
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CLINICAL STUDY PROTOCOL article
Front. Neurol. , 27 March 2019
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00293
 Paul Lingor1,2*
Paul Lingor1,2* Markus Weber3
Markus Weber3 William Camu4Tim Friede5Reinhard Hilgers5Andreas Leha5Christoph Neuwirth3
William Camu4Tim Friede5Reinhard Hilgers5Andreas Leha5Christoph Neuwirth3 René Günther6,7Michael Benatar8Magdalena Kuzma-Kozakiewicz9Helen Bidner10Christiane Blankenstein10Roberto Frontini11
René Günther6,7Michael Benatar8Magdalena Kuzma-Kozakiewicz9Helen Bidner10Christiane Blankenstein10Roberto Frontini11 Albert Ludolph12
Albert Ludolph12 Jan C. Koch2 and the ROCK-ALS Investigators
Jan C. Koch2 and the ROCK-ALS InvestigatorsObjectives: Disease-modifying therapies for amyotrophic lateral sclerosis (ALS) are still not satisfactory. The Rho kinase (ROCK) inhibitor fasudil has demonstrated beneficial effects in cell culture and animal models of ALS. For many years, fasudil has been approved in Japan for the treatment of vasospasm in patients with subarachnoid hemorrhage with a favorable safety profile. Here we describe a clinical trial protocol to repurpose fasudil as a disease-modifying therapy for ALS patients.
Methods: ROCK-ALS is a multicenter, double-blind, randomized, placebo-controlled phase IIa trial of fasudil in ALS patients (EudraCT: 2017-003676-31, NCT: 03792490). Safety and tolerability are the primary endpoints. Efficacy is a secondary endpoint and will be assessed by the change in ALSFRS-R, ALSAQ-5, slow vital capacity (SVC), ECAS, and the motor unit number index (MUNIX), as well as survival. Efficacy measures will be assessed before (baseline) and immediately after the infusion therapy as well as on days 90 and 180. Patients will receive a daily dose of either 30 or 60 mg fasudil, or placebo in two intravenous applications for a total of 20 days. Regular assessments of safety will be performed throughout the treatment period, and in the follow-up period until day 180. Additionally, we will collect biological fluids to assess target engagement and evaluate potential biomarkers for disease progression. A total of 120 patients with probable or definite ALS (revised El Escorial criteria) and within 6–18 months of the onset of weakness shall be included in 16 centers in Germany, Switzerland and France.
Results and conclusions: The ROCK-ALS trial is a phase IIa trial to evaluate the ROCK-inhibitor fasudil in early-stage ALS-patients that started patient recruitment in 2019.
Up to now only riluzole and edaravone have been approved as disease-modifying treatments for amyotrophic lateral sclerosis (ALS), but their efficacy is limited. Thus, there is an urgent need to identify more efficient disease-modifying drugs to improve function and prolong survival for this devastating motoneuron disorder.
Rho kinase (ROCK) has recently emerged as a novel therapeutic target for neurodegenerative disorders [reviewed in (1)]. ROCK is a serine/threonine kinase with two isoforms: while ROCK1 is expressed preferentially in peripheral tissue, ROCK2 is highly expressed in the central nervous system (CNS). Axonal growth inhibitory molecules (e.g., Nogo, MAG, OMgp, ephrins, semaphorins) bind to specific extracellular receptors and signal via ROCK to trigger axonal degeneration, growth cone collapse, and impaired axonal regeneration. In non-neuronal structures, regulation of the actin cytoskeleton plasticity by ROCK also mediates vasoconstriction and vascular remodeling (2). Interestingly, ROCK also regulates cell survival via Akt and mTOR (3–5).
Levels of ROCK increase with age and tissue of ALS patients show increased levels of ROCK2 as well as its downstream targets LIMK1 and cofilin (6, 7). Moreover, in the SOD1 (G93A) mouse model of ALS, increased ROCK activity results in higher levels of phosphorylated adducin as well as activation of phosphatase and tensin homolog (PTEN) and decreased Akt activity, suggesting a deactivation of these cell survival pathways (5). These findings validate the known effects of ROCK activation on pathways regulating the plasticity of the actin cytoskeleton and neuronal survival. Adducin is phosphorylated by ROCK and promotes the interaction between actin and spectrin resulting in actin bundling and stabilization of the cytoskeleton (8). PTEN activation by ROCK is important for its effects on neuronal survival. Since PTEN inhibits phosphatidylinositol (3,4,5) tris-phosphate, which is a central activator of Akt/PKB signaling, PTEN activation by ROCK also exerts negative effects on cell growth, proliferation and metabolism. Via this pathway active ROCK not only to inhibits Akt but also the mammalian target of rapamycin (mTOR) complex 1, which is also an important positive regulator of protein synthesis, cell growth and regeneration (9).
