 Alexander Kilzheimer
Alexander Kilzheimer Thomas Hentrich
Thomas Hentrich Simone Burkhardt
Simone Burkhardt Julia M. Schulze-Hentrich
Julia M. Schulze-Hentrich- Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany
Parkinson's disease (PD) is the most common neurodegenerative movement disorder that affects extensive regions of the nervous system. Its current clinical diagnosis is based on motor symptoms that appear late during disease progression when substantial proportions of the nigrostriatal dopaminergic neuron population are lost already. Although disturbances in sleep and other biofunctions often surface years prior to motor impairments and point to a long prodromal phase, these phenotypic signs in a person's midlife lack predictive power. They do, however, signal the unfolding of the disease and suggest molecular correlates that begin deviating early on. Revealing such trajectories, hence, promises not only a better understanding of prodromal PD but may also enable a much-needed earlier diagnosis. A nexus that may harbor such molecular trajectories is the epigenome as key etiological factors of PD—genetics, age, and environment—influence this substrate. An earlier diagnosis would also allow earlier interventions and lifestyle adjustments to improve brain function and reduce symptoms. In this review, we describe the challenges of diagnosing PD early on and highlight the opportunities that may arise from steering research efforts towards comprehensive interrogations of molecular layers during the long-time neglected midlife phase. In particular, we emphasize how existing cohorts of at-risk individuals, available animal models, and suitable markers may come together and aid in revealing molecular trajectories that offer diagnostic utility for PD in its prodromal stage.
Introduction
Neurodegenerative disorders are a growing health threat in demographically aging societies. The complexity of many of these disorders is stereotypically exemplified in Parkinson's disease (PD), the second most common neurodegenerative disorder after Alzheimer's disease (1–4). Numbers of PD patients globally have more than doubled between 1990 and 2015 to an estimated 6.2 million and are predicted to affect more than 14 million individuals by 2040 (5–7).
The pathological hallmark of PD are misfolded alpha-synuclein protein (aSyn) structures accumulating in cellular inclusions known as Lewy bodies (8). Identifying neurons with aSyn inclusions in post-mortem pathological examinations of the brain is considered the gold standard for diagnosing PD (7, 9).
While about 10% of PD cases are familial and can be traced to specific genetic defects either in the SNCA locus that encodes aSyn (10) or in few other genes such as LRRK2 (11), the vast majority derives from an unclear etiology. For these idiopathic cases, an unresolved interplay of genetic predisposition, age-related processes, and environmental as well as behavioral factors is assumed to be responsible for disease manifestation (12, 13). While several environmental and lifestyle factors, including exposure to pesticides, intake of beta blockers, or head trauma seemingly increase risk for disease onset and progression (14), age is the single most influential risk factor for PD (15, 16).
The prevalence rate of PD increases with age and rises from about 1% in people aged 60+ to 3.5% in individuals of 85–89 years of age (16–18). Prior to clinical diagnosis, typically at the age of 55–65, most idiopathic patients experience years or sometimes decades of unspecific symptomology, indicating the existence of a long prodromal phase. While these prodromal symptoms lack diagnostic specificity (19), they signal an early unfolding of pathology and, hence, are likely accompanied by molecular correlates that deviate from the healthy norm. Revealing such deviating molecular trajectories would greatly complement phenotypic markers and help to arrive at an earlier diagnosis than with today's criteria based primarily on motor symptoms. Earlier diagnosed patients would then benefit from engaging in neuroprotective lifestyle adjustments as well as optimized pharmacological treatments.
In this review, we stress the need and opportunity for diagnosing PD in its prodromal phase and summarize existing findings in order to identify neglected aspects and steer future research that might allow advancing from late- to early-stage PD diagnostics.
Clinical Parkinson's Disease
PD is a progressive neurodegenerative disease that manifests in selective loss of dopaminergic neurons starting in early disease stages (20) and affects an array of motor and non-motor functions (21, 22). This loss is most prominent in the substantia nigra pars compacta, from which important afferent projections for the basal ganglia originate (23). Within the substantia nigra, the caudal and ventrolateral tier that project into the dorsal putamen of the striatum are usually affected most (24). The successive dopamine deficiency particular in this area is most likely causal for the development of characteristic motor features (25). However, neuronal loss in PD is not restricted to the basal ganglia, but also takes place in other brain regions including the hypothalamus, amygdala, locus coeruleus, raphe nucleus, nucleus basalis of Meynert, pedunculopontine nucleus, and dorsal motor nucleus of the vagus nerve (26). In addition, molecular hallmarks of PD can be found in non-brain compartments of the nervous system, such as the spinal cord, vagus nerve, sympathetic ganglia, cardiac plexus, enteric nervous system, adrenal medulla, and cutaneous nerves (27–30). Aside from dopamine, non-dopaminergic pathways seem to be affected by PD, too (31).
Similarly, clinical symptoms of PD are as diverse as the affected tissues, with motor impairments being the most prominent and characterizing deficits. Specifically, resting tremor, bradykinesia, rigidity, and postural instability are regarded as indicative features of PD (32, 33), with bradykinesia having the strongest correlation with the degree of dopamine deficiency (34). Clinically, PD is diagnosed primarily on motor symptoms (35) that surface in advanced disease stages when substantial proportions of the nigrostriatal dopaminergic neurons are lost (36–38).
Prodromal Parkinson's Disease
In addition to motor impairments, symptoms of PD typically involve a plethora of non-motor features that range from sensory disturbances, to neuropsychiatric and cognitive symptoms, to sleep disorders, and to autonomic dysfunction (39–41). Olfactory impairment/hyposmia, rapid-eye movement sleep behavior disorder (RBD), constipation, impairments in memory retrieval and decision making as well as depression (42–45) often have a greater impact on the patient's quality of life than motor deficits (46). A number of these symptoms precede motor deficits by years or even decades (Figure 1) (23, 47–49), but due to the lack of specificity, diagnostic possibilities based on these markers are still limited (19). However, for research purposes, many markers have been summarized in the Movement Disorder Society (MDS) research criteria for prodromal Parkinson's disease (50), in order to estimate the probability of prodromal PD. Among them are risk markers such as sex, caffeine consumption, smoking behavior, genetic mutations, and a positive family history, as well as prodromal markers, including RBD, olfactory loss, depression, and erectile dysfunction.
