
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 20 November 2019
Sec. Neurogenetics
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.01219
This article is part of the Research TopicNeurogenetics Editor's Pick 2021View all 10 articles
 Christiane D. Much1Konrad Schwefel1Dariush Skowronek1
Christiane D. Much1Konrad Schwefel1Dariush Skowronek1 Loay Shoubash2
Loay Shoubash2 Felix von Podewils3
Felix von Podewils3 Miriam Elbracht4Stefanie Spiegler1Ingo Kurth4Agnes Flöel3Henry W. S. Schroeder2Ute Felbor1
Miriam Elbracht4Stefanie Spiegler1Ingo Kurth4Agnes Flöel3Henry W. S. Schroeder2Ute Felbor1 Matthias Rath1*
Matthias Rath1*Autosomal dominant cerebral cavernous malformation (CCM) represents a genetic disorder with a high mutation detection rate given that stringent inclusion criteria are used and copy number variation analyses are part of the diagnostic workflow. Pathogenic variants in either CCM1 (KRIT1), CCM2 or CCM3 (PDCD10) can be identified in 87–98% of CCM families with at least two affected individuals. However, the interpretation of novel sequence variants in the 5′-region of CCM2 remains challenging as there are various alternatively spliced transcripts and different transcription start sites. Comprehensive genetic and clinical data of CCM2 patients with variants in cassette exons that are either skipped or included into alternative CCM2 transcripts in the splicing process can significantly facilitate clinical variant interpretation. We here report novel pathogenic CCM2 variants in exon 3 and the adjacent donor splice site, describe the natural history of CCM disease in mutation carriers and provide further evidence for the classification of the amino acids encoded by the nucleotides of this cassette exon as a critical region within CCM2. Finally, we illustrate the advantage of a combined single nucleotide and copy number variation detection approach in NGS-based CCM1/CCM2/CCM3 gene panel analyses which can significantly reduce diagnostic turnaround time.
Cerebral cavernous malformations (CCMs; MIM: 116860, 603284, 603285) are mulberry-like vascular lesions that are formed by thin-walled and densely packed endothelial channels (1, 2). These low-flow sinusoidal convolutes are characterized by an impaired blood-brain-barrier due to a complex dysfunction of the lining endothelial cells. As a result of recurrent bleeding events, CCM patients often present with non-specific headaches, seizures and stroke-like symptoms (3). Over the last years, however, CCMs have increasingly been identified as incidental findings due to the widespread use of high-resolution magnetic resonance brain imaging (4). Since targeted therapies have not yet been approved, conservative treatment still focuses on the control of CCM-related complications. Surgical resection can be an option for easily accessible symptomatic CCMs, cavernous lesions that cause epilepsy or for deep CCMs that are either symptomatic or have already led to intracerebral hemorrhage. Treatment of CCM patients with brainstem CCMs is often difficult and the risk of post-operative morbidity and mortality always needs to be critically discussed before the indication for surgical resection is made (4).
CCMs may occur sporadically or in an autosomal dominant familial form. The prevalence of symptomatic hereditary CCM has been estimated to be in the range of 1:5,400 to 1:6,200 in the general population (5). Heterozygous germline loss-of-function variants in one of the three genes CCM1/KRIT1 (MIM 604214), CCM2/OSM/Malcavernin (MIM 607929), or CCM3/PDCD10 (MIM 609118) can be found in the vast majority of familial CCM cases (5). The identification of a pathogenic variant is not only important for the index case but also crucial for genetic counseling of at-risk family members. The mutation detection rate of current molecular genetic CCM1/CCM2/CCM3 analyses is 87–98% for families with two or more CCM patients and up to 57–75% for index cases with multiple CCMs but a negative family history (4–6). However, these high mutation detection rates can only be realized with stringent inclusion criteria and a comprehensive workflow for the detection of single nucleotide variants (SNVs), small indels, and copy-number variants (CNVs).
CCM2 nonsense, frameshift, and splice mutations can be identified in about 13% of CCM cases that meet the inclusion criteria (6). Furthermore, various CCM2 CNVs have been reported for sporadic and familial CCM cases (7–14). One of these-the deletion of exons 2 to 10 of CCM2 (LRG_664t2)-is actually a founder mutation in the US population (10, 11). The multiplex ligation-dependent probe amplification (MLPA) technique has long been used as gold standard for copy number quantification but the implementation of gene panel analyses has opened new perspectives for detection of CNVs from NGS data.