ROCK can be inhibited by fasudil, an isochinoline derivative that was originally developed as a vasodilatory drug and licensed in Japan in 1995 for the treatment of vasospasm following subarachnoid hemorrhage (SAH). Several thousands of patients have been treated with fasudil since. Fasudil has also been tested in numerous clinical trials for other applications, most frequently in cardiovascular disease, such as angina pectoris, Raynaud's syndrome, pulmonary hypertension and arterial hypertension (2). In the CNS, the effects of fasudil were assessed in a phase III trial in patients with acute ischemic stroke, where fasudil treatment significantly improved clinical outcome (10). Significant amounts of fasudil are taken up in the brain (11). Although only intravenous formulations of fasudil are licensed in Japan and China, oral (tablet) formulations, including extended release capsules, were used in clinical trials in humans. The longest published exposure to fasudil in humans is 8 and 12 weeks for angina pectoris and pulmonary arterial hypertension, respectively. Side effects included allergic skin reactions, a slight drop in systolic blood pressure and reversible renal impairment without major safety concerns arguing against longer dosing as required for the treatment of ALS (12, 13). Because of ample clinical experience and a well-known safety profile, fasudil represents an excellent candidate for repurposing as a disease-modifying therapy in ALS. Most importantly, in the SOD1 (G93A) mouse model of ALS, fasudil prolonged survival and improved motor function, which was independently reproduced by two groups (5, 14, 15). Fasudil also improved the regenerative response in the neuromuscular junction, which was accompanied by a modulation of microglial activity. ROCK inhibition not only influenced the morphology of microglia, but also attenuated the lipopolysaccharide-induced release of chemokines and cytokines (14, 16). Other groups showed beneficial effects in the SMN1 knockdown model of spinal muscular atrophy (17). These preclinical data, therefore, suggests ROCK as a novel drugable target with disease-modifying effects in ALS.
The primary objective of this phase IIa study (proof-of-concept study) is to evaluate the safety and tolerability of intravenous fasudil in two different doses. Participants will be treated for 20 days and followed up for a period of 6 months. Secondary objectives include the survival time and the change of revised ALS Functional Rating Scale (ALSFRS-R), ALS Assessment Questionnaire (ALSAQ-5), Edinburgh Cognitive and Behavioral ALS Screen (ECAS), Motor Unit Number Index (MUNIX) and slow vital capacity (SVC) from baseline to 4 weeks, 3 months, and 6 months after treatment initiation. A further secondary aim is to determine safety and tolerability until the end of the 4 weeks infusion period.
ROCK-ALS is a multi-center, international, randomized, double-blind, placebo-controlled, prospective, dose-finding Phase IIa trial. During its design we considered the “Guideline on clinical investigation of medicinal products for the treatment of amyotrophic lateral sclerosis” of the European Medicines Agency (EMA) where appropriate and the protocol adheres to the “Standard Protocol Items for Randomized Trials” (SPIRIT). It has been registered with the European Clinical Trials Database (Eudra-CT, https://eudract.ema.europa.eu/) under the number 2017-003676-31, with the German Clinical Trials Register (DRKS) under the number DRKS00013948, and on clinicaltrials.gov (NCT03792490). Figure 1 shows the design of the trial.
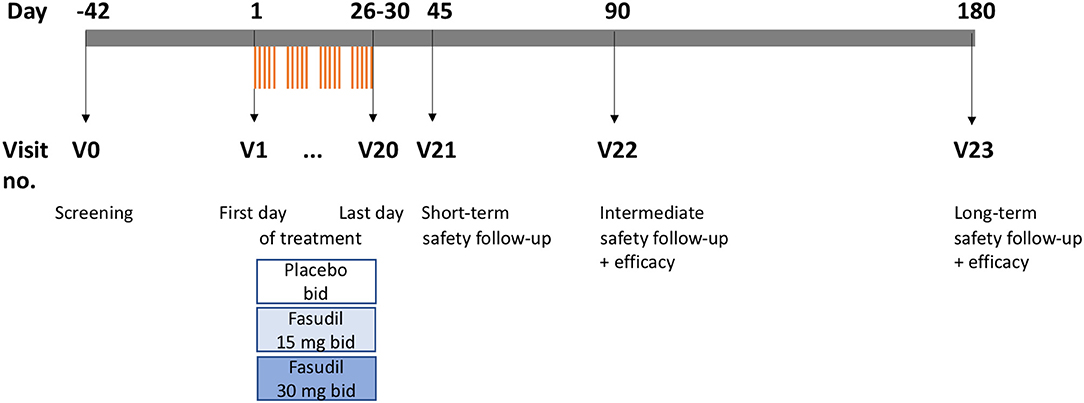
Figure 1. Scheme of trial (drawn to time scale). V, visit; bid, two times daily.
Patients with clinical probable, probable laboratory supported or definite ALS and a disease duration of more than 6 and less than 18 months (disease onset defined as date of first muscle weakness, excluding fasciculations and cramps) are eligible for participation in the ROCK-ALS trial. A co-medication with riluzole is compulsory. A previously started treatment with edaravone should be continued throughout the trial. The slow vital capacity must be at least 65% of normal. Main exclusion criteria are previous tracheostomy, continuous assisted ventilation, gastrostomy, severe arterial hypotension, and liver or kidney insufficiency. Importantly, there should be no personal and family history of intracerebral aneurysms or Moyamoya disease due to a theoretical risk of intracranial hemorrhage associated with fasudil treatment. The full list of inclusion and exclusion criteria is depicted in Table 1.

Table 1. Inclusion and exclusion criteria.
Patients will be recruited and assessed for eligibility by the ALS outpatient clinics of the highly specialized academic centers taking part in the study, where several hundred ALS patients are seen each year on average. Referring centers and established neurologists will be informed about the ROCK-ALS trial by direct contact and information letters. Patient information sheets and information leaflets for medical professionals (primary care physicians and neurologists) will be issued and distributed to national patient organizations of the participating countries which will be asked to make the information available on their websites. A website for the ROCK-ALS trial has been launched to deliver information for patients and medical professionals (http://rock-als.uni-goettingen.de).
This trial has three parallel groups: fasudil 15 mg twice daily, fasudil 30 mg twice daily, and matching placebo. Patients are randomized to treatment using an allocation ratio of 1:1:1. The randomization list will be centrally generated using a computerized system stratified by geographical region and type of onset (bulbar, spinal). Patients will be assigned to the bulbar or spinal stratum according to the location of the earliest experienced ALS symptom (defined by the first muscle weakness or in the case of bulbar onset, by the presence of dysarthria and/or dysphagia). In the case of a patient with simultaneous onset of spinal and bulbar symptoms, onset will be defined as bulbar. Cervical and respiratory onsets are stratified to the spinal onset stratum. The randomization list will be generated by the Biometry and Bioinformatics Core using a pseudo-random number generator.
