
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol. , 19 June 2019
Sec. Neurodegeneration
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00652
 Nattakarn Limphaibool*
Nattakarn Limphaibool* Piotr IwanowskiMarte Johanne Veilemand Holstad
Piotr IwanowskiMarte Johanne Veilemand Holstad Dominik KobylarekWojciech Kozubski
Dominik KobylarekWojciech KozubskiExtensive research in recent decades has expanded our insights into the pathogenesis of Parkinson's disease (PD), though the underlying cause remains incompletely understood. Neuroinflammation have become a point of interest in the interconnecting areas of neurodegeneration and infectious diseases. The hypothesis concerning an infectious origin in PD stems from the observation of Parkinson-like symptoms in individuals infected with the influenza virus who then developed encephalitis lethargica. The implications of infectious pathogens have later been studied in neuronal pathways leading to the development of Parkinsonism and PD, through both a direct association and through synergistic effects of infectious pathogens in inducing neuroinflammation. This review explores the relationship between important infectious pathogens and Parkinsonism, including symptoms of Parkinsonism following infectious etiologies, infectious contributions to neuroinflammation and neurodegenerative processes associated with Parkinsonism, and the epidemiologic correlations between infectious pathogens and idiopathic PD.
Parkinson's disease (PD) is a debilitating neurodegenerative disorder manifesting as reduced facilitation of voluntary movements. It affects 1% of the population above the age of 60 years, with an annual incidence of 15 per 100,000 (1). A substantial growth in the prevalence of PD worldwide is predicted as a result of population aging and increases in life expectancy (2). Extensive research over the past few decades, including epidemiologic and genetic studies and post-mortem analysis, has expanded our insights into the pathogenesis of the disease. For the vast majority of cases, the underlying cause of PD remains incompletely understood.
It has been suggested that PD's complex and multifactorial etiology results from environmental contributions in genetically predisposed individuals (3). Genetic links have been identified by recent genome-wide association studies as causes or risk factors for PD development (4, 5). However, the sporadic nature of the occurrences suggests interactions between additional factors that has largely remained enigmatic.
Parkinsonian symptoms refer to PD-like manifestations, such as rapidly progressing rigidity, bradykinesia, postural instability, cognitive, and oculomotor abnormalities, but which do not lead to a firm diagnosis of PD. The role of bacterial and viral infections in the etiology of Parkinsonism and idiopathic PD has been indicated by recent studies, although a clear correlation is yet to be established. Parkinsonism arising from the loss of dopaminergic neurons as a consequence of an infectious process occurs rapidly, contrary to the late-onset and progressive course of idiopathic PD (6). Although infection-dependent Parkinsonism and idiopathic PD are distinct entities, the role of infectious pathogens have been implicated in both pathologies whether in the disease pathogenesis or through epidemiologic correlations.
The original hypothesis of an infectious origin in PD stems from the observation of Parkinson-like symptoms in individuals infected with the influenza virus who developed encephalitis lethargica (7). A “dual-hit hypothesis” was later formulated concerning the pathogenesis of idiopathic PD. This hypothesis describes a neurotropic pathogen which enters the nervous system through the nasal mucosa (via the olfactory pathways) and intestinal mucosa (via enteric plexuses and preganglionic vagal fibers), ultimately leading to a cascade of neurodegenerative events in the substantia nigra pars compacta (SNpc) (8).
This review highlights the association between important infectious pathogens and Parkinsonism, including symptoms of Parkinsonism following infectious etiologies, infectious contributions to neuroinflammation and neurodegenerative processes associated with Parkinsonism, and the epidemiologic correlation of infectious pathogens to idiopathic PD (Tables 1, 2).
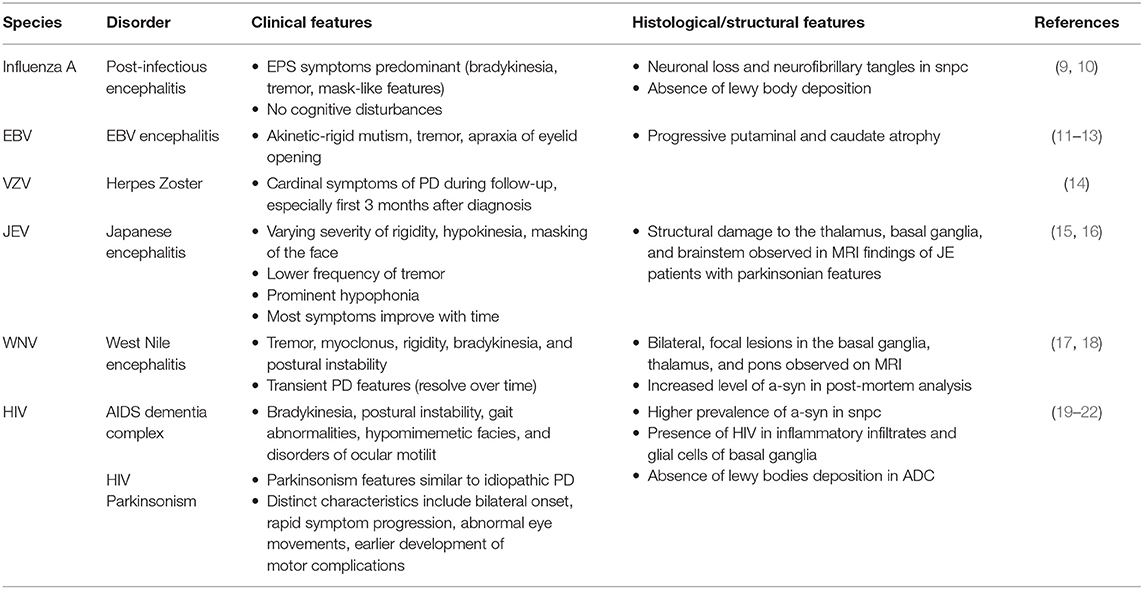
Table 1. Clinical, histological, and structural features of Parkinsonism in infectious diseases.
