 Shanglin Li
Shanglin Li Lin Wang1
Lin Wang1 Yingmai Yang
Yingmai Yang Jun Ma
Jun Ma Xinhua Wan
Xinhua Wan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 07 February 2020
Sec. Movement Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.01351
Background: Dystonia-24 (DYT24) is a monogenic autosomal dominant dystonia caused by mutations in the gene ANO3, which has shown phenotypic and genotypic heterogeneity according to previous reports.
Objective: To screen and identify ANO3 mutations in a cohort of patients with dystonia in China and to expand the spectrum of DYT24.
Methods: This study screened ANO3 mutations in 187 Chinese dystonia patients using next-generation sequencing (NGS). In silico investigations were conducted in detected ANO3 variants and co-segregation analysis was carried out if applicable. The effects of identified variants were classified according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG).
Results: Four different variants were identified in four unrelated dystonia patients, including three missense variants [c.1789G>C (p.V600L), c.182A>C (p.E61A), c.787A>G (p.M263V)] and one splice site change (c.1714-3T>C). The novel missense mutation c.1798G>C (p.V600L), identified in a teenaged girl with generalized dystonia, showed high pathogenicity and was classified as “likely pathogenic” according to ACMG guidelines. Of note, she responded well to deep brain stimulation.
Conclusion: Our study helps expand the mutational and clinical spectrum of DYT24 due to ANO3 mutations by further reporting four variants. Rare ANO3 variants appear to represent an uncommon cause of dystonia in China.
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal often repetitive movements, postures, or both, and is classified based on two distinct axes: clinical features and etiology (1). Etiological categorization based on clinical features is difficult since dystonia has a remarkable degree of phenotypic variability, with frequent overlap with different syndromes. The advent of next-generation sequencing (NGS) technology facilitates the discovery of new pathogenic genes, and different types of dystonia have been identified according to their responsible genes. Dystonia-24 (DYT24) is a monogenic autosomal dominant dystonia due to pathogenic mutations in ANO3 (NM_031418, encoding for anoctamin 3), which was first identified by Charlesworth et al. (2) with the clinical phenotype of craniocervical dystonia. To date, about 26 different variants (1 from our center in China) have been reported, with a variable clinical spectrum including craniocervical dystonia, myoclonus, limb dystonia, and generalized dystonia, with the age of onset ranging from childhood to the sixth decade of life (3–11). The phenotypic heterogeneity indicates that it is insufficient to just assess ANO3 mutations in focal dystonia, and emerging sporadic ANO3 variants raise attention to patients without family history. Thus, screening ANO3 mutations in dystonia patients without focal dystonia or family histories of dystonia may assist in expanding the diagnostic spectrum of DYT24. With the emerging application of NGS screening technologies in movement disorders, this study aims to screen ANO3 mutations in inherited or idiopathic dystonia by NGS to identify ANO3 variants in Chinese patients with dystonia and to expand the phenotypic and genotypic spectrum of DYT24.
In this single-center cohort, patients referred to our movement disorders center in the Department of Neurology at Peking Union Medical College Hospital from September 2016 to July 2019 were included according to the following inclusion and exclusion criteria. Inclusion criteria were patients (a) with dystonia as the predominant movement disorder identified by a movement disorder specialist, (b) who were clinically suspected of inherited or idiopathic dystonia with or without family history, and (c) who agreed to genetic testing. Patients meeting the following criteria were excluded: (a) clinical or paraclinical findings suggestive of an acquired cause, or (b) a TOR1A or THAP mutation identified by Sanger sequencing. Most patients underwent common biochemical testing (e.g., ceruloplasmin) and brain imaging. Demographic clinical data were collected as well as family histories. This study was approved by the ethics committee of Peking Union Medical College Hospital and written informed consent was obtained from all participants or their legal guardians.
Genomic DNA was exacted from blood samples of enrolled patients. Targeted gene capture sequencing designed for movement disorders including 148 genes (53 cases from September 2016 to August 2017) or whole exome sequencing (WES; 134 cases from September 2017 to July 2019) was carried out. Detailed methods have been described in the Supplementary Materials. Only missense, nonsense, splice site, stop-loss, in-frame insertion/deletion, and frameshift variants with a minor allele frequency (MAF) <0.5% (according to reference public databases including 1,000 Genomes Project, Exome Aggregation Consortium, and Genome Aggregation Database) were analyzed. In silico analyses were conducted to help evaluate pathogenicity using SIFT (http://sift.jcvi.org), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2), and MutationTaster (http://www.mutationtaster.org). The pathogenicity of identified variants was classified according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) (12).
