
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 21 May 2019
Sec. Neurogenetics
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00434
This article is part of the Research TopicNeurogenetics in neurology: from molecular neuroscience to precision medicineView all 13 articles
 Michelle Demos1*†
Michelle Demos1*† Ilaria Guella2†
Ilaria Guella2† Conrado DeGuzman1
Conrado DeGuzman1 Marna B. McKenzie2
Marna B. McKenzie2 Sarah E. Buerki1,3
Sarah E. Buerki1,3 Daniel M. Evans2
Daniel M. Evans2 Eric B. Toyota1
Eric B. Toyota1 Cyrus Boelman1Linda L. Huh1Anita Datta1Aspasia Michoulas1
Cyrus Boelman1Linda L. Huh1Anita Datta1Aspasia Michoulas1 Kathryn Selby1Bruce H. Bjornson1
Kathryn Selby1Bruce H. Bjornson1 Gabriella Horvath4Elena Lopez-Rangel5Clara D. M. van Karnebeek6,7
Gabriella Horvath4Elena Lopez-Rangel5Clara D. M. van Karnebeek6,7 Ramona Salvarinova4Erin Slade1Patrice Eydoux8,9
Ramona Salvarinova4Erin Slade1Patrice Eydoux8,9 Shelin Adam10
Shelin Adam10 Margot I. Van Allen10Tanya N. Nelson8,9
Margot I. Van Allen10Tanya N. Nelson8,9 Corneliu Bolbocean11,12
Corneliu Bolbocean11,12 Mary B. Connolly1
Mary B. Connolly1 Matthew J. Farrer2
Matthew J. Farrer2Targeted whole-exome sequencing (WES) is a powerful diagnostic tool for a broad spectrum of heterogeneous neurological disorders. Here, we aim to examine the impact on diagnosis, treatment and cost with early use of targeted WES in early-onset epilepsy. WES was performed on 180 patients with early-onset epilepsy (≤5 years) of unknown cause. Patients were classified as Retrospective (epilepsy diagnosis >6 months) or Prospective (epilepsy diagnosis <6 months). WES was performed on an Ion Proton™ and variant reporting was restricted to the sequences of 620 known epilepsy genes. Diagnostic yield and time to diagnosis were calculated. An analysis of cost and impact on treatment was also performed. A molecular diagnoses (pathogenic/likely pathogenic variants) was achieved in 59/180 patients (33%). Clinical management changed following WES findings in 23 of 59 diagnosed patients (39%) or 13% of all patients. A possible diagnosis was identified in 21 additional patients (12%) for whom supporting evidence is pending. Time from epilepsy onset to a genetic diagnosis was faster when WES was performed early in the diagnostic process (mean: 145 days Prospective vs. 2,882 days Retrospective). Costs of prior negative tests averaged $8,344 per patient in the Retrospective group, suggesting savings of $5,110 per patient using WES. These results highlight the diagnostic yield, clinical utility and potential cost-effectiveness of using targeted WES early in the diagnostic workup of patients with unexplained early-onset epilepsy. The costs and clinical benefits are likely to continue to improve. Advances in precision medicine and further studies regarding impact on long-term clinical outcome will be important.
Epilepsy is a common pediatric neurological disorder associated with an increased risk of developmental delay, autism and psychiatric illness; and for which treatment is ineffective in 30–40% of patients. High-throughput sequencing (HTS) has become a widespread diagnostic tool in various genetic conditions, including epilepsy (1), vastly improving molecular diagnosis. Its clinical utility has been proven in epileptic encephalopathies and in mixed epilepsy cohorts (2–11); and in neurodevelopmental disorders (12–14) in which epilepsy is a comorbid feature. The diagnostic yield ranges between 10 and 60%, settling around 25% in large studies with broader inclusion criteria, and with comparable diagnostic rates between gene panels and WES (1). A genetic diagnosis of epilepsy may enable more accurate counseling regarding prognosis and recurrence risk, avoids unnecessary medical investigations and may change care. It also allows families to connect with the same genetic condition and/or join support groups. Recent studies have demonstrated the potential cost savings of WES in the diagnostic work-up of children with suspected monogenic disorders (15–22). However, in Canada, access to such technology in clinical care is variable. In this British Columbia study, we assess the effectiveness of using WES by comparing diagnostic yield, time to diagnosis, and cost to current clinical practices. The potential treatment impact of a genetic diagnosis is also described.
One-hundred and eighty patients with epilepsy (23) were enrolled between December 2014 and September 2018. All had seizure onset at ≤5 years of undefined cause after clinical evaluation, EEG, brain MRI and chromosome microarray investigations. Seizure types and electroclinical syndromes were classified according to the International League Against Epilepsy (ILAE) (24). Patients with self-limiting benign electroclinical syndromes, such as Childhood Absence Epilepsy (onset >4 years), were excluded as they most likely have multifactorial inheritance. Patients were classified as Retrospective (n = 127), defined as an epilepsy diagnosis >6 months before study enrollment with a standard clinical approach to genetic testing (variable genetic tests which include gene-by-gene approach using Sanger sequencing, small epilepsy gene panels using high-throughput sequencing, and/or mitochondrial DNA sequencing; or Prospective (n = 53), which included an epilepsy diagnosis <6 months before study enrollment date and having limited to no genetic testing. Varying degrees of screening tests for inborn errors of metabolism; such as plasma amino acids, lactate and ammonia, were also performed in both groups. Clinical data was recorded using a secure Research Electronic Data Capture (REDCap) (25) information system hosted at the Child and Family Research Institute.