In this study, we report two novel and one previously published variants within a cassette exon of CCM2 and provide evidence for their pathogenicity. In addition, we describe the natural history of CCM disease in mutation carriers and illustrate the advantages of a combined NGS-based SNV/CNV detection workflow in CCM1/CCM2/CCM3 gene panel analyses.
Genetic analyses were performed with written informed consent of all study participants according to the German Gene Diagnostics Act and approval of the local ethics committee of the University Medicine Greifswald (No. B119/10). Genomic DNA was isolated from peripheral blood lymphocytes using the NucleoSpin Blood L Kit (Macherey-Nagel, Düren, Germany). Thirty-one probands were analyzed for pathogenic CCM1, CCM2, or CCM3 variants by hybridization capture-based target enrichment and next generation sequencing and another 2 patients were analyzed by Sanger sequencing as described previously (6). All coding exons and exon-intron-boundaries (± 20 bp) of CCM1 (exons 5 to 20 according to reference sequence LRG_650t1), CCM2 (exons 1 to 10 according to LRG_664t2 = exons 1 and 3 to 11 according to LRG_664), and CCM3 (exons 3 to 9 according to LRG_651t1) were defined as target regions in both approaches. Sequencing libraries were prepared with Nextera Rapid Capture (Panel ID: 113402, Illumina, San Diego, USA) or Agilent SureSelectQXT (Panel ID: 3152261, Agilent Technologies, Santa Clara, USA) custom enrichment kits according to the manufacturers' instructions and sequenced on a MiSeq instrument with 2 × 150 cycles (Illumina, San Diego, USA). FASTQ files were generated with the MiSeq reporter software v2.6.2 (MSR; Illumina, San Diego, USA). The SeqNext module of the Sequence Pilot software (v5.0.0, JSI Medical Systems, Ettenheim, Germany) was used for read mapping, alignment and variant calling. Sanger sequencing data were analyzed with the SeqPatient module of the Sequence Pilot software. The Human Splicing Finder 3.1 (http://www.umd.be/HSF3/), Berkeley Drosophila Genome Project (http://www.fruitfly.org/), NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/), MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), and ASSP (http://wangcomputing.com/assp/) tools were used for in silico splice predictions.
The SeqNext module (v5.0.0, JSI Medical Systems, Ettenheim, Germany) was used for CNV analyses in a read-depth approach based on a previous report for other target genes (15). In brief, all exons of CCM1, CCM2, and CCM3, as well as 17 control fragments located on different chromosomes were defined as regions of interest (ROIs). Sequencing data were mapped and aligned to the target region with the SeqNext module. In a first normalization step, the specific read depth of each target exon and the average read depth of all control fragments were used to calculate the relative product coverage (RPC) for each CCM1, CCM2, and CCM3 exon. In addition to the patient sample, at least five control samples from the same sequencing run were analyzed in the same way. In a next step, each RPC of the patient sample was normalized to the corresponding average reference RPC that had been calculated from all control samples. Thresholds of ≤ 70 and ≥ 130% were used for deletion and duplication calling, respectively. The Salsa MLPA Kits P130-A3 and P131-B1 were used according to the manufacturer's instructions to validate the CCM1, CCM2, and CCM3 deletions (MRC-Holland, Amsterdam, The Netherlands).
RNA was isolated from peripheral blood lymphocytes using the PAXgene Blood RNA Kit (PreAnalytiX, Hombrechtikon, Switzerland). Three hundred nanogram of total RNA were reverse described into cDNA using SuperScript™ III Reverse Transcriptase (Thermo Fisher Scientific, Waltham, USA). The exons 1, 3, 4, and 5 (exon numbering according to LRG_664) of the CCM2 transcript LRG_664t2 were amplified using specific forward (5′-GCGGCGATATGGAAGAGG-3′) and reverse (5'-GCACCCTGAGGATGATATC-3′) primers (7, 16). PCR products were size-separated by agarose gel electrophoresis and visualized on a Gel Doc™ EZ Imager (Bio-Rad, Hercules, USA). The excised fragments were purified with the Zymoclean™ Gel DNA Recovery Kit (Zymo Research, Freiburg, Germany) and analyzed by Sanger sequencing.
