
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 05 February 2019
Sec. Epilepsy
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00064
 Yu Jia1,2
Yu Jia1,2 Yicong Lin1,2Jing Li1,2Mingyu Li1,2Yifan Zhang1,2Yue Hou1,2Aihua Liu1,2Liping Zhang3Liping Li1,2Peng Xiang1,2
Yicong Lin1,2Jing Li1,2Mingyu Li1,2Yifan Zhang1,2Yue Hou1,2Aihua Liu1,2Liping Zhang3Liping Li1,2Peng Xiang1,2 Jing Ye1,2*†
Jing Ye1,2*† Zhaoyang Huang1,2*†
Zhaoyang Huang1,2*† Yuping Wang1,2
Yuping Wang1,2Mutations in the Potassium channel subfamily T member 1 (KCNT1) gene have been reported in a range of epileptic encephalopathies. Here we report the case of a 12-year-old male suffering from multiple types of epileptic seizures and cognitive decline from the age of 10. The patient had four types of epileptic seizures, including tonic seizures, atypical absence seizures, myoclonic seizures, and generalized tonic-clonic seizures. The electroencephalogram showed generalized slow spike-and-slow-waves, mutiple-spike-and-slow-waves, as well as short-term fast rhythms bursts. Thus, he was diagnosed with Lennox-Gastaut syndrome. The patient had failed to control seizures after using five first-line antiepileptic drugs. Whole exome sequencing revealed a missense KCNT1 mutation (c.625 C>T). Previous studies revealed that quinidine could block the KCNT1 channel. Therefore, we assumed that quinidine might be effective for him. Add-on treatment with quinidine was started when the patient was 12 years old. After an 8-month treatment, the frequency of seizures and epileptiform discharges were significantly reduced. In conclusion, quinidine therapy may offer a new choice for the treatment of Lennox-Gastaut syndrome with KCNT1 mutations.
Potassium channel subfamily T member 1 (KCNT1), also known as Slack, is a member of the Slo-type sub family of potassium channel genes (1–3). It has been reported that KCNT1 mutations were detected in many early onset epileptic encephalopathies, such as epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (4–7). Previous studies demonstrated that KCNT1 channels are expressed in neurons and interneurons in the cortex and the CA3 region of the hippocampus (8, 9). Electrophysiological studies have revealed that KCNT1 mutations enhance the channel-mediated potassium conductance and increase the K+ currents in neurons and interneurons, which result in the imbalance between neuronal excitation and inhibition (10, 11). In vitro functional studies have shown that these mechanisms may be responsible for epileptogenesis associated with KCNT1 mutations (10).
Quinidine has been used as a class I antiarrhythmic drug to prevent ventricular arrhythmias. Recent works revealed that quinidine could block the KCNT1 channel (10, 12). Thus, quinidine is expected to be effective in improving electrophysiological abnormalities caused by KCNT1 mutations. Recently, there have been several reports about the quinidine treatment of KCNT1-related epileptic encephalopathies, such as EIMFS, ADNFLE, and West syndrome (10, 13–16). Quinidine has become a new method for the treatment of KCNT1-related epilepsy syndromes.
Here we report a patient suffering from Lennox-Gastaut syndrome with a missense mutation in KCNT1 (c.625C>T) treated with quinidine. We describe the improvement of the clinical symptoms, the adverse effects, and the dosage adjustment of quinidine during the treatment. Then we review the literature on quinidine treatment of the epilepsy syndrome with KCNT1 mutation.
The patient was a 12-year-old male who had his first seizure attack at the age of 10, and had four types of epileptic seizures. The first type of seizure was tonic axial seizures characterized by flexion of the neck and body and the extension of four extremities for several seconds. The tonic seizures could last several seconds. This type of seizure occurred predominantly at night and the frequency was about 4–5 times per week. The second type was atypical absence seizures, which manifested as a sudden loss of consciousness and the resuming of normal activity right after the seizure. This type of seizure could last about 15 s and the frequency was 4–5 times per week. The third type was the myoclonic seizure, presented as prominent myoclonic jerks of bilateral upper limbs. The frequency of this type of seizures was 2–3 times per day. The last type of seizure was the generalized tonic-clonic seizure, which could last about 5 min. The mean frequency of this type of seizure was <1 time per week.