A total of 187 patients were included in the study, with 88 males (47.3%) and 99 females (52.7%). The mean age of onset of the disease was 26.5 years old (range, 1–65). The average age at study inclusion was 31.3 years old (range, 5–67) with a mean disease duration of 4.7 years.
This study identified four variants in ANO3, including three missense variants: c.1789G>C (p.V600L), c.787A>G (p.M263V), and c.182A>C (p.E61A) separately in unrelated Patients 1–3, and one splicing variant (c.1714-3T>C) in Patient 4. None of these variants were found in the 1,000 Genomes Project or gnomAD databases. All identified variants were species conservative. Segregation analysis was conducted in two patients, which revealed a de novo mutation in Patient 1 and a paternal inheritance in Patient 3. Unfortunately, no family members of the proband were available for further segregation analysis in patients with variants c.787A>G and c.1714-3T>C, either because of social circumstances or because the relatives did not wish to take part in the study. The in silico analysis assessments and detailed phenotypic and genotypic results were summarized in Table 1.
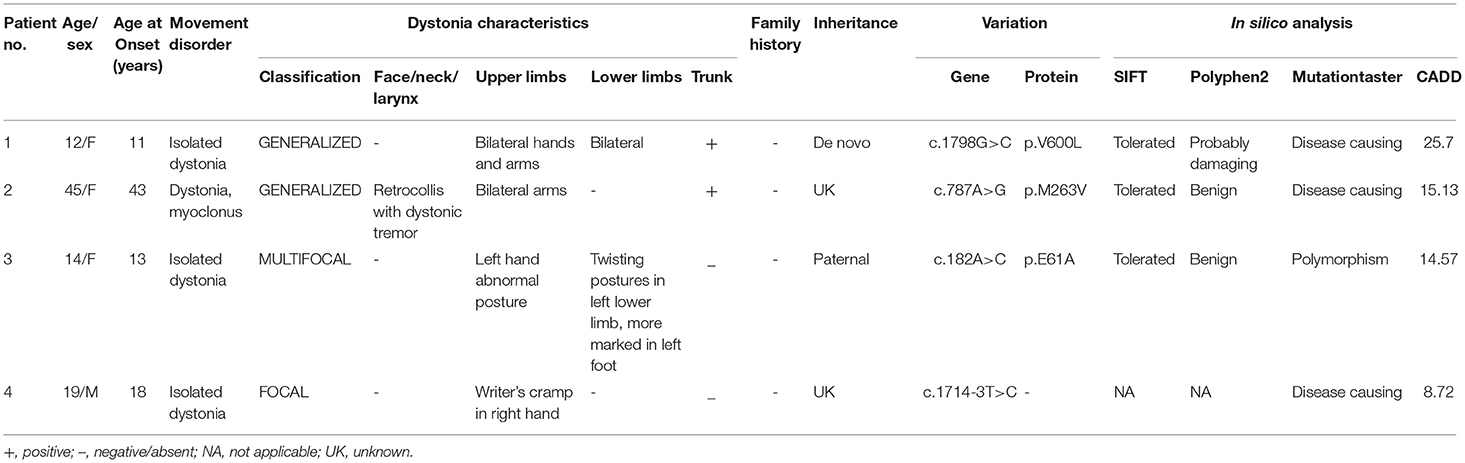
Table 1. Description of ANO3 variants identified in 187 dystonia cases, disease characteristics, and predictive pathogenicity.