This study was approved by the BC Children's Hospital and University of British Columbia Ethics Board (protocol number H14-01531). Informed consent and/or assent were obtained before study inclusion in accordance with the Declaration of Helsinki.
Genomic DNA was extracted from peripheral blood lymphocytes following standard protocols. Exonic regions were captured using the Ion AmpliSeq Exome Kit (57.7 Mb) and WES was performed on an Ion Proton™ according to manufacturers' recommendations (Life Technologies Inc., CA) within 2 weeks of receiving samples. Reads were aligned against the human reference genome hg19. Variant annotation was performed with ANNOVAR (26) integrating data from PHAST PhyloP (27), SIFT (28), Polyphen2 (29), LRT (30), and MutationTaster (31) algorithms, Combined Annotation Dependent Depletion (CADD) scores (32), dbSNP (www.ncbi.nlm.nih.gov/SNP/), the Genome Aggregation Database (gnomAD; gnomad.broadinstitute.org) and ClinVar (33) (www.ncbi.nlm.nih.gov/clinvar). Additionally, variants were compared to an in-house database containing more than 900 exomes to exclude platform artifacts and common variants not present in public databases.
Analysis was restricted to 620 genes previously implicated in epilepsy (Supplementary Table 1), using a gene-reporting pipeline developed in-house. The gene list was compiled through the combination of a comprehensive literature search (Pubmed, OMIM) and clinically available epilepsy panels. Annotation was limited to exonic non-synonymous and splicing (±5 bp) substitutions. Homozygous variants, potential compound heterozygous variants (defined as genes with >1 variant locus per individual) with a minor allele frequency (MAF) <1% and heterozygous variants with MAF <0.01% were reported. All samples were required to meet minimum quality standards, with a WES average coverage >80X.
Sanger sequencing, performed as previously described (34), was used on a case-specific basis in a few individuals with very specific clinical phenotypes to complete regions of poor coverage in genes related to the patient's phenotype when no candidate variants were identified, or when a heterozygous and potentially pathogenic variant was identified in gene previously implicated in autosomal recessive disease. No additional variants were identified though post-WES Sanger sequencing.
Cases were reviewed at a bi-weekly meeting by a multi-disciplinary genomic team. Variant prioritization was performed based on: (1) frequency in public databases; (2) predicted protein impact; (3) disease inheritance, and; (4) correlation of patient phenotype and candidate gene literature. Up to 3 putative causative variants were validated by Sanger sequencing in patient and parental samples. Clinical Sanger sequencing confirmation and interpretation in accordance with ACMG guidelines (35) allowed disclosure to families and management adjustments when indicated. Two time intervals were measured for the first 50 patients: (1) from a clinical diagnosis of epilepsy to Sanger validation of a putative pathogenic variant; and (2) from enrollment with genetic counseling to Sanger validation of the same.
Exome data was periodically reanalyzed in patients, if no genetic diagnosis was identified in the initial analysis. Moreover, trio WES was performed in 27 patients (22 Retrospective and 5 Prospective) with no diagnosis.
Pre- and post-test genetic counseling was performed for each patient/family. As only a limited set of 620 genes related to seizure disorders were annotated, and only in affected probands, reporting related to incidental (secondary) findings was uncommon (36). Genetic disorders with specific therapeutic implications (47 genes) were defined as conditions in which current literature supports a preferred antiepileptic medication and/or approach (37–39).
For the first 50 patients, resource use data were retrospectively acquired from electronic health records and medical charts. Cost estimates in Canadian dollars were based on micro-cost information from the British Columbia Provincial Medical Service Plan Index (2015), Canadian Interprovincial Reciprocal Billing Rates (2014/2015), Children's and Women's Health Center of British Columbia Internal Fee Schedule (2015) and the internal accounting system. Diagnostic costs included: biochemical tests, imaging tests, genetic tests, neurophysiological tests, and biopsies (a complete list of tests is provided in Appendix 1). Academic and/or hospital pricing is used throughout. Inpatient hospitalization costs, outpatient visits such as clinic visits, and indirect costs such as parental time off work for medical visits related to their child's epilepsy were not included. All categorical and quantitative variables were analyzed using STATA (Release 13, College Station, TX).
Targeted WES was performed on 180 subjects and clinical features are summarized (Supplementary Table 2-first 50 patients are indicated in bold). Detailed clinical information has previously been reported for subjects 002, 013, 044, 073, and 144 (40–43). The average age of epilepsy onset was 18 months (range 0.03–60 months), 16 months for Prospective cases (n = 53) and 18 months for Retrospective cases (n = 127) (Tables 1, 2). Of the 620 genes, 87% had at least 80% of their consensus-coding region sequenced with >20X coverage (Supplementary Table 1).