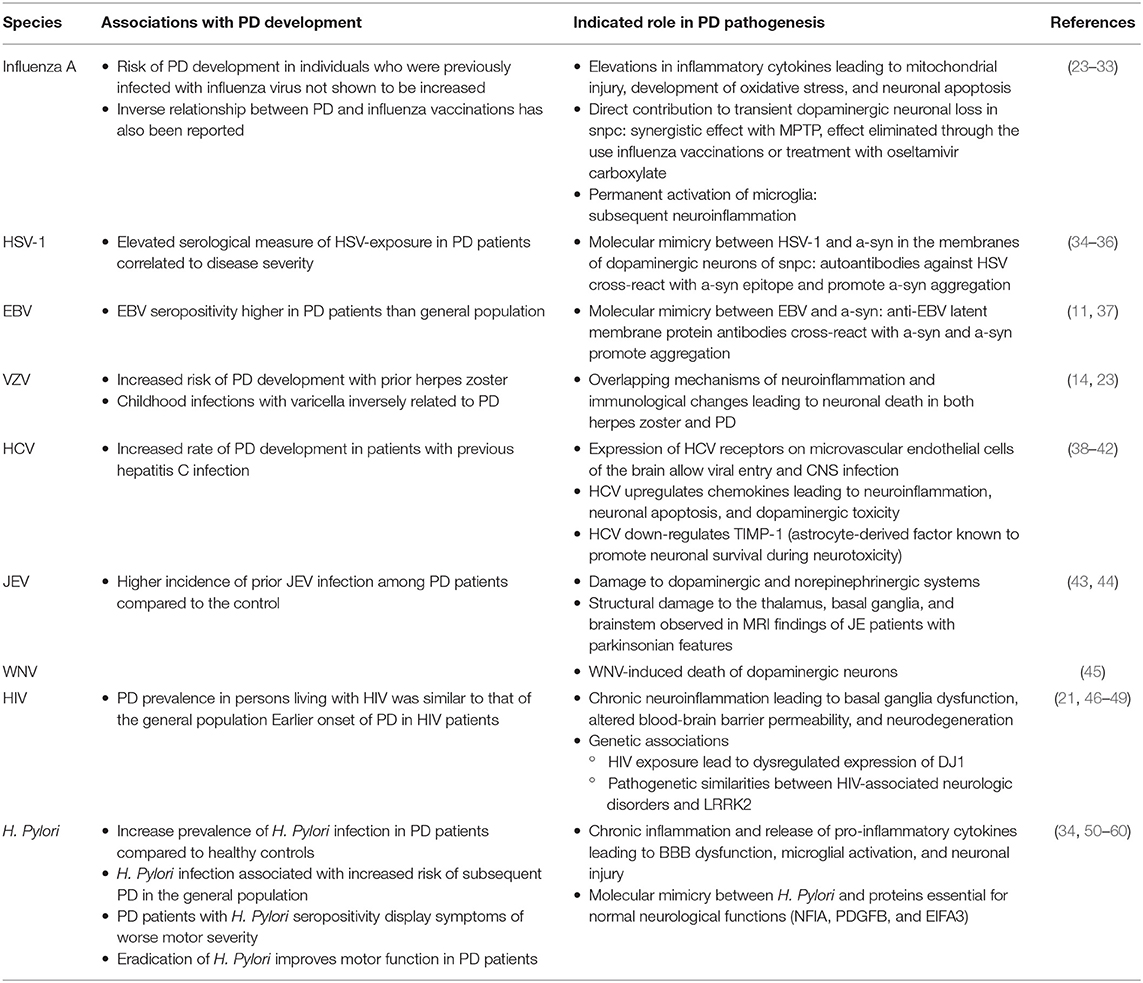
Table 2. Association of infectious pathogens in PD development and pathogenesis.
The premise of a causative association between influenza virus and PD stems from the outbreak of encephalitic lethargica and postencephalitic Parkinsonism (PEP) which occurred in the aftermath of the 1918 influenza pandemic. Although the two events are temporally coincidental, influenza virus has not been confirmed as a direct causation to encephalitic lethargica and PEP. PEP is clinically and pathologically distinct from idiopathic PD. The overlapping clinical features include the classic extrapyramidal symptoms of bradykinesia, tremor, and “mask-like” features. However, patients affected with PEP do not exhibit cognitive disturbances such as aphasia and apraxia. Pathological evidence of neuronal loss and neurofibrillary tangles were similarly observed in the substantia nigra. Unlike in idiopathic PD, there is an absence of Lewy bodies deposition on histological samples from patients with PEP (9).
Although reports of PEP have become extremely rare in the last decade when compared to almost 50% of all diagnosed cases of Parkinsonism between 1925 and 1938, clinical research has suggested the role of influenza A virus in the processes of neuroinflammation and neurodegeneration contributing to the development of Parkinsonism. Transient neurological sequelae (including tremor and gait disturbance) have been reported in association with influenza infections, particularly within the first few weeks of diagnosis (10). A significant link between severe influenza and PD, as well as an inverse relationship between PD and influenza vaccinations has also been reported (23). Although the association of influenza and symptoms of Parkinsonism has been indicated, the risk of developing idiopathic PD in individuals who were previously infected with influenza virus was not shown to be increased (10).
The increasing risk of developing Parkinsonism is associated with increasing number of influenza attacks, suggesting that influenza-associated neuronal injury may be a cumulative inflammatory process (10). Individuals with susceptible genetic makeup, may suffer from immunologically mediated mitochondrial injury and development of neuronal oxidative stress subsequent to influenza-induced pyrexia and increased inflammatory cytokines, ultimately resulting to neuronal apoptosis. This is supported by findings of increased pro-inflammatory mediators, including interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α), elevated levels of cytochrome C, a marker of mitochondrial injury, and reactive oxygen species (ROS) production in infected individuals, which point to the underlying immunological mechanisms in the pathophysiology of PEP (24–26).
On the other hand, a study on animal models found that the H5N1 influenza virus, upon its progression to the central nervous system (CNS) from the peripheral nervous system, is able to activate the innate immune response in the brain and cause the degeneration of dopaminergic neurons in the SNpc (27). Though this transient dopaminergic neuronal loss was found to be mostly restored within 90 days of infection, a long-lasting inflammatory response—permanent activation of microglia—persisted (28). The sustained activation of microglial cells was also reported after H1N1 infection, suggested to be a non-neurotropic virus, supporting the possibility that the virus may initiate inflammatory signals via direct microglia activation, contributing to disorders of protein aggregation, and neurodegeneration pathologies in the CNS (29). Synergistic effects of influenza and the parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) have been observed in animal models infected with H1N1, in which the cumulative effects induced a greater loss of SNpc dopaminergic neurons than either insult alone. This loss of dopaminergic neurons is shown to be eliminated through the use of influenza vaccinations or treatment with oseltamivir carboxylate (30). These findings of synergistic effects from multiple insults supports the “multiple hit hypothesis,” where the combination of toxic stress and an inhibition of neuroprotective response lead to neuronal death (31, 32). Despite recent histologic evidence in animal models, unclear conclusions can be drawn from these findings as no significant neuropathological evidence of direct viral invasion in the CNS has been presented in infected individuals (33).
The role of herpes simplex virus 1 (HSV-1) in the development and progression of neuropsychiatric disorders has been reported. Of particular interest is its pathological effect in provoking beta-amyloid deposition, tau phosphorylation, and demyelination, leading to cognitive deficits observed in neurodegenerative disorders including Alzheimer's disease and multiple sclerosis (61, 62). With regards to PD, the serological measure of exposure to HSV-1 amongst other common infectious pathogens such as CMV and EBV is shown to be elevated. The level of this infectious burden was additionally found to correlate to the severity of clinical symptoms and higher levels of serum inflammatory cytokines and alpha-synuclein (α-syn), supporting the role of infection in the etiology of PD (34).
The involvement of autoimmunity in PD's pathogenesis and the hypothesis that HSV-1 infections may lead to progression of the disease has been investigated. Recent findings indicate a mechanism of molecular mimicry between HSV-1 and α-syn in membranes of dopaminergic neurons of the SNpc. A difference in the level of autoantibodies recognizing HSV-1-Ul4222−36 was observed in PD patients compared to healthy controls. The antibodies were able cross-react with the homologous α-syn100−114 epitope, potentially promoting α-syn aggregation. These results suggest that HSV-1 may play a role in triggering an autoimmune response in PD, leading to dopaminergic neuronal destruction (35, 36).
Statistical evidence suggests that Epstein-Barr virus (EBV) seropositivity in patients with PD was found to be higher than that of the general population. Rare incidences of Parkinsonism in EBV infection, specifically EBV encephalitis, comprises the clinical symptoms of akinetic-rigid mutism, tremor, and apraxia of eyelid opening. Structural brain changes including progressive putaminal and caudate atrophy were reported with one case showing direct acute neutropic effect of EBV on nigral dopaminergic cells (11–13). Similar to HSV-1, the evidence for molecular mimicry with α-syn, although currently speculative, has been indicated in EBV infection and PEP. Specifically, anti-EBV latent membrane protein antibodies targeting the critical repeat region cross reaction with the homologous epitope on the α-syn and induce its oligomerization (11, 37).
Herpes zoster results from the reactivation of latent varicella-zoster virus (VZV) as a result of a decline in human cell-mediated immunity (63). A recent population-based cohort study in Taiwan found an increased risk of PD development among individuals with prior diagnosis of herpes zoster (age > 65 years) (14). Based on the overlapping mechanisms of neuroinflammation, immunological changes and resulting neuronal death in the two conditions, patients with herpes zoster may present with cardinal symptoms of PD during their follow-up period. These symptoms, particularly in the first 3 months after herpes zoster diagnosis, can lead to an earlier detection of PD (14).