A 12-year-old girl presented with abnormal postures in her left lower limb at the age of 11, which were aggravated when walking but almost normal during sleep. She was the only child of her non-consanguineous parents with no family history, or past or perinatal history. The progressing gait problems attracted her parents' attention and they took her to our clinic. Neurological examination showed a normal range of intelligence and generalized dystonia. On enrollment, she demonstrated slight dystonic trunk posturing, abnormal gait, and involuntary movements and abnormal posture in bilateral hands, which were more marked when walking (Supplementary Video 1). Common biochemical testing (e.g., peripheral smear, ceruloplasmin), blood metabolic screening, and brain magnetic resonance imaging (MRI) were unremarkable. WES of the patient and her unaffected biological parents revealed a novel de novo heterozygous missense variant in ANO3, rated as damaging by PolyPhen2 and MutationTaster. The girl partially responded to benzhexol for 1 month initially. However, over time, the efficacy dwindled, and her gait problems progressed despite multiple titrations in medication. After bilateral globus pallidus interna (Gpi) deep brain stimulation, a substantial improvement of dystonia was obtained, with a self-rated dystonia severity decreasing from 6 (on a scale of 0–10) pre-operation to 2 three months post-operation in a recent follow-up.
A 45-year-old woman had suffered from abnormal postures and involuntary movements in bilateral upper limbs, neck, and trunk for 2 years. The initial symptoms were myoclonic and dystonic movements in the shoulders, which ceased when lying in bed. Concurrently, her trunk started tilting to the left with progressive retrocollis. She had hyperthyroidism for 20 years with normal thyroid function tests 1 year before disease onset. She denied any significant family history. On examination, she demonstrated generalized dystonia including pronounced retrocollis with dystonic head tremor, and bilateral dystonic arms with occasional myoclonus in shoulders associated with abnormal trunk posturing. Cognitive function, muscle strength, and cerebellar function were normal. Brain imaging was unremarkable. Other tests such as autoimmune tests, metabolic screening, cerebrospinal fluid tests of oligoclonal band or cytology, and paraneoplastic markers were in normal ranges. WES revealed the missense mutation c.970A>G (p.M324V) in ANO3, predicted as damaging by PolyPhen2 and MutationTaster separately.
A 14-year-old girl developed progressive abnormal posturing in the left hand and leg for 1 year. She was diagnosed with scoliosis at 11 years. About 1 year prior to clinic admission, the girl noticed involuntary movement and abnormal posturing of her left hand when playing piano. Two months later, a slight strange posture of her left foot occurred and developed progressively in the following 10 months, resulting in gait problems. Administration of benzhexol showed slight benefits. She was the only child of her non-consanguineous parents with negative family history. A physical exam showed torsion scoliosis and twisting postures of the left foot and hand associated with an abnormal gait. No additional neurological signs such as tremor, spasticity, ataxia, weakness, or Kayser–Fleischer rings were present. Biochemical tests were unremarkable. A brain MRI showed static subtle T2 hyperintensity associated with T1 hypointensity in the right putamen. A novel mutation [c.182A>C (p.E61A)] in ANO3 was identified by WES and was predicted to be tolerated by SIFT, a polymorphism by MutationTaster, and a CADD (Combined Annotation Dependent Deletion) score of 19.07. Co-segregation analysis revealed that her unaffected father carried the same variant.
A 19-year-old male patient complained of moderate abnormal posture and involuntary movements only in the right hand while writing. Other tasks using the right hand were unaffected. Past history and family history were normal. Upon examination, isolated focal dystonia of writer's cramp in the right hand was present with no additional abnormal neurological signs. Tests including metabolic screening, ceruloplasmin, and brain MRI showed no significant findings. After a trial of levodopa with no benefit, the patient refused to take medicine and began writing using his left hand. WES detected a novel c.1714-3T>C mutation in ANO3, predicted as “disease causing” by MutationTaster. As it was a splicing variant, analyses by SIFT and PolyPhen2 were not applicable. Samples of his parents were not available since they lived far away.
DYT24 is an autosomal dominant isolated dystonia with typical clinical features of craniocervical dystonia caused by mutations in ANO3, which was first identified by Charlesworth et al. (2). ANO3 (27 exons) is located in chromosome 11p14.3-11p.14.2, encodes anoctamin 3 (also known as TMEM16C), and is mainly expressed in the striatum, hippocampus, cortex, and dorsal root ganglion (DRG) (13). TMEM16C belongs to the transmembrane protein (TMEM16) family, a group of 10 homologous membrane-spanning proteins, which are related to genetic diseases such as asthma, dystonia, limb-girdle muscular dystrophy, Miyoshi myopathy, Scott syndrome, and cerebellar ataxia (13). Since the identification of the ANO3 gene in craniocervical dystonia families, subsequent work confirmed the role of this gene in dystonia, which was summarized in Table 2. To date, 26 variants have been reported. In the majority of patients, ANO3 variants were confirmed in familial torticollis patients. However, sporadic variants with different clinical features of segmental, multifocal, or generalized dystonia have also been reported, with some even accompanied by other neurological signs such as tremor and myoclonus.