This study includes double-blind (participant- and investigator blind) treatment. Study medication will be packed and blinded by the Pharmacy at University of Leipzig Medical Center, Germany, according to the randomization list. Each patient medication package will be sent together with the sealed unblinding codes to the sites. The investigator at the site will take care that each patient is provided with the study medication box of the correct randomization number. The randomization list will be kept in safe and confidential custody at Pharmacy at University of Leipzig Medical Center, Germany. For all patients, emergency codes will be available to the investigator. A code, which reveals the treatment group for a specific study patient, may be opened during the study only if the choice of treatment depends on the study subject's therapy assignment. During the study, emergency unblinding should occur only by accessing the study patient's emergency code.
Fasudil hydrochloride hydrate will be delivered as medicinal product to the Pharmacy at University of Leipzig Medical Center, Germany, and the trial medication as well as the according placebo will be produced, packaged, stored, blinded, labeled, and shipped to the trial sites in accordance with the regulations of the participating country and good manufacturing practice (GMP) (Annex 13 of the EU Guideline for GMP).
Trial medication will be supplied as fasudil hydrochloride hydrate solution 30 mg for IV application (2 × 1 ml of 15 mg/ml fasudil hydrochloride hydrate), or fasudil hydrochloride hydrate solution 15 mg for IV application (1 ml of 15 mg/ml fasudil hydrochloride hydrate and 1 ml NaCl 0.9%). The appropriate Placebo to fasudil hydrochloride solution will be supplied as 2 × 1 ml NaCl 0.9%. Placebo solution is identical in appearance to the study medication (clear, colorless fluid), but does not contain the active ingredient.
The trial drug or placebo has to be diluted in 100 ml NaCl 0.9% prior to administration and is administered two times daily IV as infusion over 45 min using a CE-certified infusion pump. The second application starts 7 ± 1 h after the start of the first application.
The trial is structured into 24 visits (V0–V23). V0 is the screening visit and may take place up to 42 days before the first treatment. Treatments are then performed twice daily from V1 to V20 on work days, excluding week-ends and holidays. Subsequently, trial participants are seen at three follow-up visits, V21, V22, and V23, at 45, 90, and 180 days after baseline, respectively. The detailed trial plan is shown in Table 2.
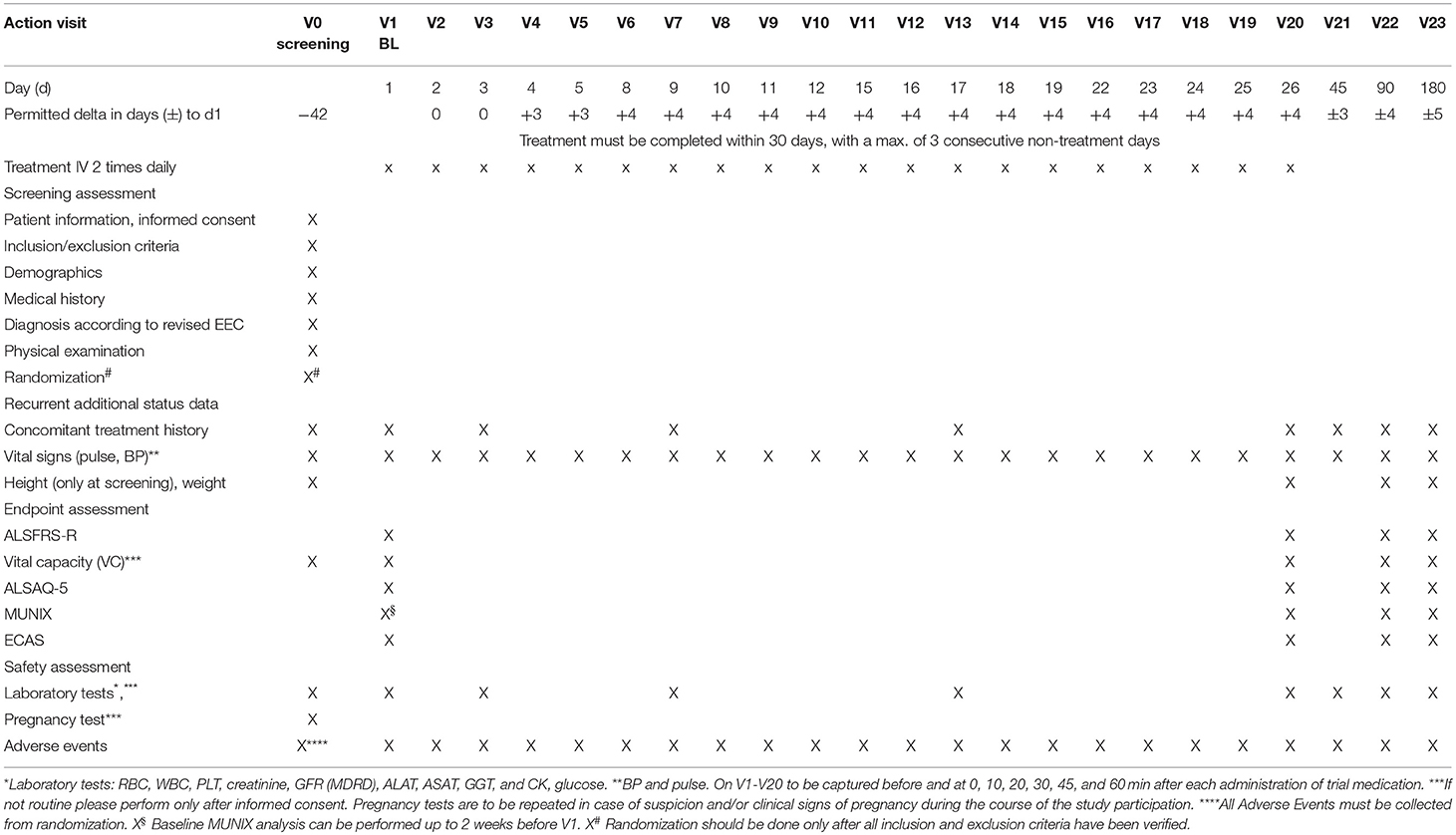
Table 2. Trial visits schedule.