This potential link was initially proposed by Ragozzino et al. (64), although the results were deemed equivocal due to limitations of the study (64). Findings reported that varicella, on the other hand, was not associated with PD (65). In fact, childhood infections with chicken pox were found to be inversely related to PD, suggesting a possible protective mechanism (23). This may be due to the “hygiene hypothesis,” in which immune challenges in early childhood are believed to support the development of a strong immune system later in life, though more evidence is necessary to develop a stronger connection (66).
Hepatitis C virus (HCV) primarily infect hepatocytes, leading to progressive liver diseases. Its association with a number of CNS abnormalities including cognitive dysfunction, fatigue, and depression has also been well-documented (38). The development for HCV-associated neuropathology was described through the findings of Fletcher et al. in which the expression of functional HCV receptors (CD81, claudin-1, occluding, LDLR, scavenger receptor-B1) were found on microvascular endothelial cells of the brain, paving a pathway for HCV entry and replication into the CNS (39).
A recent meta-analysis demonstrated an increased risk of subsequent PD in patients with and hepatitis C (40). As there are no observed associations between autoimmune and chronic hepatitis to PD, it has been suggested that a specific aspect of viral hepatitis, rather than the general hepatic inflammatory process, contributes to these findings (41). HCV-induced upregulation of chemokines (including sICAM-1 and RANTES) in animal models which mediate mechanisms of neuroinflammation, neuronal apoptosis, and dopaminergic toxicity, was found to be similar to that of toxicity caused by 1-methyl-4-phenylpyridinium (MPP+) (42). In addition, HCV was shown to down-regulate TIMP-1, one of the neuroprotectants derived from astrocytes known to promote neuronal survival subsequent to toxic effects during neuroinflammation.
Japanese encephalitis virus (JEV) and West Nile virus (WNV) are two important examples of zoonotic viral encephalitides. The clinical spectrum of these flaviviruses ranges from self-limiting flu-like illness to severe fatal meningoencephalitis, often with parkinsonian features (17, 67). Japanese encephalitis presents a wide spectrum of movement disorders including hypokinesia, tremor, rigidity, and dystonia. A transient form of parkinsonian syndrome, characterized by varying severity of rigidity, hypokinesia, masking of the face, lower frequency of tremor and prominent hypophonia, was observed in the acute stage of the illness after 1–4 weeks after the disease onset. Hypophonia was striking in most patients and was an important sequelae after the substantial regression of other manifestations in the subsequent months (15). Reports of Parkinsonism as a long-term sequela of JEV was also presented in patients 3–5 years after acute JE infection with associated lesions in the substantia nigra observed on MRI (16).
Burdwan, India has witnessed several outbreaks of JEV over the past two decades. Das et al. reported a higher incidence of prior JEV infection among PD patients within this region, compared to the control subject, suggesting the viral association to PD development (68). The analysis of JEV-induced Parkinsonism model rats showed decreased dopamine levels and neuropathological changes resembling those with idiopathic PD (43). Evaluation of cerebrospinal fluid in JEV-infected patients with movement disorders showed lower concentrations of norepinephrine, dopamine, and homovanillic acid, with respect to patients with non-JEV movement disorders. This observed decrease in catecholamine levels may be a result of damage to the dopaminergic and norepinephrinergic systems (44). Structural damage to the thalamus, basal ganglia, and brainstem observed in magnetic resonance imaging (MRI) findings of JE patients with parkinsonian features may contribute to this damage (44).
Features of Parkinsonism in acute WNV illness, including tremor myoclonus, rigidity, bradykinesia, and postural instability, are prominently reported and appear in most cases to be transient and resolve over time (18). These manifestations are uncommon among WNV-seronegative patients. Parkinsonian features were present in patients both in the presence and absence of MRI abnormalities showing bilateral, focal lesions in the basal ganglia, thalamus, and pons (17). Post-mortem analysis showed an increased level of α-syn in WNV-infected individuals. An increase in α-syn expression was also observed subsequent to WNV infection of primary neurons in vitro. The introduction of WNV into α-syn-knockout mice models were conducted, in which a 10-fold increase in viral production, increased neuronal injury, and a more rapid mortality was observed. This suggests the potential role of α-syn in inhibiting viral infection, rather than incurring CNS damage following viral infection. This data implies that the acute onset of parkinsonian features during WNV encephalitis is a result of the WNV-induced death of dopaminergic neurons (45).
Parkinsonism is a common movement disorder in human immunodeficiency virus (HIV), occurring in up to 5% of infected individuals in the context of neuroleptic drugs exposure, cerebral opportunistic infections or AIDS dementia complex (ADC) (69). Parkinsonism may occur early in HIV infection reflecting viral infection within the basal ganglia or late in the disease course in combination with ADC. The basal ganglia and dopamine-rich brain regions are a vulnerable target to HIV, and Parkinsonism may develop as a result of underlying chronic neuroinflammation leading to basal ganglia dysfunction, altered blood-brain barrier (BBB) permeability, and neurodegeneration (19).
Bradykinesia, postural instability, gait abnormalities, hypomimemetic facies, and disorders of ocular motility, are common parkinsonian manifestations of ADC, a collective of neuropsychiatric complications in HIV (20–22). The clinical features of HIV Parkinsonism are similar to idiopathic PD. Several distinctions include bilateral onset, rapid symptom progression, abnormal eye movements, and earlier development of motor complications (69). The introduction of highly active antiretroviral therapy (HAART) has contributed to the evident decrease in AIDS-related Parkinsonism, while dopaminergic medication may also help to alleviate some symptoms of Parkinsonism and could be used in selected cases in which the benefits outweigh the medication's adverse effects (20, 70).
PD prevalence in persons living with HIV was similar to that of the general population. Earlier symptom onset (before the age of 60) was noted (21, 46). Studies of brain tissue from postmortem autopsies showed a higher prevalence of α-syn in the SNpc in individuals with HIV infection compared to healthy control samples. The presence of HIV preferentially in inflammatory infiltrates and glial cells of the basal ganglia including the substantia nigra were also observed (47). In ADC, however, typical pathological features in brains of patients with PD, such as Lewy bodies, have not yet been reported.
From a genetic viewpoint, HIV has been shown to affect the levels of PD-associated proteins, including DJ1 and Leucine-rich repeat kinase 2 (LRRK2). DJ1, a gene linked to early-onset PD, is a key regulator of dopamine transmission and ROS production in neuronal cells. Acute and chronic HIV exposure has been found to play a role in its dysregulated expression (48). LRRK2, a common genetic cause of familial and sporadic PD, shares pathogenetic and neurologic similarities with HIV-associated neurologic disorders. Pathologic LRRK2 activation was found to be an important mediator of neuroinflammation and neuronal damage in in-vitro and in-vivo models of HIV-associated neurologic disorders (49).
Helicobacter pylori (H. pylori) is a bacterium on the luminal surface of the gastric epithelium that induces chronic inflammation of the underlying mucosa (71). It is found to be prevalent in the gastric mucosa of over half of the global human population (50, 72, 73). Ever since the initial observation of a high prevalence of gastric ulcers in addition to the gastrointestinal symptoms which preceded motor manifestations in PD patients, the link between H. pylori infection and the pathogenesis of PD has been extensively explored (51, 74). Stemming from the initial hypothesis of PD's origin within the gastrointestinal tract, the bidirectional interactions between the central and the enteric nervous system (namely, the gut-brain axis) is of particular interest (8). Research has supported the premise that H. pylori may play a role in the development of PD via several pathways. This centers around its involvement in neuronal damage, through the production of neurotoxic bacterial products and the disruption of the BBB. Another possible link is H. pylori's disruption of the intestinal microbiome, leading to altered inflammatory mediators that predispose to PD as part of the brain-gut axis interactions (75, 76).