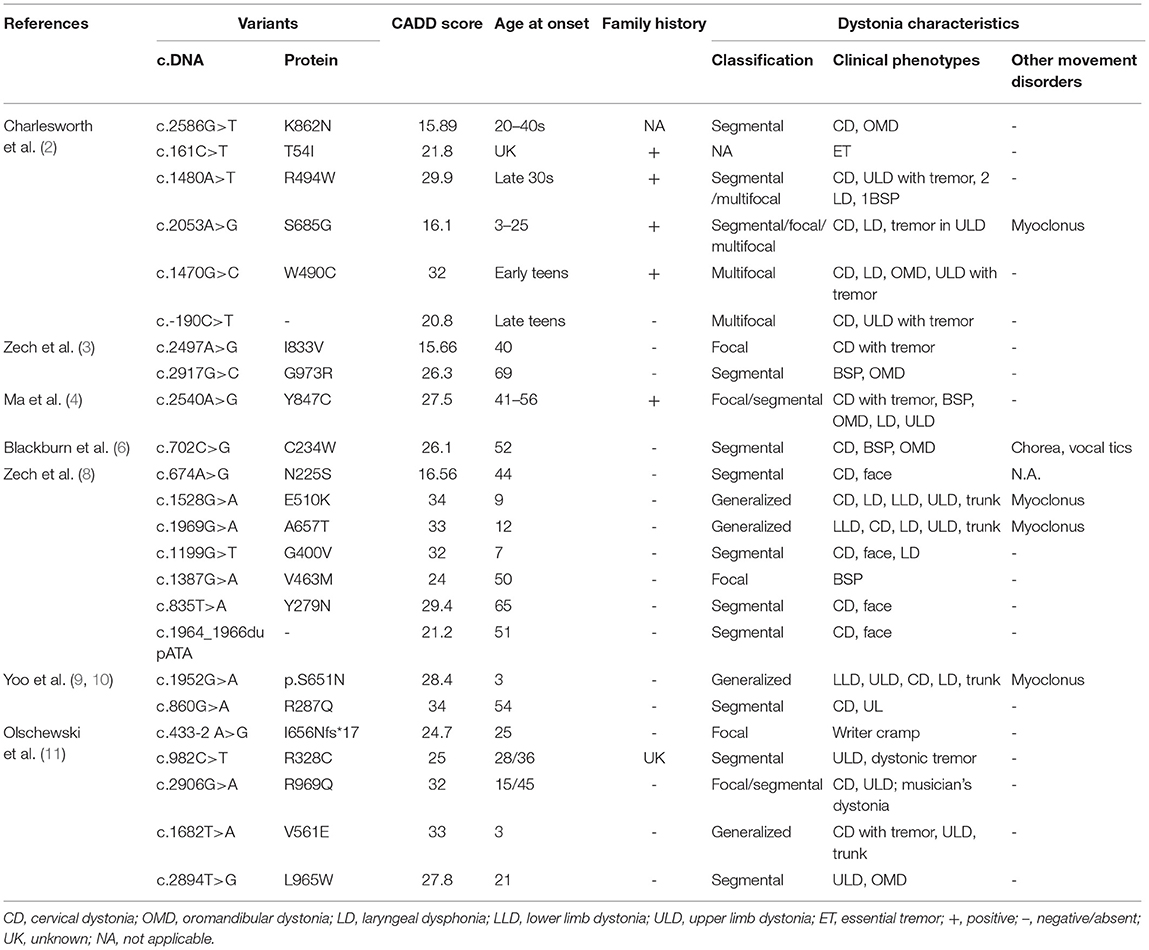
Table 2. Previously described genetic and clinical characteristics of ANO3 variants that are likely associated with disease.