In our present study, we included 33 CCM index cases with multiple CCMs and/or a positive family history that had been analyzed for pathogenic variants in CCM1, CCM2 or CCM3 between 2017 and 2019 (Table 1). As expected, most patients with an autosomal dominant CCM disease were heterozygous for a pathogenic or likely pathogenic CCM1 variant (n = 18) (Table S1). Another four patients had pathogenic CCM2 germline variants and two patients were CCM3 mutation carriers (Table S1). The unexpected observation that three of the four CCM2 mutations clustered in a cassette exon that can be skipped in the splicing process prompted us to focus on these CCM2 variants that had been identified in families 1–3 in more detail (Table 2).
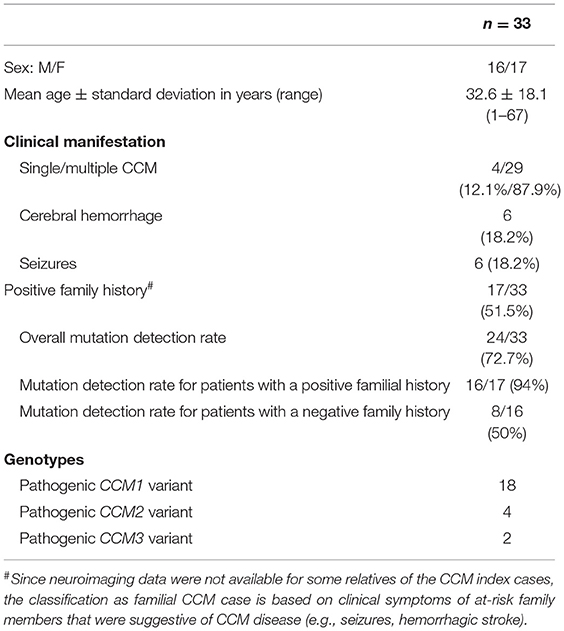
Table 1. Overview of the CCM cohort.

Table 2. Overview of the pathogenic CCM2 SNVs and indel variants reported in our present study.
The index patient of family 1 (III/4, Figure 1A) was first admitted to neurosurgery at the age of 25. He reported a 6-year history of headaches and focal seizures with intermittent numbness and tingling in his right arm and leg. Magnetic resonance imaging (MRI) identified two small CCMs in his left frontal and right occipital lobe as well as a large cavernoma in his left parietal lobe (20 × 18 × 20 mm). The latter was resected by minimally invasive microsurgery. Histological evaluation confirmed CCM diagnosis. At the age of 28, the frequency of the index patient's seizures increased again, and the anti-epileptic therapy was intensified. However, it was not possible to prevent seizures with levetiracetam at maximum dosage of 2 × 1,500 mg. Since an increase in the size of the left frontal CCM (Figures 1B,C) was noticed, it was finally resected by neurosurgery at the age of 33. At last follow-up at the age of 35 years, he still reported epileptic seizures and two novel CCMs were identified in his right frontal lobe and cerebellum. Anti-epileptic therapy was modified to a combination of brivaracetam, lacosamide, perampanel, and eslicarbazepine acetate.
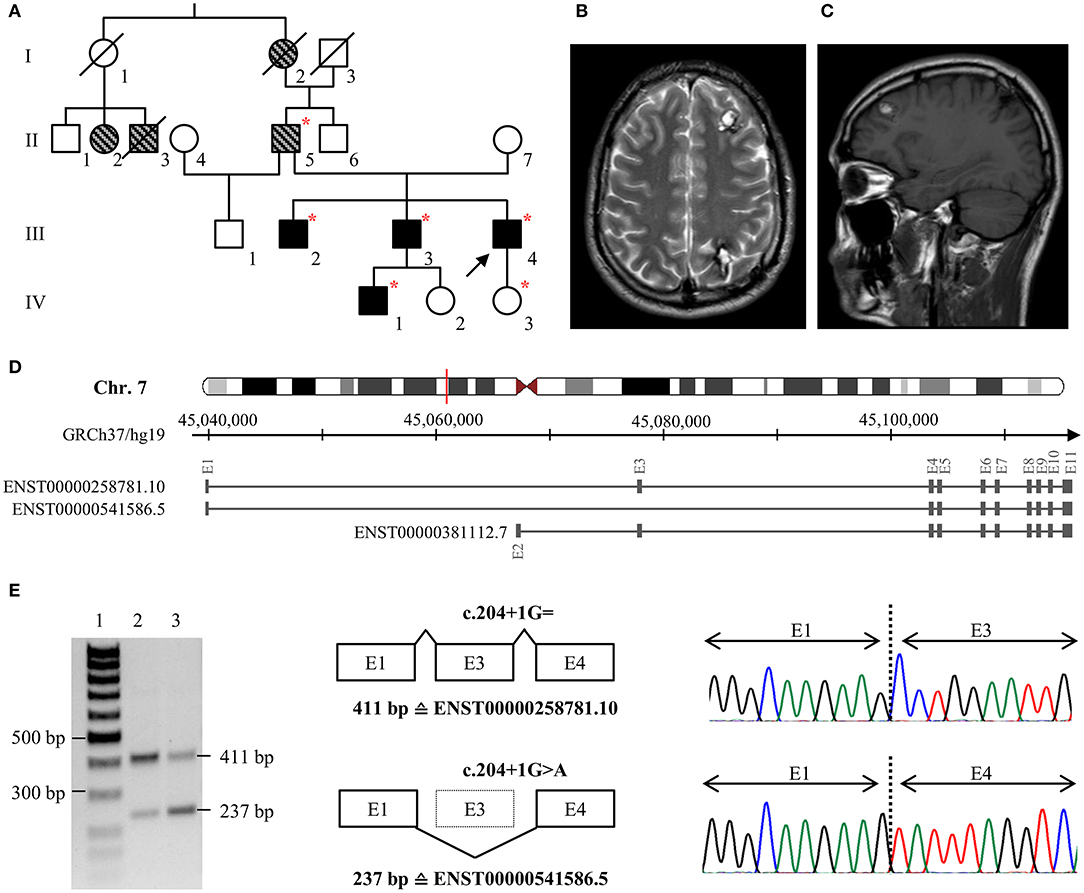
Figure 1. (A) Pedigree of family 1. The index case (III/4) is marked with an arrow. Black filled symbols represent patients with CCMs, gray striped symbols indicate relatives with neurological symptoms that are suggestive for CCM. Deceased family members are crossed out. Red stars indicate heterozygous carriers of the pathogenic CCM2 splice variant c.204+1G>A. (B,C) Representative axial T2-weighted (B) and sagittal