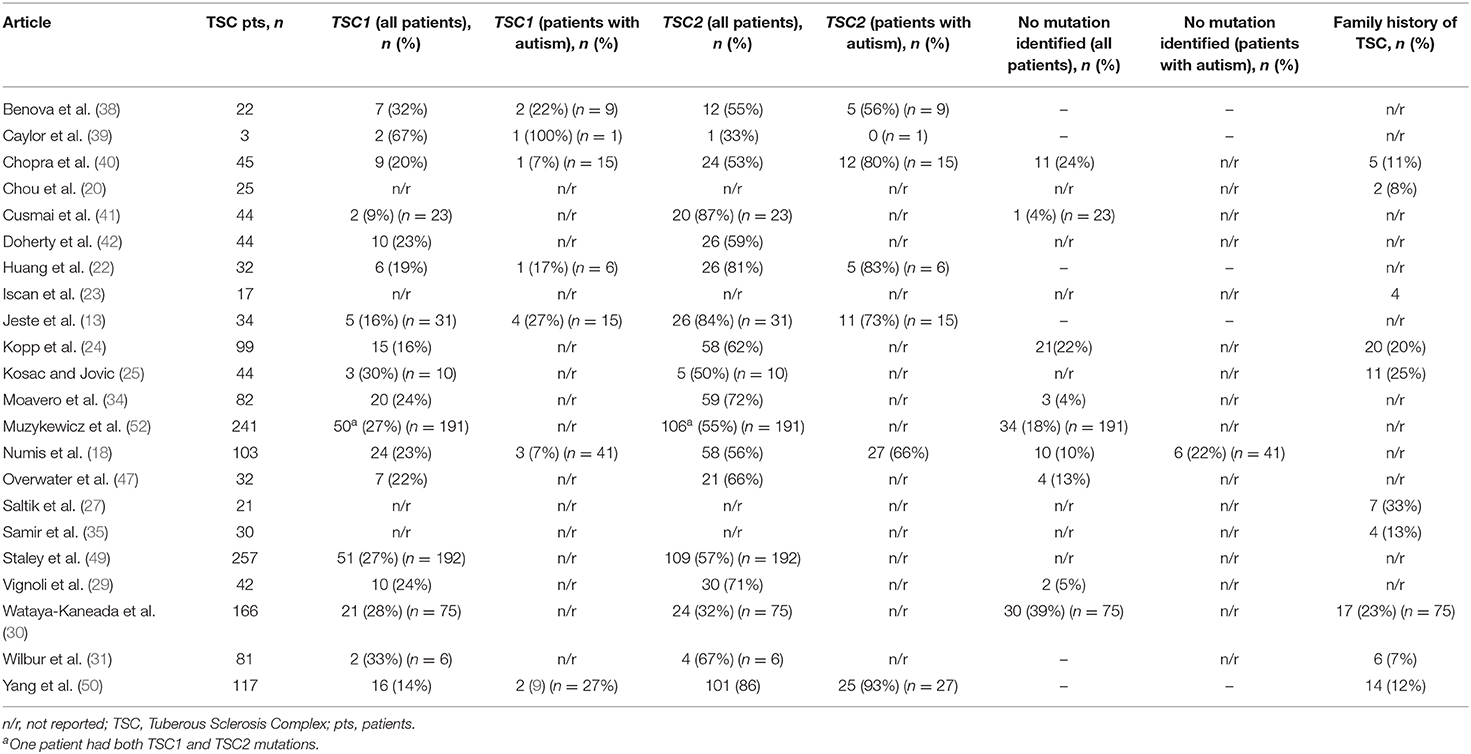
Table 4. Summary of family history of Tuberous Sclerosis Complex (TSC) and genetic mutations in patients with TSC.
A total of 841 abstracts were identified in the original search. Of these, 673 were duplicates or congress abstracts only. The remaining abstracts and articles were reviewed for inclusion/exclusion criteria, and a total of 36 articles were considered suitable for inclusion (Figure 1). Included articles are presented in Table 1. In total, 2,666 patients with TSC were included in this review, with a mean age of 15.9 years (range 1.94–30.3 years). TSC populations included within the selected articles were predominantly male; males represented 52.5% of overall participants, ranging from 41 to 75% of patients in articles (Table 1).