Primary endpoints are safety and tolerability. The treatment with fasudil is considered safe for an individual patient if no drug-related serious adverse events (SAE) is recorded through to visit V23. The treatment with fasudil is considered tolerable if participants do not discontinue treatment due to suspected drug-related adverse events (AE). The proportions of patients for whom the treatment is tolerable/safe are derived for each treatment group.
Secondary endpoints are the survival time and the change of ALSFRS-R, ALSAQ-5, ECAS, MUNIX (18), and SVC from baseline to visits V20, V22 and V23. Secondary safety endpoint is safety until V20.
As part of the therapeutic trial we will collect biosamples which will be used for the analysis of pharmacodynamics, disease progression, genotyping and potentially for future studies. EDTA-blood, serum, plasma, urine, and saliva will be collected from all individuals at specified time points. At V20 (or exceptionally on V19 or V18), a CSF sample and a blood plasma sample will be collected to determine the peak concentration of fasudil and hydroxyfasudil as well as ROCK activity (Table 3). Individual alterations of the biosample collection and analysis plan will not be classified as violations of the general study protocol. All biomaterial samples will be stored at the Biobank of the University Medical Center of Göttingen, except for samples that will be analyzed immediately.
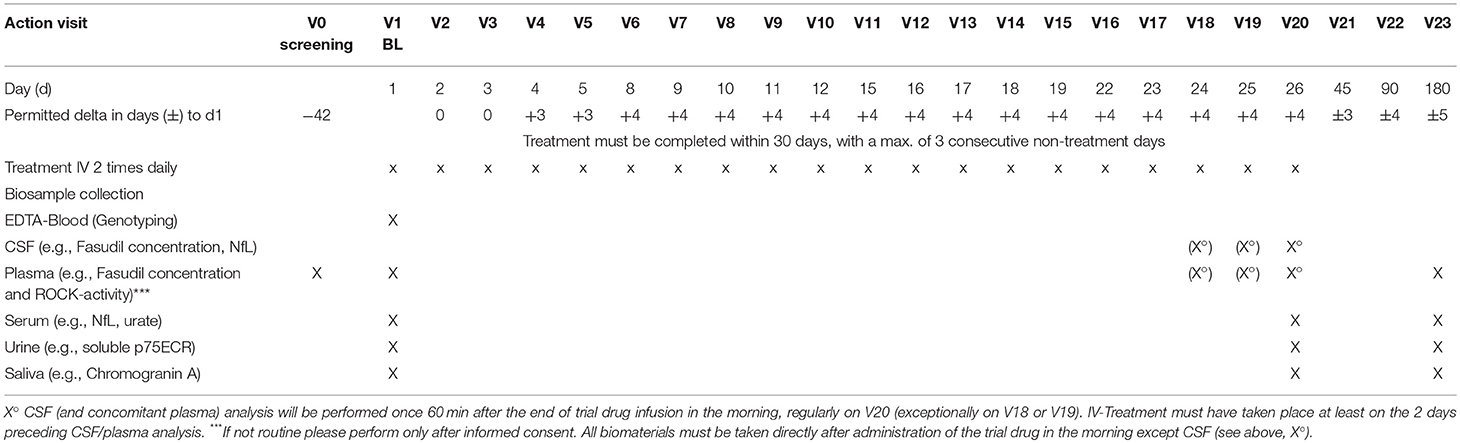
Table 3. Biosample collection plan.
During the study, the definitions of the directive 2001/20/EC are effective. All AE, irrespective of seriousness, will be collected from the day of randomization to the follow-up visit/end of trial visit at the time points specified in the trial visits schedule. The investigator should report any AE occurring in this time period that is believed to be related to study drug or protocol-specified procedure. All SAE for which a causal relationship with the administration of study medication may not be ruled out (serious adverse reactions, SAR) need to be defined and reported as suspected unexpected serious adverse reactions (SUSAR). Once an AE is detected, it should be followed until its resolution or stabilization and assessment should be made at each visit (or more frequently, if necessary) of any changes in severity, the suspected relationship to the study, the interventions required to treat it and the outcome. All SAE will be reviewed by an independent medical monitor, who will be independent from the reporting investigator, the trial sponsor and the coordinating investigator. The investigator must carry out a causality assessment for all AEs. The relationship of an AE to the study treatment regimen has to be recorded on the CRF and defined as not related, unlikely, possible related, probable related or highly probable/definite.
Expedited reporting applies if the AE is considered serious, unexpected, and drug related (SUSAR). This type of SAE must be reported by the CRO to the appropriate national health authority and ethics committees within 15 days; fatal or life-threatening events must be reported within 7 days. All person-related data will always be transmitted pseudonymized/de-identified. The CRO will immediately, within 15 days after it becomes known, report all circumstances that require a revision of the risk-benefit analysis to the relevant ethics committees and the federal authorities. This especially includes:
• Singular cases of expected SAE with an unexpected outcome.