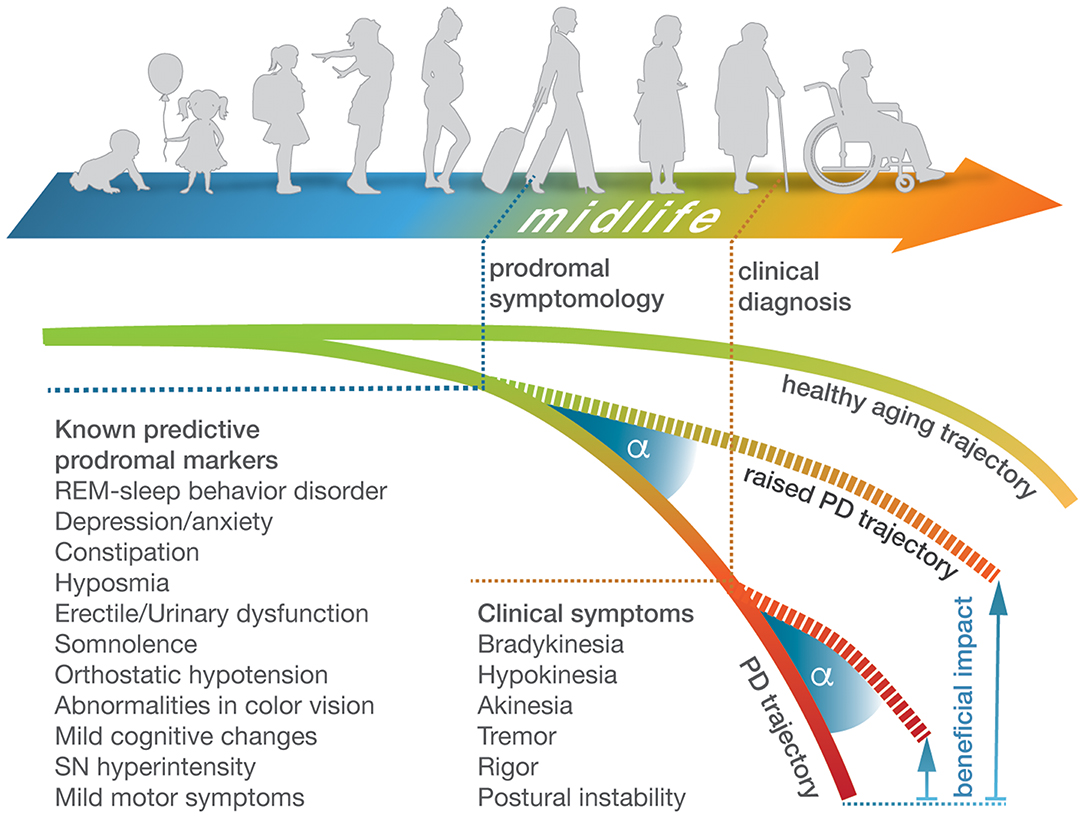
Figure 1. Prodromal and clinical symptomology of Parkinson's disease over the course of life. Schematic representation of the course of life from early childhood to old age and associated brain health trajectories that deviate in PD patients from the healthy norm (upper and lower curves). Current clinical diagnosis of PD is based on hallmark motor phenotypes that occur in advanced disease stages. They are preceded by prodromal symptomology that surfaces years to decades earlier during midlife but is less specific and varies considerably with respect to timing and features between individuals (44). Tracing deviating molecular trajectories from clinical stages into midlife would enable a prodromal diagnosis of PD, thereby widening the beneficial impact (shown as angle under raised brain health trajectories) of neuroprotective lifestyle adjustments or treatment options.
The Need in Research to Focus on Midlife in Parkinson's Disease
The prodromal stage of PD coincides with midlife, approximately between 40 and 60 years of age (51). Midlife is a highly relevant period for biopsychosocial development with high interindividual heterogeneity and long-term impact for health in later life phases (51). In particular, the health status of the nervous system is influenced by lifestyle choices during midlife. Extensive stress during midlife, for example, correlates with self-care disability in older age (52). In contrast, individuals who exercise regularly in midlife have higher speed of cognitive processing, better memory and executive functions, and reduced risk of dementia later in life (53, 54). In addition, supportive social relationships and positive control beliefs in midlife support functional health and cognitive skills and curtail age-related health decline (55, 56). Regarding behavioral influences specific to PD, a physically and mentally active lifestyle in midlife is associated with a lower disease incidence and capable to reduce risk for developing PD by up to 40% (57–60). Hence, diagnosing PD in midlife would allow taking advantage of the neuroprotective potential of beneficial behavioral and environmental factors, most importantly by promoting physical activity. Pharmacological interventions might profit from a wider therapeutic time window that may lead to increased efficacy, too (61).
The role of midlife in PD, however, has largely been neglected by research so far (Figure 2A). Despite publication shares on PD and related neurodegenerative disorders like Alzheimer's and Huntington's disease indicate a focus on old age, the Alzheimer's field already shows rising awareness for midlife as increasing publication shares suggest (Figure 2B). In PD, this paradigm shift is not obvious yet but essential to embrace in order to derive a comprehensive understanding of the prodrome in PD.