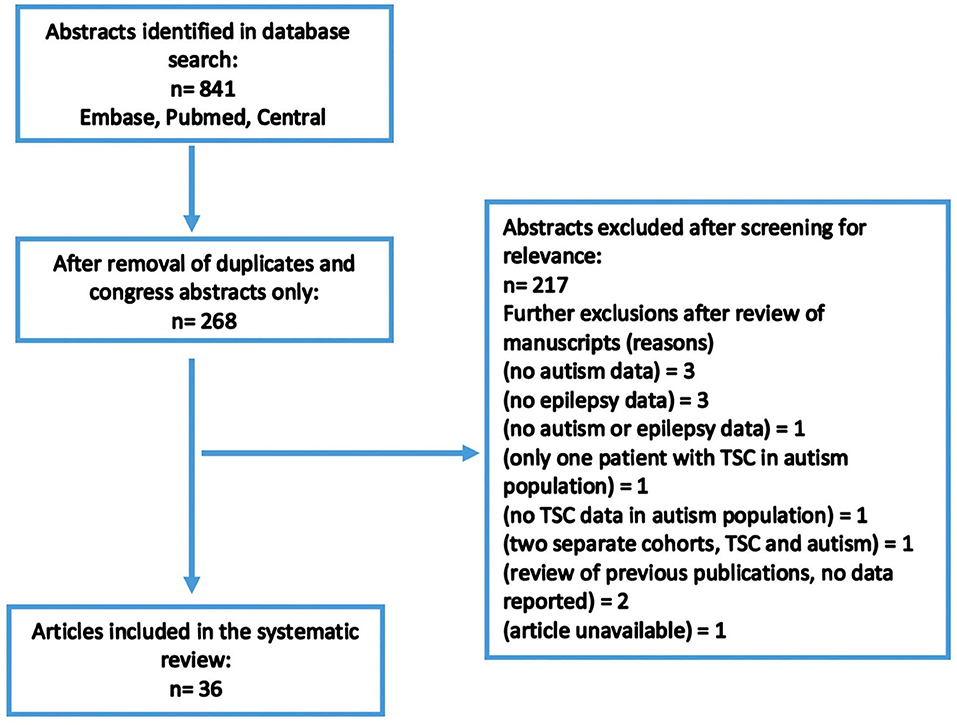
Figure 1. Search strategy.
Of the patients with TSC included with available data in this review, the overall percentage of patients with autism was 29.8% (732 of 2,458 patients with available data), ranging from 6% (23) to 69% (28, 45), and those with epilepsy/seizures was 88.2% (2,352 of 2,666 patients), ranging from 59 to 100% (21, 26, 27, 29, 35, 39, 41–45) (Table 1). Patients with epilepsy and autism are also reported where available (Table 1), with the overall percentage being 33.7% (279 of 828 patients with available data) and ranging from 10% (27) to 69% (45).
The mean age for onset of epilepsy was below 33 months; however, data were available for 859 patients only. Infantile spasms were reported in 42.8% of TSC populations studied (Table 2), ranging from 20% (34) to 67% (44). Other epilepsy types were less frequently reported within the articles reviewed, but Huang et al. (22) suggested that focal seizures were also frequent in infants with TSC under 1 year of age. Reports of refractory epilepsy in patients with TSC ranged from 14% to 100% (44), although the latter specifically focused on 6 TSC patients with refractory epilepsy.
The relationship between epilepsy, ASD, and TSC is complex. Autism, the onset of seizures in children <2 years of age and with >3 tubers (21, 31) have all been associated with an increased risk of refractory epilepsy in TSC patients. However, the direction of the relationship is unclear because a history of epilepsy (33) or infantile spasms (31, 35) in patients with TSC is also associated with an increased likelihood of ASD. Patients with TSC and autism showed more frequent seizures than TSC patients without autism (18) and an earlier age of onset of epilepsy has been associated with ASD (18, 28, 31), delayed language, intellectual disability (ID), and poor cognitive flexibility (28). In Table 3 are reported the epilepsy features in patients with TSC and autism.
Clinically significant behavioral problems and social withdrawal are common in young children with TSC (28). Conditions including mood disorder, anxiety, ADHD, and aggressive behavior were reported in 66% of a pediatric population with TSC (n = 241) (52). Aggressive behavior was associated with both increased severity of epilepsy and features of autism/pervasive developmental disorder (PDD) (52).
Early identifiers of autism or autistic-like features in patients with TSC include early DD or a slowing in nonverbal cognition (13, 38). Studies of very young infants with TSC suggest early delay in visual reception (12) and under-developed fine-motor skills to be markers of the development of autism traits (12, 34). Deficits across all domains of the Bayley Scales of Infant Development (BSDI) at 1 year of age were predictive of higher autism traits on the Autism Diagnostic Observation Schedule (ADOS) at 2 years within a prospective study of infants with TSC (n = 82) in 10 sites across Europe and Australia (34). ID is often more common in TSC patients with ASD than those with TSC alone (31). Behavioral problems have been reported to be exacerbated by seizure frequency and a mixed seizure profile (24). Results of a study exploring the relationship between cognitive delay and clinical features of TSC in Egypt reported that the age of seizure onset (p = 0.044) and number of brain tubers (p = 0.06) increased the odds for cognitive delay in 24 children with TSC (32). Similarly, ID has been associated with early onset of seizures, infantile spasms (35), and intractable epilepsy (38). Early seizure onset was the most significant predictor of DD at 2 years of age in a longitudinal prospective analysis of developmental outcomes in infants (0–3 years) with TSC (51). Since the data in most of the reported studies did not specify infantile spasms, many of the early onset seizures could have been infantile spasms. The neurologic symptoms of TSC, refractory epilepsy, ASD, and ID have all shown an interrelationship (30), making specific relationships between ASD, ID, and epilepsy difficult to discern.
Self-injurious behavior in patients with TSC was associated with a history of infantile spasms and seizures, ID, ASD, and TSC2 mutations (49). Aggressive behavior was also associated with ID and TSC2 mutations (52), suggesting a potential genetic link.
Data on severity of autism and developmental delay were sparse and therefore not reported.