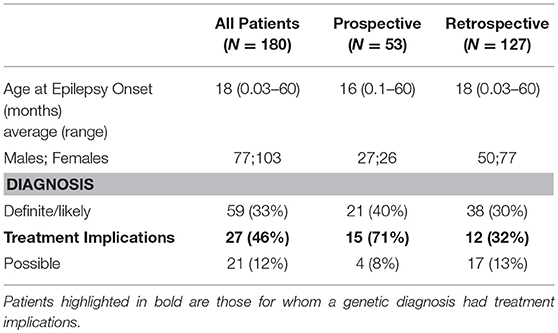
Table 1. Results: Demographics and diagnostic yield.
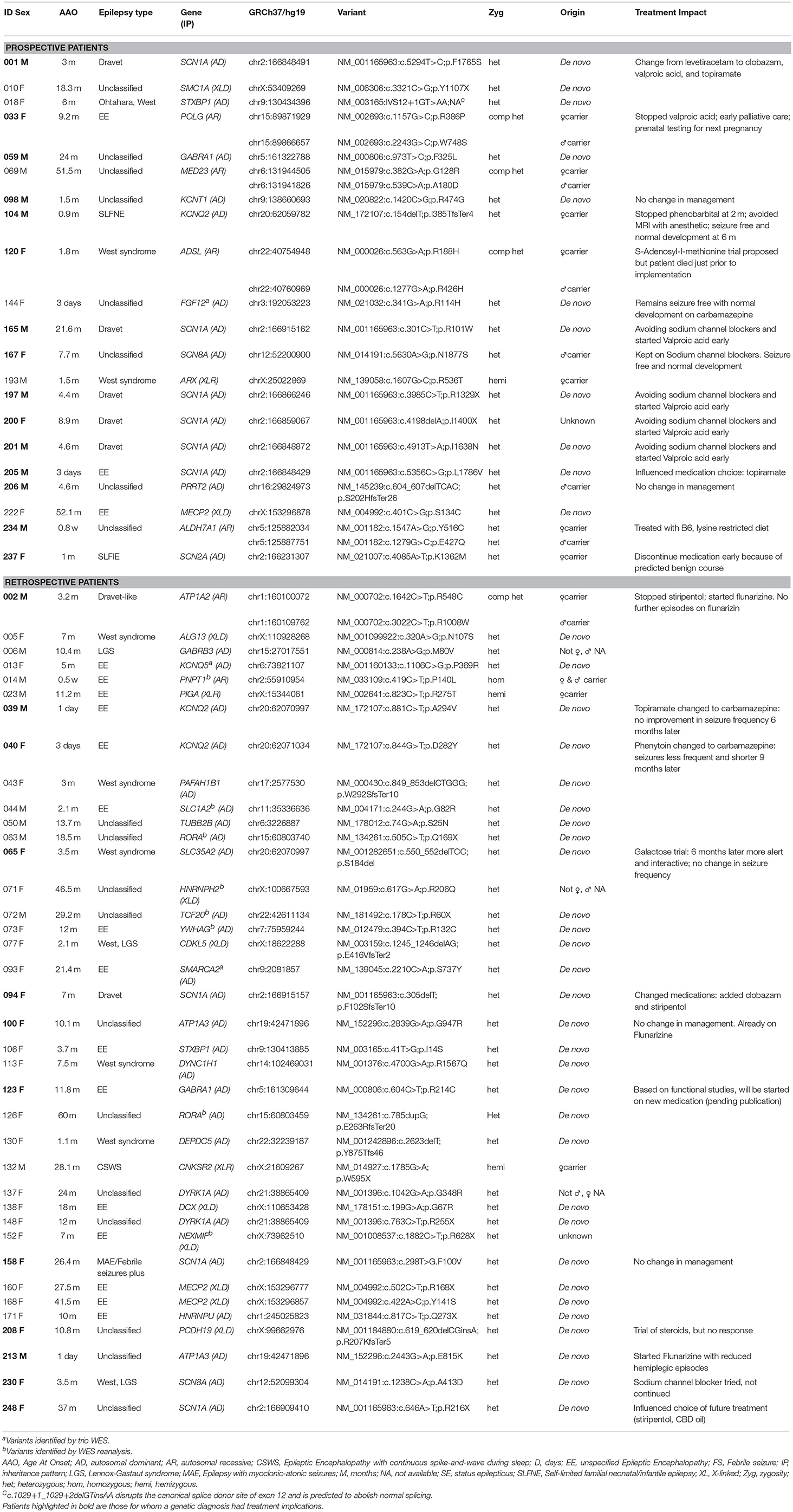
Table 2. Patients with definite/likely diagnosis and treatment impact.
A molecular diagnosis was established in 59/180 patients (33%) (Table 1). Pathogenic/likely pathogenic variants were identified in 41 genes. The majority of the diagnosed patients (41/59) had an autosomal-dominant disorder; of these 33 had a de-novo variant and remaining inherited from an affected parent and/or inheritance status unknown for one or more parent. Five patients had an autosomal-recessive disorder and the remaining 13 patients had an X-linked disorder (10 patients with X-linked dominant variants and 3 male patients with maternally inherited X-linked recessive disorders). In addition, a variant of uncertain significance (VUS) possibly explaining the clinical symptoms of the index patient was identified in 21 cases (12%) (Supplementary Table 3).