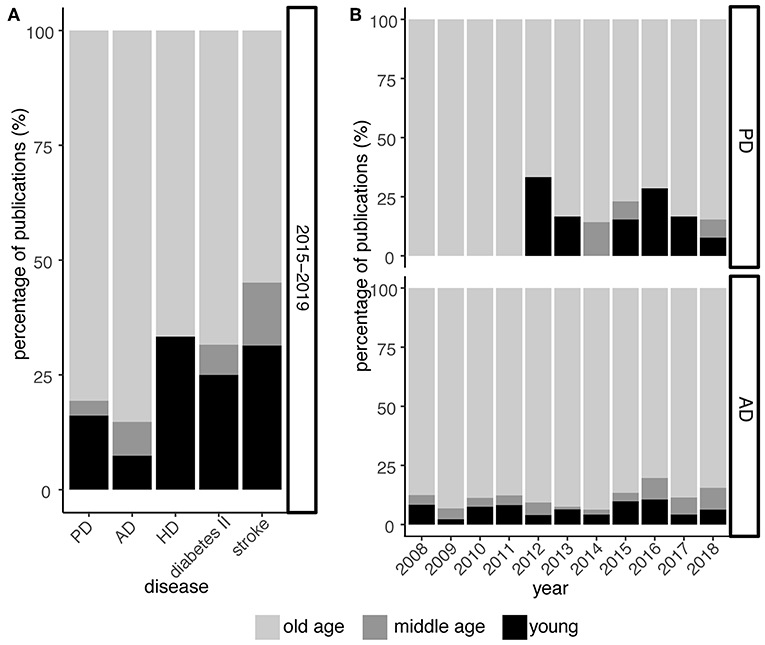
Figure 2. Midlife is still largely neglected in PD research. (A) Relative publication shares over the past 5 years (2015–2019) examining PD, Alzheimer's disease (AD), Huntington's disease (HD), type II diabetes, and stroke with respect to young, middle, and old age. (B) As in (A) but focusing on PD and AD at greater temporal resolution for the past 10 years. While research on the role of midlife in context of AD is continuously increasing, this trend is less obvious and seemingly lagging behind for PD. Numbers are derived from PubMed searches, querying publication titles and abstracts. For diseases, the terms [parkinson*] for PD, [alzheimer*] for AD, [huntington*] for HD, [maturity-onset diabetes, noninsulin-dependent diabetes mellitus, diabetes mellitus type II, type 2 diabetes mellitus] for type II diabetes, and [ischemic stroke, cerebrovascular accident, cerebral infarction] for stroke were used. For the “young” phase, the terms [adolescen*, youth*, juven*, young adult*, early*life, early adulthood, childhood], for the “middle age” phase, the terms [mid*life, middle*age*, mid*adulthood, middle*adulthood] and for the “old age” phase, the terms [late life, old*age*, senescence, elder*, senior*, geriatr*, aged] were used, respectively.
Molecular Markers of Parkinson's Disease
Research has explored a number of potential molecular markers for PD that center on measures that directly or indirectly assess the degree of mitochondrial dysfunction, oxidative stress, Lewy bodies formation, neuroinflammation, and other core pathological characteristics of PD in readily accessible biofluids or biopsies (62). Their utility in diagnosing or prognosing the disease early on, however, has remained limited (63, 64). Even for aSyn and its pivotal role in the etiology of familial and sporadic PD, its protein level in cerebrospinal fluid shows no correlation with progression of clinical motor symptoms (65). Similarily in saliva, in which aSyn can be detected, its concentration does not differ between PD patients and healthy controls (66). While even Lewy bodies have been found in salivary glands of PD patients (30), they too lack sensitivity and specificity (67). And neither in blood, for which aSyn (68), EGF (69), and several cytocines (70) have been considered, tests are conclusive. Interestingly, a growing amount of data suggests that aSyn may also accumulate in skin nerves of patients with PD and related diseases such as dementia with Lewy bodies and idiopathic RBD (71–75). These observations are currently being followed up with respect to their utility as potential biomarkers.
Despite these research efforts of the last decades that identified a wealth of candidate molecular readouts, there is no single reliable biomarker available yet that provides clinical utility for diagnosing or prognosing PD (76). This issue may largely result from the relatively low prevalence of PD on which sensitivity, specificity, and the predictive values of these measures are dependent (77) and has been discussed before (78–80).
The Promising Field of Epigenetic Biomarkers
While suggestions exist to combine multiple markers in order to improve their diagnostical accuracy and to better capture the complexity of the disease (62), other approaches seek to increase the predictive power by focusing on molecular substrates on which several etiological or pathological aspects converge. With respect to the latter line of research, the epigenome has gained increasing research attention as major etiological contributors of the disease—genetics, age, and environment—are known to influence epigenetic modifications such as DNA methylation (81). The metastable nature of some epigenetic marks is both dynamic and yet sufficiently stable to capture and record environmental influences and behavioral stimuli that allow tracing disease pathology (82, 83).
Also with respect to the passage of time, the epigenome undergoes distinctive changes (84) and allows intriguingly accurate readouts of biological age through so-called epigenetic clocks that are based on DNA methylation at particular CpG sites in the genome (85–87). The accuracy and robustness of some of these clocks with respect to sex and tissue type (87, 88) led to their consideration as diagnostic readout for biological age in the context of PD, given that the disease has been suggested to represent a form of accelerated aging (89). Indeed, the epigenetic clock in blood of clinical PD cases indicates advanced age (90), although the deviation between chronological and biological age compared to effect sizes for related neurodegenerative diseases is much smaller (91). Nevertheless, genes near such clock sites are also often linked to PD and other neurodegenerative diseases (92).
These and other findings support the idea that epigenetic regulation is highly relevant in the context of complex neurodegenerative disease, especially in PD (93, 94), and that the epigenome represents a nexus which allows identifying epigenetic traces of early molecular deviations long before protein aggregation, widespread neuronal loss, and other characterizing features of advanced disease stages dominate molecular disturbances (95).
Nevertheless, while the epigenome may yield diagnostic markers for PD, it also harbors challenges and limitations. A critical challenge is that the magnitude of epigenetic changes in patients has to exceed the variation within populations and cell composition of the investigated tissue. Standardization has to be improved in order to reduce technical noise and evaluate the most suitable of the available interrogation technologies.
Longitudinal Studies of at-risk Individuals
In order to reveal epigenetic traces as a proxy for deviating molecular trajectories during prodromal PD stages, comprehensive interrogations of epigenomic layers are required. While there is rich molecular data available covering the transcriptome, the epigenome, and even the metabolome for advanced disease stages (96–98), similar datasets for preclinical and prodromal stages of PD are still limited. Closing this gap, hence, requires a research shift from molecular interrogations in clinically diagnosed PD patients in advanced disease stages to early disease stages and, most importantly, at-risk individuals with prodromal symptomology in midlife. To address this need, some cohorts have already been established over the past few years (7). Among others, there are the:
• Parkinson's Progression Markers Initiative/PPMI (99): The study consists of 400 newly diagnosed PD patients and 200 healthy subjects that are being assessed for clinical, imaging, and molecular markers in urine, DNA, serum, plasma, RNA, whole blood, and cerebrospinal fluid.
• Prospective Validation of Risk Factors for the Development of Parkinsonian Syndromes/PRIPS (100): 1847 at baseline PD-free participants stratified by age, sex, family history, olfaction, motor functions, and substantia nigra echogenicity.