Increased prevalence of H. pylori infection and higher titers of antibodies to H. pylori are observed among patients with PD compared to healthy controls. Accumulating evidence suggests that H. pylori infection is associated with increased risk of subsequent PD development in the general population. A positive correlation has been reported between worse motor severity and H. pylori seropositivity among patients (51–55). This is speculated to be a result of chronic inflammation exacerbating the neurodegenerative process or due to reduced absorption of anti-parkinsonian medication secondary to H. pylori-related gastroduodenitis. The improvement in clinical status, particularly symptoms of bradykinesia, subsequent to the eradication of H. pylori further supports a significant correlation (50, 53, 56–58).
H. pylori infection has been indicated to trigger neuroinflammation, neurotoxicity, and apoptosis related to the pathogenesis of PD. Chronic inflammation with H. pylori infection is associated with the substantial release of pro-inflammatory cytokines, which may lead to the disruption of the BBB, microglial activation and ultimately, neuronal injury (34, 59, 60). A subset of patients infected with H. pylori have been found to have elevated levels of autoantibodies against proteins essential for normal neurological functions (Nuclear factor 1 A-type (NFIA), Platelet Derived Growth Factor Subunit B (PDGFB), eukaryotic translation initiation factor 4A3 (EIFA3), suggesting a mechanism of molecular mimicry contributing to increased PD motor severity in H. pylori seropositive individuals (77).
Nocardia asteroides is one of the bacterial influences that has been studied in PD pathoetiology over the past decades, initially stemming from the study by Kohbata et al. (78) which observed the development of motor abnormalities in animal models infected by a strain of Nocardia asteroides. This L-dopa-responsive movement disorder was found to be accompanied by neuronal inclusions which resembled lewy bodies (78). Nocardiae were reported to be able to propagate through neuroglia to neurons in mouse models. They are seen to multiply within the astroglia, through which they may invade midbrain neurons and induce neuronal loss and lewy body formation (79).
The association remains, however, inconclusive, as there is a lack of evidence for Nocardia asteroides in brain specimens from lewy body-containing disorders (80). Because spheroplasts found in the SNpc of PD patients have been shown to not be Nocardia, researchers have postulated that the bacterium known to form spheroplasts may alternatively be Mycobacterium avium ss. Paratuberculosis (MAP). MAP has been proposed to be the “unidentified enteric pathogen” that triggers α-syn aggregation (81).
Recent research has explored the relationship between gut dysbiosis and the onset and aggravation of PD. The interaction between intestinal microbiota and the autonomic and CNS has been indicated via diverse pathways including the enteric nervous system and the vagal nerve (82–84). In a recent study, Proteus mirabilis (commonly increased in the gut microbiota of PD mouse models) has been shown to directly induce PD-related pathological changes and motor deficits. This includes the induction of dopaminergic neuronal damage and inflammation within the substantia nigra and the striatum, and the stimulation of α-syn aggregation in the brains and colons of PD mice (85). Lipopolysaccharide (LPS), a virulence factor of P. mirabilis, has been implicated in these pathological changes. Increased intestinal permeability leading to a greater exposure of intestinal neuronal tissue to pro-inflammatory products have been suggested to result in oxidative stress and neuronal pathological α-syn aggregates (85, 86).
A number of bacterial infections have been explored in association with PD, including Chlamydia pneumoniae (34, 87), Bordetella pertussis (65, 88), Streptococcus pyogenes (65, 89), and Borrelia burgdoferi (34). The varying results amongst epidemiological and laboratory studies are primarily suggestive, as there is inadequate evidence to support their role in PD development. The relationship between these bacterial etiologies and PD requires more comprehensive studies in the future.
Neuroinflammation is a common feature of neurodegenerative disorders of the CNS, characterized by augmented numbers of activated and primed microglia, increased level of inflammatory cytokines and decreased levels of anti-inflammatory molecules. Neuroinflammation might initiate from the periphery and relative data suggest that peripheral conditions through the disrupted BBB notably influence various pathologic processes in the brain (60). Subsequent to its access to the CNS through the systemic circulation, the olfactory pathways, and the gastrointestinal tract, infectious pathogens may induce neuroinflammation through the induction of pro-inflammatory cytokines in microglia (27, 29).
The activation of microglia and release of inflammatory factors promote damage to DA neurons. Infectious pathogens have been shown to be associated with the release of large amounts of proinflammatory mediators, including TNF-α, IL-6, and IL-1β which may lead to disruption of the BBB, microglial activation and, ultimately, neuronal injury and death (90). Such relationships between the innate autoimmune response and infectious pathogens has also been investigated by other literature such as Caggiu et al. (91).
This mechanism of microglial activation and hypercytokinemia can cause further activation and clustering of microglia around DA neurons, leading to a continuous cycle of chronic inflammation and neuronal damage. Chronic activation of microglia and release of cytokines have been shown to cause extensive damage to the dopaminergic neurons in the SNpc, contributing significantly to neuronal death in PD (92, 93) (Figure 1).
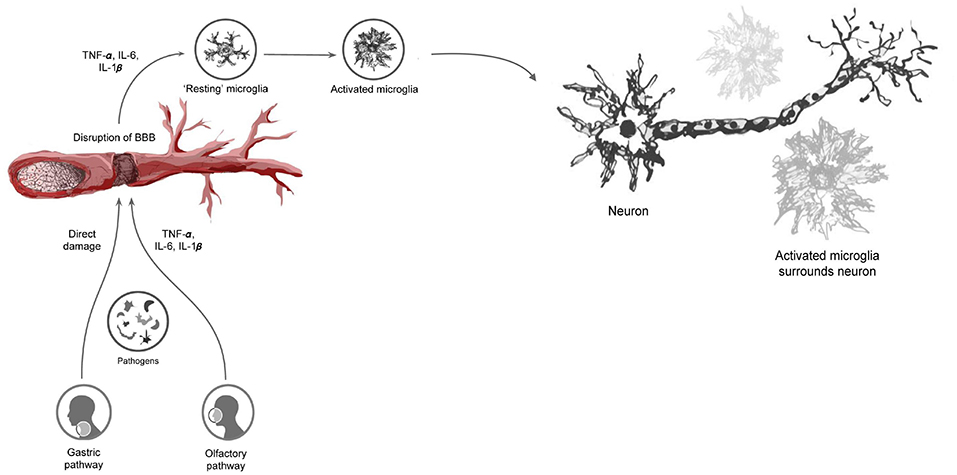
Figure 1. Infectious pathogens inciting the neuroinflammatory process and subsequent blood-brain barrier disturbance through the release of pro-inflammatory cytokines. This ultimately leads to the activation of microglia and subsequent clustering around neuronal cells, resulting in neuronal damage.
From another view point, viral agents through their replication and subsequent cell lysis may result in direct neuronal damage (30, 94). Similarly, vascular inflammation due to hypercytokinemia can lead to apoptosis of endothelial cells and loss of vascular integrity, which result in increased permeability of cerebral vessels and cerebral edema and ultimately neuronal damage.
A-syn is a presynaptic neuronal protein involved in neurotransmitter release that is thought to play a significant role in the initiation and progression of PD. A-syn is also a major constituent of Lewy bodies and Lewy neurites, which are well-known pathological hallmarks of the disease. Recent studies suggest that oligomers or protofibrils of α-syn, rather than the fibrils, are responsible for the toxic effects causing neurodegeneration (95–97). Oligomerized α-syn is hypothesized to have toxic effects on synaptic transmission (98), cause membrane disruption (99), impairment of protein degradation, as well as impairment of several organelles including the mitochondria and endoplasmic reticulum (100, 101). The ability of α-syn to cause membrane disruption and organelle impairment has also been hypothesized to give the protein antimicrobial properties. This was demonstrated in a study by Park et al. in which recombinant a-syn proteins were used in antimicrobial assays against various bacterial and fungal strains (102). This antimicrobial effect could suggest a protective mechanism of the post-infectious increase in α-syn oligomers.