T1-weighted (C) MR images of the index case at the age of 32. (D) Schematic exon-intron structure of CCM2 and selected protein-coding CCM2 transcripts that are listed in the ENSEMBL database. ENST00000258781.10 and ENST00000541586.5 are both expressed in blood lymphocytes but ENST00000541586.5 in which exon 3 is skipped is less abundant in brain or blood vessels. (E) Skipping of exon 3 on the c.204+1G>A CCM2 allele was confirmed by RT-PCR and cDNA sequencing. Lane 1: size marker, lane 2: control sample, lane 3: patient III/4. E, exon.
Patient III/4 reported a positive family history for CCMs. His eldest brother (III/2) has had trigeminal neuralgia since the age of 23, and six cavernous lesions were identified on his MRI. The index patient's second brother (III/3) became symptomatic with three focal to bilateral tonic-clonic seizures at the age of 32. One cavernous lesion of 25 × 25 × 20 mm in his left frontal lobe demonstrated signs of an acute hemorrhage and was resected by neurosurgery. In addition, at least seven small CCMs were found in both hemispheres. Two cavernous malformations were also identified in his yet asymptomatic 10-year old son (IV/1) at the age of five. The index patient's father (II/5) had a stroke at the age of 33 and reported chronic headaches. Because of his pacemaker, no MRI data were available for him. Furthermore, the index patient's paternal grandmother (I/2) had chronic headaches, and two cousins of his father (II/2, II/3) had seizures.
The 46-year-old female index patient of family 2 (II/2, Figure 2A) reported a 11/2-year history of chronic headaches and recurrent attacks of vertigo that had lasted up to 1 min. MRI identified two CCMs located in her cerebellum and frontal lobe. Since her paternal aunt (I/1, Figure 2A) had a CCM bleeding, she was classified as familial CCM case. The male index patient of family 3 (II/1, Figure 2B) had five CCMs located in both hemispheres and became symptomatic at the age of 30 with focal to bilateral tonic-clonic seizures. A sufficient seizure control could be achieved by levetiracetam monotherapy. The patient appears to be a sporadic CCM case since no neurological symptoms that would have been indicative for a CCM disease were reported for his maternal and paternal lineage. However, a cryptic familial case cannot be excluded since his father died of colorectal cancer at the age of 75 without molecular genetic analysis and the index patient's 71-year-old asymptomatic mother decided against predictive genetic testing or MRI.