The diagnostic yield was higher in the Prospective (40%) than Retrospective group (30%). Patients in whom a diagnosis was made had earlier onset epilepsy (mean 13.2 vs. 21.9 months), and an epileptic encephalopathy was more common (Table 3). Of 82 patients with epileptic encephalopathy a definite or likely pathogenic variant was identified in 36 (44%) (Supplementary Table 4).
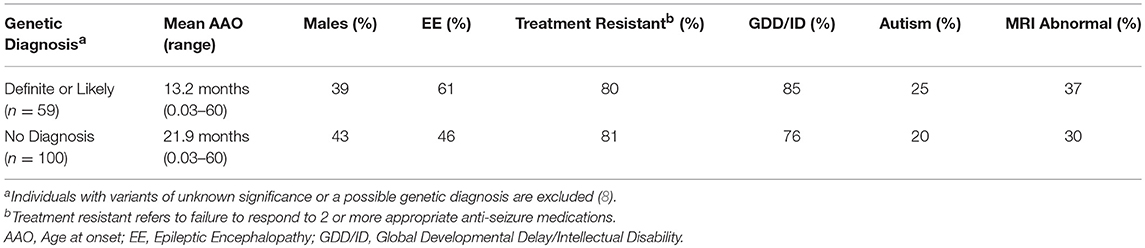
Table 3. Clinical features in patients with and without a genetic diagnosis.
A genetic disorder with specific therapeutic implications was diagnosed in 27 patients and management was influenced and/or altered in 23 (12 Prospective and 11 Retrospective). Clinical information, treatment changes, and impact are summarized (Table 2).
The mean time to genetic diagnosis from study enrolment with genetic counseling to research validation of the variant was 38 days (20–70) for the Prospective group, 54 days (22–105) for the Retrospective group, and 47 days (20–105) overall. The mean time from epilepsy diagnosis to research validation of genetic diagnosis was 145 days (42–242) for the Prospective group and 2,882 days (429–7,686) or ~8 years for the Retrospective group.
Point estimates and 95% confidence intervals based on bootstrapped standard errors (1,000 times with replacement) for each category of diagnostic test by cohorts were calculated (Table 4). All cost estimates use rates effective for the 2014–2015 fiscal year. The mean total cost related to the diagnosis of epilepsy was $4,524 (range $1,223–$7,852) for the Prospective cohort and $8,344 (range $3,319–$17,579) for the Retrospective cohort. Diagnostic imaging and electrophysiological tests comprise >60% of total epilepsy-related diagnostic costs. The mean for diagnostic imaging testing constituted $1,391 and $3,276, for Prospective and Retrospective cohorts, respectively. The mean for electrophysiological testing constituted $1,353 and $2,731, for Prospective and Retrospective cohorts, respectively. Our alternative scenario for diagnostic testing is MRI, EEG, chromosome microarray (CMA) and WES testing with Sanger sequencing validation, which amounts to $3,234 per patient (Supplementary Table 5). The difference in mean total cost related to the diagnosis of epilepsy for Prospective ($4,524) and Retrospective ($8,344) groups, exceeds the cost of our diagnostic alternative ($3,234). The potential average savings of targeted WES in the diagnostic workup constitute $1,290 per Prospective patient and $5,110 per Retrospective patient.
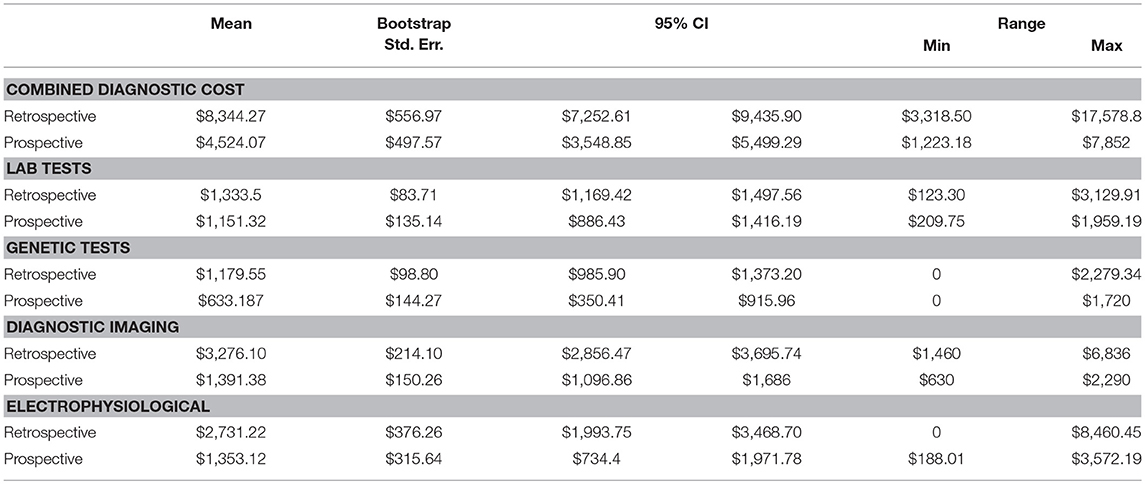
Table 4. Average diagnostic investigation cost per patient.