Bidirectional relationships have been proposed between α-syn and microglia through the activation and release of inflammatory mediators, potentiating each other and causing dopaminergic neuronal loss and neurodegeneration (103). A similar relationship is also seen between α-syn and mitochondria, in which α-syn oligomers cause disruption of mitochondrial function and the resultant oxidative stress in turn furthers the oligomerization of α-syn. It has been proposed that aggregated α-syn may function as a messenger molecule for immune responses against infectious pathogens by inducing microglial activation. This is similar to the proinflammatory cytokines' incitation of antimicrobial responses within the intestinal epithelium (83, 104–106). Together with its antimicrobial and antifungal effect, this supports a potential immunoprotective role of α-syn. Viral agents have been shown through these mechanisms of molecular mimicry to induce the aggregation and oligomerization of α-syn, leading to deposition in neuronal cells and further damage (35, 36) (Figure 2).
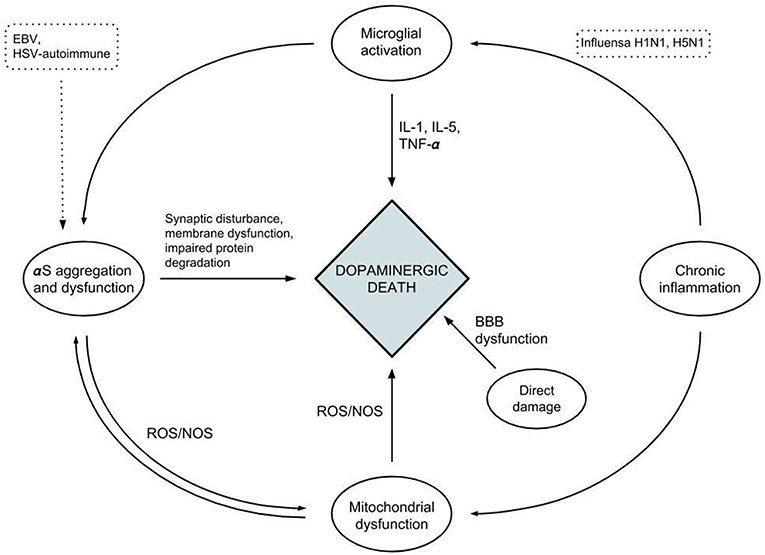
Figure 2. A diagram depicting the relationships between the different factors affecting neuroinflammation and neuronal cell death.
Currently, the diagnosis of PD relies on the clinical presence of the disease cardinal features, including resting tremor, rigidity, bradykinesia, and loss of postural reflexes. The efficacy of this diagnostic method is limited during the early stages of the disease, permitting PD progression before any therapeutic intervention. The positive correlation of PD development amongst patients with underlying chronic infection suggests the benefit of PD screening before progressive symptom onset. Neurological examinations may be beneficial to detect the earlier stages of PD in, for example, anti-HCV positive groups, which would be an important clinical implication for high endemic HCV areas. Conversely, PD patients may benefit from an evaluation for underlying infection as clinical improvement of motor symptoms have been shown subsequent to the eradication of disease, for example as in chronic H. pylori infection (50, 53, 56–58). Whether the clinical deterioration observed in these cases is a result of chronic inflammation exacerbating the neurodegenerative process or due to reduced absorption of anti-parkinsonian medication secondary to H. pylori-related infection is a topic for further investigation. On the other hand, anti-parkinsonian drugs may have possible therapeutic antiviral potential. For example, Parkinsonian drugs (L-dopa, Isatin, Amantadine) have shown in-vitro to significantly inhibit WNV multiplication as well as reduced viral RNA levels (Amantidine) (107).
Another significant limitation in diagnosis is that the neurodegenerative process associated with PD precedes the onset of clinical symptoms, in which up to 70% of neurons in the SNpc are lost before the appearance of motor features (108, 109). A-syn oligomers could serve as a biomarker that would allow the identification of at-risk individuals before clinical diagnosis. Detection of α-syn oligomers may additionally be useful in monitoring disease progression and response to treatments. Emerging information on the pathomechanism of diseases such as loss of BBB integrity may also lead to formulating strategies to protect BBB and to prevent and treat PD and other BBB-related neurodegenerative disorders (110, 111).
Accumulating evidence in recent years implicates the role of infectious etiologies in neuronal pathways leading to the development of Parkinsonism and PD. Studies have demonstrated the independent association between specific pathogens and PD. The synergistic effect of infectious pathogens in inducing neuroinflammation leading to the PD development have also been observed. Neuroinflammation may not only play a part in the precipitation of PD but may also be a sustaining factor in its progression. However, the pathomechanism of dopaminergic neuronal loss in infection-induced Parkinsonism cannot explain the pathogenesis of idiopathic PD. The conclusion that all cases of PD are associated with increased inflammation and underlying chronic infection cannot be established. This is due multiple factors, including the consideration that not all PD patients have consistent evidence of inflammatory cytokine dysregulation and that the state of increased inflammation in individuals do not always lead to PD development. Parkinsonism development may be part of a response to an underlying immune system dysregulation in individuals with genetic predisposition to the disease.
Further research is necessary to examine the involvement and extent to which pathogens and inflammatory cytokines play in the pathomechanism of PD. Confounding factors, including the roles of genetics and exposure to environmental insults, have to be taken into account in further investigation. Studies should explore the possibility of a subtype of PD that is characteristically associated with pro/anti-inflammatory cytokines and the reason why certain individuals develop PD in the context of elevated inflammatory markers. Through further understanding these mechanisms, we may be able to classify variants of PD, use biological markers to aid in the diagnosis and prediction of treatment response and adjust treatment for an improved prognosis for patients.
NL: conception and design of the study, acquisition and analysis of data, drafting the manuscript and figures, project administration, supervision, validation. PI: conception and design of the study, supervision, validation. MH: acquisition and analysis of data, drafting the manuscript and figures. DK: drafting the manuscript and figures. WK: supervision, validation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer PB and handling Editor declared their shared affiliation at the time of review.