• Tübingen Evaluation of Risk Factors for Early Detection of Neurodegeneration/TREND (101): At baseline, this study enlists about 1,200 healthy individuals with no or at least one of three prodromal PD symptoms from depression, rapid eye movement behavior disorder, and hyposmia.
• The Parkinson's Associated Risk Study/PARS (102): A cohort of 303 hyp- and normosmic subjects without baseline diagnosis of PD.
These cohorts provide rich prior-to-diagnosis data from at-risk individuals that may develop PD during the observation period or that have just been diagnosed. Studies based on these cohorts focus on developments of prodromal symptomology and epidemiologic characteristics, but many of them also collect biological samples for molecular interrogations and profiling. Intensifying these efforts towards a better understanding of the molecular prodrome is crucial in order to tell apart typical age-related adaptations from disease-relevant molecular changes that might point at distinctive markers.
Modeling Parkinson's Disease in Animals
Any studies on these promising cohorts, however, will still remain restricted to measures available through biofluids or biopsies. Inferring disease progression indirectly from these measures and not being able to directly profile disease unfolding on molecular level in the brain remains a fundamental limitation in neurodegenerative disease research. That is why animal models are inevitable as they uniquely enable studying any disease-relevant tissue directly and longitudinally over disease progression. In addition, they allow assessing environmental and behavioral influences on etiologically complex diseases such as PD through standardization and fine-grained monitoring of living conditions. However, this approach comes with the complex question of what exactly is being modeled. While it is possible to induce aSyn overexpression, Lewy bodies-like structures, or motor-deficient phenotypes, they only partially recapitulate the underlying molecular mechanism, histopathology, and phenotype of the disease. As with any model, of course, this reduction leads to comparability problems between the genuine disease and its models (103).
Despite these limitations, there are commonly accepted animal models of PD for a wide range of organisms along the evolutionary tree from worms and flies to non-human primates (104–106). Among them, rodent models lend themselves to research due to their relatively short lifespan but still sufficiently high organismal complexity.
Toxin-Induced Animal Models of Parkinson's Disease
For PD rodent models, a common approach to induce pathology is to specifically target dopaminergic neurons with exogenous neurological toxins. Compounds such as 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administered over days to weeks lead to loss of dopaminergic neurons and developments of PD-like phenotypes (107). The severity and extent of symptoms depend on dosage, mode of administration, and choice of rodent strain, thus allowing a certain combinatorial flexibility to mimic different and rather advanced stages of the disease (108, 109). As toxin-induced models typically lack aSyn inclusions (110) and starkly differ in their rapid loss of dopaminergic neurons from the much slower neuronal demise in patients, their utility for understanding prodromal PD remains limited (111, 112).
Transgenic Models of Parkinson's Disease
Complementing toxin-induced models, transgenic animals that carry genetic defects linked to familial forms of PD represent an alternative approach to study the disease. Besides models with mutations in key loci including LRRK2, PINK1, PRKN, or DJ-1 (113), those with defects in SNCA belong to the most commonly used ones. The human wildtype SNCA-overexpressing rat model (114), for example, represents a system that displays aSyn inclusions as well as a loss of dopaminergic neurons in an age-dependent manner accompanied by phenotypic impairments in sense of smell, increased anxiety-like behavior (115), and motor deficits later on (114). Genome-wide molecular investigations in a mouse model carrying the same transgenic construct suggest distinctive disturbance modes of deviating gene expression trajectories that underlie the earliest stages of SNCA-related pathology (116) and their modulation through environmental conditions (117, 118). By capturing early characteristics of pathology, these rodent models are also suited to investigate epigenetic mechanisms that presumably play a role in integrating and transducing external stimuli into the genomic regulatory program and form the basis of disease-modulating gene-environment interactions.
Lastly, combinations of toxin-induced and transgenic models exist, too. Examples are the LPS-treated Parkin-deficient mouse model (119) or the rotenone-treated LRRK2R1441G knock-in mice line (120), which seek to unite the advantages of both modeling approaches and are geared to reflect larger parts of PD pathogenesis.
Discussion
Taken together, the current clinical diagnosis of PD is primarily based on motor symptoms that occur at advanced disease stages when neuroprotective and risk-mitigating lifestyle adjustments are capped because of wide-spread neuronal loss (36–38). Distinctive phenotypic changes, however, already surface up to 20 years earlier and suggest unfolding of early pathogenic processes in midlife. Yet, these phenotypic characteristics alone lack discriminative power to diagnose PD in its so-called prodromal stage (19). To overcome this hurdle and substantially advance PD diagnostics, the underlying molecular prodrome in midlife requires greater research attention. Given that sequencing technology allows genome-wide molecular interrogations of unprecedented depth and breadth down to the level of single cells at ever-decreasing cost, research needs to fully embrace the paradigm shift and look prospectively at this alleged old-age disease. Cohorts enriched for at-risk individuals in tandem with animal models that facilitate paired measurements of brain and peripheral pathology in controlled environmental conditions have great potential to tackle the complexity of the disease from a new angle and to identify novel biomarkers. With respect to the latter, the epigenome should receive particular attention as individual aetiological contributors are already known to influence epigenetic marks. If this holds true also in context of early disease stages, distinctive epigenetic signatures might be the sought-after molecular complement to the phenotypic prodromal symptomology. Together, they may translate into a pheno-molecular tool of clinical utility that not only allows diagnosing prodromal PD but also offers a monitorable readout for disease progression and assessment of pharmacological treatments.
Author Contributions
AK drafted the manuscript. SB did the PubMed data analysis. AK, TH, and JS-H wrote, reviewed, and edited the manuscript. JS-H conceived and coordinated the work. All authors have approved the final version.