High-throughput sequencing has been proven to be a great diagnostic tool in clinical practice in a variety of genetic conditions, and if used early in the diagnostic workup can lead to a reduction of costs associated with obtaining a molecular diagnosis (22). Gene panel sequencing is often favored over WES based on diagnostic yield, higher coverage and cost-savings (1). However, direct comparisons of diagnostic yield and/or costs of gene panel testing are limited. A comparative coverage analysis restricted to disease-causing variants identified through panels demonstrated that targeted WES detects ≥98.5% of those mutations (44), and targeted WES has been recently shown to have a higher diagnostic yield compared to gene panels (45). A major advantage of WES over panels is the ability to sequence the entire coding genome. Such comprehensive assessment can facilitate re-analysis for novel genes as they are implicated. In the course of this study several new epilepsy genes were identified by other research groups and published in the medical literature. We were able to go back to the original WES data and examine those genes in patients without a diagnosis. Re-analysis of WES data identified new diagnoses in eight patients (014, 044, 063, 071, 072, 073, 126, 152) and three additional diagnosis (013, 093, 144) were identified by trio WES, overall increasing the diagnostic rate from 27 to 33%. Given its static nature, panel sequencing is unable to include such contemporary targets. Newly discovered genes cannot be added to the test without re-design and validation, ultimately reducing the cost-effectiveness of gene panel testing.
The clinical utility of targeted WES with Sanger validation (limited ≤3 variants/exome) is supported by the identification of a definite or likely diagnosis in 59/180 (33%) patients and a possible diagnosis in an additional 21/180 (12%) (Table 1). A higher yield was found in the Prospective group with new-onset epilepsy and supports earlier testing, though the number of patients is small. However, this may also reflect increased severity of the disease, survival and potentially some bias in case referral. The Retrospective group had already undergone extensive clinical testing that was non-diagnostic. Nevertheless, our ability to still identify a genetic diagnosis supports the technology's superior resolution, while related data on phenotypes, management and outcomes may yet inform clinical practice.
The diagnostic yield in our study is comparable to previous findings (2–11). Most variants were de-novo and the genetic causes identified were heterogeneous. However, multiple variants were identified in several genes with the most common being SCN1A, followed by KCNQ2 and MECP2 (Table 2). In a comparable cohort, positive results were identified by WES in 112/293 (38.2%) epilepsy patients (3). We concur that the diagnostic yield is likely affected by the characteristics of the group studied, sample size, platform used (gene panel or WES) and the timing of the study, given ongoing gene discoveries in epilepsy. In our study, patients with a genetic diagnosis were younger and more likely to have an epileptic encephalopathy when compared to patients in which no genetic cause was found. Similar to a prior study (3), patients with an epileptic encephalopathies had a high rate of positive findings (44%).
Our results support the feasibility of targeted WES to rapidly provide clinically-confirmed genetic diagnoses in early-onset epilepsy. Time to Sanger sequencing validation from enrollment averaged 7 weeks which is similar to the 6–8 week turn-around-time quoted by most commercial testing labs. However, this estimate did not include the additional time required to obtain provincial government approval, on a case-by-case basis, to fund WES.
A timely genetic diagnosis is important when considering the potential for treatment impact and optimization of patient outcomes. Twenty-seven of the fifty nine patients with a genetic diagnosis (46%) had a disorder with specific treatment implications; for 23 patients an immediate change in medical management was made (Table 2). The number of genetic disorders identified to have specific treatments implications is likely to grow with ongoing advances in precision medicine.
In British Columbia, the average savings are estimated to be between $1,290 and $5,110 per patient, depending on whether they are new Prospective referrals or Retrospective. Of note, price estimates reflect academic and/or hospital costs rather than commercial costs which are 1–5X higher. The Canadian sequencing costs cited are comparable to previous reports but will decrease as even higher throughput sequencing technologies become accessible (15–18). Current healthcare cost estimates are also conservative as patients without a genetic diagnosis will undoubtedly require additional clinic visits and inpatient hospital stays, including epilepsy monitoring unit admissions related to finding the cause of their condition. Of note, a targeted WES approach did not lead to a substantial increase in referrals for incidental findings. Overall, our findings show targeted WES may provide an effective end to an otherwise invasive, time consuming and costly diagnostic odyssey, with societal and economic benefits. Our results also support WES implementation beyond early-onset epileptic encephalopathies as we have examined a larger and more diverse group of children (18).