• Increased incidence of expected SAE that are judged as being clinically relevant.
• SUSARs which occur after termination of the clinical trial (6 months) after termination or exclusion)
• Events related to study procedures or development of the study medication, which could affect a subject's safety.
The study will be monitored by an independent safety monitoring board (SMB), which will come together once before the start of the trial and then hold telephone conferences approximately every 3 months during the trial in order to review trial progress, safety data, and adherence to protocol, with the frequency of meetings depending on the rate of recruitment and safety issues.
The documentation of the study data in adherence to the Good Clinical Practice (GCP)-guidelines and the clinical trial protocol is the responsibility of the local investigator. Original data (source documents) remain in hospital medical records and information on the eCRF must be traceable and consistent with the original data. Source documents are e.g., laboratory results, ALSFRS-R measurements, vital capacity measurements, and quality of life questionnaire. Original written informed consent signed by the patient is kept by the investigator and a signed copy will be given to the patient. No information in source documents about the identity of the patients will be disclosed. All data collected in this study must be entered in an eCRF which has to be completed by the investigator or authorized trial personnel and signed by the principle investigator. This also applies for those patients who do not complete the study. If a patient withdraws from the study, the reason must be recorded in the eCRF. The investigator is responsible for ensuring the accuracy, completeness, and timeliness of all data reported to the sponsor in the eCRFs and in all required reports.
After database lock, the principal investigator will receive data on an electronic device that includes the investigational site data for archiving in the Investigator Site File (ISF). Data are processed by data management of the CRO with the support of a study database (eCRF) according to the SOPs of the CRO. The evaluation of the data takes place by programmed validity and consistency checks.
Monitoring activities are performed to ensure that the trial is conducted in accordance with the trial protocol, the principles of GCP, and local legislation. A monitoring manual describing in detail the scope of the monitoring activities will be prepared. A monitoring visit report is prepared for each visit describing the progress of the clinical trial and all identified problems.
This trial will be conducted in accordance with the current ICH-GCP-guidelines. GCP is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected and consistent with the principles that have their origin in the Declaration of Helsinki and that the clinical trial data are credible. Prior to study start the following documents will be obtained:
• Approval of ethics committees (lead and local ethics committees)
• Approval of national competent authorities: Bundesamt für Arzneimittel und Medizinprodukte (BfArM), Schweizerisches Heilmittelinstitut (SwissMedic), Agence nationale de sécurité du médicament et des produits de santé (ANSM)
• Notification to applicable regional authorities
• Informed Consent
• Insurance
• Data privacy and confidentiality.
Irrespective of study outcome, the results will be presented during scientific symposia and published on www.clinicaltrials.gov and in a peer-reviewed, international journal after written approval by the involved parties and respecting the privacy of the participants.
In total, 16 recruiting study centers participate in the ROCK-ALS trial at the time of writing. In Germany, all centers (Berlin, Dresden, Essen, Göttingen, Halle (Saale), Hannover, Jena, Munich, Münster, Ulm, Würzburg) belong to the German Network for Motor Neuron Diseases (MND-NET) and represent specialized referral centers for motoneuron disorders. The Swiss center in St. Gallen is the largest ALS clinic in Switzerland. In France, four highly dedicated ALS referral centers participate in the trial (Marseille, Montpellier, Nice, Tours).
It is expected that each center
• randomizes 8–10 patients (in France and Switzerland 6 per center), administers the treatment, performs all required clinical examinations and takes scores, collects the biospecimens, and enters all data into the eCRF
• contributes at least one principle investigator, one clinical neurologist as study physician and one study nurse dedicated to the trial
• trains sufficient personnel to perform MUNIX assessments.
In addition, an associated center in Warsaw (without explicit funding through E-Rare and thus outside of the formal trial protocol) will recruit patients with the same inclusion and exclusion criteria for a biosample collection, but without treatment.
Safety analyses will be performed at the local laboratories at each center. Drug levels (riluzole, fasudil, and hydroxyfasudil) will be determined at the Medizinische Labor Bremen (MLB). Multiple other research labs are foreseen for the evaluation of liquid biomarkers (e.g., neurofilament, soluble p75ECR, chromogranin A, and urate). In addition, we will attempt to genotype each patient by whole genome sequencing, which will be performed in collaboration with the CReATe Consortium.
The trial will be supported and monitored by the Munich Trial Center (MSZ), an academic CRO with long-standing experience in the conduct of clinical trials, particularly IITs.
A sample size of 102 patients (i.e., 34 patients per treatment group) yields sufficiently narrow confidence intervals for the difference in proportions between the placebo group and the treatment groups for both primary endpoints, the proportion of patients without drug-related SAEs and the proportion of patients without treatment intolerability. Under the assumption of no difference between the treatment groups and the placebo group the half-width of the 95% confidence interval for the difference in proportions is at most 0.24. Expected are high proportions of tolerability and safety in which case the confidence interval becomes narrower. If the proportion is for instance 0.9 the half-width reduces to 0.14, which is considered sufficiently narrow. Calculations were carried out using the software nQuery 4.0. Adjusting for dropout of 15% we aim to recruit a total number of 120 patients (i.e., 40 patients per treatment group).
The study will yield information about safety, tolerability, and efficacy. The main focus of the primary analysis is to determine whether either one or both doses are safe and tolerable. Each patient will be treated with fasudil or placebo over 45 min twice a day for 20 days. Significant drug intolerance will result in stopping the infusion and termination of the trial participation for this patient.