Refractory epilepsy (38), ID (24), and autism in TSC patients have all been associated with the TSC2 genotype (29, 40, 50). The TSC2 genotype was more common than TSC1 genotype among TSC patients overall (Table 4), with the exception of one study that focused on individuals from three families, in which three individuals had a diagnosis of TSC: two with the TSC1 genotype and one with the TSC2 genotype (39). Overall, 73.2% of TSC individuals had the TSC2 genotype—ranging from 32% (30) to 89% (41)—and 26.8% had the TSC1 genotype—ranging from 9% (41) to 67% (39) (Table 4). Among patients with TSC and autism, 85.6% had the TSC2 genotype. Autistic behavior correlated with nonsense mutations in the TSC2 gene group in a retrospective review of medical records from patients with TSC in Taiwan (n = 32) (22).
A magnetic resonance imaging (MRI) study including 25 children (aged >2 years) reported that lesion load within the left temporal lobe was positively correlated with the neurological severity score (r = 0.609; p = 0.001). This finding was supported in an electroencephalogram (EEG) study that found greater interictal epileptiform features in the left temporal lobe only (18).
Two studies exploring potential impact of TSC proteins on white-matter tract pathways have identified abnormal diffusion characteristics, which are believed to arise from abnormal neuronal and axonal organization and hypomyelination (36, 37). Furthermore, these effects were each associated with TSC, epilepsy, and autism (36, 37). In a diffusion MRI study exploring the directionality of water movement [fractional anisotropy (FA)], TSC alone was related to lower callosal FA values than controls—and this difference was greater in the TSC patients with autism than without—when comparing study groups of TSC patients with either epilepsy (with and without comorbid autism; n = 19 and n = 32, respectively) or autism alone (n = 46) with a healthy control group (n = 89) (37). A positron emission tomography (PET) study comparing TSC patients with and without a cerebellar lesion (n = 20 vs. n = 57, respectively) reported that the group with cerebellar lesions had higher overall autistic symptomology (i.e., social isolation and communicative/developmental disturbance) and that these deficits were associated with right-sided cerebellar lesions (43).
The size, number, and anatomical location of tubers have all independently been linked to autism and/or epilepsy in TSC (22, 26, 35, 38, 42), although this relationship has not always been established (55). The number of tubers is strongly associated with infantile spasms (42) and ASD (35). Tubers of larger size were associated with increased likelihood of seizures and autism (26), and higher prevalence of cyst-like tubers was associated with ASD (22). ASD and intractable epilepsy were both predicted by a higher number of areas with dysplastic features (38). ASD or PDD have been linked with tubers in the frontal areas of the brain (35), increased tuber count in the occipital lobe (42), cystic-like tubers, and tubers in insular and temporal areas (22). Infantile spasms are more likely to occur in children with cortical tubers in the parietal lobes (55).
Data relating to ASM use was not commonly provided in the studies included in this review. Where reported, the mean number of ASMs per patient with TSC ranged from 1.46 to 3.95 (12, 13, 18, 38). Combination treatment with two ASMs or more was common and, where reported, the number of TSC patients using polytherapy ranged from 52 to 100% (20, 21, 25, 29, 38). Common ASMs included valproic acid, carbamazepine, topiramate, lamotrigine, and vigabatrin (25, 38, 41). Only two of the reviewed studies reported individual use of ASMs among TSC patients, and these data are summarized in Figure 2.
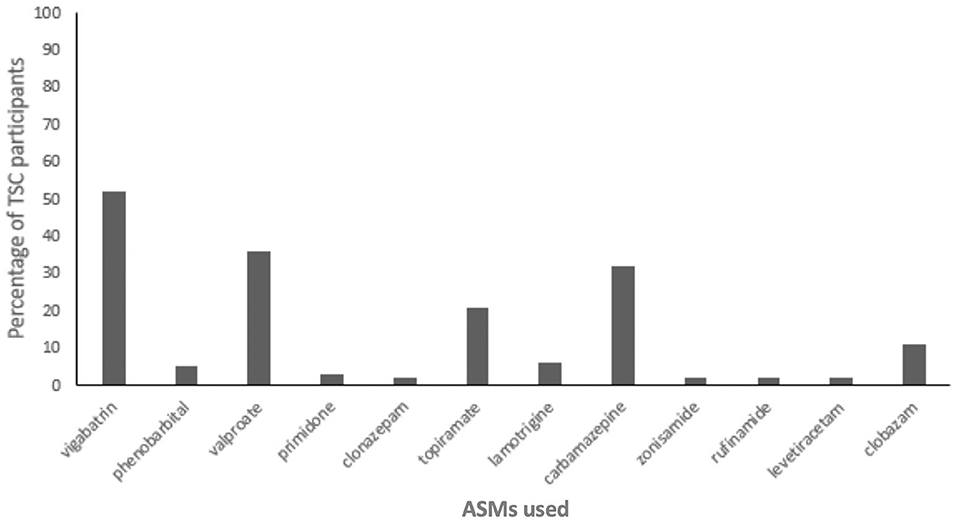
Figure 2. Proportion of Tuberous Sclerosis Complex (TSC) participants using antiseizure medications (ASMs) (n = 66) (38, 41).