In our study, ANO3 variants were found in 2.1% of clinically suspected idiopathic or inherited dystonia patients, which indicated that this type of dystonia might be rare in Chinese patients. However, the exact prevalence of ANO3 mutations still needs to be evaluated in large sample populations. In addition, we observed heterogeneity in both genotypes and phenotypes in DYT24 caused by ANO3 mutations in Chinese dystonia patients. In terms of phenotypes, all patients in our study were the only affected ones in their family pedigree. Previous studies have noted two peaks of age of onset in DYT24, one in childhood and one in mid-adulthood (40–49 years). Among the four patients with ANO3 mutations, three were affected under 18 years of age whereas one developed symptoms in her 40s. Interestingly, only one patient had cervical dystonia, accompanied by dystonia and myoclonus in other parts including the trunk and bilateral upper limbs, which was considered generalized dystonia. One patient presented with generalized dystonia involving the trunk and bilateral lower limbs and hands. Of note, she responded well to deep brain stimulation surgery in line with three previously reported successful operations (8, 9, 11). Similar to a recent study (11), writer's cramp was also observed in one patient with a different missense ANO3 mutation. Despite a negative past history and the missense mutation in ANO3 in the patient presenting with dystonia in the left lower limb and hand, we were cautious in diagnosing DYT24 because her brain MRI showed subtle abnormal signals in the contralateral putamen and her unaffected father carried the same variant. However, due to a gradual progression of dystonia despite subtle static abnormal signals in the brain MRI, and a possible reduced penetrance in ANO3 (5), it is difficult to draw a definite diagnosis of etiology, making further functional studies required.
Regarding the genotypes, three missense variants and one splice site change were found in unrelated patients (Figure 1). According to ACMG guidelines, the variant c.1798G>C (p.V600L) was rated “likely pathogenic” because this variant was proven to be de novo by co-segregation analysis [strong pathogenic criterion 2 (PS2)], it was absent from population databases [moderate pathogenic criterion 2 (PM2)], and it was predicted to be deleterious by multiple computational methods [supporting pathogenic criterion 3 (PP3)]. The interpretation of the other three variants is difficult and they are rated as “variants of unknown reason” for now, needing additional data such as segregation analysis and functional studies to elucidate their role in DYT24.
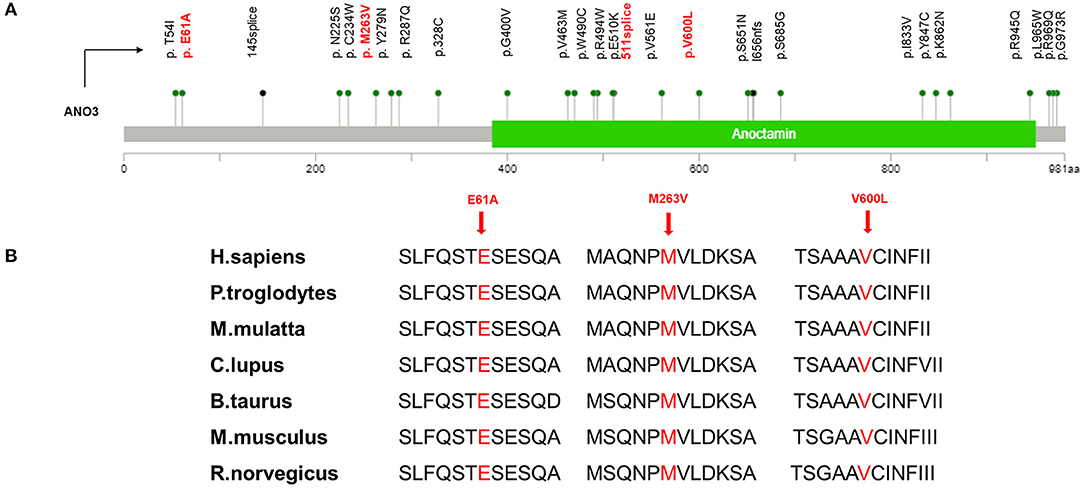
Figure 1. (A) Mutation mapper of previously identified ANO3 mutations related to dystonia were marked in black and variants in our study were marked in red. (B) High evolutionary conservation of three missense mutation.
However, it remains unclear how ANO3 mutations cause dystonia at a mechanistic level. Early studies showed that the ANO3 protein (or TMEM16C) functions as a calcium-activated chloride channel, like its family members TMEM16A and TMEM16B (2). However, a recent study by Huang et al. (14) has demonstrated that TMEM16C acts indirectly by regulating sodium-activated potassium channel trafficking in the dorsal root ganglion, which functions in the regulation of pain signaling. Further functional studies are needed to better understand the role of ANO3 in dystonia.