The proportions of patients for whom the treatment is tolerable/safe are derived for each treatment group. For both active treatment groups and for both, safety and tolerability, separately the difference in proportions to the placebo group will be calculated with its 95% confidence interval. Subsequent analyses will model tolerability and safety in logistic regressions adjusting for randomization stratification factors and important prognostic factors assessed at baseline. Treatment group differences will be reported in terms of odds ratios with 95% confidence intervals.
Primary analyses of safety and tolerability will be carried out on the ITT population. For the purpose of the tolerability analyses, subjects who discontinue during the treatment period will be considered as worst case, i.e., no drug tolerability. If a number of patients withdraw from the study following completion of the treatment period, there observations will be dealt with as independent right censoring and the time to first drug-related SAE will be considered as primary endpoint. The difference in proportions free of any drug-related SAE at V23 will be assessed using Kaplan-Meier estimates and corresponding 95% confidence intervals. Subsequent analyses will model time to first drug-related SAE in Cox proportional hazard regressions adjusting for randomization stratification factors and important prognostic factors assessed at baseline. Treatment group differences will be reported in terms of hazard ratios with 95% confidence intervals. The ITT analyses will be complemented with a per-protocol (PP) analysis. Differences between both analyses will be reported and evaluated in detail.
Efficacy outcomes including ALSFRS-R, ALSAQ-5, SVC, and MUNIX through to visit V23 will be analyzed by means of Gaussian linear model for repeated measures (so-called MMRM) with treatment group, time (visits V20, V22, V23), treatment-by-time interaction, region, and stratum of onset as factors and baseline measurements of the outcome as covariate. The error terms are assumed to follow a multivariate normal distribution with unstructured covariance. Least squares mean changes from baseline will be reported for the treatment groups with 95% confidence interval (CI) as well as the difference between the least squares treatment group means with 95% CI and p-value testing the null hypothesis of no treatment effect. The analysis will be primarily performed on the ITT population, but complemented by PP analysis.
Survival time will be used as a secondary endpoint. The Kaplan-Meier method will be applied to estimate the survival probabilities in each group. The 95% CIs will be calculated with the variance derived according to Greenwoods' formula. Subjects will be right-censored at end of their follow-up. Pairwise group comparisons against placebo will be analyzed using exact log rank tests. Cox proportional hazards regression will be carried out if there are a sufficient number of events.
Disease-modification is an urgent need for the treatment of ALS. Although a multitude of different pathomechanisms are thought to contribute to disease progression, only the glutamate antagonist riluzole and the antioxidant edaravone have been approved so far, both with limited therapeutic potential. Novel drugs should thus target different, preferentially yet unaddressed mechanisms and ideally act in an additive or even synergistic manner with both licensed substances. ROCK inhibition appears as a promising novel approach in this respect acting on multiple pathomechanisms: it has been shown to increase axonal regeneration, attenuate neuronal cell death, modulate microglial activity and beneficially affect survival and motor function in mouse models of ALS. Several ROCK inhibitors with different chemical backbones have been developed (19), but only fasudil and ripasudil have been licensed for clinical use so far. Ripasudil has only been licensed as local treatment for glaucoma, but fasudil is used as systemic drug for intravenous application and is therefore the ROCK inhibitor of choice employed in this trial.
Because of the long-standing clinical use of fasudil for the treatment of subarachnoid hemorrhage-induced vasospasms, a phase I dose-escalation trial is not required. However, since there are no published data on safety and tolerability of fasudil in ALS patients, a phase IIa trial has been designed. Because the drug is only licensed as IV formulation and the maximally approved dosage is 1.260 mg over 14 days (3 × 30 mg per day), a compromise in the treatment duration was made: the treatment duration of 20 days (until V20) is long enough to estimate safety and tolerability of fasudil IV in ALS patients and to expect a long-lasting regulation of motoneuron survival pathways and alteration of microglial activity. Long-lasting effects of ROCK inhibition have been observed after a circumscribed drug application in animal models (20). At the same time, 20 treatment days are short enough to expect reasonable patient adherence for an IV therapy and to keep trial costs in a financially feasible range.
Results of this phase IIa study will allow an estimation if a longer treatment period could be favorable and if an oral formulation (e.g., with extended release tablets) can be implemented for subsequent phase IIb and phase III studies. Insufficient tolerance or significant safety concerns after application of fasudil IV in both doses will argue against a subsequent phase IIb or III study. On the other hand, lack of significantly improved efficacy readouts will per se not argue against a follow-up study, because we can only estimate the magnitude of improvement at this stage and the study may be underpowered to detect a significant improvement.
In order to achieve a long-lasting disease-modifying effect in a chronic neurodegenerative disorder, such as ALS, a future treatment most likely will have to be administered in a continuous way, which is best achieved by an oral medication. In the case of satisfactory tolerance and safety of IV fasudil in ALS patients, we plan to propose a follow-up phase IIb study (extended dose-finding) exploring the safety, tolerability and clinical effects of oral fasudil over 52 weeks in three different dosages. Subsequently, an international, multi-site, randomized phase III study will have to be performed aiming at an efficacy readout in a larger trial population.
Concomitant treatment with riluzole is considered standard therapy; in order to reduce bias, patients need to be treated with riluzole for at least 4 weeks. Most ALS patients in the participating countries receive riluzole and in most participating countries it is the only licensed drug, which therefore will not be withheld. For ethical reasons, patients who have started on edaravone therapy shall continue edaravone treatment. Edaravone treatment must not be discontinued for reasons of trial participation.
Fasudil is approved for the treatment of vasospasms following subarachnoid hemorrhage by Japanese authorities since 1995. Several thousand patients have been treated with this drug and thus the safety profile of fasudil in humans is well-known. In patients with ischemic stroke, fasudil also showed a beneficial effect on functional outcome and was well-tolerated in the study population (10). Numerous clinical trials for other disease conditions, mainly cardiovascular disorders, have been performed and did not raise major safety issues. Therefore, it is considered justified and safe to assess the effect of fasudil in patients with ALS.