Our study has several limitations including small sample size although our diagnostic yield is comparable to previous studies. Incomplete coverage of the 620 genes analyzed was partially addressed as outlined in the methods. Proband-parent trio-based WES analyses were not used primarily for financial reasons. Analysis was restricted to 620 epilepsy genes, rather than the entire exome, to identify a genetic diagnosis as quickly as possible and to minimize secondary findings. Assessing relevance of secondary findings and proving pathogenicity of variants in novel candidate epilepsy genes is costly; thus, this approach was taken to maximize patient care and minimize cost. WES data from patients with initial negative results continues to be periodically reviewed for variants in newly described epilepsy genes. In subsequent WES trio analysis, a subset of families has helped identify novel genetic etiologies (41). All rare variants were considered however the Ion Proton™ sequencing platform tends to make mis-leading variant calling errors with small in/dels and homo-polymer stretches, with an excess of between 58 and 76% false positive calls (46). We have also identified instances of negative calls. Illumina sequencing platforms have better performance but assessing exon dosage from exome data is challenging at this sequencing depth. Panel sequencing, designed for more uniform coverage and greater depth, would be advantageous if the candidate gene was included or whole genome sequencing given the ability to detect all variants albeit for greater cost at lower coverage. Limited demographic information was provided for on the majority of patients and may be helpful in assessing variant frequencies in specific ethnic groups. Unfortunately, the ability to infer ancestry information from exome data is rather limited, in contrast to high-density SNP arrays, and not entirely reliable.
Although a significant number of genetic diagnoses had potential treatment implications, the long-term impact on clinical outcome following genetically-informed therapeutic interventions is unknown. Early diagnosis and early intervention are important, but advances in precision medicine are also required.
The methods employed for cost analysis cannot replace a prospective randomized controlled trial (RCT) and may not have accurately assessed or included all healthcare costs related to an epilepsy diagnosis. However, an RCT assessing the effect of WES testing on healthcare costs is not yet a practical consideration. Our estimates are not a perfect or a complete description of the current diagnostic work-up, as test records are scattered across different electronic health records systems and paper charts. Data collation within an accessible unified health electronic record would help identify where additional savings are possible. In this study, indirect costs, and the psychosocial impact on the child and family were not measured.
Targeted WES with limited Sanger sequencing validation is a rapid and minimally invasive test with potential to save costs within the Canadian healthcare system. An early genetic diagnosis may improve a patient's clinical outcome and quality of life. Further research on larger cohorts is warranted to inform diagnosis, clinical outcome and precision medicine. Acknowledging the limitations of our study, targeted WES with Sanger sequencing validation substantially improves current practice and is recommended as the dominant diagnostic strategy in early onset epilepsy. Minimally, as high-throughput sequencing costs continue to fall, trio-based whole exome sequencing reporting (and potentially re-reporting if negative), averting the need for Sanger validation for de novo variants, should be implemented as a first-line test strategy in British Columbia.
This study was carried out in accordance with the recommendations of BC Children's Hospital and University of British Columbia Ethics Board with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the BC Children's Hospital and University of British Columbia Ethics Board (protocol number H14-01531).
MD contributed to study conception and design, data analysis and interpretation, review of patients clinically, obtaining funding, drafting the manuscript. IG contributed to genetic data acquisition, analysis and interpretation, revised the manuscript. CD contributed to data acquisition, analysis and interpretation, and revised the manuscript. MM contributed to genetic data acquisition and to data analysis. CyB contributed to clinical assessments, data acquisition and interpretation, revised the manuscript. DE performed bioinformatics analysis. SB designed REDCap database, contributed to data acquisition, analysis and interpretation, revised the manuscript. ET contributed to data acquisition and organization. LH, AD, AM, KS, EL-R, and BB contributed to clinical assessments and data acquisition/interpretation. GH and RS performed biochemical clinical assessments, contributed to data interpretation. CvK contributed to study design and variant interpretation. ES assisted preparation of execution of study. PE performed cytogenetic assessment. SA performed genetic counseling, contributed to data acquisition and literature review. MV contributed to genetic counseling, review of results and literature, and revising manuscript. TN contributed to data analysis and interpretation, performed clinical variant interpretation, revised the manuscript, negotiated funding for variant clinical validation. CoB performed economics data acquisition, analysis and interpretation; drafted economics section of manuscript. MC performed electroclinical phenotyping, revised the manuscript, obtained funding. MF contributed to study conception and design, data analysis and interpretation, obtained funding, and revised the manuscript.