In summary, our study expands the mutational and clinical spectrum of DYT24 due to ANO3 mutations in dystonia. Rare ANO3 variants seem to represent an uncommon cause of dystonia in China. The interpretation of variants in ANO3 remains difficult, thus demanding further functional studies to better understand the role of ANO3 in dystonia.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by Institutional Review Board of Peking Union Medical College Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
SL: conception of the work, data acquisition, statistical analysis, and writing of the first draft. XW: design and organization of the work, manuscript review, and critique. LW, YY, and JM: data acquisition, manuscript review, and critique.
This study was supported by the National Key Research and Development Program of China (2018YFC1314700).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank all the study participants and their family members.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.01351/full#supplementary-material
1. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. (2013) 28:863–73. doi: 10.1002/mds.25475
2. Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. (2012) 91:1041–50. doi: 10.1016/j.ajhg.2012.10.024
3. Zech M, Gross N, Jochim A, Castrop F, Kaffe M, Dresel C, et al. Rare sequence variants in ANO3 and GNAL in a primary torsion dystonia series and controls. Mov Disord. (2014) 29:143–7. doi: 10.1002/mds.25715
4. Ma LY, Wang L, Yang YM, Feng T, Wan XH. Mutations in ANO3 and GNAL gene in thirty-three isolated dystonia families. Mov Disord. (2015) 30:743–4. doi: 10.1002/mds.26190
5. Stamelou M, Charlesworth G, Cordivari C, Schneider SA, Kagi G, Sheerin UM, et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov Disord. (2014) 29:928–34. doi: 10.1002/mds.25802
6. Blackburn PR, Zimmermann MT, Gass JM, Harris KG, Cousin MA, Boczek NJ, et al. A novel ANO3 variant identified in a 53-year-old woman presenting with hyperkinetic dysarthria, blepharospasm, hyperkinesias, and complex motor tics. BMC Med Genet. (2016) 17:93. doi: 10.1186/s12881-016-0354-7
7. Miltgen M, Blanchard A, Mathieu H, Kreisler A, Jean Pierre D, Salgado D, et al. Novel heterozygous mutation in ANO3 responsible for craniocervical dystonia. Mov Disord. (2016) 31:1251–2. doi: 10.1002/mds.26717
8. Zech M, Boesch S, Jochim A, Weber S, Meindl T, Schormair B, et al. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Mov Disord. (2017) 32:549–59. doi: 10.1002/mds.26808
9. Yoo D, Kim HJ, Lee JS, Lee S, Kim SY, Choi M, et al. Early-onset generalized dystonia starting in the lower extremities in a patient with a novel ANO3 variant. Parkinsonism Relat Disord. (2018) 50:124–5. doi: 10.1016/j.parkreldis.2018.02.012
10. Yoo HS, Lee H, Chung SJ, Lee JS, Hong SK, Lee PH, et al. A novel heterozygous ANO3 mutation with basal ganglia dysfunction in a patient with adult-onset isolated segmental dystonia. J Clin Neurol. (2018) 14:596–7. doi: 10.3988/jcn.2018.14.4.596
11. Olschewski L, Jesus S, Kim HJ, Tunc S, Lons S, Junker J, et al. Role of ANO3 mutations in dystonia: a large-scale mutational screening study. Parkinsonism Relat Disord. (2019) 62:196–200. doi: 10.1016/j.parkreldis.2018.12.030
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
13. Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev. (2014) 94:419–59. doi: 10.1152/physrev.00039.2011
Keywords: ANO3, DYT24, dystonia, Chinese, NGS - next generation sequencing
Citation: Li S, Wang L, Yang Y, Ma J and Wan X (2020) ANO3 Mutations in Chinese Dystonia: A Genetic Screening Study Using Next-Generation Sequencing. Front. Neurol. 10:1351. doi: 10.3389/fneur.2019.01351
Received: 18 September 2019; Accepted: 09 December 2019;
Published: 07 February 2020.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Juan Carlos Martinez Castrillo, Ramón y Cajal University Hospital, SpainCopyright © 2020 Li, Wang, Yang, Ma and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinhua Wan, eGh3YW5wdW1jaEBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.