The patient was a full-term infant with no history of perinatal asphyxia, head injury, encephalitis, and febrile convulsions. His family history was unremarkable. He had a mild degree of intellectual impairment and learning disability after the onset of the disease. The physical examinations were normal. Auxiliary examinations, including blood routine examination, serum biochemical examination, thyroid function, autoimmunity antibody, and blood ammonia, were normal. The electrocardiogram was normal and the QT interval (QTc) was 372 ms. There was no lesion on the brain MRI. The Wechsler Intelligence Scale showed a borderline cognition impairment. The electroencephalogram (EEG) before treatment showed that there was a large number of 3 to 5 Hz slow waves with middle and high amplitude in the anterior region in the background. Fast rhythms bursts of 16 to 20 Hz and multiple-spike-and-slow-waves of 0.5 to 1 Hz were observed during the sleep period. Slow spike-and-slow-waves of 1.5 to 2.5 Hz were observed during awake time (Figure 1).
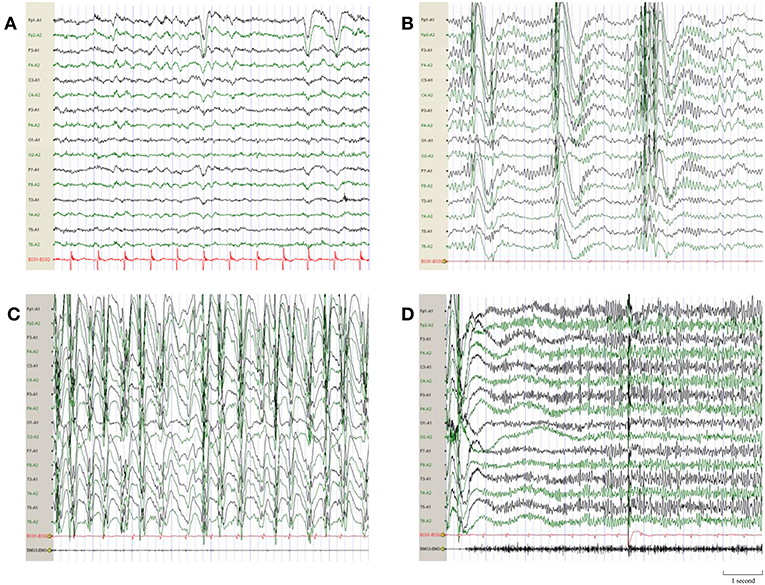
Figure 1. EEG demonstrations before quinidine therapy. (A) Interictal EEG before quinidine therapy showed a large number of 3–5 Hz slow waves with middle and high amplitude in the anterior region in background. (B) Multiple-spike-and-slow-waves of 0.5–1 Hz were observed during the sleep period. (C) Slow spike-and-slow-waves of 1.5–2.5 Hz were observed when the patient was awake. (D) Ictal EEG of the tonic seizure showed short-term fast rhythms burst of 16–20 Hz.
The patient was diagnosed with Lennox-Gastaut syndrome after considering his multiple types of epileptic seizures, mental retardation, and typical electroencephalographic features. He was refractory to a multiple anti-epileptic drugs treatment, including sodium valproate (8 mg/kg/day), levetiracetam (50 mg/kg/day), clonazepam (0.0375 mg/kg/day), topiramate (3.75 mg/kg/day), and lamotrigine (2.5 mg/kg/day). Whole exome sequencing (WES) identified a novel heterozygous KCNT1 mutation (chr9:138649026; c.625C>T; p.Arg209Cys) inherited from his father. This missense mutation was highly likely to cause the dysfunction of the KCNT1 channel and led to a gain-of-function phenotype. This alteration had not been previously reported and was not found in the ExAC database (http://exac.broadinstitute.org/), and was predicted to be likely pathogenic.