Early treatment with ASMs may be of importance, since better long-term epileptic encephalopathy outcomes were reported in those treated early in a randomized trial of early vs. later treatment with vigabatrin (41). In general, studies should distinguish between early and later treatment of epilepsy in TSC, considering that later treatment of seizures in TSC is often disappointing and research reports that the development of ID is predicted by the number of ASMs used (potentially related to delay in effective treatment) to treat epilepsy in children with TSC (38).
The studies in our review with data on individuals with uncontrolled epilepsy reported these to represent 14–100% of patients with a history of epilepsy, with the majority of studies reporting >40% of the epilepsy population still having seizures (Table 2). Although these data suggest a greater proportion of TSC patients with difficult-to-treat epilepsy than is typical of a general population, the bias in study participation remains a caveat to such speculation.
Three studies evaluated the effects of an mTOR kinase inhibitor, everolimus, which can be used to reduce tumor size (44, 45, 47, 48). The first, a three-armed randomized trial in Japan (n = 29), reported adjunctive everolimus treatment to significantly reduce seizure frequency in TSC patients with refractory epilepsy, with a trend for improvements in ASD symptoms (45). A similar finding was reported in a small case study evaluating everolimus for refractory epilepsy in six TSC patients with refractory epilepsy (44). This second study also reported improvement in ASD symptoms, such as social contact, language, and repetitive behavior (44). However, the third study—a recent randomized controlled trial conducted in the Netherlands including 32 children with TSC—found no benefit of everolimus on cognitive or neuropsychological functioning, or autism traits, in comparison with placebo (47). In this third study, age at enrollment was high—the median age was 11.5 years for patients on placebo and 12.2 years for patients on everolimus—therefore, firm conclusions cannot be drawn (47). However, early treatment with everolimus might be required for improvement in features such as social contact, language and repetitive behavior; there is a need for formal studies to determine whether this is the case.
Based on the 36 articles included in this review, our findings were consistent with previous reports of high rates of epilepsy in patients with TSC (5). Interestingly, epilepsy was reported in 83.6% of patients with TSC in an international TuberOus SClerosis registry to increase disease Awareness (TOSCA); however, data on the prevalence of ASD in this population were not reported (56). The prevalence of autism in patients with TSC in the subjects included in this review is high, but is consistent with previous estimates of syndromic ASD in TSC (14, 15, 57).
The risk of autism is increased by early onset seizures (18, 28, 31, 35) and by DD and ID (28), which in turn have been associated with early onset epilepsy and infantile spasms (28, 31, 32, 35, 38). Existence of phenotypic variability should be acknowledged: TSC is also associated with high-functioning autism, normal intelligence, hypercalculia, and drug-resistant epilepsy with an EEG pattern characterized by hypsarrhythmia and electrical status epilepticus during sleep (58).
The relationship between TSC, epilepsy, and ASD is highly complex. A poor prognosis of epilepsy outcomes is largely reported to be exacerbated by ASD (18, 21, 31). An additive neuroanatomical impact of TSC, epilepsy, and autism has been proposed that is predominantly evident in white-matter pathways (36, 37), supporting the association between autism, epilepsy, and DD/ID in patients with TSC.
Evidence suggests both epilepsy and autism are linked with mutations on the TSC1 and TSC2 genes. Mutations in the TSC2 gene are more prevalent in association with epilepsy and autism (18, 22, 29, 38, 40, 50). Early genotyping may, therefore, help identify TSC patients at increased risk of poorer long-term outcomes.
In terms of autism, neuroimaging studies report that tuber features, such as larger size or increased number of cyst-like tubers, are associated with increased risk (22, 26). It is also demonstrated that diffusion imaging abnormalities correlate with reduced myelination in TSC patients (59) and the effect of mTOR overactivation on white matter might be modified by pharmacological inhibition (60). Moreover, TSC patients with autism have been documented to have a reduction of fractional anisotropy in different white-matter regions, and this happens over the first 2 years of life (61). Since size, type, and location of tubers influence the longer-term risk of autism and epilepsy in TSC, early characterization of such features could assist in determining the focus of early intervention.
Cells in the central nervous system express TSC1 and TSC2 proteins throughout childhood and into adulthood. These proteins help regulate myelination, axon guidance, and dendritic arborization, promoting normal synaptic formation and function (37). Dysregulation of the neurotransmission of GABA, resulting from genetic mutations of TSC, has previously been argued to underlie development of epilepsy and autism in this population (5). Limited evidence suggests that treatment with everolimus, particularly if commenced early, may improve epilepsy outcomes and reduce the risk of autism in TSC patients (44, 45). Data coming from the EXIST-3 trial confirm that adjunctive everolimus might reduce seizure frequency in pediatric patients with treatment-refractory seizures associated with tuberous sclerosis complex also in patients younger than 6 years (62). However, these findings were based on few trials and contradictory evidence also exists (47). Additional research into alternative treatment strategies and an increased focus on the longer-term outcomes would help elucidate whether size, type, and location of tubers influence the longer-term risk of autism and epilepsy in TSC.
Early treatment with ASMs to control epilepsy is reported to improve longer-term epilepsy outcomes (41), and controlled epilepsy is associated with reduced symptoms of autism (44, 45). ID and DD are in turn associated with increased presence of autism (31, 34), so the number and choice of ASMs in infants with TSC needs to be managed with care. Figure 3 is a diagrammatic overview of the complex relationship between the phenotypic features of TSC and polytherapy treatment with ASMs based on the evidence reviewed here.