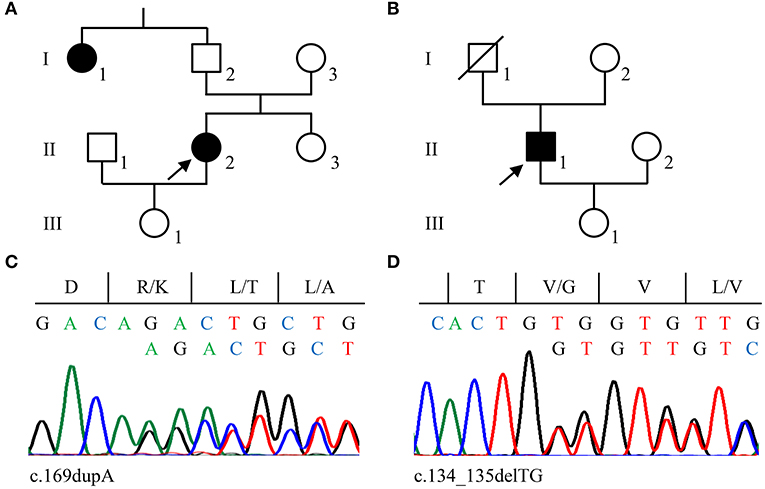
Figure 2. Pedigrees of families 2 (A) and 3 (B). Arrows indicate the index cases in each family. (C,D) Sanger sequencing validation of pathogenic CCM2 variants identified in family 2 (c.169dupA; C) and family 3 (c.134_135delTG; D). Nucleotide and protein sequences are shown above each chromatogram.
A novel substitution at position +1 of the highly conserved donor splice site of exon 3 was identified in family 1 [LRG_664t2: c.204+1G>A]. In silico predictions indicated disruption of normal CCM2 splicing. Therefore, we decided to perform additional transcript analyses. RT-PCR demonstrated skipping of exon 3 on the mutant CCM2 allele (Figures 1D,E). Moreover, a heterozygous SNP in exon 3 that was located in cis with the CCM2 splice donor variant could not be identified by cDNA sequencing indicating complete exon skipping. The loss of exon 3 leads to an in-frame deletion of 58 amino acids on protein level [LRG_664p2: p.(Pro11_Lys68del)]. It is noteworthy to mention that this alteration involves the first ten amino acids of the PTB domain of CCM2. Targeted CCM2 sequence analyses of blood samples from multiple affected family members (III/2; III/3; II/5; IV/1) demonstrated co-segregation of the splice site variant with CCM disease in family 1. According to the latest ACMG guidelines for the interpretation of sequence variants (18), the splice site variant was classified as pathogenic.
CCM2 loss-of-function variants were also found in families 2 and 3. A novel pathogenic 1-bp duplication [LRG_664t2: c.169dupA; LRG_664p2: p.(Arg57Lysfs*8)] was detected in the blood sample of the index patient of family 2 (Figure 2C) and a 2-bp deletion [LRG_664t2: c.134_135delTG; LRG_664p2: p.(Val45Glyfs*6)] was identified in family 3 (Figure 2D). The latter had previously been described in an Italian CCM family (17) and was listed as likely pathogenic in the ClinVar database (ClinVar-ID: 590648). Unfortunately, a blood sample from the maternal aunt of the index patient in family 2 was not available to test for the CCM2 variant. Of note, neither the two frameshift variants in exon 3 nor the CCM2 splice site variant that had been identified in family 1 are listed in the Genome Aggregation Database (gnomAD) which contains 125,748 whole-exome and 15,708 whole-genome datasets.
Besides SNVs and small indels that can be found in exon 3 of CCM2, a complete in-frame deletion of this exon has been reported in the literature (10, 16). Therefore, we analyzed the available NGS data of the CCM index cases of our cohort in a combined workflow for SNVs and CNVs. Notably, we did not detect a deletion of exon 3 of CCM2 but three other CNVs in 31 evaluated samples. In particular, we found a heterozygous deletion of the exons 4 and 5 of CCM2 (exon numbering according to LRG_664) in a 40-year-old symptomatic CCM patient (Figures 3A,B). The relative product coverage ratios (RPC ratios) of both exons were reduced to 52 and 49%, respectively. In addition, heterozygous deletions of the exons 1 to 6 of CCM1 and of the exons 8 and 9 of the CCM3 gene (5) were detected in two other CCM patients. RPC ratios were reduced to 49–63 and 41–43%, respectively (Figures S1A-C). All three CNVs were verified by MLPA analysis. Our data demonstrate that apparently mutation-negative CCM patients always have to be analyzed for CNVs and that a combined NGS-based SNV/CNV detection workflow can reduce the diagnostic turnaround time since it renders time-consuming MLPA analyses unnecessary.
