 Young-Kook Kim
Young-Kook Kim Kwang Il Nam3*
Kwang Il Nam3* Juhyun Song
Juhyun Song
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 31 October 2018
Sec. Sleep Disorders
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00867
The glymphatic system has emerged as an important player in central nervous system (CNS) diseases, by regulating the vasculature impairment, effectively controlling the clearance of toxic peptides, modulating activity of astrocytes, and being involved in the circulation of neurotransmitters in the brain. Recently, several studies have indicated decreased activity of the glymphatic pathway under diabetes conditions such as in insulin resistance and hyperglycemia. Furthermore, diabetes leads to the disruption of the blood-brain barrier and decrease of apolipoprotein E (APOE) expression and the secretion of norepinephrine in the brain, involving the impairment of the glymphatic pathway and ultimately resulting in cognitive decline. Considering the increased prevalence of diabetes-induced dementia worldwide, the relationship between the glymphatic pathway and diabetes-induced dementia should be investigated and the mechanisms underlying their relationship should be discussed to promote the development of an effective therapeutic approach in the near future. Here, we have reviewed recent evidence for the relationship between glymphatic pathway dysfunction and diabetes. We highlight that the enhancement of the glymphatic system function during sleep may be beneficial to the attenuation of neuropathology in diabetes-induced dementia. Moreover, we suggest that improving glymphatic system activity may be a potential therapeutic strategy for the prevention of diabetes-induced dementia.
The relationship between type 2 diabetes mellitus (T2DM) and dementia, also called “type 3 diabetes”, has emerged as a critical health issue across the world as it is fast increasing in incidence (1). There are several studies supporting the relationship between diabetes and dementia. A recent meta-analysis indicated that individuals with T2DM have a 65% increased risk for Alzheimer's disease (AD) (2). A population-based longitudinal study has reported a 16% increased risk for dementia in T2DM patients compared with nondiabetic patients (3). Furthermore, many T2DM patients have cognitive impairments (4) and even patients in the prediabetic stage of insulin resistance exhibit a decrease in memory function and dysfunction of cognitive flexibility and cognitive control (5). Moreover, patients with chronic hyperinsulinemia exhibit insulin resistance and cognitive dysfunction (6). Taking into consideration the previous evidence for the relationship between diabetes conditions, including insulin resistance and hyperglycemia, and cognitive decline (5), the neurological changes in diabetes-induced dementia should be investigated to elucidate the underlying mechanisms and thus support the design of appropriate therapy and prevention in clinical practice.
Current studies highlight the role of the glymphatic system in neurodegenerative diseases such as AD, suggesting that it influences the clearance of amyloid-beta (Aβ) peptide (7). In addition, the roles of the glymphatic system in the central nervous system (CNS) include the regulation of astrocyte activity (8), neurohormones (9), and glucose metabolism (10); modulation of apolipoprotein E (APOE) circulation (11); and regulation of insulin resistance in the brain (12, 13). Diabetes has been shown to contribute to the impairment of glymphatic activity, leading to declined cognitive function (14). Sleep has been known to promote the activity of the glymphatic pathway, subsequently enhancing the efficiency of the brain-clearance system (15), and ultimately improving memory function and synaptic plasticity (16). Here, we have reviewed recent evidence for the relationship between glymphatic pathway dysfunction and diabetes-induced dementia. Furthermore, we have suggested a therapeutic approach for alleviating the neuropathological symptoms of diabetes-induced dementia through the improvement of glymphatic system function.
In the CNS, approximately 68% of the total water volume is within the intracellular compartment, whereas the remaining 32% of the water exists in the extracellular compartment (8). The extracellular fluid is distributed into the interstitial fluid (ISF), cerebrospinal fluid (CSF), and blood compartments (8). In the CNS, a variety of nutrients circulate through the brain, but it is also essential to remove efficiently the metabolites (17). The CSF produced by the choroid plexus contributes to the delivery of nutrients to the brain parenchyma and the clearance of interstitial toxic waste (18, 19). The CSF is absorbed inside the CNS via filtration and reabsorption of water through the capillaries into the ISF of the surrounding brain regions (20, 21). The CSF enters the para-arterial spaces, mixes with the ISF, and is finally removed from the brain through the paravenous spaces (18, 22). The ISF with the toxic peptide wastes enters the lymphatic circulation through the paravenous space (7, 23). The ISF drained into the paravenous space can eliminate the solutes from the interstitial space similar to the function of the lymphatic system outside the brain (18).
The perivascular space of cerebral blood vessels is the place where the CSF/ISF exchange occurs (18, 24). In addition, the glial cells that provide the outer boundary of the perivascular space have an important function in the clearance and waste turnover (25, 26). Iliff et al. demonstrated that fluorescent dextran injected into the cisterna magna in mice was found in the basement membrane of parenchymal capillaries and in the perivascular spaces of caliber draining veins. This paravascular clearance system was named as the glymphatic system by merging the words “glial” and “lymphatic”, because of its dependence on glial cells and also due to its functional resemblance to the peripheral lymphatic system (18). In other words, the glymphatic system is a paravascular pathway that lies in the space between the vascular adventitia and the vascular end-feet of astrocytes (27). The glymphatic system has greater vascular permeability and contributes to the clearance of macromolecules from the brain parenchyma (27). Moreover, the glymphatic pathway contributes to CSF influx into the brain parenchyma through para-arterial spaces to exchange solutes with the ISF (28). Previous studies about the glymphatic pathway have fundamentally altered the traditional model of CSF hydrodynamics (18, 29) and shown that CSF can be recycled back into the brain and exchanged with ISF (29–31). Current studies have reported that the glymphatic pathway involves the dural lymphatic vessels that finally drain toward the cervical lymph nodes (23, 32, 33).