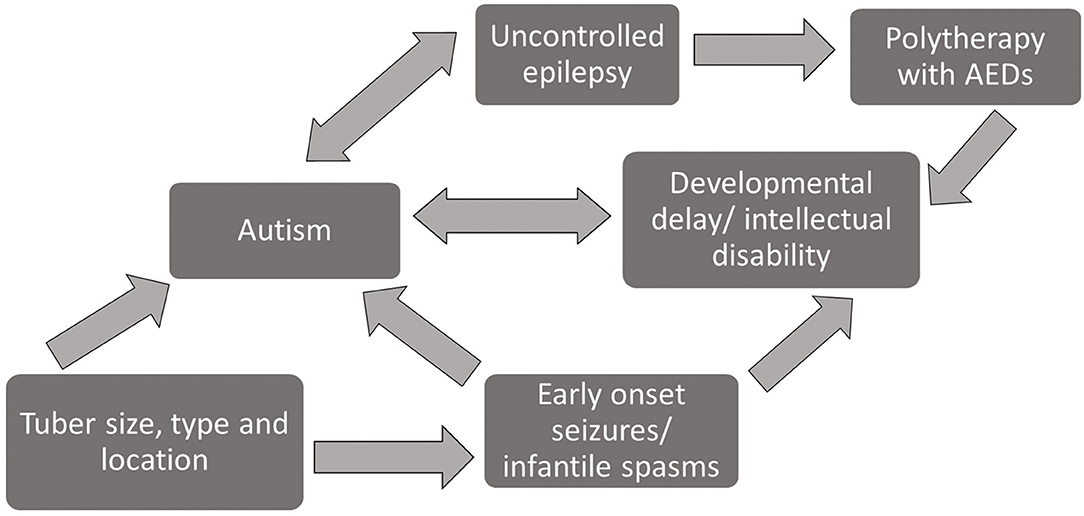
Figure 3. Diagram of interrelationship between phenotypic profiles and prognostic risk factors among patients with TSC.
Although we identified 36 articles reporting autism and epilepsy in TSC, only approximately half of these articles indicated which patients were experiencing either of these comorbid conditions. Very few of the included studies summarized the potential prognostic features of patients with all three conditions (TSC, epilepsy, and autism). This review has therefore identified a need for future studies to focus on common associative factors.
A second limitation was that, because the studies were conducted in different settings across different countries, practices were not standardized with respect to identification of TSC, epilepsy, and—above all—autism. Different diagnostic criteria were used to identify patients with TSC according to the clinical practice of the country or region. Likewise, the tools used to define the presence of autism varied considerably. In some cases, the diagnosis of autism was not confirmed, but relied on reports from parents and caregivers. In populations that only focused on very young infants, in whom a clinical diagnosis of autism was not possible, the conclusions regarding risk of autism were based on autistic features, which do not necessarily indicate a later clinical outcome.
The methodological approaches of the included articles also varied widely and ranged from small clinical series to large retrospective studies, each with differing strengths and limitations. One of the challenges of establishing a representative sample of individuals with TSC is the rarity of the disease. The TSC populations within the included articles ranged from infants to adults, sometimes within the same study. Consequently, the core features of TSC and age of onset of the conditions may not have been reliable.
Lastly, the quality of the available data does not allow a meaningful review to be performed.
Early onset epilepsy, frequently represented by epileptic encephalopathy, can be considered one of the risk factors for ID in TSC patients. However, the role of genetic variations should be highlighted as the major player in determining both epilepsy and intellectual disability due to mTOR overactivation (63).
In terms of further defining the prognostic features of epilepsy and autism within TSC, large prospective studies, such as TACERN or those conducted by the EPISTOP group (34, 51, 64), are helping to identify early biomarkers for treatment.
The prevalence of autism and epilepsy in TSC is much higher than that in the general population, both alone and as comorbid features. We summarized the phenotypic, genetic, and neurological risk factors for the association of autism and epilepsy in TSC patients from available data, but the inherent limitations of the source studies should be noted.
The relationship between these three conditions is complex. Early identification of the risk factors, together with early use of m-TOR inhibitors might be a priority to optimize favorable outcomes in this vulnerable population.
NS conceptualized and designed the study, drafted the initial manuscript, supervised data collection, and reviewed and revised the manuscript. NP, MT, and LD designed the data collection instruments, collected data, carried out the initial analyses, and reviewed and revised the manuscript. AF and RM collected data, carried out the initial analyses, and reviewed and revised the manuscript. FV and PC conceptualized and designed the study and critically reviewed the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Medical writing support was provided by Dr. Brenda J. Meyer. English text editing was provided by Dr. David Macari. Paolo Curatolo participated in the EPISTOP study (www.epistop.eu) funded under the European Community's 7th Framework Programme under Grant Agreement No. 602391.
NS has received support from Livanova and Biomarin, and has served as a paid consultant for Livanova. PC has served as a paid consultant for Novartis. FV has served as paid consultant for Zogenix, Eisai, and GW Pharma. MT has served as paid consultant for Biomarin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ADC, apparent diffusion coefficient; ASM, antiseizure medication; ASD, Autism Spectrum Disorder; EEG, electroencephalogram; DD, developmental delay; FA, fractional anisotropy; GABA, gamma-amino-butyric acid; ID, intellectual disability; MRI, magnetic resonance imaging; PDD, pervasive developmental disorder; QUIPS, Quality in Prognosis Strategy; TSC, Tuberous Sclerosis Complex.
1. Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. (2015) 14:733–45. doi: 10.1016/S1474-4422(15)00069-1
2. Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. (2016) 2:16035. doi: 10.1038/nrdp.2016.35
3. O'Callaghan FJ, Shiell AW, Osborne JP, Martyn CN. Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet. (1998) 351:1490. doi: 10.1016/S0140-6736(05)78872-3
4. European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. (1993) 75:1305–5. doi: 10.1016/0092-8674(93)90618-Z
5. Moavero R, Bombardieri R, Marciano S, Cerminara C, Curatolo P. Epilepsy in tuberous sclerosis complex. J Pediatr Epilepsy. (2015) 6:15–23. doi: 10.1055/s-0035-1570070
6. Maenner MJ, Shaw KA, Baio J. Prevalence of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveill Summ. (2020) 69:1–12. doi: 10.15585/mmwr.ss6904a1
7. Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG. The epidemiology and global burden of autism spectrum disorders. Psychol Med. (2015) 45:601–13. doi: 10.1017/S003329171400172X
8. Centers for Disease Control and Prevention. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. In Morbidity and Mortality Weekly Report. Surveillance Summaries (2018).
9. Jones EJ, Venema K, Earl R, Lowy R, Barnes K, Estes A. Reduced engagement with social stimuli in 6-month-old infants with later autism spectrum disorder: a longitudinal prospective study of infants at high familial risk. J Neurodev Disord. (2016) 8:7. doi: 10.1186/s11689-016-9139-8
10. Rogers SJ, Vismara L, Wagner AL, McCormick C, Young G, Ozonoff S. Autism treatment in the first year of life: a pilot study of infant start, a parent-implemented intervention for symptomatic infants. J Autism Dev Disord. (2014) 44:2981–95. doi: 10.1007/s10803-014-2202-y
11. Rague L, Caravella K, Tonnsen B, Klusek J, Roberts J. Early gesture use in fragile X syndrome. J Intellect Disabil Res. (2018) 62:625–36. doi: 10.1111/jir.12498
12. Spurling Jeste S, Wu JY, Senturk D, Varcin K, Ko J, McCarthy B, et al. Early developmental trajectories associated with ASD in infants with tuberous sclerosis complex. Neurology. (2014) 83:160–8. doi: 10.1212/WNL.0000000000000568
13. Jeste SS, Varcin KJ, Hellemann GS, Gulsrud AC, Bhatt R, Kasari C, et al. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology. (2016) 87:766–72. doi: 10.1212/WNL.0000000000003002
14. Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. (2008) 23:520–5. doi: 10.1177/0883073807309788
15. Leclezio L, de Vries PJ. Advances in the treatment of tuberous sclerosis complex. Curr Opin Psychiatry. (2015) 28:113–20. doi: 10.1097/YCO.0000000000000136
16. de Vries PJ, Whittemore VH, Leclezio L, Byars AW, Dunn D, Ess KC, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. (2015) 52:25–35. doi: 10.1016/j.pediatrneurol.2014.10.004
17. Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. (2010) 14:146–9. doi: 10.1016/j.ejpn.2009.03.003
18. Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. (2011) 76:981–7. doi: 10.1212/WNL.0b013e3182104347
19. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
20. Chou IJ, Lin KL, Wong AM, Wang HS, Chou ML, Hung PC, et al. Neuroimaging correlation with neurological severity in tuberous sclerosis complex. Eur J Paediatr Neurol. (2008) 12:108–12. doi: 10.1016/j.ejpn.2007.07.002
21. Gül Mert G, Altunbaşak S, Hergüner Ö, Incecik F, Övetti HC, Özcan N, et al. Factors affecting epilepsy prognosis in patients with tuberous sclerosis. Childs Nerv Syst. (2019) 35:463–8. doi: 10.1007/s00381-019-04066-7
22. Huang CH, Peng SS, Weng WC, Su YN, Lee WT, National Taiwan University Hospital Tuberous Sclerosis Complex Study Group. The relationship of neuroimaging findings and neuropsychiatric comorbidities in children with tuberous sclerosis complex. J Formos Med Assoc. (2015) 114:849–54. doi: 10.1016/j.jfma.2014.02.008
23. Iscan A, Kurul S, Dirik E. Clinical findings of tuberous sclerosis in Turkish children. J Pediatr Neurol. (2015) 3:95–102. doi: 10.1055/s-0035-1557249
24. Kopp CM, Muzykewicz DA, Staley BA, Thiele EA, Pulsifer MB. Behavior problems in children with tuberous sclerosis complex and parental stress. Epilepsy Behav. (2008) 13:505–10. doi: 10.1016/j.yebeh.2008.05.010
25. Kosac A, Jovic N. Tuberous sclerosis complex, Serbian referral center experience. Vojnosanitetski Pregled. (2017) 76:170. doi: 10.2298/VSP170410170K
26. Pascual-Castroviejo I, Hernandez-Moneo JL, Pascual-Pascual SI, Viaño J, Gutiérrez-Molina M, Velazquez-Fragua R, et al. Significance of tuber size for complications of tuberous sclerosis complex. Neurologia. (2013) 28:550–7. doi: 10.1016/j.nrleng.2013.10.013