This study was approved by the human research ethic committees of Xuanwu hospital capital medical university. Written informed consent was obtained from all participants and guardians of minors for the quinidine therapy and the publication of this study. Additive quinidine therapy to our patient was initiated at 12 years of age. The doses of the above anticonvulsants remained unchanged. In the month before quinidine therapy, the patient had 16 tonic seizures, 12 atypical absence seizures, 10 myoclonic seizures, and 1 generalized tonic-clonic seizures.
After admission, the quinidine therapy was initiated with 5 mg/kg/day in 3 divided doses under electrocardiographic (ECG) monitoring. The QTc ranged from 361 to 415 ms, with an average of 378 ms (the normal limit of QTc is within 450 ms). After 1 month of treatment, the dose of quinidine was titrated to 10 mg/kg/day and he had 13 tonic seizures during this month. The dose was maintained during the following 2 months. QTc was in normal range (391–436 ms). There was also no other adverse effect of quinidine. At the fourth month, the dose of quinidine was increased to 13.75 mg/kg/day in 3 divided doses. The frequency of tonic seizures ranged between 4 and 6 times per month. The mean QTc interval was 383 ms. As no adverse effects were experienced, the dose of quinidine was maintained to 13.75 mg/kg/day during the following 4 months. The patient had 4 tonic seizures per month. The frequency of tonic seizures subsided by 75% (Figure 2A), whereas the frequency of the other types of seizures was not reduced significantly.
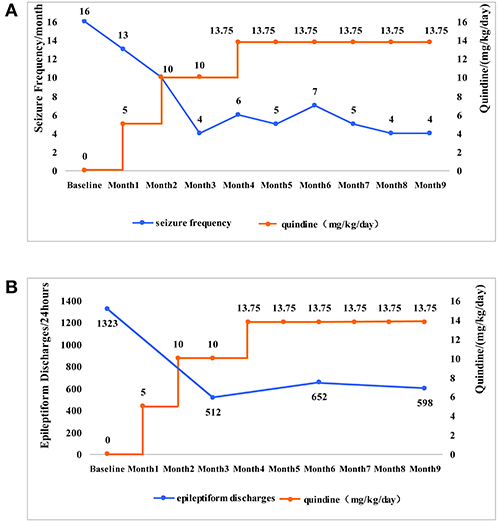
Figure 2. The changes of the seizure frequency and epileptiform discharges along with the changes of quinidine dosages. (A) The patient was given quinidine at a starting dosage of 5 mg/kg/day. The dose slowly increased and maintained at 13.75 mg/kg/day. As the dosage of quinidine increased, the frequency of tonic seizures (the main type of seizures in the patient) gradually reduced from 16 times per month to 4 times per month. (B) The epileptiform discharge of EEG was 1,323 times per 24 h before taking the quinidine. The epileptiform discharge was 512 times per 24 h in the 3rd month, 652 times per 24 h in the 6th month, 598 times per 24 h in the 9th month. With the increase of dosages, the epileptiform discharge showed a significant reduction.
Video EEG (VEEG) was performed regularly during treatment and the number of epileptic discharges was counted by four expert technicians who had not seen the patient's clinical information. One slow spike-and-wave complex or one episode of paroxysmal fast rhythms in EEGs were counted as one epileptiform discharge. 1323 epileptiform discharges were recorded during a 24-h video EEG before the treatment of quinidine. We subsequently applied the 24-h VEEG every 3 months to evaluated the efficacy of the quinidine treatment. The total number of epileptic discharges was 512, 652, and 598 in the next 3, 6, and 9 months, respectively. The epileptiform discharges decreased by 54.80% (Figure 2B).
In this case, the patient had four types of epileptic seizures, including tonic seizures, atypical absence seizures, myoclonic seizures, and generalized tonic-clonic seizures. The EEG before treatment showed generalized slow spike-and-slow-waves, multiple-spikes-and-slow-waves, and short-term fast rhythms bursts. He was diagnosed with Lennox-Gastaut syndrome. The WES identified a missense KCNT1 mutation (c.625 C>T). Quinidine was reported to be effective in patients with KCNT1 mutations. Thus, we applied this drug to our patient. After a treatment period of 8 months, the frequency of tonic seizures and epileptiform discharges decreased significantly.