MD has received research support from the Rare Disease Foundation and the Alva Foundation. TN has received research support from the BCCH Foundation and Genome BC. MC has received research grants and/or speakers honoraria from UCB, Novartis, Biocodex, Eisai, and Sage Therapeutics. All honoraria are donated to the Epilepsy Research and Development Fund. She has also received research grants from CIHR (Canadian Institute for Health Research) and The Alva Foundation. She is Co-Chair of the Canadian Pediatric Epilepsy Network. MF was a founding partner Neurocode Labs Inc., that currently provides commercial exome sequencing, but has since relinquished any ownership stake in that company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Djavad Mowafaghian Foundation generously enabled this molecular research to take place at the Djavad Mowafaghian Centre for Brain Health. The work was also supported by Canada Excellence Research Chair and Leading Edge Endowment funds, the Rare Disease Foundation, Grocholski Foundation and the Alva Foundation. We thank the children and families that took part in this study. We gratefully acknowledge Dr. Suzanne Vercauteren MD, PhD (Director), Dr. William Gibson MD, PhD (Chair and Biospecimen Advisory Committee member), Tamsin Tarling (Biobank Administrative Manager), and the technical expertise of Katelin Townsend of the BC Children's Hospital BioBank, which is supported by Mining for Miracles through the BC Children's Hospital Foundation. We also thank the BC Children's Hospital Department of Pathology and Laboratory Medicine and the BC Children's Hospital EEG (Electroencephalogram) Department. We are also grateful to Dr. Vesna Popovska MD, Katie Pezarro, and Giselle Hunt BSc, from the Neurology Research Team at BC Children's Hospital; and Dr. Hilary Vallance MD and Dr. Graham Sinclair PhD, from the Department of Pathology, Biochemical Genetics Laboratory at BC Children's Hospital, for their assistance on this project.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00434/full#supplementary-material
1. Dunn P, Albury CL, Maksemous N, Benton MC, Sutherland HG, Smith RA, et al. Next generation sequencing methods for diagnosis of epilepsy syndromes. Front Genet. (2018) 9:20. doi: 10.3389/fgene.2018.00020
2. Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. (2012) 53:1387–98. doi: 10.1111/j.1528-1167.2012.03516.x
3. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. (2016) 18:898–905. doi: 10.1038/gim.2015.186
4. Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SDJ, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. (2016) 57:e12–17. doi: 10.1111/epi.13250
5. Della Mina E, Ciccone R, Brustia F, Bayindir B, Limongelli I, Vetro A, et al. Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur J Hum Genet. (2015) 23:354–62. doi: 10.1038/ejhg.2014.92
6. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. (2015) 56:707–16. doi: 10.1111/epi.12954
7. de Kovel CGF, Brilstra EH, van Kempen MJA, van‘t Slot R, Nijman IJ, Afawi Z, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med. (2016) 4:568–80. doi: 10.1002/mgg3.235
8. Perucca P, Scheffer IE, Harvey AS, James PA, Lunke S, Thorne N, et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. (2017) 131:1–8. doi: 10.1016/j.eplepsyres.2017.02.001
9. Peng J, Pang N, Wang Y, Wang X-L, Chen J, Xiong J, et al. Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci Ther. (2018) 25:14–20. doi: 10.1111/cns.12869
10. Yang L, Kong Y, Dong X, Hu L, Lin Y, Chen X, et al. Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet Med. (2018) 21:564–71. doi: 10.1038/s41436-018-0091-8
11. Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. (2017) 38:216–25. doi: 10.1002/humu.23149
12. Evers C, Staufner C, Granzow M, Paramasivam N, Hinderhofer K, Kaufmann L, et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol Genet Metab. (2017) 121:297–307. doi: 10.1016/J.YMGME.2017.06.014
13. Chérot E, Keren B, Dubourg C, Carré W, Fradin M, Lavillaureix A, et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin Genet. (2018) 93:567–76. doi: 10.1111/cge.13102
14. Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. (2016) 89:700–7. doi: 10.1111/cge.12732
15. Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. (2014) 6:265ra168. doi: 10.1126/scitranslmed.3010076
16. Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center's experience. Front Pediatr. (2015) 3:67. doi: 10.3389/fped.2015.00067
17. Stark Z, Schofield D, Alam K, Wilson W, Mupfeki N, Macciocca I, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. (2017) 19:867–74. doi: 10.1038/gim.2016.221
18. Joshi C, Kolbe DL, Mansilla MA, Mason SO, Smith RJH, Campbell CA. Reducing the cost of the diagnostic odyssey in early onset epileptic encephalopathies. Biomed Res Int. (2016) 2016:1–8. doi: 10.1155/2016/6421039
19. Payne K, Gavan SP, Wright SJ, Thompson AJ. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat Rev Genet. (2018) 19:235–46. doi: 10.1038/nrg.2017.108
20. Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KLI, Geenen JW, Hövels AM, et al. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur J Hum Genet. (2018) 26:1566–71. doi: 10.1038/s41431-018-0203-6
21. Dragojlovic N, Elliott AM, Adam S, van Karnebeek C, Lehman A, Mwenifumbo JC, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med. (2018) 20:1013–21. doi: 10.1038/gim.2017.226