27. Saltik S, Karatoprak EY, Tasel B. Characteristics and the clinical prognosis of epilepsy in patients with a diagnosis of tuberous sclerosis complex. Turk Arch Pediatr. (2013) 48:123–30. doi: 10.4274/tpa.116
28. Toldo I, Brasson V, Miscioscia M, Pelizza MF, Manara R, Sartori S, et al. Tuberous sclerosis-associated neuropsychiatric disorders: a paediatric cohort study. Dev Med Child Neurol. (2019) 61:168–73. doi: 10.1111/dmcn.14055
29. Vignoli A, La Briola F, Peron A, Turner K, Vannicola C, Saccani M, et al. Autism spectrum disorder in tuberous sclerosis complex: searching for risk markers. Orphanet J Rare Dis. (2015) 10:154. doi: 10.1186/s13023-015-0371-1
30. Wataya-Kaneda M, Tanaka M, Hamasaki T, Katayama I. Trends in the prevalence of tuberous sclerosis complex manifestations: an epidemiological study of 166 Japanese patients. PLoS ONE. (2013) 8:e63910. doi: 10.1371/journal.pone.0063910
31. Wilbur C, Sanguansermsri C, Chable H, Anghelina M, Peinhof S, Anderson K, et al. Manifestations of tuberous sclerosis complex: the experience of a provincial clinic. Can J Neurol Sci. (2017) 44:35–43. doi: 10.1017/cjn.2016.311
32. Metwellay KA, Farghaly HS, Darweesh AEM, Hamed SA. Cognitive delay in children with tuberous sclerosis in a developing country: Clinical correlations. J Pediatr Epilepsy. (2012) 1:221–8. doi: 10.3233/PEP-12034
33. de Vries PJ, Hunt A, Bolton PF. The psychopathologies of children and adolescents with tuberous sclerosis complex (TSC): a postal survey of UK families. Eur Child Adolesc Psychiatry. (2007) 16:16–24. doi: 10.1007/s00787-006-0570-3
34. Moavero R, Benvenuto A, Emberti Gialloreti L, Siracusano M, Kotulska K, Weschke B, et al. Early clinical predictors of autism spectrum disorder in infants with tuberous sclerosis complex: results from the EPISTOP Study. J Clin Med. (2019) 8:788. doi: 10.3390/jcm8060788
35. Samir H, Ghaffar HA, Nasr M. Seizures and intellectual outcome: clinico-radiological study of 30 Egyptian cases of tuberous sclerosis complex. Eur J Paediatr Neurol. (2011) 15:131–7. doi: 10.1016/j.ejpn.2010.07.010
36. Baumer FM, Song JW, Mitchell PD, Pienaar R, Sahin M, Grant PE, et al. Longitudinal changes in diffusion properties in white matter pathways of children with tuberous sclerosis complex. Pediatr Neurol. (2015) 52:615–23. doi: 10.1016/j.pediatrneurol.2015.02.004
37. Baumer FM, Peters JM, Clancy S, Prohl AK, Prabhu SP, Scherrer B, et al. Corpus callosum white matter diffusivity reflects cumulative neurological comorbidity in tuberous sclerosis complex. Cereb Cortex. (2018) 28:3665–72. doi: 10.1093/cercor/bhx247
38. Benova B, Petrak B, Kyncl M, Jezdik P, Maulisova A, Jahodova A, et al. Early predictors of clinical and mental outcome in tuberous sclerosis complex: a prospective study. Eur J Paediatr Neurol. (2018) 22:632–41. doi: 10.1016/j.ejpn.2018.03.001
39. Caylor RC, Grote L, Thiffault I, Farrow EG, Willig L, Soden S, et al. Incidental diagnosis of tuberous sclerosis complex by exome sequencing in three families with subclinical findings. Neurogenetics. (2018) 19:205–13. doi: 10.1007/s10048-018-0551-y
40. Chopra M, Lawson JA, Wilson M, Kennedy SE, Taylor P, Buckley MF, et al. An Australian tuberous sclerosis cohort: are surveillance guidelines being met? J Paediatr Child Health. (2011) 47:711–6. doi: 10.1111/j.1440-1754.2011.02038.x
41. Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. (2011) 22:735–9. doi: 10.1016/j.yebeh.2011.08.037
42. Doherty C, Goh S, Young Poussaint T, Erdag N, Thiele EA. Prognostic significance of tuber count and location in tuberous sclerosis complex. J Child Neurol. (2005) 20:837–41. doi: 10.1177/08830738050200101301
43. Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. (2006) 21:846–51. doi: 10.1177/08830738060210100301
44. Kilincaslan A, Kok BE, Tekturk P, Yalcinkaya C, Ozkara C, Yapici Z. Beneficial effects of everolimus on autism and attention-deficit/hyperactivity disorder symptoms in a group of patients with tuberous sclerosis complex. J Child Adolesc Psychopharmacol. (2017) 27:383–8. doi: 10.1089/cap.2016.0100
45. Mizuguchi M, Ikeda H, Kagitani-Shimono K, Yoshinaga H, Suzuki Y, Aoki M, et al. Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain Dev. (2019) 41:1–10. doi: 10.1016/j.braindev.2018.07.003