Up to now, the exact mechanism of KCNT1-related epilepsy is still unknown. Previous studies suggested that this mutation resulted in a gain of function in this potassium channel, and the magnitude of gain of function correlated with the clinical severity (10). It has been confirmed that quinidine can block KCNT1 channels in vitro (10, 12, 17, 18). In our study, the patient was treated with quinidine for 8 months and the frequency of seizures and epileptiform discharges were significantly reduced. Thus, quinidine therapy was effective for our patient. Our study suggested that quinidine therapy might offer a new method for the treatment of KCNT1-related epilepsy syndromes.
We summarized a total number of 16 unrelated cases [15 sporadic patients and 5 patients of a pedigree (13–16, 19–24)] with KCNT1-related epilepsy syndrome treated with the quinidine reported previously (Table 1). The mutation sites were c.1887G>C; p.K629N, c.1283G>A; p.R428Q (recurrent in two unrelated patients), c.808C>C/G; p.Q270E, c.2965G>T, c.2849G>A; p.R950Q, c.1955G>T; p.G652V, p.R428Q, c.1421G>A, c.2386T>C; p.Y796H, c.1193G>A, c.820C>A, p.L274I, c.2849G>A; p.R950Q, c.2677G>A; p.E893K and c.2782C>T; p.R928C. All KCNT1 mutations reported were heterozygous missense mutations except for those not mentioned in the literature. In these 16 cases, the treatments were effective in 8 cases (13–16, 19) and the others were ineffective (Effective response was defined as >50% reduction in seizure frequency) (14, 20–24).
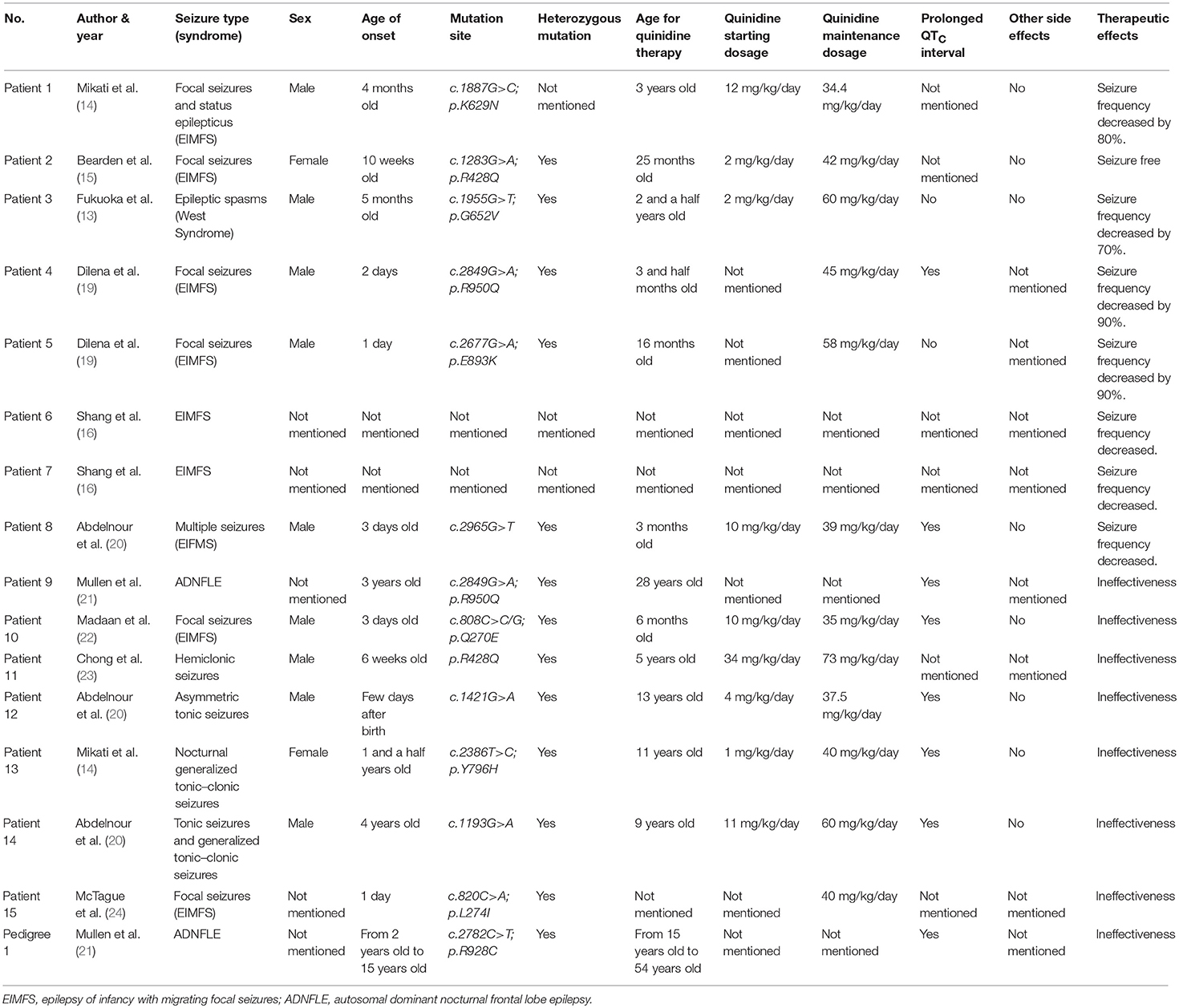
Table 1. Summary of quinidine in the treatment of KCNT1 gene mutation related-epilepsy.
There were 9 previously reported patients who suffered from EIMFS with KCNT1 mutation and treated by quinidine. The treatment was effective in 7 cases and ineffective in 2 cases. Interestingly, patient 2 and patient 11 had the same KCNT1 mutation (p.R428Q), but the therapeutic effects were completely opposite. Patient 2 of Bearden et al. (15) was a 3-year-old female patient of EIMFS, who became seizure-free after treatment. Patient 11 of Chong et al. (23) was a 5-year-old male patient who suffered from an unclassified