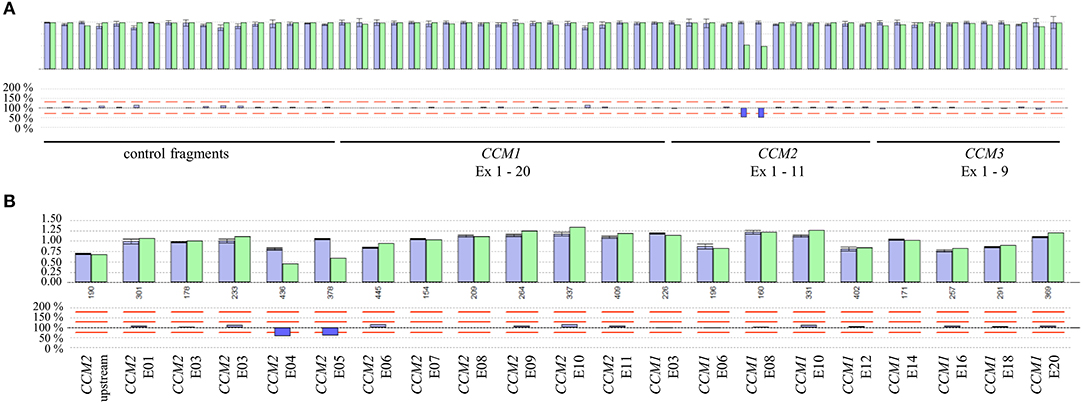
Figure 3. (A) Identification of a deletion of exons 4 and 5 of the CCM2 gene by NGS-based CNV analysis. The ROIs and their relative product coverages are shown in the upper histogram for control samples (blue) and the sample of the CCM proband (green). The ratios for the relative coverage of each ROI are shown in the lower panel. Red lines indicate the detection threshold for deletions and duplications. As some exons of CCM1-3 have been joined to combined ROIs and alternative exons are also included in the CNV analysis, the number of analyzed ROIs is not equal to the number of exons of CCM1-3. (B) Verification of the heterozygous CCM2 deletion by MLPA analysis.
We here present comprehensive clinical and genetic data of three CCM2 families with pathogenic variants in a cassette exon of CCM2, provide further evidence for the classification of exon 3 as a critical region within CCM2, and describe our procedure for NGS-based CNV analyses.
Familial CCM can have many clinical faces. Pathogenic CCM1, CCM2, and CCM3 variants are associated with incomplete and locus-specific penetrance of 63, 55, and 68%, respectively (19). In addition, expressivity even between family members sharing the same pathogenic variant can be extremely variable. CCM2 mutation carriers reported in this study mainly became symptomatic in their third to fifth decade of life. The mean age at first clinical symptoms of the four symptomatic CCM2 mutation carriers of family 1 (II/5; III/2; III/3; III/4) and the two index patients of family 2 and 3 was 30.2 years (range 19–44). Hence, it was slightly lower than that described for other CCM2 patients before. The mean age of onset reported by Denier and colleagues was 34.9 years (range 1–69) (19) and our group has observed a mean age at referral to genetic testing of 40.8 years (range 16–71) (6). The mean number of cavernous lesions in our symptomatic CCM cases was also slightly lower (mean number: 5.3; range 2 to 8) than the mean number of 6.6 CCM lesions identified on T2-weighted MRIs of 37 other CCM2 mutation carriers described in the literature (19).