46. Kingswood C, Bolton P, Crawford P, Harland C, Johnson SR, Sampson JR, et al. The clinical profile of tuberous sclerosis complex (TSC) in the United Kingdom: a retrospective cohort study in the Clinical Practice Research Datalink (CPRD). Eur J Paediatr Neurol. (2016) 20:296–308. doi: 10.1016/j.ejpn.2015.11.011
47. Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW, et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. (2019) 93:e200–9. doi: 10.1212/WNL.0000000000007749
48. Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol. (2015) 52:281–9. doi: 10.1016/j.pediatrneurol.2014.10.028
49. Staley BA, Montenegro MA, Major P, Muzykewicz DA, Halpern EF, Kopp CM, et al. Self-injurious behavior and tuberous sclerosis complex: frequency and possible associations in a population of 257 patients. Epilepsy Behav. (2008) 13:650–3. doi: 10.1016/j.yebeh.2008.07.010
50. Yang G, Shi ZN, Meng Y, Shi XY, Pang LY, Ma SF, et al. Phenotypic and genotypic characterization of Chinese children diagnosed with tuberous sclerosis complex. Clin Genet. (2017) 91:764–8. doi: 10.1111/cge.12920
51. Capal JK, Bernardino-Cuesta B, Horn PS, Murray D, Byars AW, Bing NM, et al. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. (2017) 70:245–52. doi: 10.1016/j.yebeh.2017.02.007
52. Muzykewicz DA, Newberry P, Danforth N, Halpern EF, Thiele EA. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy Behav. (2007) 11:506–13. doi: 10.1016/j.yebeh.2007.07.010
53. Wong V, Khong PL. Tuberous sclerosis complex: correlation of magnetic resonance imaging (MRI) findings with comorbidities. J Child Neurol. (2006) 21:99–105. doi: 10.1177/08830738060210020901
54. Campbell JM. Diagnostic assessment of Asperger's disorder: a review of five third-party rating scales. J Autism Dev Disord. (2005) 35:25–35. doi: 10.1007/s10803-004-1028-4
55. Wong V. Study of the relationship between tuberous sclerosis complex and autistic disorder. J Child Neurol. (2006) 21:199–204.
56. Nabbout R, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. Epilepsy in tuberous sclerosis complex: findings from the TOSCA Study. Epilepsia Open. (2018) 4:73–84. doi: 10.1002/epi4.12286
57. de Vries PJ, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. TSC-associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet J Rare Dis. (2018) 13:157. doi: 10.1186/s13023-018-0901-8
58. Pacheva I, Panov G, Gillberg C, Neville B. A girl with tuberous sclerosis complex presenting with severe epilepsy and electrical status epilepticus during sleep, and with high-functioning autism and mutism. Cogn Behav Neurol. (2014) 27:88–95. doi: 10.1097/WNN.0000000000000026
59. Peters JM, Prohl A, Kapur K, Nath A, Scherrer B, Clancy S, et al. Longitudinal effects of everolimus on white matter diffusion in tuberous sclerosis complex. Pediatr Neurol. (2018) 90:24–30. doi: 10.1016/j.pediatrneurol.2018.10.005
60. Peters JM, Struyven RR, Prohl AK, Vasung L, Stajduhar A, Taquet M, et al. White matter mean diffusivity correlates with myelination in tuberous sclerosis complex. Ann Clin Transl Neurol. (2019) 6:1178–90. doi: 10.1002/acn3.793
61. Prohl AK, Scherrer B, Tomas-Fernandez X, Davis PE, Filip-Dhima R, Prabhu SP, et al. Early white matter development is abnormal in tuberous sclerosis complex patients who develop autism spectrum disorder. J Neurodev Disord. (2019) 11:36. doi: 10.1186/s11689-019-9293-x
62. Curatolo P, Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. (2018) 2:495–504. doi: 10.1016/S2352-4642(18)30099-3
63. Curatolo P, Aronica E, Jansen A, Jansen F, Kotulska K, Lagae L, et al. Early onset epileptic encephalopathy or genetically determined encephalopathy with early onset epilepsy? Lessons learned from TSC. Eur J Paediatr Neurol. (2016) 20:203–11. doi: 10.1016/j.ejpn.2015.12.005
Keywords: tuberous sclerosis complex, epilepsy, autism spectrum disorder, prognostic factors, age at onset, genetic, TSC1, TSC2
Citation: Specchio N, Pietrafusa N, Trivisano M, Moavero R, De Palma L, Ferretti A, Vigevano F and Curatolo P (2020) Autism and Epilepsy in Patients With Tuberous Sclerosis Complex. Front. Neurol. 11:639. doi: 10.3389/fneur.2020.00639
Received: 11 March 2020; Accepted: 29 May 2020;
Published: 11 August 2020.
Edited by:
Brahim Tabarki Melaiki, University of Sousse, TunisiaReviewed by:
Iliyana Pacheva, Plovdiv Medical University, BulgariaCopyright © 2020 Specchio, Pietrafusa, Trivisano, Moavero, De Palma, Ferretti, Vigevano and Curatolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Specchio, bmljb2xhLnNwZWNjaGlvQG9wYmcubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.