1. Tysnes OB, Storstein A. Epidemiology of parkinson's disease. J Neural Transm (Vienna). (2017) 124:901–5. doi: 10.1007/s00702-017-1686-y
2. Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. (2007) 68:384–6. doi: 10.1212/01.wnl.0000247740.47667.03
3. Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of parkinson's disease: a review of the evidence. Eur J Epidemiol. (2011) 26(Suppl. 1):S1–58. doi: 10.1007/s10654-011-9581-6
4. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying parkinson's disease. Nat Genet. (2009) 41:1308–12. doi: 10.1038/ng.487
5. International Parkinson Disease Genomics C, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, et al. Imputation of sequence variants for identification of genetic risks for parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. (2011) 377:641–9. doi: 10.1016/S0140-6736(10)62345-8
6. Jang H, Boltz DA, Webster RG, Smeyne RJ. Viral Parkinsonism. Biochim Biophys Acta. (2009) 1792:714–21. doi: 10.1016/j.bbadis.2008.08.001
7. Maurizi CP. Was a neurovirulent influenza virus the cause of amyotrophic lateral sclerosis and Parkinsonism -dementia on Guam? Med Hypotheses. (1987) 23:325–6. doi: 10.1016/0306-9877(87)90024-7
8. Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. (2007) 33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x
9. Rail D, Scholtz C, Swash M. Post-encephalitic Parkinsonism: current experience. J Neurol Neurosurg Psychiatry. (1981) 44:670–6. doi: 10.1136/jnnp.44.8.670
10. Toovey S, Jick SS, Meier CR. Parkinson's disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir Viruses. (2011) 5:328–33. doi: 10.1111/j.1750-2659.2011.00232.x
11. Espay AJ, Henderson KK. Postencephalitic Parkinsonism and basal ganglia necrosis due to Epstein-Barr virus infection. Neurology. (2011) 76:1529–30. doi: 10.1212/WNL.0b013e318217e7dd
12. Noyce AJ, Treacy C, Budu C, Fearnley J, Lees AJ, Giovannoni G. The new Bradykinesia Akinesia Incoordination (BRAIN) test: preliminary data from an online test of upper limb movement. Mov Disord. (2012) 27:157–8. doi: 10.1002/mds.23947
13. Hsieh JC, Lue KH, Lee YL. Parkinson-like syndrome as the major presenting symptom of Epstein-Barr virus encephalitis. Arch Dis Child. (2002) 87:358. doi: 10.1136/adc.87.4.358
14. Lai SW, Lin CH, Lin HF, Lin CL, Lin CC, Liao KF. Herpes zoster correlates with increased risk of parkinson's disease in older people: a population-based cohort study in Taiwan. Medicine (Baltimore). (2017) 96:e6075. doi: 10.1097/MD.0000000000006075
15. Misra UK, Kalita J. Prognosis of Japanese encephalitis patients with dystonia compared to those with parkinsonian features only. Postgrad Med J. (2002) 78:238–41. doi: 10.1136/pmj.78.918.238
16. Murgod UA, Muthane UB, Ravi V, Radhesh S, Desai A. Persistent movement disorders following Japanese encephalitis. Neurology. (2001) 57:2313–5. doi: 10.1212/WNL.57.12.2313
17. Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. (2003) 290:511–5. doi: 10.1001/jama.290.4.511
18. Sejvar JJ. Clinical manifestations and outcomes of West Nile Virus Infection. Viruses. (2014) 6:606–23. doi: 10.3390/v6020606
19. Berger JR, Nath A, Greenberg RN, Andersen AH, Greene RA, Bognar A, et al. Cerebrovascular changes in the basal ganglia with HIV dementia. Neurology. (2000) 54:921–6. doi: 10.1212/WNL.54.4.921
20. Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Riederer P. Parkinsonism in HIV dementia. J Neural Transm (Vienna). (2002) 109:767–75. doi: 10.1007/s007020200063
21. Dehner LF, Spitz M, Pereira JS. Parkinsonism in HIV infected patients during antiretroviral therapy - data from a Brazilian tertiary hospital. Braz J Infect Dis. (2016) 20:499–501. doi: 10.1016/j.bjid.2016.05.008
22. Tisch S, Brew B. Parkinsonism in hiv-infected patients on highly active antiretroviral therapy. Neurology. (2009) 73:401–3. doi: 10.1212/WNL.0b013e3181b04b0d
23. Harris MA, Tsui JK, Marion SA, Shen H, Teschke K. Association of parkinson's disease with infections and occupational exposure to possible vectors. Mov Disord. (2012) 27:1111–7. doi: 10.1002/mds.25077
24. Fazekas T, Wiesbauer P, Schroth B, Pötschger U, Gadner H, Heitger A. Selective IgA deficiency in children with recurrent parotitis of childhood. Pediatr Infect Dis J. (2005) 24:461–2. doi: 10.1097/01.inf.0000160994.65328.dd
25. Yamanaka G, Kawashima H, Suganami Y, Watanabe C, Watanabe Y, Miyajima T, et al. Diagnostic and predictive value of CSF d-ROM level in influenza virus-associated encephalopathy. J Neurol Sci. (2006) 243:71–5. doi: 10.1016/j.jns.2005.11.029
26. Sun G, Ota C, Kitaoka S, Chiba Y, Takayanagi M, Kitamura T, et al. Elevated serum levels of neutrophil elastase in patients with influenza virus-associated encephalopathy. J Neurol Sci. (2015) 349:190–5. doi: 10.1016/j.jns.2015.01.017
27. Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA. (2009) 106:14063–8. doi: 10.1073/pnas.0900096106
28. Jang H, Boltz D, McClaren J, Pani AK, Smeyne M, Korff A, et al. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci. (2012) 32:1545–59. doi: 10.1523/JNEUROSCI.5123-11.2012
29. Sadasivan S, Zanin M, O'Brien K, Schultz-Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE. (2015) 10:e0124047. doi: 10.1371/journal.pone.0124047
30. Sadasivan S, Sharp B, Schultz-Cherry S, Smeyne RJ. Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. NPJ Parkinsons Dis. (2017) 3:18. doi: 10.1038/s41531-017-0019-z
31. Sulzer D. Multiple hit hypotheses for dopamine neuron loss in parkinson's disease. Trends Neurosci. (2007) 30:244–50. doi: 10.1016/j.tins.2007.03.009
32. Schirinzi T, Martella G, Pisani A. Double hit mouse model of parkinson's disease. Oncotarget. (2016) 7:80109–10. doi: 10.18632/oncotarget.13460
33. Takahashi M, Yamada T, Nakashita Y, Saikusa H, Deguchi M, Kida H, et al. Influenza virus-induced encephalopathy: clinicopathologic study of an autopsied case. Pediatr Int. (2000) 42:204–14. doi: 10.1046/j.1442-200x.2000.01203.x
34. Bu XL, Wang X, Xiang Y, Shen LL, Wang QH, Liu YH, et al. The association between infectious burden and parkinson's disease: a case-control study. Parkinsonism Relat Disord. (2015) 21:877–81. doi: 10.1016/j.parkreldis.2015.05.015
35. Caggiu E, Paulus K, Arru G, Piredda R, Sechi GP, Sechi LA. Humoral cross reactivity between alpha-synuclein and herpes simplex-1 epitope in parkinson's disease, a triggering role in the disease? J Neuroimmunol. (2016) 291:110–4. doi: 10.1016/j.jneuroim.2016.01.007
36. Caggiu E, Paulus K, Galleri G, Arru G, Manetti R, Sechi GP, et al. Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in parkinson's disease. J Neuroimmunol. (2017) 310:26–31. doi: 10.1016/j.jneuroim.2017.06.004
37. Woulfe J, Hoogendoorn H, Tarnopolsky M, Munoz DG. Monoclonal antibodies against Epstein-Barr virus cross-react with -synuclein in human brain. Neurology. (2000) 55:1398–401. doi: 10.1212/WNL.55.9.1398
38. McAndrews MP, Farcnik K, Carlen P, Damyanovich A, Mrkonjic M, Jones S, et al. Prevalence and significance of neurocognitive dysfunction in hepatitis C in the absence of correlated risk factors. Hepatology. (2005) 41:801–8. doi: 10.1002/hep.20635