early onset epileptic encephalopathy. In this latter case, the seizure frequency did not decrease significantly. In addition, Mullen et al. (21) studied the effectiveness of quinidine for six patients (Patient 7 and five patients of the pedigree) who suffered from ADNFLE with KCNT1 mutations. They found that the seizure frequency of these six patients decreased by less than 50%. It was reported that one patient who suffered from West Syndrome with KCNT1 mutation benefited from quinidine therapy. Thus, compared to ADNFLE and other epileptic syndromes, quinidine therapy tended to be more effective in EIMFS and West syndrome in patients who carried the KCNT1 mutation. Therefore, we assumed that the epilepsy phenotype was likely associated with the therapeutic effect.
Moreover, in a recent study by Abdelnour et al. (20) we noticed that the response of quinidine therapy may be age-dependent; younger patients may respond better to this therapy (20). In 16 unrelated cases reported, the mean age of 8 cases responded, except for those whose ages were not identified in the literature, was 2.11 ± 1.18 years old (the range was between 3 months and 3-and-a-half years old) (13–16, 19), and the mean age of the other cases not responded was 21.13 ± 15.92 years old (the range was between 6 months and 54 years old) (14, 20–24). Using a cut-off of 4 years of age, 6/7 patients < 4 years of age responded, and 0/11 patients > 4 years responded. These data revealed that efficacy of quinidine for KCNT1-related epilepsy might also depend on the age at quinidine therapy initiation. In our case, the patient was diagnosed as suffering from Lennox-Gastaut syndrome, and quinidine treatment was initiated at 12 years of age. The quinidine therapy proved to be significantly effective. The therapeutic effect of quinidine may be influenced by multiple factors. The epilepsy phenotype, initiation age of therapy, and prior neuronal injury, may all play a role in the efficacy of quinidine therapy. Henceforth, randomized controlled trials (RCT) should be performed to identify the relationship between the influencing factors and the efficacy of quinidine therapy.