The glymphatic pathway has important diverse roles. Some studies have highlighted the role of the glymphatic pathway in waste drainage. Indeed, it was demonstrated that the glymphatic pathway could be the first step of the brain drainage system (34). Another study reported that the CSF containing toxic waste circulates in the arachnoid space and flows to the dural venous sinuses (7, 35). Moreover, the role of the glymphatic system in nutrient delivery was reported by demonstrating that lipoproteins and small molecules could be delivered from the CSF to the brain parenchyma via the glymphatic pathway (36). Other researchers have shown that the glymphatic system is crucial for the distribution of nutrients throughout the whole brain (37, 38). Additionally, several studies identified the role played by the glymphatic pathway in hormone circulation and signal transduction. It was reported that the glymphatic pathway is not only involved in the volume transmission and the paracrine system, but also in the activation of astrocytic Ca2+ signaling within the cortex (30) and in the opening of N-methyl-D-aspartate (NMDA) receptors in cultured astrocytes (39). Furthermore, the glymphatic pathway could be involved in regulating the circulation of norepinephrine, the major neuromodulator of arousal (40) that is related with cognitive decline (9), and which plays a role in AD neuropathology (41). Moreover, the glymphatic pathway influences not only the CNS, but also other organs via the circulatory system (42). In this view, current anatomical studies support the fact that the glymphatic pathway is connected with the peripheral system through glymphatic efflux sites, including arachnoid granulations, perineural spaces of cranial and spinal nerves (43), and meningeal lymphatics (23). Based on these observations, the function of the glymphatic pathway is deemed indispensable, and further studies are necessary for the identification of its relationship with various neurological changes.
As mentioned earlier, the glymphatic system acts as an effective waste-clearance pathway for the brain (28). Previous studies demonstrated that dysfunctions of the glymphatic system aggravate neuropathological symptoms of various neurological diseases such as stroke and AD (8, 44). One magnetic resonance imaging (MRI) study indicated that alterations of the glymphatic system could be used as disease risk indicators for neurodegenerative disorders, including AD (45). Impairment of the glymphatic pathway can be the result of abnormal changes in CSF influx dependent on arterial pulsatility (8, 29). The AD is characterized by several neuropathologies, including Aβ accumulation and the tau tangle formation in various brain regions (46). Blood-brain barrier (BBB) breakdown increases the accumulation of Aβ in the blood plasma, ISF, and CSF (47), causing synaptic dysfunction in the brain. Furthermore, BBB disruption causes inflammation that also contributes to glymphatic dysfunction, suppresses CSF-to-ISF turnover, and impairs glymphatic clearance (48, 49). Amyloid-beta peptide exists in the normal brain, circulating blood, and CSF (50). While the normal brain is able to control Aβ influx and efflux through glymphatic drainage, the AD brain cannot control this process. Therefore, toxic Aβ accumulates in the brain parenchyma and vascular structures (51), and ultimately triggers BBB disruption and vasculature impairment (16).
Amyloid-beta clearance via BBB transport depends on the glymphatic pathway (52), as toxic Aβ can be transported across the BBB through specific transporters such as the low-density lipoprotein receptor-related protein-1 (53). However, when the amount of Aβ exceeds the capacity of the efflux transporter, Aβ is cleared via ISF flow in the glymphatic system (54, 55). The glymphatic system drains over 60% of the brain Aβ to the lymph nodes using the convective flow caused by arterial pulsations (56). The increased permeability of the BBB triggers glymphatic pathway impairment and ultimately leads to the defective clearance of Aβ by BBB transport in dementia (42, 57). Thus, the glymphatic system is considerably relevant to AD progression by transporting Aβ and other metabolites out of the brain (15).
The dysfunction of the glymphatic pathway increases the accumulation of toxic waste products in the brain (58), and is associated with impaired cognitive function recorded in behavioral tests (59, 60). Moreover, it is associated with the dysregulation of water transport into astrocytes (8). Aquaporin-4 (AQP4), a water transport channel expressed in the astrocytic end-feet near the capillaries, is considered to be critical for water movement between the cellular and ventricular compartments (61). Loss of AQP4 results in impairment of CSF influx and CSF-to-ISF turnover (62), aggravating glymphatic pathway dysfunction (28). The loss of AQP4 polarization has been related to glymphatic dysfunction in the brains of mice and considered to be a predictor of AD in humans (63). Additionally, the decrease in AQP4 expression contributes to reduced Aβ clearance (63–65) and tau clearance (66) through the glymphatic system (67), and has been shown to impair water permeability in vitro (68). Moreover, AQP4 is associated with the modulation of neurotrophic factor-dependent synaptic plasticity (69), and its absence results in defects in memory consolidation (70, 71). Collectively, the glymphatic system is affected by AQP4 expression in astrocytes and associated with AD progression.
Furthermore, a recent study suggested that an increase in CNS norepinephrine levels and ISF secretion are the results of reduced glymphatic influx in AD mouse models (9). Noradrenergic neurons located in the locus coeruleus supply norepinephrine to various brain regions (72). Elevated norepinephrine levels result in the contraction of the extracellular volume fraction, reduction of CSF influx, and brain ISF (15). Locus coeruleus-derived norepinephrine increases BBB permeability by elevating Na+/K+-ATPase activity, leading to augmentation of ISF secretion, and subsequently contributing to the glymphatic function (73). The noradrenergic system in the brain has critical roles in cognitive activities, including attention, perception, and memory function (41, 74). Loss of locus coeruleus neurons and abnormal levels of CSF norepinephrine were observed in the AD brain (75, 76). In addition, several subtypes of adrenergic receptors have been shown to control the production of Aβ (77) or mediate Aβ toxicity (78), involved in AD pathogenesis. Altogether, norepinephrine contributes to the function of the glymphatic system and is implicated in the neuropathology of AD including memory loss.