Funding
AK was funded through the Interdisciplinary Center of Clinical Research Tübingen (IZKF Promotionskolleg). TH and JS-H are members of the decipherPD transnational consortium on Epigenomics of Complex Diseases (BMBF grant number 01KU1503). The research group of JS-H was supported by the Interdisciplinary Center of Clinical Research Tübingen (IZKF, 2053-0-0). We acknowledge support through the Deutsche Forschungsgemeinschaft and the Open Access Publishing Fund of the University of Tübingen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Heinzel S, Roeben B, Ben-Shlomo Y, Lerche S, Alves G, Barone P, et al. Prodromal markers in Parkinson's disease: limitations in longitudinal studies and lessons learned. Front Aging Neurosci. (2016) 8:147. doi: 10.3389/fnagi.2016.00147
2. Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. (2007) 68:384–6. doi: 10.1212/01.wnl.0000247740.47667.03
3. Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci. (2015) 9:124. doi: 10.3389/fncel.2015.00124
4. Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, et al. Past, present, and future of Parkinson's disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. (2017) 32:1264–310. doi: 10.1002/mds.27115
5. Bach J-P, Ziegler U, Deuschl G, Dodel R, Doblhammer-Reiter G. Projected numbers of people with movement disorders in the years 2030 and 2050. Mov Disord. (2011) 26:2286–90. doi: 10.1002/mds.23878
6. Dorsey ER, Bloem BR. The Parkinson pandemic—a call to action. JAMA Neurol. (2018) 75:9–10. doi: 10.1001/jamaneurol.2017.3299
7. Rees RN, Acharya AP, Schrag A, Noyce AJ. An early diagnosis is not the same as a timely diagnosis of Parkinson's disease. F1000Res. (2018) 7:1106. doi: 10.12688/f1000research.14528.1
8. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. (2013) 9:13–24. doi: 10.1038/nrneurol.2012.242
9. Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease. Neurology. (2016) 86:566–76. doi: 10.1212/WNL.0000000000002350
10. Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a008888. doi: 10.1101/cshperspect.a008888
11. Rui Q, Ni H, Li D, Gao R, Chen G. The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol. (2018) 16:1348–57. doi: 10.2174/1570159X16666180222165418
12. Kieburtz K, Wunderle KB. Parkinson's disease: evidence for environmental risk factors. Mov Disord. (2013) 28:8–13. doi: 10.1002/mds.25150
13. Racette BA, Willis AW. Time to change the blind men and the elephant approach to Parkinson disease? Neurology. (2015) 85:190–6. doi: 10.1212/WNL.0000000000001739
14. Bellou V, Belbasis L, Tzoulaki I, Evangelou E, Ioannidis JPA. Environmental risk factors and Parkinson's disease: an umbrella review of meta-analyses. Parkinsonism Relat Disord. (2016) 23:1–9. doi: 10.1016/j.parkreldis.2015.12.008
15. Emamzadeh FN, Surguchov A. Parkinson's disease: biomarkers, treatment, and risk factors. Front Neurosci. (2018) 12:612. doi: 10.3389/fnins.2018.00612
16. de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. (2006) 5:525–35. doi: 10.1016/S1474-4422(06)70471-9
17. de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, et al. Prevalence of parkinsonism and Parkinson's disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson's disease. J Neurol Neurosurg Psychiatry. (1997) 62:10–5. doi: 10.1136/jnnp.62.1.10
18. Twelves D, Perkins KSM, Counsell C. Systematic review of incidence studies of Parkinson's disease. Mov Disord. (2003) 18:19–31. doi: 10.1002/mds.10305
19. Berg D, Postuma RB. From prodromal to overt Parkinson's disease: towards a new definition in the year 2040. J Parkinsons Dis. (2018) 8:S19–23. doi: 10.3233/JPD-181457
20. Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. (2013) 136:2419–31. doi: 10.1093/brain/awt192
21. Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. (1986) 9:357–81. doi: 10.1146/annurev.ne.09.030186.002041
22. Lang AE, Lozano AM. Parkinson's disease. N Engl J Med. (1998) 339:1044–53. doi: 10.1056/NEJM199810083391506
23. Kalia LV, Lang AE. Parkinson's disease. Lancet. (2015) 386:896–912. doi: 10.1016/S0140-6736(14)61393-3
24. Jellinger KA. Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord. (2012) 27:8–30. doi: 10.1002/mds.23795
25. Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. (2009) 8:1150–7. doi: 10.1016/S1474-4422(09)70238-8
26. Dickson DW. Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. (2012) 2:a009258. doi: 10.1101/cshperspect.a009258
27. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White CL III, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. (2010) 119:689–702. doi: 10.1007/s00401-010-0664-3
28. Fumimura Y, Ikemura M, Saito Y, Sengoku R, Kanemaru K, Sawabe M, et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J Neuropathol Exp Neurol. (2007) 66:354–62. doi: 10.1097/nen.0b013e3180517454
29. Iwanaga K, Wakabayashi K, Yoshimoto M, Tomita I, Satoh H, Takashima H, et al. Lewy body-type degeneration in cardiac plexus in Parkinson's and incidental Lewy body diseases. Neurology. (1999) 52:1269–71. doi: 10.1212/WNL.52.6.1269
30. Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson's disease. Acta Neuropathol. (2010) 119:703–13. doi: 10.1007/s00401-010-0665-2
31. van der Heeden JF, Marinus J, Martinez-Martin P, van Hilten JJ. Importance of nondopaminergic features in evaluating disease severity of Parkinson disease. Neurology. (2014) 82:412–8. doi: 10.1212/WNL.0000000000000087
32. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. (2008) 79:368–76. doi: 10.1136/jnnp.2007.131045
33. Chaudhuri KR, Healy DG, Schapira AH, National Institute for Clinical Excellence. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. (2006) 5:235–45. doi: 10.1016/S1474-4422(06)70373-8