So far, there is still no consensus on the effective dosage of quinidine for KCNT1-related epilepsy. The therapeutic dosage of quinidine in the treatment of pediatric cardiac disease is 15–60 mg/kg/day, and the maximum daily dose is 3,000–4,000 mg (14, 25, 26). In our case, we used the initial amount of 5 mg/kg/day. The dosage was slowly increased to 13.75 mg/kg/day. No adverse effects of quinidine were reported, and the frequency of tonic seizures and epileptiform discharges decreased significantly. Thus, we maintained this dose for treatment. Further studies are needed to determine the suitable dosage of quinidine for the treatment of KCNT1-related epilepsy.
Previously, it was believed that the penetration was complete in patients with KCNT1 mutations. However, it was reported that there was one individual from an ADNFLE family with a KCNT1 mutation without causing a phenotype (7). The authors proposed that this individual's unaffected status was most likely due to incomplete penetrance or non-penetrance (7). For our patient, the WES identified a missense KCNT1 mutation. He was inferred to inherit a KCNT1 mutation from his unaffected father. We assumed that his father's unaffected status might be due to incomplete penetrance or non-penetrance. Because his grandparental samples were unavailable, we could not confirm the origin of the mutations.
In summary, we used quinidine to treat a patient with KCNT1-related Lennox-Gastaut syndrome, and the seizure frequency and epileptiform discharges were significantly reduced. Quinidine provided valuable clinical experience for the individualized treatment for epilepsy patients with KCNT1 mutations. Nevertheless, there are too few cases reported with quinidine treatment for KCNT1-related epilepsy syndromes. Thus, further studies are required to confirm the effectiveness and the suitable dosage of quinidine therapy for KCNT1-related epilepsy syndromes.
All procedures were approved by the ethics committee of Xuanwu Hospital. The parents of our patient provided written informed consent.
YJ, ZH, and YL were the major contributors in writing the manuscript. YW, JY, LZ, and AL contributed to the diagnosis and treatment of the patient. LL contributed to the EEG analysis. JL, ML, PX, and YZ contributed to counting the number of epileptic discharges. JY, ZH, and YH contributed to the analysis of genetic examination. JY and ZH contributed to checking the manuscript. All authors read and approved the final manuscript.
This work was supported by Beijing Municipal Science & Technology Commission, Grant No. Z161100002616001, Beijing Municipal Education Commission, Grant No. TJSH20161002502 and Capital's Funds for Health Improvement and Research, Grant No. 2016-2-2015.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
EIMFS, epilepsy of infancy with migrating focal seizures; ADNFLE, autosomal dominant nocturnal frontal lobe epilepsy; EEG, electroencephalogram;WES, whole exome sequencing; PMA, phorbol-12-myristate-13-acetate.
1. Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+. Trends Neurosci. (2005) 28:422–8. doi: 10.1016/j.tins.2005.06.003
2. Yuan A, Santi CM, Wei A, Wang ZW, Pollak K, Nonet M, et al. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron (2003) 37:765–73. doi: 10.1016/S0896-6273(03)00096-5
3. Kim GE, Kaczmarek LK. Emerging role of the KCNT1 slack channel in intellectual disability. Front Cell Neurosci. (2014) 8:209. doi: 10.3389/fncel.2014.00209
4. Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. (2012) 44:1188–90. doi: 10.1038/ng.2440
5. Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. (2012) 44:1255–9. doi: 10.1038/ng.2441
6. Coppola G. Malignant migrating partial seizures in infancy: an epilepsy syndrome of unknown etiology. Epilepsia (2009) 50(Suppl. 5):49–51. doi: 10.1111/j.1528-1167.2009.02121.x
7. Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia (2015) 56:e114–20. doi: 10.1111/epi.13071
8. Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the slack potassium channel in the rat central nervous system. J Comp Neurol. (2002) 454:241–54. doi: 10.1002/cne.10439
9. Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, et al. Formation of intermediate-conductance calcium-activated potassium channels by interaction of slack and slo subunits. Nat Neurosci. (1998) 1:462–9. doi: 10.1038/2176
10. Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. (2014) 75:581–90. doi: 10.1002/ana.24128
11. Kim GE, Jack K, Giulia B, Quraishi IH, Martin HC, Edward B, et al. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. (2014) 9:1661–72. doi: 10.1016/j.celrep.2014.11.015
12. Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, et al. Pharmacological activation and inhibition of slack (Slo2.2) channels. Neuropharmacology (2006) 51:896–906. doi: 10.1016/j.neuropharm.2006.06.003
13. Fukuoka M, Kuki I, Kawawaki H, Okazaki S, Kim K, Hattori Y, et al. Quinidine therapy for west syndrome with KCNT1 mutation: a case report. Brain Dev Jpn. (2016) 39:80–3. doi: 10.1016/j.braindev.2016.08.002
14. Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. (2016) 78:995–9. doi: 10.1002/ana.24520
15. Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. (2014) 76:457–61. doi: 10.1002/ana.24229
16. Shang K, Zhang Y, Yang X, Liu A, Yang Z, Liu X, et al. Clinical features and gene mutations in epilepsy of infancy with migrating focal seizures. Zhonghua Er Ke Za Zhi. (2016) 54:735–9. doi: 10.3760/cma.j.issn.0578-1310.2016.10.005
17. Santi CM, Ferreira G, Yang B, Gazula VR, Butler A, Wei A, et al. Opposite regulation of slick and slack K+ channels by neuromodulators. J Neurosci. (2006) 26:5059–68. doi: 10.1523/JNEUROSCI.3372-05.2006
18. Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. (2003) 23:11681–91. doi: 10.1523/JNEUROSCI.23-37-11681.2003
19. Dilena R, DiFrancesco JC, Soldovieri MV, Giacobbe A, Ambrosino P, Mosca I, et al. Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics (2018) 15:1112–26. doi: 10.1007/s13311-018-0657-9
20. Abdelnour E, Gallentine W, McDonald M, Sachdev M, Jiang Y, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature. Seizure (2018) 55:1–3. doi: 10.1016/j.seizure.2017.11.017
21. Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, et al. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine. Neurology (2017) 90:e67–2. doi: 10.1212/WNL.0000000000004769
22. Madaan P, Jauhari P, Gupta A, Chakrabarty B, Gulati S. A quinidine non responsive novel KCNT1 mutation in an Indian infant with epilepsy of infancy with migrating focal seizures. Brain Dev. (2018) 40:229–32. doi: 10.1016/j.braindev.2017.09.008
23. Chong PF, Nakamura R, Saitsu H, Matsumoto N, Kira R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. (2016) 79:502–3. doi: 10.1002/ana.24598
24. McTague A, Nair U, Malhotra S, Meyer E, Trump N, Gazina EV, et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology (2018) 90:e55–66. doi: 10.1212/WNL.0000000000004762
25. Cohen IS, Jick H, Cohen SI. Adverse reactions to quinidine in hospitalized patients: findings based on data from the boston collaborative drug surveillance program. Prog Cardiovasc Dis. (1977) 20:151–63. doi: 10.1016/0033-0620(77)90004-4
Keywords: quinidine therapy, Lennox-Gastaut syndrome, KCNT1 mutation, sodium-activated potassium channel, epileptiform discharges
Citation: Jia Y, Lin Y, Li J, Li M, Zhang Y, Hou Y, Liu A, Zhang L, Li L, Xiang P, Ye J, Huang Z and Wang Y (2019) Quinidine Therapy for Lennox-Gastaut Syndrome With KCNT1 Mutation. A Case Report and Literature Review. Front. Neurol. 10:64. doi: 10.3389/fneur.2019.00064
Received: 19 October 2018; Accepted: 17 January 2019;
Published: 05 February 2019.
Edited by:
Udaya Seneviratne, Monash Medical Centre, AustraliaReviewed by:
Maurizio Taglialatela, University of Naples Federico II, ItalyCopyright © 2019 Jia, Lin, Li, Li, Zhang, Hou, Liu, Zhang, Li, Xiang, Ye, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Ye, eWVqaW5nXzAxQHNpbmEuY29t
Zhaoyang Huang, ZHJodWFuZ3p5QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.