39. Fletcher NF, Wilson GK, Murray J, Hu K, Lewis A, Reynolds GM, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. (2012) 142:634–43 e6. doi: 10.1053/j.gastro.2011.11.028
40. Wijarnpreecha K, Chesdachai S, Jaruvongvanich V, Ungprasert P. Hepatitis C virus infection and risk of parkinson's disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol. (2018) 30:9–13. doi: 10.1097/MEG.0000000000000991
41. Pakpoor J, Noyce A, Goldacre R, Selkihova M, Mullin S, Schrag A, et al. Viral hepatitis and Parkinson disease: a national record-linkage study. Neurology. (2017) 88:1630–3. doi: 10.1212/WNL.0000000000003848
42. Wu WY, Kang KH, Chen SL, Chiu SY, Yen AM, Fann JC, et al. Hepatitis C virus infection: a risk factor for parkinson's disease. J Viral Hepat. (2015) 22:784–91. doi: 10.1111/jvh.12392
43. Hamaue N, Ogata A, Terado M, Ohno K, Kikuchi S, Sasaki H, et al. Brain catecholamine alterations and pathological features with aging in Parkinson disease model rat induced by Japanese encephalitis virus. Neurochem Res. (2006) 31:1451–5. doi: 10.1007/s11064-006-9197-5
44. Misra UK, Kalita J, Pandey S, Khanna VK, Babu GN. Cerebrospinal fluid catecholamine levels in Japanese encephalitis patients with movement disorders. Neurochem Res. (2005) 30:1075–8. doi: 10.1007/s11064-005-7414-2
45. Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, et al. Alpha-Synuclein expression restricts RNA viral infections in the brain. J Virol. (2015) 90:2767–82. doi: 10.1128/JVI.02949-15
46. Moulignier A, Gueguen A, Lescure FX, Ziegler M, Girard PM, Cardon B, et al. Does HIV infection alter Parkinson disease? J Acquir Immune Defic Syndr. (2015) 70:129–36. doi: 10.1097/QAI.0000000000000677
47. Khanlou N, Moore DJ, Chana G, Cherner M, Lazzaretto D, Dawes S, et al. Increased frequency of alpha-synuclein in the substantia nigra in human immunodeficiency virus infection. J Neurovirol. (2009) 15:131–8. doi: 10.1080/13550280802578075
48. Roy U, Atluri VS, Agudelo M, Yndart A, Huang Z, Nair M. DJ1 expression downregulates in neuroblastoma cells (SK-N-MC) chronically exposed to HIV-1 and cocaine. Front Microbiol. (2015) 6:749. doi: 10.3389/fmicb.2015.00749
49. Puccini JM, Marker DF, Fitzgerald T, Barbieri J, Kim CS, Miller-Rhodes P, et al. Leucine-rich repeat kinase 2 modulates neuroinflammation and neurotoxicity in models of human immunodeficiency virus 1-associated neurocognitive disorders. J Neurosci. (2015) 35:5271–83. doi: 10.1523/JNEUROSCI.0650-14.2015
50. Mridula KR, Borgohain R, Chandrasekhar Reddy V, Bandaru V, Suryaprabha T. Association of Helicobacter pylori with parkinson's disease. J Clin Neurol. (2017) 13:181–6. doi: 10.3988/jcn.2017.13.2.181
51. Shen X, Yang H, Wu Y, Zhang D, Jiang H. Meta-analysis: association of Helicobacter pylori infection with parkinson's diseases. Helicobacter. (2017) 22:e12398 doi: 10.1111/hel.12398
52. Hashim H, Azmin S, Razlan H, Yahya NW, Tan HJ, Manaf MR, et al. Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with parkinson's disease. PLoS ONE. (2014) 9:e112330. doi: 10.1371/journal.pone.0112330
53. Tan AH, Mahadeva S, Marras C, Thalha AM, Kiew CK, Yeat CM, et al. Helicobacter pylori infection is associated with worse severity of parkinson's disease. Parkinsonism Relat Disord. (2015) 21:221–5. doi: 10.1016/j.parkreldis.2014.12.009
54. Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B. Treatment for Helicobacter pylori infection and risk of parkinson's disease in Denmark. Eur J Neurol. (2012) 19:864–9. doi: 10.1111/j.1468-1331.2011.03643.x
55. Huang HK, Wang JH, Lei WY, Chen CL, Chang CY, Liou LS. Helicobacter pylori infection is associated with an increased risk of parkinson's disease: a population-based retrospective cohort study. Parkinsonism Relat Disord. (2018) 47:26–31. doi: 10.1016/j.parkreldis.2017.11.331
56. Pierantozzi M, Pietroiusti A, Brusa L, Galati S, Stefani A, Lunardi G, et al. Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology. (2006) 66:1824–9. doi: 10.1212/01.wnl.0000221672.01272.ba
57. Liu H, Su W, Li S, Du W, Ma X, Jin Y, et al. Eradication of Helicobacter pylori infection might improve clinical status of patients with parkinson's disease, especially on bradykinesia. Clin Neurol Neurosurg. (2017) 160:101–4. doi: 10.1016/j.clineuro.2017.07.003
58. Dobbs SM, Dobbs RJ, Weller C, Charlett A, Bjarnason IT, Lawson AJ, et al. Differential effect of Helicobacter pylori eradication on time-trends in brady/hypokinesia and rigidity in idiopathic Parkinsonism. Helicobacter. (2010) 15:279–94. doi: 10.1111/j.1523-5378.2010.00768.x
59. Kountouras J, Zavos C, Polyzos SA, Deretzi G, Vardaka E, Giartza-Taxidou E, et al. Helicobacter pylori infection and parkinson's disease: apoptosis as an underlying common contributor. Eur J Neurol. (2012) 19:e56. doi: 10.1111/j.1468-1331.2012.03695.x
60. Deretzi G, Kountouras J, Polyzos SA, Zavos C, Giartza-Taxidou E, Gavalas E, et al. Gastrointestinal immune system and brain dialogue implicated in neuroinflammatory and neurodegenerative diseases. Curr Mol Med. (2011) 11:696–707. doi: 10.2174/156652411797536660
61. Kobayashi N, Nagata T, Shinagawa S, Oka N, Shimada K, Shimizu A, et al. Increase in the IgG avidity index due to herpes simplex virus type 1 reactivation and its relationship with cognitive function in amnestic mild cognitive impairment and Alzheimer's disease. Biochem Biophys Res Commun. (2013) 430:907–11. doi: 10.1016/j.bbrc.2012.12.054
62. Wozniak MA, Itzhaki RF. Intravenous immunoglobulin reduces beta amyloid and abnormal tau formation caused by herpes simplex virus type 1. J Neuroimmunol. (2013) 257:7–12. doi: 10.1016/j.jneuroim.2013.01.005
63. Johnson BH, Palmer L, Gatwood J, Lenhart G, Kawai K, Acosta CJ. Annual incidence rates of herpes zoster among an immunocompetent population in the United States. BMC Infect Dis. (2015) 15:502. doi: 10.1186/s12879-015-1262-8
64. Ragozzino MW, Kurland LT, Rajput AH. Investigation of the Association between Herpes zoster and parkinson's Disease. Neuroepidemiology. (1983) 2:89–92 doi: 10.1159/000110514
65. Vlajinac HD, Sipetic SB, Maksimovic JM, Marinkovic JM, Dzoljic ED, Ratkov IS, et al. Environmental factors and parkinson's disease: a case-control study in Belgrade, Serbia. Int J Neurosci. (2010) 120:361–7. doi: 10.3109/00207451003668374
66. Okada H, Kuhn C, Feillet H, Bach JF. The 'hygiene hypothesis' for autoimmune and allergic diseases: an update. Clin Exp Immunol. (2010) 160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x
67. Yin Z, Wang X, Li L, Li H, Zhang X, Li J, et al. Neurological sequelae of hospitalized Japanese encephalitis cases in Gansu province, China. Am J Trop Med Hyg. (2015) 92:1125–9. doi: 10.4269/ajtmh.14-0148
68. Das K, Ghosh M, Nag C, Nandy SP, Banerjee M, Datta M, et al. Role of familial, environmental and occupational factors in the development of parkinson's disease. Neurodegener Dis. (2011) 8:345–51. doi: 10.1159/000323797
69. Bhidayasiri R, Tarsy D. HIV-induced Parkinsonism. In: Movement Disorders: A Video Atlas. Current Clinical Neurology. Totowa, NJ: Humana Press (2012). p. 44–5.