The APOE gene, the only strongly confirmed genetic risk factor for AD, has been associated with cognitive impairment (79), lipid metabolism, and various brain pathologies (80). The CSF is a major source of APOE for ISF because it circulates through the brain parenchyma via the glymphatic pathway (15, 81). The CSF contributes to the delivery of APOE to the brain via the glymphatic system, for molecules including Aβ (9), lipophilic molecules (30), and tau (82). Apolipoprotein E has been known to regulate transport and metabolism of cholesterol in the periphery and CNS (83), and is further associated with neurite growth, synaptic plasticity, and cognitive function (84). The APOE polymorphisms influence the structure and function of the glymphatic pathway (85) and there is a strong correlation between the ε4 allele and neurodegeneration (86).
In conclusion, dysfunctions of the glymphatic pathway and subsequent impairment of metabolite circulation aggravate the onset and development of AD. Further studies on the implication of the glymphatic system in AD are necessary for the development of effective therapeutic strategies for AD.
Diabetes has been known to be a risk factor for various complications including hypertension, cardiovascular diseases, and neurological diseases, such as stroke and AD (87, 88). Recently, diabetes-induced dementia has been highlighted in CNS studies, showing that diabetes features, including insulin resistance and hyperglycemia, can trigger impairment of memory function, neuronal cell damage, and neuroinflammation (18, 89–91). A recent study demonstrated synaptic dysfunction through the loss of synaptic proteins in hyperglycemia-induced dementia (92). Moreover, in line with the diagnosis of diabetes in patients with cortical embolism due to atherothrombosis and stroke, diabetes conditions may alter the arteriolar structure and influence the perivascular space in the brain (93). A previous study demonstrated that Aβ plaques were accumulated in the brains of diabetes patients affecting cognitive function (94). Other experimental studies showed that diabetes induced by high-fat and/or high-sugar diets triggered Aβ accumulation in the brain (95–98). Therefore, more study on the mechanisms linking diabetes and dementia is necessary for understanding of the onset and progression of diabetes-induced dementia.
Type 2 diabetes mellitus is characterized by enhanced glymphatic CSF influx and a slowing of the interstitial solute clearance, leading to cognitive decline (13). Several studies have reported diabetes-associated cerebrovascular dysfunctions, neurodegenerative processes, and cognitive impairments following abnormal glycemia and insulinemia (5, 10, 12, 99). Another study has reported that hyperglycemia could result in cerebral neurovascular dysfunction, neurotoxicity, and impairment of neural insulin metabolism, leading to cognitive impairment (100). Chronic microvascular dysfunction caused by hyperglycemia can also cause severe cognitive dysfunction in diabetes patients (5, 10, 99).
Moreover, diabetes is associated with vascular pathology. It contributes to the development of small blood vessel disease and triggers the impairment of glymphatic activity, leading to cognitive dysfunction (14, 101, 102). Several studies have shown that BBB integrity was compromised and permeability was dramatically increased in the brain of diabetes patients (103). The BBB protects the brain against toxic components that may cause synaptic dysfunction or generate neurotoxins and maintains homeostasis (104, 105).
Hyperglycemia caused by diabetes is also related to neuronal pathogenesis by inducing the generation of excessive reactive oxygen species (ROS) and microvascular complications (106) and by suppressing the supply of vitamin C as an antioxidant and scavenger of free radicals into the brain, subsequently promoting oxidative stress in the brains of diabetes patients (107, 108). In addition, diabetes results in abnormal cerebral neovascularization and neurovascular remodeling (109). Brain endothelial cells are vulnerable to hyperglycemic stress in diabetes (110) and diabetes-induced hyperglycemia is associated with neurodegenerative diseases such as AD (111). Hyperglycemia promotes the generation of excessive superoxide species and boosts the activation of the protein kinase C (PKC) and advanced glycation end products (AGE) pathway, leading to increased BBB permeability mediated by the disruption of tight junction proteins and increased vascular endothelial growth factor (VEGF) expression (112, 113).
Moreover, one study demonstrated that the correlation between diabetes and AD depends on the APOEε4 allele, which was involved in lipid homeostasis in diabetes (114). In diabetes patients, the increase of APOEε4 increases the risk for AD compared with nondiabetic patients (115).
In conclusion, diabetes triggers the disruption of the BBB and increase of APOE and ultimately aggravates cognitive decline through metabolite imbalance due to glymphatic pathway dysfunction. Thus, the investigation and the understanding of the role of the glymphatic system in diabetes-induced dementia are necessary for the development of an efficient treatment for diabetes-induced dementia.
Sleep is necessary for the bulk flow of brain ISF and clearance of solutes, and is also involved in memory function and synaptic plasticity through several mechanisms, including Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling (16, 116–118). It was reported that astrocytes undergo contraction during sleep, and subsequently, the extracellular space is enlarged and the flow of ISF is enhanced. These processes promoted the clearance of macromolecular metabolites from the brain, during sleep (15). The disturbance of glymphatic transport due to inadequate sleep may mediate neuropathologies in AD, given that sleep disruption aggravates the assembly of Aβ plaques and tangles (119, 120). Recent studies have focused on the effect of sleep deprivation on synaptic plasticity and on structural changes of brain regions related to learning and memory (121, 122), and the progression of AD (123–125). Moreover, impaired glymphatic transport results in a 40% decrease in the clearance of Aβ in the brain of mice (54). Sleep disturbance is also associated with the deterioration of diabetes conditions such as insulin resistance (126), the dysregulation of energy and glucose homeostasis in healthy adults (127, 128), and the dysregulation of body weight (129). Thus, the influence of sleep on glymphatic transport is an important aspect of manipulation to control glymphatic system dysfunction in diabetes-induced dementia.
During sleep deprivation, norepinephrine secretion is increased (130), while glymphatic fluid transport is reduced (131). Diabetes conditions, such as hyperglycemia, also cause changes in the CSF concentrations of norepinephrine (132–134). Norepinephrine results in vasoconstriction of the pial arteries (135) and leads to the reduction of CSF inflow during sleep deprivation (9). In addition, several studies have demonstrated that the impairment of glymphatic pathway activity caused by sleep deprivation triggered APOE-related neuronal dysfunction in AD, leading to cognitive decline (11, 16). Hence, the modulation of norepinephrine secretion and other related pathways may enhance the function of the glymphatic system and ameliorate memory in diabetes-induced dementia.