The identification of a pathogenic variant within a CCM family allows predictive testing for at-risk family members. The exclusion of a known familial variant relieves relatives of anxieties and renders special clinical surveillance unnecessary. However, interpretation of novel CCM2 variants-especially of those in exon 3-can be challenging since there are several alternatively spliced CCM2 transcripts. The full-length CCM2 protein with its 444 amino acids has two major functional domains. While the phosphotyrosine binding (PTB) domain is thought to be essential for binding to CCM1 (20), the function of the harmonin-homology domain (HHD) at the C-terminus has not yet been fully elucidated. However, another CCM2 transcript (ENST00000541586.5, Figure 1D) without exon 3, which encodes for part of the PTB domain, is highly expressed in blood lymphocytes and spleen but less abundant in brain or blood vessels. Of note, in vitro studies have demonstrated that deletion of the amino acids encoded by exon 3 abrogates formation of a ternary CCM1/CCM2/CCM3 protein complex (16). Thus, the alternative CCM2 transcript seems to be unable to compensate for loss of the full-length CCM2 protein. To the best of our knowledge, at least ten pathogenic CCM2 SNVs and small indel variants in exon 3 have been described so far (Table S2). These mutations account for ~15% of all known CCM2 nonsense, frameshift and splice mutations (HGMD Professional 2019.1). Taken together, these data implicate that only the full-length CCM2 protein but not an alternative isoform without the 58 amino acids encoded by exon 3 can maintain cerebrovascular stability and quiescence in brain endothelial cells. Therefore, exon 3 can be defined to encode for a “well-established functional domain” according to the ACMG guidelines for variant interpretation (18).
Molecular genetic analyses of the CCM1/CCM2/CCM3 genes have long been performed in a step-wise approach. Sequencing for identification of SNVs was followed by MLPA analyses for CNV detection (6). CNVs have been defined as duplications and deletions of at least 1 kb and belong-together with inversions of the same size-to structural genome variants (21). In a modern diagnostic sense, single exon deletions and duplications (> 50 bp) are also classified as CNVs (22). More than 60 different single- and multi-exon deletions and duplications in CCM1, CCM2, and CCM3 are currently listed in the Human Gene Mutation Database (HGMD Professional 2019.1). The widespread use of NGS gene panel analyses and the further development of bioinformatic algorithms nowadays allow SNV and CNV detection in a single NGS pipeline. The advantage of such a comprehensive approach is the significant reduction of costs and turnaround time when compared to sequential Sanger sequencing and MLPA analyses. Additionally, NGS-based CNV analyses enable a higher resolution and more precise breakpoint detection (23). However, current gene panel analyses also have limitations. We have recently identified a 24 kb inversion by whole genome sequencing that includes exon 1 of CCM2 in a family with multiple CCM cases (24). The detection of such copy-neutral structural genomic rearrangements in targeted gene panel approaches is extremely challenging and often even impossible.
In conclusion, the results of our study demonstrate that accurate phenotyping and genotyping are crucial for variant interpretation in CCM1/CCM2/CCM3 gene panel sequencing and that CNV calling from NGS data can help to make the analyses more effective.
All datasets analyzed for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Ethics committee of the University Medicine Greifswald (No. B119/10). Written informed consent of the study participants to publish all data and information that are presented in our current manuscript was obtained.
CM, MR, and UF contributed to the intellectual conception and the design of the study. CM, KS, SS, and DS performed most of the experiments. CM, KS, ME, SS, DS, IK, MR, and UF contributed to interpretation of the genetic results. LS, FP, ME, IK, AF, and HS cared for the patients and contributed to interpretation of the clinical and neuroimaging results, CM, UF, and MR drafted the manuscript. All authors contributed to writing.