The administration of placebo in our study population is acceptable since both placebo and fasudil are given as add-on to the standard therapy riluzole, which is an inclusion criterion. If a treatment with edaravone has been started prior the trial, it should be continued throughout the study.
A direct interaction of the two substances fasudil and riluzole leading to adverse effects has not been described yet and is not expected based on their different mechanism of action. Research in www.drugbank.ca (global drug properties), http://dgidb.genome.wustl.edu/ (drug-gene interactions), http://medicine.iupui.edu/clinpharm/ddis/index (Cytochrome P450 drug interaction table), and http://lmmd.ecust.edu.cn:8000 (chemical ADMET properties) did not reveal any known or expected drug interactions on the enzymatic or genetic level. To detect a putative influence of fasudil on riluzole levels, riluzole levels will be assessed.
In addition to its neuroprotective and pro-regenerative effects, the kinase inhibitor fasudil was also shown to affect microglial activity. The mechanism of action of riluzole is thought to be in part related to its stabilizing effect on sodium channels and the resulting reduction of presynaptic glutamate release. It can thus be hypothesized that the neuroprotective effect of riluzole—the standard therapy for patients with ALS—could be increased by the neuroprotective effect of fasudil in an additive manner.
Hemorrhage, particularly intracerebral bleedings, are described as a side effect of fasudil. This, however, was assessed in a population of patients with SAH and the incidence of hemorrhage was not significantly different in the placebo-treated group. Thus, hemorrhage is not expected to be a risk for our patient population. Moreover, patients with a personal or family history of intracranial bleeding, known intracerebral aneurysms or Moyamoya disease will be excluded from the trial.
Irrespective of the trial treatment longtime benefit may be limited, yet cannot be ruled out. Furthermore, all patients will contribute to the generation of valid data from a prospective, placebo-controlled clinical study. In addition, exploratory assessments will help to identify markers that indicate prognosis and activity of disease.
In conclusion, the ROCK-ALS trial will yield important data on safety, tolerability, and efficacy of a novel therapeutic strategy, ROCK inhibition, as disease-modifying treatment of ALS. The biomarker collection linked to this trial will provide additional information on target engagement and the usefulness of selected molecular markers as indicators of progression.
PL and JK developed the trial concept, wrote the trial protocol, wrote and edited the manuscript. MW, WC, CN, MK-K, MBe, HB, CB, RF, and ALu provided input on trial design, and critically edited the manuscript for content. TF, RH, and ALe detailed the statistical aspects of the study in the study protocol, and critically reviewed the manuscript. RG contributed to the preclinical data. SA, MBä, MBoe, NB, IC, PC, MD, TG, JG, RG, AH, JK, TL, FM, TM, SP, YR, JeS, JoS, M-HS, JS, JW, and EZ contributed to and corrected the manuscript, actively participating study center. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Shahram Attarian (Reference center for neuromuscular disorders and ALS, University Hospital la Timone, Marseille, France), Mathias Bähr (Department of Neurology, University Medical Center Göttingen, Germany), Matthias Boentert (Institute for Sleep Medicine and Neuromuscular Disorders, Muenster University Hospital, Münster, Germany), Nathalie Braun (Neuromuscular Diseases Unit/ALS Clinic, Kantonsspital St. Gallen, St. Gallen Switzerland), Philippe Corcia (Reference Center for ALS and other Motoneuron Disorders, University Hospital Bretonneau, Tours, France), Isabell Cordts (Department of Neurology, Technical University of Munich, Munich, Germany), Marcus Deschauer (Department of Neurology, Technical University of Munich, Munich, Germany), Thorsten Grehl (Department of Neurology, Alfried Krupp Hospital, Essen, Germany), Julian Grosskreutz (Department of Neurology, Jena University Hospital, Jena, Germany), Andreas Hermann (Department of Neurology, Technical University of Dresden, Dresden, Germany; German Center for Neurodegenerative Diseases (DZNE) Dresden, Dresden, Germany), Josua Kuttler (Department of Neurology, University Medical Center Göttingen, Germany), Teresa Lengenfeldt (Department of Neurology, University Medical Center Göttingen, Germany), Fabian Maass (Department of Neurology, University Medical Center Göttingen, Germany), Thomas Meyer (Center for ALS and other motor neuron disorders, Charité–Universitätsmedizin Berlin, Germany), Susanne Petri (Department of Neurology, Hannover Medical School, Hannover, Germany), Yvonne Remane (Pharmacy at the University of Leipzig Medical Center, Leipzig, Germany), Jens Schmidt (Department of Neurology, University Medical Center Göttingen, Germany), Joachim Schuster (Department of Neurology, University of Ulm, Ulm, Germany), Marie-Hélène Soriani (Center for ALS, University Hospital Pasteur, CHU de Nice, France), Jeffrey Statland (University of Kansas Medical Center, Kansas City, KS, United States), Jochen Weishaupt (Department of Neurology, University of Ulm, Ulm, Germany), Daniel Zeller (Department of Neurology, University of Würzburg, Würzburg, Germany), Eirini Zielke (Department of Neurology, University Medical Center Göttingen, Germany).