Sleep is influenced by the hormone melatonin, which is mainly produced in the pineal gland, which receives input from the suprachiasmatic nucleus in the hypothalamus (136, 137). Melatonin is the major hormone regulating the circadian rhythm (138) and is also known to regulate memory function by acting on hippocampal neurons involved in memory formation (139, 140). Several studies have reported that melatonin could control hippocampal synaptic plasticity by binding to the melatonin specific receptor (141) and alter synaptic transmission and long-term potentiation in the hippocampus (142). In addition, melatonin could regulate calcium influx by controlling the conductance of voltage-gated Ca2+ ion channels and NMDA receptors (143, 144) in gamma-aminobutyric acid (GABAergic) neurons (145). Based on these studies, supplementation with melatonin has been considered as an effective method to alleviate sleep onset latency and to improve sleep quality in children (146) and adults (147, 148). Furthermore, the increase of melatonin secretion during sleep improves the sleep quality by enhancing the amplitude of circadian oscillations through melatonin receptors MT1 and MT2 (149).
Dysregulation of melatonin results in cognitive impairment by synaptic dysfunction. Recently, two studies have suggested that impairments of the sleep/wake cycle owing to sleep disturbances increase T2DM risk (128, 150). A genome-wide study revealed the relationship between single nucleotide polymorphisms in the MTNR1B gene (encoding MT2) and T2DM (151). In addition, decreased serum melatonin levels have been found in both diabetes mouse models and diabetes patients with hyperinsulinemia (152). Oral administration of melatonin alleviated hyperglycemia, hyperinsulinemia, and hyperlipidemia in T2DM rats (153, 154). Melatonin suppressed the levels of cytosolic cyclic adenosine monophosphate and/or cytosolic guanosine monophosphate and regulated insulin secretion via these receptors (155). Moreover, a study demonstrated that sleep disturbance in AD is related to the physiological changes in melatonin function (156). Furthermore, given that melatonin administration could attenuate the rate of AD progression, inhibit the accumulation of Aβ (157, 158), decrease neuronal cell death (159, 160), and reduce insulin resistance (152), the decrease in melatonin levels due to sleep disturbance might be associated with the impairment of the glymphatic system in diabetes-induced dementia. Diabetes triggers memory dysfunction in rats, which can be alleviated by melatonin treatment (161). Considering previous evidence, we suggest that melatonin administration should be considered as an approach to reduce neuropathology in diabetes-induced dementia.
A recent study demonstrated that impaired sleep duration was recorded in hyperglycemia patients (162). Epidemiological studies also showed that short duration and poor quality of sleep increase the risk of diabetes in adults (163, 164). Consequently, sleep impairment is strongly related to diabetes pathologies such as hyperglycemia (165).
The study reported that the lateral decubitus body position during sleep leads to an enhanced influx of a fluorescent CSF tracer into the cerebrum with a reduction of interstitial solute retention and an increase of clearance efficiency (43). Smooth glymphatic flow during sleep contributes to the improvement of paracrine signaling, whereas the decline in glymphatic flow suppresses the perivascular lipid transport, the astrocytic Ca2+ signaling within the cortex (30), and the opening of NMDA receptors (166). Moreover, a recent study suggested that AQP4-mediated glymphatic pathway improvement could be used as a therapeutic treatment for AD patients (167). The AQP4 gene could regulate the progression of cognitive dysfunction in AD, and this was related to poor sleep and Aβ burden (168). The genetic variation in AQP4 was also identified to be a factor correlating sleep and Aβ accumulation in the brain (169).
Based on the previous evidence stated, the sleep-induced metabolite clearance through the glymphatic system has a critical role in neuropathological features, including excessive accumulation of Aβ in brains with diabetes-induced dementia. Thus, we highlight that the improvement of glymphatic system function by the regulation of sleep may be a promising and effective strategy to reduce the neuropathological symptoms observed in diabetes-induced dementia.
In diabetes-induced dementia, the glymphatic system dysfunction characterized by the failure of interstitial solute clearance leads to extracellular solute accumulation and cognitive decline. Even though there is no experimental approach providing a direct relationship between sleep and the glymphatic system, many studies have implicated their relationship. Thus, we highlight the necessity for further exploration of the improvement of glymphatic system through sleep modulation toward attenuation of the neuropathology in diabetes-induced dementia. Here, we have reviewed the dysregulation of the glymphatic pathway in diabetes-induced dementia, the effects of sleep on glymphatic system function, including the improvement of toxic peptide clearance, the enhancement of melatonin secretion, the regulation of APOE expression, the improvement of synaptic plasticity, the regulation of norepinephrine levels, and the alleviation of insulin resistance (11, 120, 128, 130, 170, 171) (Figure 1).