34. Vingerhoets FJ, Schulzer M, Calne DB, Snow BJ. Which clinical sign of Parkinson's disease best reflects the nigrostriatal lesion? Ann Neurol. (1997) 41:58–64. doi: 10.1002/ana.410410111
35. Zhang T-M, Yu S-Y, Guo P, Du Y, Hu Y, Piao Y-S, et al. Nonmotor symptoms in patients with Parkinson disease: a cross-sectional observational study. Medicine. (2016) 95:e5400. doi: 10.1097/MD.0000000000005400
36. Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Cli Morphol Neurochem Correlations J Neurol Sci. (1973) 20:415–55. doi: 10.1016/0022-510X(73)90175-5
37. Marek K, Innis R, van Dyck C, Fussell B, Early M, Eberly S, et al. [123I]beta-CIT SPECT imaging assessment of the rate of Parkinson's disease progression. Neurology. (2001) 57:2089–94. doi: 10.1212/WNL.57.11.2089
38. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. (2004) 318:121–34. doi: 10.1007/s00441-004-0956-9
39. Rodríguez-Violante M, Zerón-Martínez R, Cervantes-Arriaga A, Corona T. Who can diagnose Parkinson's disease first? Role of pre-motor symptoms. Arch Med Res. (2017) 48:221–7. doi: 10.1016/j.arcmed.2017.08.005
40. Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson's disease in primary care: a case-control study. Lancet Neurol. (2015) 14:57–64. doi: 10.1016/S1474-4422(14)70287-X
41. Stern MB, Siderowf A. Parkinson's at risk syndrome: can Parkinson's disease be predicted? Mov Disord. (2010) 25(Suppl 1):S89–93. doi: 10.1002/mds.22719
42. Chen H, Zhao EJ, Zhang W, Lu Y, Liu R, Huang X, et al. Meta-analyses on prevalence of selected Parkinson's nonmotor symptoms before and after diagnosis. Transl Neurodegener. (2015) 4:1. doi: 10.1186/2047-9158-4-1
43. Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The onset of nonmotor symptoms in Parkinson's disease (The ONSET PDStudy). Mov Disord. (2015) 30:229–37. doi: 10.1002/mds.26077
44. Postuma RB, Berg D. Prodromal Parkinson's disease: the decade past, the decade to come. Mov Disord. (2019) 34:665–75. doi: 10.1002/mds.27670
45. Siderowf A, Lang AE. Premotor Parkinson's disease: concepts and definitions. Mov Disord. (2012) 27:608–16. doi: 10.1002/mds.24954
46. Sjödahl Hammarlund C, Hagell P, Nilsson MH. Motor and non-motor predictors of illness-related distress in Parkinson's disease. Parkinsonism Relat Disord. (2012) 18:299–302. doi: 10.1016/j.parkreldis.2011.10.015
47. Castillo-Barnes D, Ramírez J, Segovia F, Martínez-Murcia FJ, Salas-Gonzalez D, Górriz JM. Robust ensemble classification methodology for I123-ioflupane SPECT images and multiple heterogeneous biomarkers in the diagnosis of Parkinson's disease. Front Neuroinform. (2018) 12:53. doi: 10.3389/fninf.2018.00053
48. Pellicano C, Benincasa D, Pisani V, Buttarelli FR, Giovannelli M, Pontieri FE. Prodromal non-motor symptoms of Parkinson's disease. Neuropsychiatr Dis Treat. (2007) 3:145–52. doi: 10.2147/nedt.2007.3.1.145
49. Sveinbjornsdottir S. The clinical symptoms of Parkinson's disease. J Neurochem. (2016) 139:318–24. doi: 10.1111/jnc.13691
50. Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord. (2015) 30:1600–11. doi: 10.1002/mds.26431
51. Lachman ME, Teshale S, Agrigoroaei S. Midlife as a pivotal period in the life course. Int J Behav Dev. (2015) 39:20–31. doi: 10.1177/0165025414533223
52. Kulmala J, von Bonsdorff MB, Stenholm S, Törmäkangas T, von Bonsdorff ME, Nygård C-H, et al. Perceived stress symptoms in midlife predict disability in old age: a 28-year prospective cohort study. J Gerontol Ser A. (2013) 68:984–91. doi: 10.1093/gerona/gls339
53. Chang M, Jonsson PV, Snaedal J, Bjornsson S, Saczynski JS, Aspelund T, et al. The effect of midlife physical activity on cognitive function among older adults: AGES—Reykjavik Study. J Gerontol Ser A. (2010) 65A:1369–74. doi: 10.1093/gerona/glq152
54. Defina LF, Willis BL, Radford NB, Gao A, Leonard D, Haskell WL, et al. The association between midlife cardiorespiratory fitness levels and later-life dementia: a cohort study. Ann Intern Med. (2013) 158:162–8. doi: 10.7326/0003-4819-158-3-201302050-00005
55. Lachman ME, Agrigoroaei S. Promoting functional health in midlife and old age: long-term protective effects of control beliefs, social support, and physical exercise. PLoS ONE. (2010) 5:e13297. doi: 10.1371/journal.pone.0013297
56. Sabia S, Singh-Manoux A, Hagger-Johnson G, Cambois E, Brunner EJ, Kivimaki M. Influence of individual and combined healthy behaviours on successful aging. C Can Med Assoc J. (2012) 184:1985–92. doi: 10.1503/cmaj.121080
57. Goodwin VA, Richards SH, Taylor RS, Taylor AH, Campbell JL. The effectiveness of exercise interventions for people with Parkinson's disease: a systematic review and meta-analysis. Mov Disord. (2008) 23:631–40. doi: 10.1002/mds.21922
58. Hamer M, Chida Y. Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence. Psychol Med. (2009) 39:3–11. doi: 10.1017/S0033291708003681
59. Thacker EL, Chen H, Patel AV, McCullough ML, Calle EE, Thun MJ, et al. Recreational physical activity and risk of Parkinson's disease. Mov Disord. (2008) 23:69–74. doi: 10.1002/mds.21772
60. Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, et al. Physical activities and future risk of Parkinson disease. Neurology. (2010) 75:341–8. doi: 10.1212/WNL.0b013e3181ea1597
61. Zeuner KE, Schäffer E, Hopfner F, Brüggemann N, Berg D. Progress of pharmacological approaches in Parkinson's disease. Clin Pharmacol Ther. (2019) 105:1106–20. doi: 10.1002/cpt.1374
62. He R, Yan X, Guo J, Xu Q, Tang B, Sun Q. Recent advances in biomarkers for Parkinson's disease. Front Aging Neurosci. (2018) 10:305. doi: 10.3389/fnagi.2018.00305