22. Howell KB, Eggers S, Dalziel K, Riseley J, Mandelstam S, Myers CT, et al. A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia. (2018) 59:1177–87. doi: 10.1111/epi.14087
23. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
24. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/epi.13670
25. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)-A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. (2009) 42:377–81. doi: 10.1016/j.jbi.2008.08.010
26. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
27. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. (2010) 20:110–21. doi: 10.1101/gr.097857.109
28. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
29. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
30. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. (2009) 19:1553–61. doi: 10.1101/gr.092619.109
31. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
32. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–15. doi: 10.1038/ng.2892
33. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. (2016) 44:D862–8. doi: 10.1093/nar/gkv1222
34. Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol. (2015) 77:458–68. doi: 10.1002/ana.24346
35. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–23. doi: 10.1038/gim.2015.30
36. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. (2017) 19:249–55. doi: 10.1038/gim.2016.190
37. Quilichini PP, Chiron C, Ben-Ari Y, Gozlan H. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABAA-receptor channels. Epilepsia. (2006) 47:704–16. doi: 10.1111/j.1528-1167.2006.00497.x
38. Stewart JD, Horvath R, Baruffini E, Ferrero I, Bulst S, Watkins PB, et al. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. (2010) 52:1791–6. doi: 10.1002/hep.23891
39. Klepper J, Scheffer H, Leiendecker B, Gertsen E, Binder S, Leferink M, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. (2005) 36:302–8. doi: 10.1055/s-2005-872843
40. Wilbur C, Buerki SE, Guella I, Toyota EB, Evans DM, McKenzie MB, et al. An infant with epilepsy and recurrent hemiplegia due to compound heterozygous variants in ATP1A2. Pediatr Neurol. (2017) 75:87–90. doi: 10.1016/j.pediatrneurol.2017.06.003
41. Lehman A, Thouta S, Mancini GMS, Naidu S, van Slegtenhorst M, McWalter K, et al. Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am J Hum Genet. (2017) 101:65–74. doi: 10.1016/j.ajhg.2017.05.016
42. Guella I, McKenzie MB, Evans DM, Buerki SE, Toyota EB, Van Allen MI, et al. De novo mutations in YWHAG cause early-onset epilepsy. Am J Hum Genet. (2017) 101:300–10. doi: 10.1016/j.ajhg.2017.07.004
43. Guella I, Huh L, McKenzie MB, Toyota EB, Bebin EM, Thompson ML, et al. De novo FGF12 mutation in 2 patients with neonatal-onset epilepsy. Neurol Genet. (2016) 2:e120. doi: 10.1212/NXG.0000000000000120
44. LaDuca H, Farwell KD, Vuong H, Lu H-M, Mu W, Shahmirzadi L, et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS ONE. (2017) 12:e0170843. doi: 10.1371/journal.pone.0170843
45. Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Gaff C, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. (2018) 26:644–51. doi: 10.1038/s41431-018-0099-1
46. Seo H, Park Y, Min BJ, Seo ME, Kim JH. Evaluation of exome variants using the Ion Proton Platform to sequence error-prone regions. PloS ONE. (2017) 12:e0181304. doi: 10.1371/journal.pone.0181304
List of Relevant Tests:
Laboratory Services/Tests:
• Bloodspot acylcarnitines
• Plasma amino acids
• Plasma total homocysteine
• Serum ceruloplasmin
• Serum copper
• Ammonia Serum
• Lactate Serum/plasma
• Lactate whole blood
• Acylcarnitine Serum
• Homocysteine total from Plasma
• Plasma amino-acids
• Serum copper
• Serum coeruloplasmin
• Plasma very long chain fatty acids
• Plasma cholesterol
• Urine creatine metabolites
• Urine glycosaminoglycans
• Urine oligosaccharides
• Urine organic acids
• Urine purines and pyrimidines
• Amino acids-urine
• Urine creatine metabolites
• Urine mucopolysaccharides
• CSF Protein
• CSF Protein
• CSF Glucose
• CSF Cell
• CSF Lactate
• CSF Amino-Acids
• CSF Neurotransmitters
• Plasma Vit B12
Genetic Tests:
• Chromosome microarray
• Fluorescence in situ Hybridization
• Single Gene testing
• Gene Panel/HTS testing
• Mitochondrial DNA analysis
Diagnostic Imaging Tests:
• MRI
• CT
• PET
• PET/CT
• Ultrasound Electrophysiological Tests:
• EEG
• EMG
Biopsies:
• Skin biopsy
• Muscle biopsy
Keywords: targeted WES, early-onset epilepsy, diagnostic yield, cost estimation, Canada
Citation: Demos M, Guella I, DeGuzman C, McKenzie MB, Buerki SE, Evans DM, Toyota EB, Boelman C, Huh LL, Datta A, Michoulas A, Selby K, Bjornson BH, Horvath G, Lopez-Rangel E, van Karnebeek CDM, Salvarinova R, Slade E, Eydoux P, Adam S, Van Allen MI, Nelson TN, Bolbocean C, Connolly MB and Farrer MJ (2019) Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front. Neurol. 10:434. doi: 10.3389/fneur.2019.00434
Received: 17 November 2018; Accepted: 09 April 2019;
Published: 21 May 2019.
Edited by:
Giovanni Stevanin, INSERM U1127 Institut du Cerveau et de la Moelle épinière, FranceReviewed by:
Gaetan Lesca, Université Claude Bernard Lyon 1, FranceCopyright © 2019 Demos, Guella, DeGuzman, McKenzie, Buerki, Evans, Toyota, Boelman, Huh, Datta, Michoulas, Selby, Bjornson, Horvath, Lopez-Rangel, van Karnebeek, Salvarinova, Slade, Eydoux, Adam, Van Allen, Nelson, Bolbocean, Connolly and Farrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle Demos, bWRlbW9zQGN3LmJjLmNh
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.