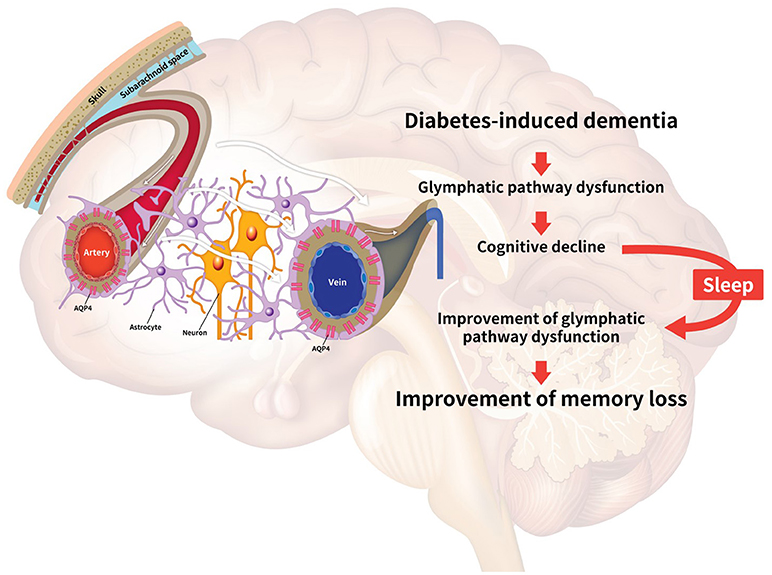
Figure 1. Schematic image illustrating the relationship between the glymphatic system and sleep. Diabetes conditions trigger the impairment of the glymphatic pathway. The decrease of glymphatic activity results in reduced efficiency for clearing toxic peptides, dysregulation of glucose metabolism, insulin resistance, and dysregulation of apolipoprotein E (APOE) circulation in the brain, leading to cognitive decline. Sleep improves the clearance of toxic amyloid-beta (Aβ), the secretion of norepinephrine, and the circulation of APOE and melatonin in the brain. Collectively, sleep is associated with the alleviation of the impaired glymphatic function, leading to the enhancement of memory in diabetes-induced dementia. The white arrows indicate the glymphatic flow.
Hence, we suggest that the improvement of glymphatic function by sleep regulation may be a novel target for attenuating neuropathological symptoms such as memory loss in diabetes-induced dementia, through the enhancement of the circulation of melatonin, APOE and norepinephrine, and reduction of Aβ aggregation in the brain.
Y-KK and JS wrote the manuscript. JS and KN prepared the figure.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Basic Research Program through the National Research Foundation of Korea, funded by grant NRF-2016R1D1A1B03930394 (JS), NRF-2018R1A2B6001104 (Y-KK). This study was also financially supported by Chonnam National University (2017–2859) (JS).
1. Mittal K, Katare DP. Shared links between type 2 diabetes mellitus and Alzheimer's disease: a review. Diabetes Metab Syndr. (2016) 10(2 Suppl. 1):S144–9. doi: 10.1016/j.dsx.2016.01.021
2. Gudala K, Bansal D, Schifano F, Bhansali A. Diabetes mellitus and risk of dementia: a meta-analysis of prospective observational studies. J Diabetes Investig. (2013) 4:640–50. doi: 10.1111/jdi.12087
3. Haroon NN, Austin PC, Shah BR, Wu J, Gill SS, Booth GL. Risk of dementia in seniors with newly diagnosed diabetes: a population-based study. Diabetes Care (2015) 38:1868–75. doi: 10.2337/dc15-0491
4. Shaik MA, Chan QL, Xu J, Xu X, Hui RJ, Chong SS, et al. Risk factors of cognitive impairment and brief cognitive tests to predict cognitive performance determined by a formal neuropsychological evaluation of primary health care patients. J Am Med Dir Assoc. (2016) 17:343–7. doi: 10.1016/j.jamda.2015.12.007
5. McCrimmon RJ, Ryan CM, Frier BM. Diabetes and cognitive dysfunction. Lancet (2012) 379:2291–9. doi: 10.1016/S0140-6736(12)60360-2
6. Vanhanen M, Koivisto K, Kuusisto J, Mykkanen L, Helkala EL, Hanninen T, et al. Cognitive function in an elderly population with persistent impaired glucose tolerance. Diabetes Care (1998) 21:398–402. doi: 10.2337/diacare.21.3.398
7. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. (2015) 11:457–70. doi: 10.1038/nrneurol.2015.119
8. Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The Glymphatic system: a beginner's guide. Neurochem Res. (2015) 40:2583–99. doi: 10.1007/s11064-015-1581-6
9. Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. (2016) 93:215–25. doi: 10.1016/j.nbd.2016.05.015
10. Moheet A, Mangia S, Seaquist ER. Impact of diabetes on cognitive function and brain structure. Ann N Y Acad Sci. (2015) 1353:60–71. doi: 10.1111/nyas.12807
11. Yang G, Lai CS, Cichon J, Ma L, Li W, Gan WB. Sleep promotes branch-specific formation of dendritic spines after learning. Science (2014) 344:1173–8. doi: 10.1126/science.1249098
12. Baglietto-Vargas D, Shi J, Yaeger DM, Ager R, LaFerla FM. Diabetes and Alzheimer's disease crosstalk. Neurosci Biobehav Rev. (2016) 64:272–87. doi: 10.1016/j.neubiorev.2016.03.005
13. Kondo H, Shirotake S, Okabe T, Makino S, Nishimoto K, Oyama M. Clinical impact of consolidative and salvage radiotherapy for lymph node metastasis in upper urinary tract urothelial carcinoma. Case Rep Urol. (2018) 2018:1471839. doi: 10.1155/2018/1471839
14. Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. (2013) 12:483–97. doi: 10.1016/S1474-4422(13)70060-7
15. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science (2013) 342:373–7. doi: 10.1126/science.1241224
16. Jackson ML, Gunzelmann G, Whitney P, Hinson JM, Belenky G, Rabat A, et al. Deconstructing and reconstructing cognitive performance in sleep deprivation. Sleep Med Rev. (2013) 17:215–25. doi: 10.1016/j.smrv.2012.06.007
17. Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke (2013) 44(6 Suppl. 1):S93–95. doi: 10.1161/STROKEAHA.112.678698
18. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. (2012) 4:147ra111. doi: 10.1126/scitranslmed.3003748
19. Matsumae M, Atsumi H, Hirayama A, Hayashi N, Takizawa K, Sano F, et al. A new look at cerebrospinal fluid motion. No Shinkei Geka (2016) 44:909–24. doi: 10.11477/mf.1436203402.