63. Delenclos M, Jones DR, McLean PJ, Uitti RJ. Biomarkers in Parkinson's disease: advances and strategies. Parkinsonism Relat Disord. (2016) 22:S106–10. doi: 10.1016/j.parkreldis.2015.09.048
64. Miller DB, O'Callaghan JP. Biomarkers of Parkinson's disease: present and future. Metabolism. (2015) 64:S40–6. doi: 10.1016/j.metabol.2014.10.030
65. Stewart T, Liu C, Ginghina C, Cain KC, Auinger P, Cholerton B, et al. Cerebrospinal fluid α-synuclein predicts cognitive decline in parkinson disease progression in the DATATOP cohort. Am J Pathol. (2014) 184:966–75. doi: 10.1016/j.ajpath.2013.12.007
66. Devic I, Hwang H, Edgar JS, Izutsu K, Presland R, Pan C, et al. Salivary α-synuclein and DJ-1: potential biomarkers for Parkinson's disease. Brain. (2011) 134:e178. doi: 10.1093/brain/awr015
67. Folgoas E, Lebouvier T, Leclair-Visonneau L, Cersosimo M-G, Barthelaix A, Derkinderen P, et al. Diagnostic value of minor salivary glands biopsy for the detection of Lewy pathology. Neurosci Lett. (2013) 551:62–4. doi: 10.1016/j.neulet.2013.07.016
68. Ravina B, Tanner C, Dieuliis D, Eberly S, Flagg E, Galpern WR, et al. A longitudinal program for biomarker development in Parkinson's disease: a feasibility study. Mov Disord. (2009) 24:2081–90. doi: 10.1002/mds.22690
69. Lim NS, Swanson CR, Cherng H-R, Unger TL, Xie SX, Weintraub D, et al. Plasma EGF and cognitive decline in Parkinson's disease and Alzheimer's disease. Ann Clin Transl Neurol. (2016) 3:346–55. doi: 10.1002/acn3.299
70. Choi C, Jeong J-H, Jang JS, Choi K, Lee J, Kwon J, et al. Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer's disease by color-coded bead technology. J Clin Neurol. (2008) 4:84. doi: 10.3988/jcn.2008.4.2.84
71. Donadio V, Incensi A, Rizzo G, Capellari S, Pantieri R, Stanzani Maserati M, et al. A new potential biomarker for dementia with Lewy bodies: Skin nerve α-synuclein deposits. Neurology. (2017) 89:318–26. doi: 10.1212/WNL.0000000000004146
72. Donadio V, Incensi A, El-Agnaf O, Rizzo G, Vaikath N, Del Sorbo F, et al. Skin α-synuclein deposits differ in clinical variants of synucleinopathy: an in vivo study. Sci Rep. (2018) 8:14246. doi: 10.1038/s41598-018-32588-8
73. Donadio V, Doppler K, Incensi A, Kuzkina A, Janzen A, Mayer G, et al. Abnormal α-synuclein deposits in skin nerves: intra- and inter-laboratory reproducibility. Eur J Neurol. (2019) 26:1245–51. doi: 10.1111/ene.13939
74. Doppler K, Jentschke H-M, Schulmeyer L, Vadasz D, Janzen A, Luster M, et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson's disease. Acta Neuropathol. (2017) 133:535–45. doi: 10.1007/s00401-017-1684-z
75. Gibbons CH, Garcia J, Wang N, Shih LC, Freeman R. The diagnostic discrimination of cutaneous α-synuclein deposition in Parkinson disease. Neurology. (2016) 87:505–12. doi: 10.1212/WNL.0000000000002919
76. Rizek P, Kumar N, Jog MS. An update on the diagnosis and treatment of Parkinson disease. CMAJ. (2016) 188:1157–65. doi: 10.1503/cmaj.151179
77. Brenner H, Gefeller O. Variation of sensitivity, specificity, likelihood ratios and predictive values with disease prevalence. Stat Med. (1997) 16:981–91. doi: 10.1002/(SICI)1097-0258(19970515)16:93.3.CO;2-E
78. Montgomery EB. Predictors of Parkinson's disease–not quite sound. Mov Disord. (2013) 28:413–5. doi: 10.1002/mds.25432
79. Montgomery EB, Koller WC, LaMantia TJ, Newman MC, Swanson-Hyland E, Kaszniak AW, et al. Early detection of probable idiopathic Parkinson's disease: I. Development of a diagnostic test battery. Mov Disord. (2000) 15:467–73. doi: 10.1002/1531-8257(200005)15:3<467::AID-MDS1007>3.0.CO;2-#
80. Montgomery EB, Lyons K, Koller WC. Early detection of probable idiopathic Parkinson's disease: II. A prospective application of a diagnostic test battery. Mov Disord. (2000) 15:474–8. doi: 10.1002/1531-8257(200005)15:3<474::AID-MDS1008>3.0.CO;2-X
81. van Dongen J, Nivard MG, Willemsen G, Hottenga J-J, Helmer Q, Dolan CV, et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun. (2016) 7:11115. doi: 10.1038/ncomms11115
82. García-Giménez JL, Mena-Mollá S, Beltrán-García J, Sanchis-Gomar F. Challenges in the analysis of epigenetic biomarkers in clinical samples. Clin Chem Lab Med. (2017) 55:1474–7. doi: 10.1515/cclm-2016-1162
83. Moore DS. Behavioral epigenetics. Wiley Interdiscip Rev Syst Biol Med. (2017) 9:e1333. doi: 10.1002/wsbm.1333
84. Booth LN, Brunet A. The aging epigenome. Mol Cell. (2016) 62:728–44. doi: 10.1016/j.molcel.2016.05.013
85. Florath I, Butterbach K, Muller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. (2014) 23:1186–201. doi: 10.1093/hmg/ddt531
86. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. (2013) 49:359–67. doi: 10.1016/j.molcel.2012.10.016
87. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. (2013) 14:R115. doi: 10.1186/gb-2013-14-10-r115
88. Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CCY, O'Donovan MC, et al. Methylomic trajectories across human fetal brain development. Genome Res. (2015) 25:338–52. doi: 10.1101/gr.180273.114
89. Collier TJ, Kanaan NM, Kordower JH. Aging and Parkinson's disease: different sides of the same coin? Mov Disord. (2017) 32:983–90. doi: 10.1002/mds.27037
90. Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients. Aging. (2015) 7:1130–42. doi: 10.18632/aging.100859
91. Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, et al. Huntington's disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging. (2016) 8:1485–512. doi: 10.18632/aging.101005
92. Lu AT, Hannon E, Levine ME, Hao K, Crimmins EM, Lunnon K, et al. Genetic variants near MLST8 and DHX57 affect the epigenetic age of the cerebellum. Nat Commun. (2016) 7:10561. doi: 10.1038/ncomms10561