As potential conflict of interest, we report that PL (together with Lars Tönges) are inventors on a patent held by the University Medical Center of Göttingen on the use of fasudil for the treatment of ALS (EP 2825175, US 9980972B2).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This project is supported by the Bundesministerium für Bildung und Forschung (BMBF), Grant-No. 01GM1704A, 01GM1704B, the Schweizer Nationalfonds (SNF), Grant-No. 32ER30_17367, and the Ministère des Affaires sociales, de la Santé et des Droits des femmes (DGOS), Grant-No. DGOS2016-SERI E-RARE under the frame of E-Rare-3, the ERA-Net for Research on Rare Diseases. Whole genome sequencing through the CReATe Consortium CReATe (U54 NS092091) is part of Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS. This consortium is funded through collaboration between NCATS, and the NINDS.
We acknowledge the contribution of the members of the independent Safety Monitoring Board (SMB): Prof. Peter Schlattmann (Jena), Prof. Claudia Trenkwalder (Kassel) and Prof. Thomas Herdegen (Kiel).
1. Koch J-C, Tatenhorst L, Roser AE, Saal KA, Tönges L, Lingor P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol Ther. (2018) 189(C):1–21. doi: 10.1016/j.pharmthera.2018.03.008
2. Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. (2010) 15:622–9. doi: 10.1016/j.drudis.2010.06.011
3. Tönges L, Koch JC, Bähr M, Lingor P. ROCKing regeneration: rho kinase inhibition as molecular target for neurorestoration. Front Mol Neurosci. (2010) 4:39. doi: 10.3389/fnmol.2011.00039
4. Tönges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego ÉM, et al. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain. (2012) 135(Pt 11):3355–70. doi: 10.1093/brain/aws254
5. Takata M, Tanaka H, Kimura M, Nagahara Y, Tanaka K, Kawasaki K, et al. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br J Pharmacol. (2013) 170:341–51. doi: 10.1111/bph.12277
6. Komagome RR, Kimura KK, Saito MM. Postnatal changes in Rho and Rho-related proteins in the mouse brain. Jpn J Vet Res. (2000) 47:127–33.
7. Conti A, Riva N, Pesca M, Iannaccone S, Cannistraci CV, Corbo M, et al. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys Acta. (2014) 1842:99–106. doi: 10.1016/j.bbadis.2013.10.013
8. Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, et al. Phosphorylation of adducin by rho-kinase plays a crucial role in cell motility. J Cell Biol. (1999) 145:347–61. doi: 10.1083/jcb.145.2.347
9. Park J, Arakawa-Takeuchi S, Jinno S, Okayama H. Rho-associated kinase con- nects a cell cycle-controlling anchorage signal to the mammalian target of rapamycin pathway. J Biol Chem. (2011) 286:23132–41. doi: 10.1074/jbc.M110.209114
10. Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E, Fasudil Ischemic Stroke Study Group. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J Neurol Sci. (2005) 238:31–9. doi: 10.1016/j.jns.2005.06.003
11. Taniguchi J, Seki C, Takuwa H, Kawaguchi H, Ikoma Y, Fujinaga M, et al. Evaluation of Rho-kinase activity in mice brain using N-[11C]methyl-hydroxyfasudil with positron emission tomography. Mol Imaging Biol. (2013) 16:395–402. doi: 10.1007/s11307-013-0695-y
12. Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina. J Am Coll Cardiol. (2005) 46:1803–11. doi: 10.1016/j.jacc.2005.07.047
13. Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. (2013) 77:2619–25. doi: 10.1253/circj.CJ-13-0443
14. Tönges L, Günther R, Suhr M, Jansen J, Balck A, Saal KA, et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia. (2014) 62:217–32. doi: 10.1002/glia.22601
15. Zhang H, Li Y, Yu J, Guo M, Meng J, Liu C, et al. Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation. (2013) 20:313–22. doi: 10.1159/000351221
16. Borrajo A, Rodriguez-Perez AI, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology. (2014) 85:1–8. doi: 10.1016/j.neuropharm.2014.05.021
17. Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. (2011) 10:24. doi: 10.1186/1741-7015-10-24
18. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, de Carvalho M, et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the motor unit number index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatry. (2015) 86:1172–9. doi: 10.1136/jnnp-2015-310509
19. Boland S, Defert O, Boland S. Rho kinase inhibitors: a patent review (2014–2016). Expert Opin Ther Pat. (2017)00:1–9. doi: 10.1080/13543776.2017.1272579
20. Lingor P, Teusch N, Schwarz K, Mueller R, Mack H, Bähr M, et al. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J Neurochem. (2007) 103:181–9. doi: 10.1111/j.1471-4159.2007.04756.x
Keywords: amyotrophic lateral sclerosis, disease-modification, clinical trial protocol, ROCK inhibition, study design
Citation: Lingor P, Weber M, Camu W, Friede T, Hilgers R, Leha A, Neuwirth C, Günther R, Benatar M, Kuzma-Kozakiewicz M, Bidner H, Blankenstein C, Frontini R, Ludolph A, Koch JC and the ROCK-ALS Investigators (2019) ROCK-ALS: Protocol for a Randomized, Placebo-Controlled, Double-Blind Phase IIa Trial of Safety, Tolerability and Efficacy of the Rho Kinase (ROCK) Inhibitor Fasudil in Amyotrophic Lateral Sclerosis. Front. Neurol. 10:293. doi: 10.3389/fneur.2019.00293
Received: 24 January 2019; Accepted: 06 March 2019;
Published: 27 March 2019.
Edited by:
Chiara Briani, University of Padova, ItalyReviewed by:
Adriano Chio, University of Turin, ItalyCopyright © 2019 Lingor, Weber, Camu, Friede, Hilgers, Leha, Neuwirth, Günther, Benatar, Kuzma-Kozakiewicz, Bidner, Blankenstein, Frontini, Ludolph, Koch and the ROCK-ALS Investigators. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Lingor, cGF1bC5saW5nb3JAdHVtLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.