This work was supported by the Deutsche Forschungs-gemeinschaft (DFG, German Research Foundation)-RA 2876/2-1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank the patients for their cooperation and are grateful to S. Göbel and K. Tominski for their excellent technical assistance. The CCM2 expression data described here were obtained from the GTEx Portal (https://www.gtexportal.org/home/gene/CCM2).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.01219/full#supplementary-material
1. Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. (2001) 71:188–92. doi: 10.1136/jnnp.71.2.188
2. Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. (2013) 19:302–8. doi: 10.1016/j.molmed.2013.02.004
3. Batra S, Lin D, Recinos PF, Zhang J, Rigamonti D. Cavernous malformations: natural history, diagnosis and treatment. Nat Rev Neurol. (2009) 5:659–70. doi: 10.1038/nrneurol.2009.177
4. Akers A, Al-Shahi Salman R, Award IA, Dahlem K, Flemming K, Hart B, et al. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: onsensus recommendations based on systematic literature review by the angioma alliance scientific advisory board clinical experts panel. Neurosurgery. (2017) 80:665–80. doi: 10.1093/neuros/nyx091
5. Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol. (2018) 9:60–9. doi: 10.1159/000486292
6. Spiegler S, Najm J, Liu J, Gkalympoudis S, Schroder W, Borck G, et al. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol Genet Genomic Med. (2014) 2:176–85. doi: 10.1002/mgg3.60
7. Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. (2004) 74:326–37. doi: 10.1086/381718
8. Labauge P, Krivosic V, Denier C, Tournier-Lasserve E, Gaudric A. Frequency of retinal cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. Arch Ophthalmol. (2006) 124:885–6. doi: 10.1001/archopht.124.6.885
9. Felbor U, Gaetzner S, Verlaan DJ, Vijzelaar R, Rouleau GA, Siegel AM. Large germline deletions and duplication in isolated cerebral cavernous malformation patients. Neurogenetics. (2007) 8:149–53. doi: 10.1007/s10048-006-0076-7
10. Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, et al. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet. (2007) 80:69–75. doi: 10.1086/510439
11. Liquori CL, Penco S, Gault J, Leedom TP, Tassi L, Esposito T, et al. Different spectra of genomic deletions within the CCM genes between Italian and American CCM patient cohorts. Neurogenetics. (2008) 9:25–31. doi: 10.1007/s10048-007-0109-x
12. Cau M, Loi M, Melis M, Congiu R, Loi A, Meloni C, et al. C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet. (2009) 52:344–8. doi: 10.1016/j.ejmg.2009.05.002
13. Riant F, Cecillon M, Saugier-Veber P, Tournier-Lasserve E. CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics. (2013) 14:133–41. doi: 10.1007/s10048-013-0362-0
14. Tsutsumi S, Ogino I, Miyajima M, Ikeda T, Shindo N, Yasumoto Y, et al. Genomic causes of multiple cerebral cavernous malformations in a Japanese population. J Clin Neurosci. (2013) 20:667–9. doi: 10.1016/j.jocn.2012.05.041
15. Schmidt AY, Hansen TVO, Ahlborn LB, Jonson L, Yde CW, Nielsen FC. Next-generation sequencing-based detection of germline copy number variations in BRCA1/BRCA2: validation of a one-step diagnostic workflow. J Mol Diagn. (2017) 19:809–16. doi: 10.1016/j.jmoldx.2017.07.003
16. Stahl S, Gaetzner S, Voss K, Brackertz B, Schleider E, Surucu O, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat. (2008) 29:709–17. doi: 10.1002/humu.20712
17. Nardella G, Visci G, Guarnieri V, Castellana S, Biagini T, Bisceglia L, et al. A single-center study on 140 patients with cerebral cavernous malformations: 28 new pathogenic variants and functional characterization of a PDCD10 large deletion. Hum Mutat. (2018) 39:1885–900. doi: 10.1002/humu.23629
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
19. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. (2006) 60:550–6. doi: 10.1002/ana.20947
20. Fisher OS, Zhang R, Li X, Murphy JW, Demeler B, Boggon TJ. Structural studies of cerebral cavernous malformations 2 (CCM2) reveal a folded helical domain at its C-terminus. FEBS Lett. (2013) 587:272–7. doi: 10.1016/j.febslet.2012.12.011
21. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. (2006) 7:85–97. doi: 10.1038/nrg1767
22. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. (2011) 12:363–76. doi: 10.1038/nrg2958
23. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. (2010) 11:685–96. doi: 10.1038/nrg2841
Keywords: cerebral cavernous malformations, novel CCM2 mutations, CCM2 transcript analyses, CNV analyses, seizures, cerebral hemorrhage
Citation: Much CD, Schwefel K, Skowronek D, Shoubash L, von Podewils F, Elbracht M, Spiegler S, Kurth I, Flöel A, Schroeder HWS, Felbor U and Rath M (2019) Novel Pathogenic Variants in a Cassette Exon of CCM2 in Patients With Cerebral Cavernous Malformations. Front. Neurol. 10:1219. doi: 10.3389/fneur.2019.01219
Received: 31 May 2019; Accepted: 01 November 2019;
Published: 20 November 2019.
Edited by:
Nobutaka Hattori, Juntendo University, JapanReviewed by:
Lucía Fernández Cardo, Cardiff University, United KingdomCopyright © 2019 Much, Schwefel, Skowronek, Shoubash, von Podewils, Elbracht, Spiegler, Kurth, Flöel, Schroeder, Felbor and Rath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Rath, bWF0dGhpYXMucmF0aEBtZWQudW5pLWdyZWlmc3dhbGQuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.