93. Feng Y, Jankovic J, Wu Y-C. Epigenetic mechanisms in Parkinson's disease. J Neurol Sci. (2015) 349:3–9. doi: 10.1016/j.jns.2014.12.017
94. Henderson-Smith A, Fisch KM, Hua J, Liu G, Ricciardelli E, Jepsen K, et al. (2019). DNA methylation changes associated with Parkinson's disease progression: outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics 14:365–382. doi: 10.1080/15592294.2019.1588682
95. Jakubowski JL, Labrie V. Epigenetic biomarkers for Parkinson's disease: from diagnostics to therapeutics. J Parkinsons Dis. (2017) 7:1–12. doi: 10.3233/JPD-160914
96. Borrageiro G, Haylett W, Seedat S, Kuivaniemi H, Bardien S. A review of genome-wide transcriptomics studies in Parkinson's disease. Eur J Neurosci. (2018) 47:1–16. doi: 10.1111/ejn.13760
97. van Heesbeen HJ, Smidt MP. Entanglement of genetics and epigenetics in Parkinson's disease. Front Neurosci. (2019) 13:277. doi: 10.3389/fnins.2019.00277
98. Shao Y, Le W. Recent advances and perspectives of metabolomics-based investigations in Parkinson's disease. Mol Neurodegener. (2019) 14:3. doi: 10.1186/s13024-018-0304-2
99. Marek K, Jennings D, Lasch S, Siderowf A, Tanner C, Simuni T, et al. The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol. (2011) 95:629–35. doi: 10.1016/j.pneurobio.2011.09.005
100. Berg D, Godau J, Seppi K, Behnke S, Liepelt-Scarfone I, Lerche S, et al. The PRIPS study: screening battery for subjects at risk for Parkinson's disease. Eur J Neurol. (2013) 20:102–8. doi: 10.1111/j.1468-1331.2012.03798.x
101. Gaenslen A, Wurster I, Brockmann K, Huber H, Godau J, Faust B, et al. Prodromal features for Parkinson's disease - baseline data from the TREND study. Eur J Neurol. (2014) 21:766–72. doi: 10.1111/ene.12382
102. Jennings D, Siderowf A, Stern M, Seibyl J, Eberly S, Oakes D, et al. Imaging prodromal Parkinson disease: the Parkinson Associated Risk Syndrome Study. Neurology. (2014) 83:1739–46. doi: 10.1212/WNL.0000000000000960
103. Montgomery EB. Reproducibility in Biomedical Research Epistemological and Statistical Problems. London, UK: Academic Press (2019).
104. Blesa J, Trigo-Damas I, del Rey NL-G, Obeso JA. The use of nonhuman primate models to understand processes in Parkinson's disease. J Neural Transm. (2018) 125:325–35. doi: 10.1007/s00702-017-1715-x
105. Cooper JF, Van Raamsdonk JM. Modeling Parkinson's disease in C. elegans. J Parkinsons Dis. (2018) 8:17–32. doi: 10.3233/JPD-171258
106. Xiong Y, Yu J. Modeling Parkinson's disease in drosophila: what have we learned for dominant traits? Front Neurol. (2018) 9:228. doi: 10.3389/fneur.2018.00228
107. Terzioglu M, Galter D. Parkinson's disease: genetic versus toxin-induced rodent models. FEBS J. (2008) 275:1384–91. doi: 10.1111/j.1742-4658.2008.06302.x
108. Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: an update. J Parkinsons Dis. (2011) 1:19–33. doi: 10.3233/JPD-2011-11023
109. Tieu K. A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harb Perspect Med. (2011) 1:a009316. doi: 10.1101/cshperspect.a009316
110. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. (2015) 11:25–40. doi: 10.1038/nrneurol.2014.226
111. Mattace Raso G, Avagliano C, Calignano A. Response to comment by Juan Segura-Aguilar: new preclinical model are required to discover neuroprotective compound in Parkinson's disease. Pharmacol Res. (2017) 119:491–2. doi: 10.1016/j.phrs.2016.11.026
112. Segura-Aguilar J. New preclinical model are required to discover neuroprotective compound in Parkinson's disease. Pharmacol Res. (2017) 119:490. doi: 10.1016/j.phrs.2016.11.034
113. Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. (2018) 21:1370–9. doi: 10.1038/s41593-018-0236-8
114. Nuber S, Harmuth F, Kohl Z, Adame A, Trejo M, Schönig K, et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain. (2013) 136:412–32. doi: 10.1093/brain/aws358
115. Kohl Z, Ben Abdallah N, Vogelgsang J, Tischer L, Deusser J, Amato D, et al. Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC α-synuclein transgenic rat model of Parkinson's disease. Neurobiol Dis. (2016) 85:206–17. doi: 10.1016/j.nbd.2015.10.021
116. Hentrich T, Wassouf Z, Riess O, Schulze-Hentrich JM. SNCA overexpression disturbs hippocampal gene expression trajectories in midlife. Aging. (2018) 10:4024–41. doi: 10.18632/aging.101691
117. Wassouf Z, Hentrich T, Samer S, Rotermund C, Kahle PJ, Ehrlich I, et al. Environmental enrichment prevents transcriptional disturbances induced by alpha-synuclein overexpression. Front Cell Neurosci. (2018) 12:112. doi: 10.3389/fncel.2018.00112
118. Wassouf Z, Hentrich T, Casadei N, Jaumann M, Knipper M, Riess O, et al. Distinct stress response and altered striatal transcriptome in alpha-synuclein overexpressing mice. Front Neurosci. (2019) 12:1033. doi: 10.3389/fnins.2018.01033
119. Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. (2008) 28:10825–34. doi: 10.1523/JNEUROSCI.3001-08.2008
Keywords: Parkinson's disease, prodromal, midlife, molecular prodrome, biomarker, diagnosis
Citation: Kilzheimer A, Hentrich T, Burkhardt S and Schulze-Hentrich JM (2019) The Challenge and Opportunity to Diagnose Parkinson's Disease in Midlife. Front. Neurol. 10:1328. doi: 10.3389/fneur.2019.01328
Received: 12 July 2019; Accepted: 02 December 2019;
Published: 17 December 2019.
Edited by:
Oscar Arias-Carrión, Hospital General Dr. Manuel Gea Gonzalez, MexicoReviewed by:
Erwin Montgomery, McMaster University, CanadaGiovanni Rizzo, University of Bologna, Italy
Copyright © 2019 Kilzheimer, Hentrich, Burkhardt and Schulze-Hentrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia M. Schulze-Hentrich, anVsaWEuc2NodWx6ZS1oZW50cmljaEB1bmktdHVlYmluZ2VuLmRl