70. Rosso AL, Mattos JP, Correa RB, Nicaretta DH, Novis SA. Parkinsonism and AIDS: a clinical comparative study before and after HAART. Arq Neuropsiquiatr. (2009) 67:827–30. doi: 10.1590/S0004-282X2009000500009
71. McColl KE. Clinical practice. Helicobacter pylori infection. N Engl J Med. (2010) 362:1597–604. doi: 10.1056/NEJMcp1001110
72. Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology. (2017) 153:420–9. doi: 10.1053/j.gastro.2017.04.022
73. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. (2014) 19(Suppl 1):1–5. doi: 10.1111/hel.12165
74. Strang RR. The association of gastro-duodenal ulceration and Parkinson's disease. Med J Aust. (1965) 1:842–3.
75. Altschuler E. Gastric Helicobacter pylori infection as a cause of idiopathic Parkinson disease and non-arteric anterior optic ischemic neuropathy. Med Hypotheses. (1996) 47:413–4. doi: 10.1016/S0306-9877(96)90223-6
76. McGee DJ, Lu XH, Disbrow EA. Stomaching the possibility of a pathogenic role for Helicobacter pylori in parkinson's disease. J Parkinsons Dis. (2018) 8:367–74. doi: 10.3233/JPD-181327
77. Suwarnalata G, Tan AH, Isa H, Gudimella R, Anwar A, Loke MF, et al. Augmentation of autoantibodies by Helicobacter pylori in parkinson's disease patients may be linked to greater severity. PLoS ONE. (2016) 11:e0153725. doi: 10.1371/journal.pone.0153725
78. Kohbata S, Beaman BL. L-dopa-responsive movement disorder caused by Nocardia asteroides localized in the brains of mice. Infect Immun. (1991) 59:181–91.
79. Kohbata S, Emura S, Kadoya C. Filterable forms of Nocardia: a preferential site of infection in the mouse brain. Microbes Infect. (2009) 11:744–52. doi: 10.1016/j.micinf.2009.04.013
80. Lu L, Camp DM, Loeffler DA, LeWitt PA. Lack of evidence for Nocardia asteroides in brain specimens from Lewy body-containing disorders. Microb Pathog. (2005) 39:205–11. doi: 10.1016/j.micpath.2005.08.001
81. Dow CT. M. paratuberculosis and parkinson's disease–is this a trigger. Med Hypotheses. (2014) 83:709–12. doi: 10.1016/j.mehy.2014.09.025
82. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to parkinson's disease and clinical phenotype. Mov Disord. (2015) 30:350–8. doi: 10.1002/mds.26069
83. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinson's disease. Cell. (2016) 167:1469–80 e12. doi: 10.1016/j.cell.2016.11.018
84. Li W, Wu X, Hu X, Wang T, Liang S, Duan Y, et al. Structural changes of gut microbiota in parkinson's disease and its correlation with clinical features. Sci China Life Sci. (2017) 60:1223–33. doi: 10.1007/s11427-016-9001-4
85. Choi JG, Kim N, Ju IG, Eo H, Lim SM, Jang SE, et al. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci Rep. (2018) 8:1275. doi: 10.1038/s41598-018-19646-x
86. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early parkinson's disease. PLoS ONE. (2011) 6:e28032. doi: 10.1371/journal.pone.0028032
87. Turkel Y, Dag E, Gunes HN, Apan T, Yoldas TK. Is there a relationship between parkinson's disease and Chlamydia pneumoniae? Niger J Clin Pract. (2015) 18:612–5. doi: 10.4103/1119-3077.154215
88. Fiszer U, Tomik B, Grzesiowski P, Krygowska-Wajs A, Walory J, Michalowska M, et al. The antibodies against Bordetella pertussis in sera of patients with parkinson's disease and other non-neurological diseases. Acta Neurol Scand. (2004) 110:113–7. doi: 10.1111/j.1600-0404.2004.00285.x
89. McKee DH, Sussman JD. Case report: severe acute Parkinsonism associated with streptococcal infection and antibasal ganglia antibodies. Mov Disord. (2005) 20:1661–3. doi: 10.1002/mds.20641
90. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in parkinson disease: a systematic review and meta-analysis. JAMA Neurol. (2016) 73:1316–24. doi: 10.1001/jamaneurol.2016.2742
91. Caggiu E, Arru G, Hosseini S, Niegowska M, Sechi G, Zarbo IR, et al. Inflammation, infectious triggers, and parkinson's disease. Front Neurol. (2019) 10:122. doi: 10.3389/fneur.2019.00122
92. Tansey MG, Goldberg MS. Neuroinflammation in parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. (2010) 37:510–8. doi: 10.1016/j.nbd.2009.11.004
93. Rohn TT, Catlin LW. Immunolocalization of influenza A virus and markers of inflammation in the human parkinson's disease brain. PLoS ONE. (2011) 6:e20495. doi: 10.1371/journal.pone.0020495
94. Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. (2010) 129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x
95. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci USA. (2011) 108:4194–9. doi: 10.1073/pnas.1100976108
96. Ingelsson M. Alpha-synuclein oligomers—neurotoxic molecules in parkinson's disease and other lewy body disorders. Front Neurosci. (2016) 10:408. doi: 10.3389/fnins.2016.00408
97. Emmanouilidou E, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging. (2010) 31:953–68. doi: 10.1016/j.neurobiolaging.2008.07.008
98. Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, et al. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci USA. (2013) 110:4087–92. doi: 10.1073/pnas.1218424110
99. Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of parkinson's disease. Biochemistry. (2001) 40:7812–9. doi: 10.1021/bi0102398
100. Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in parkinson's disease. Neurobiol Dis. (2018) 109(Pt B):249–57. doi: 10.1016/j.nbd.2017.04.004
101. Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. (2012) 32:3301–5. doi: 10.1523/JNEUROSCI.5368-11.2012
102. Park SC, Moon JC, Shin SY, Son H, Jung YJ, Kim NH, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. (2016) 478:924–8. doi: 10.1016/j.bbrc.2016.08.052
103. Zhang QS, Heng Y, Yuan YH, Chen NH. Pathological alpha-synuclein exacerbates the progression of parkinson's disease through microglial activation. Toxicol Lett. (2017) 265:30–7. doi: 10.1016/j.toxlet.2016.11.002
104. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. (2016) 535:65–74. doi: 10.1038/nature18847
105. Fitzgerald E, Murphy S, Martinson HA. Alpha-synuclein pathology and the role of the microbiota in parkinson's disease. Front Neurosci. (2019) 13:369. doi: 10.3389/fnins.2019.00369
106. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in parkinson's disease. Ageing Res Rev. (2018) 45:53–61. doi: 10.1016/j.arr.2018.04.004
107. Blazquez AB, Martin-Acebes MA, Saiz JC. Inhibition of West Nile Virus multiplication in cell culture by anti-parkinsonian drugs. Front Microbiol. (2016) 7:296. doi: 10.3389/fmicb.2016.00296
108. Ross GW, Petrovitch H, Abbott RD, Nelson J, Markesbery W, Davis D, et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol. (2004) 56:532–9. doi: 10.1002/ana.20226
109. Cheng HC, Ulane CM, Burke RE. Clinical progression in parkinson disease and the neurobiology of axons. Ann Neurol. (2010) 67:715–25. doi: 10.1002/ana.21995
110. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. (2004) 16:1–13. doi: 10.1016/j.nbd.2003.12.016
Keywords: encephalitis lethargica, infectious diseases, Parkinson's disease, neurodegeneration, neuroinflammation
Citation: Limphaibool N, Iwanowski P, Holstad MJV, Kobylarek D and Kozubski W (2019) Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 10:652. doi: 10.3389/fneur.2019.00652
Received: 17 April 2019; Accepted: 04 June 2019;
Published: 19 June 2019.
Edited by:
Gianfranco Spalletta, Fondazione Santa Lucia (IRCCS), ItalyReviewed by:
Juan Segura-Aguilar, Universidad de Chile, ChileCopyright © 2019 Limphaibool, Iwanowski, Holstad, Kobylarek and Kozubski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nattakarn Limphaibool, dG9uZ24ubGltQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.