20. Oreskovic D, Klarica M. The formation of cerebrospinal fluid: nearly a hundred years of interpretations and misinterpretations. Brain Res Rev. (2010) 64:241–62. doi: 10.1016/j.brainresrev.2010.04.006
21. Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS (2014) 11:10. doi: 10.1186/2045-8118-11-10
22. Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. (2008) 88:1277–340. doi: 10.1152/physrev.00027.2007
23. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
24. Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. (2004) 45:545–52. doi: 10.1016/j.neuint.2003.11.006
25. Smith AJ, Jin BJ, Verkman AS. Muddying the water in brain edema? Trends Neurosci. (2015) 38:331–2. doi: 10.1016/j.tins.2015.04.006
26. Jin BJ, Smith AJ, Verkman AS. Spatial model of convective solute transport in brain extracellular space does not support a “glymphatic” mechanism. J Gen Physiol. (2016) 148:489–501. doi: 10.1085/jgp.201611684
27. Iliff JJ, Goldman SA, Nedergaard M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. (2015) 14:977–9. doi: 10.1016/S1474-4422(15)00221-5
28. Pien GW, Pack AI, Jackson N, Maislin G, Macones GA, Schwab RJ. Risk factors for sleep-disordered breathing in pregnancy. Thorax (2014) 69:371–7. doi: 10.1136/thoraxjnl-2012-202718
29. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. (2013b) 33:18190–9. doi: 10.1523/JNEUROSCI.1592-13.2013
30. Rangroo Thrane V, Thrane AS, Plog BA, Thiyagarajan M, Iliff JJ, Deane R, et al. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci Rep. (2013) 3:2582. doi: 10.1038/srep02582
31. Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. (2015) 35:518–26. doi: 10.1523/JNEUROSCI.3742-14.2015
32. Eide PK, Ringstad G. MRI with intrathecal MRI gadolinium contrast medium administration: a possible method to assess glymphatic function in human brain. Acta Radiol Open (2015) 4:2058460115609635. doi: 10.1177/2058460115609635
33. Taoka T, Masutani Y, Kawai H, Nakane T, Matsuoka K, Yasuno F, et al. Evaluation of glymphatic system activity with the diffusion MR technique: diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer's disease cases. Jpn J Radiol. (2017) 35:172–8. doi: 10.1007/s11604-017-0617-z
34. Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. (2017) 8:1434. doi: 10.1038/s41467-017-01484-6
35. Nedergaard M. Neuroscience. Garbage truck of the brain Science. Science (2013) 340:1529–30. doi: 10.1126/science.1240514
36. Achariyar TM, Li B, Peng W, Verghese PB, Shi Y, McConnell E, et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener. (2016) 11:74. doi: 10.1186/s13024-016-0138-8
37. Hawkins RA, Peterson DR, Vina JR. The complementary membranes forming the blood-brain barrier. IUBMB Life (2002) 54:101–7. doi: 10.1080/15216540214541
38. Popescu BO, Toescu EC, Popescu LM, Bajenaru O, Muresanu DF, Schultzberg M, et al. Blood-brain barrier alterations in ageing and dementia. J Neurol Sci. (2009) 283:99–106. doi: 10.1016/j.jns.2009.02.321
39. Zhang CE, Wong SM, van de Haar HJ, Staals J, Jansen JF, Jeukens CR, et al. Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology (2017) 88:426–32. doi: 10.1212/WNL.0000000000003556
40. Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. (2003) 42:33–84. doi: 10.1016/S0165-0173(03)00143-7
41. Gannon M, Che P, Chen Y, Jiao K, Roberson ED, Wang Q. Noradrenergic dysfunction in Alzheimer's disease. Front Neurosci. (2015) 9:220. doi: 10.3389/fnins.2015.00220
42. Sironi L, Guerrini U, Tremoli E, Miller I, Gelosa P, Lascialfari A, et al. Analysis of pathological events at the onset of brain damage in stroke-prone rats: a proteomics and magnetic resonance imaging approach. J Neurosci Res. (2004) 78:115–22. doi: 10.1002/jnr.20219
43. Lee H, Xie L, Yu M, Kang H, Feng T, Deane R, et al. The effect of body posture on brain glymphatic transport. J Neurosci. (2015) 35:11034–44. doi: 10.1523/JNEUROSCI.1625-15.2015
44. Grandner MA, Patel NP, Jean-Louis G, Jackson N, Gehrman PR, Perlis ML, et al. Sleep-related behaviors and beliefs associated with race/ethnicity in women. J Natl Med Assoc. (2013) 105:4–15. doi: 10.1016/S0027-9684(15)30080-8
45. Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. (2013) 123:1299–309. doi: 10.1172/JCI67677
46. Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. (2010) 9:119–28. doi: 10.1016/S1474-4422(09)70299-6
47. Parashar A, Mehta V, Malairaman U. Type 2 diabetes mellitus is associated with social recognition memory deficit and altered dopaminergic neurotransmission in the amygdala. Ann Neurosci. (2018) 24:212–20. doi: 10.1159/000479637
48. Al Khatib HK, Harding SV, Darzi J, Pot GK. The effects of partial sleep deprivation on energy balance: a systematic review and meta-analysis. Eur J Clin Nutr. (2017) 71:614–24. doi: 10.1038/ejcn.2016.201
49. Verheggen ICM, Van Boxtel MPJ, Verhey FRJ, Jansen JFA, Backes WH. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci Biobehav Rev. (2018) 90:26–33. doi: 10.1016/j.neubiorev.2018.03.028
50. Grandner MA, Jackson N, Gooneratne NS, Patel NP. The development of a questionnaire to assess sleep-related practices, beliefs, and attitudes. Behav Sleep Med. (2014) 12:123–42. doi: 10.1080/15402002.2013.764530
51. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. (2016) 8:595–608. doi: 10.15252/emmm.201606210
52. Burgmans S, van de Haar HJ, Verhey FR, Backes WH. Amyloid-beta interacts with blood-brain barrier function in dementia: a systematic review. J Alzheimers Dis. (2013) 35:859–73. doi: 10.3233/JAD-122155
53. Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M. Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci. (2017) 74:2167–201. doi: 10.1007/s00018-017-2463-7
54. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. (2014) 76:845–61. doi: 10.1002/ana.24271
55. Simon MJ, Iliff JJ. Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim Biophys Acta (2016) 1862:442–51. doi: 10.1016/j.bbadis.2015.10.014
56. Morris AW, Carare RO, Schreiber S, Hawkes CA. The cerebrovascular basement membrane: role in the clearance of beta-amyloid and cerebral amyloid angiopathy. Front Aging Neurosci. (2014) 6:251. doi: 10.3389/fnagi.2014.00251
57. van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology (2016) 281:527–35. doi: 10.1148/radiol.2016152244
58. Wostyn P, Killer HE, De Deyn PP. Glymphatic stasis at the site of the lamina cribrosa as a potential mechanism underlying open-angle glaucoma. Clin Exp Ophthalmol. (2017) 45:539–47. doi: 10.1111/ceo.12915
59. Jiang Q, Zhang L, Ding G, Davoodi-Bojd E, Li Q, Li L, et al. Impairment of the glymphatic system after diabetes. J Cereb Blood Flow Metab. (2017) 37:1326–37. doi: 10.1177/0271678X16654702
60. Venkat P, Chopp M, Zacharek A, Cui C, Zhang L, Li Q, et al. White matter damage and glymphatic dysfunction in a model of vascular dementia in rats with no prior vascular pathologies. Neurobiol Aging (2017) 50:96–106. doi: 10.1016/j.neurobiolaging.2016.11.002
61. Lundgaard I, Lu ML, Yang E, Peng W, Mestre H, Hitomi E, et al. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab. (2017) 37:2112–24. doi: 10.1177/0271678X16661202
62. Howard ME, Jackson ML, Berlowitz D, O'Donoghue F, Swann P, Westlake J, et al. Specific sleepiness symptoms are indicators of performance impairment during sleep deprivation. Accid Anal Prev. (2014) 62:1–8. doi: 10.1016/j.aap.2013.09.003
63. Zeppenfeld DM, Simon M, Haswell JD, D'Abreo D, Murchison C, Quinn JF, et al. Association of perivascular localization of aquaporin-4 with cognition and alzheimer disease in aging brains. JAMA Neurol. (2017) 74:91–9. doi: 10.1001/jamaneurol.2016.4370
64. Moftakhar P, Lynch MD, Pomakian JL, Vinters HV. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J Neuropathol Exp Neurol. (2010) 69:1201–9. doi: 10.1097/NEN.0b013e3181fd252c
65. Yang W, Wu Q, Yuan C, Gao J, Xiao M, Gu M, et al. Aquaporin-4 mediates astrocyte response to beta-amyloid. Mol Cell Neurosci. (2012) 49:406–14. doi: 10.1016/j.mcn.2012.02.002
66. Xu Z, Xiao N, Chen Y, Huang H, Marshall C, Gao J, et al. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener. (2015) 10:58. doi: 10.1186/s13024-015-0056-1
67. Xiao M, Hu G. Involvement of aquaporin 4 in astrocyte function and neuropsychiatric disorders. CNS Neurosci Ther. (2014) 20:385–90. doi: 10.1111/cns.12267
68. Sorani MD, Zador Z, Hurowitz E, Yan D, Giacomini KM, Manley GT. Novel variants in human Aquaporin-4 reduce cellular water permeability. Hum Mol Genet. (2008) 17:2379–89. doi: 10.1093/hmg/ddn138
69. Skucas VA, Mathews IB, Yang J, Cheng Q, Treister A, Duffy AM, et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci. (2011) 31:6392–7. doi: 10.1523/JNEUROSCI.6249-10.2011
70. Fan Y, Liu M, Wu X, Wang F, Ding J, Chen J, et al. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct Funct. (2013) 218:39–50. doi: 10.1007/s00429-011-0373-2
71. Zhang J, Li Y, Chen ZG, Dang H, Ding JH, Fan Y, et al. Glia protein aquaporin-4 regulates aversive motivation of spatial memory in Morris water maze. CNS Neurosci Ther. (2013) 19:937–44. doi: 10.1111/cns.12191
72. Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. (1975) 163:467–505. doi: 10.1002/cne.901630406
73. Harik SI. Blood–brain barrier sodium/potassium pump: modulation by central noradrenergic innervation. Proc Natl Acad Sci USA. (1986) 83:4067–70. doi: 10.1073/pnas.83.11.4067
74. Chamberlain SR, Robbins TW. Noradrenergic modulation of cognition: therapeutic implications. J Psychopharmacol. (2013) 27:694–718. doi: 10.1177/0269881113480988
75. Elrod R, Peskind ER, DiGiacomo L, Brodkin KI, Veith RC, Raskind MA. Effects of Alzheimer's disease severity on cerebrospinal fluid norepinephrine concentration. Am J Psychiatry (1997) 154:25–30. doi: 10.1176/ajp.154.1.25
76. Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. (2003) 60:337–41. doi: 10.1001/archneur.60.3.337
77. Chen Y, Peng Y, Che P, Gannon M, Liu Y, Li L, et al. alpha(2A) adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proc Natl Acad Sci USA. (2014) 111:17296–301. doi: 10.1073/pnas.1409513111
78. Wang D, Fu Q, Zhou Y, Xu B, Shi Q, Igwe B, et al. beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem. (2013) 288:10298–307. doi: 10.1074/jbc.M112.415141
79. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:9649–53. doi: 10.1073/pnas.90.20.9649
80. Dallongeville J, Lussier-Cacan S, Davignon J. Modulation of plasma triglyceride levels by apoE phenotype: a meta-analysis. J Lipid Res. (1992) 33:447–54.
81. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. (2013) 9:106–18. doi: 10.1038/nrneurol.2012.263
82. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. (2014) 34:16180–93. doi: 10.1523/JNEUROSCI.3020-14.2014
83. Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. (2000) 1:507–37. doi: 10.1146/annurev.genom.1.1.507
84. Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci. (2004) 23:189–204. doi: 10.1385/JMN:23:3:189
85. Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. (2006) 361:932–44. doi: 10.1016/j.jmb.2006.06.080
86. Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. (2011) 10:241–52. doi: 10.1016/S1474-4422(10)70325-2
87. Giorda CB, Avogaro A, Maggini M, Lombardo F, Mannucci E, Turco S, et al. Incidence and risk factors for stroke in type 2 diabetic patients: the DAI study. Stroke (2007) 38:1154–60. doi: 10.1161/01.STR.0000260100.71665.2f
88. Janghorbani M, Hu FB, Willett WC, Li TY, Manson JE, Logroscino G, et al. Prospective study of type 1 and type 2 diabetes and risk of stroke subtypes: the Nurses' Health Study. Diabetes Care (2007) 30:1730–5. doi: 10.2337/dc06-2363
89. Dallongeville J, Selinger E, Davignon J, Lussier-Cacan S. Fish-oil supplementation reduces Ip(a) concentrations in type III dysbetalipoproteinemia. Clin Chem. (1992) 38(8 Pt 1), 1510–1.
90. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (1993) 261:921–3. doi: 10.1126/science.8346443
91. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:1977–81. doi: 10.1073/pnas.90.5.1977
92. Pintana H, Apaijai N, Kerdphoo S, Pratchayasakul W, Sripetchwandee J, Suntornsaratoon P, et al. Hyperglycemia induced the Alzheimer's proteins and promoted loss of synaptic proteins in advanced-age female Goto-Kakizaki (GK) rats. Neurosci Lett. (2017) 655:41–5. doi: 10.1016/j.neulet.2017.06.041
93. Giwa MO, Williams J, Elderfield K, Jiwa NS, Bridges LR, Kalaria RN, et al. Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology (2012) 78:167–74. doi: 10.1212/WNL.0b013e3182407968
94. Prasad S, Sajja RK, Naik P, Cucullo L. Diabetes mellitus and blood-brain barrier dysfunction: an overview. J Pharmacovigil. (2014) 2:125. doi: 10.4172/2329-6887.1000125
95. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. (2004) 18:902–4. doi: 10.1096/fj.03-0978fje
96. Yang Y, Wu Y, Zhang S, Song W. High glucose promotes Abeta production by inhibiting APP degradation. PLoS ONE (2013) 8:e69824. doi: 10.1371/journal.pone.0069824
97. Mehla J, Chauhan BC, Chauhan NB. Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits. J Alzheimers Dis. (2014) 39:145–62. doi: 10.3233/JAD-131238
98. Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, et al. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes (2014) 63:4291–301. doi: 10.2337/db14-0375
99. Mayeda ER, Whitmer RA, Yaffe K. Diabetes and cognition. Clin Geriatr Med. (2015) 31:101–15, ix. doi: 10.1016/j.cger.2014.08.021
100. Qiu C, Sigurdsson S, Zhang Q, Jonsdottir MK, Kjartansson O, Eiriksdottir G, et al. Diabetes, markers of brain pathology and cognitive function: the Age, gene/environment susceptibility-reykjavik study. Ann Neurol. (2014) 75:138–46. doi: 10.1002/ana.24063
101. Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer's disease. J Alzheimers Dis. (2009) 16:693–704. doi: 10.3233/JAD-2009-1022
102. Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke (2010) 41:450–4. doi: 10.1161/STROKEAHA.109.564914
103. Acharya NK, Levin EC, Clifford PM, Han M, Tourtellotte R, Chamberlain D, et al. Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis. (2013) 35:179–98. doi: 10.3233/JAD-122254
104. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. (2005) 57:173–85. doi: 10.1124/pr.57.2.4
105. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron (2008) 57:178–201. doi: 10.1016/j.neuron.2008.01.003
106. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
107. Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA. (2001) 98:11720–4. doi: 10.1073/pnas.171325998
108. Minamizono A, Tomi M, Hosoya K. Inhibition of dehydroascorbic acid transport across the rat blood-retinal and -brain barriers in experimental diabetes. Biol Pharm Bull. (2006) 29:2148–50. doi: 10.1248/bpb.29.2148
109. Prakash R, Johnson M, Fagan SC, Ergul A. Cerebral neovascularization and remodeling patterns in two different models of type 2 diabetes. PLoS ONE (2013) 8:e56264. doi: 10.1371/journal.pone.0056264
110. Russo VC, Higgins S, Werther GA, Cameron FJ. Effects of fluctuating glucose levels on neuronal cells in vitro. Neurochem Res. (2012) 37:1768–82. doi: 10.1007/s11064-012-0789-y
111. Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. (2007) 101:566–76. doi: 10.1111/j.1471-4159.2006.04393.x
112. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414:813–20. doi: 10.1038/414813a
113. Cipolla MJ, Huang Q, Sweet JG. Inhibition of protein kinase Cbeta reverses increased blood-brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke (2011) 42:3252–7. doi: 10.1161/STROKEAHA.111.623991
114. Peila R, Rodriguez BL, Launer LJ, Honolulu-Asia Aging S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia aging study. Diabetes (2002) 51:1256–62. doi: 10.2337/diabetes.51.4.1256
115. Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, et al. Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am J Pathol. (1998) 153:1149–55. doi: 10.1016/S0002-9440(10)65659-3
116. Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron (2004) 41:35–43. doi: 10.1016/S0896-6273(03)00814-6
117. Aton SJ, Seibt J, Dumoulin M, Jha SK, Steinmetz N, Coleman T, et al. Mechanisms of sleep-dependent consolidation of cortical plasticity. Neuron (2009) 61:454–66. doi: 10.1016/j.neuron.2009.01.007
118. Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron (2014) 81:12–34. doi: 10.1016/j.neuron.2013.12.025
119. Lucke-Wold BP, Smith KE, Nguyen L, Turner RC, Logsdon AF, Jackson GJ, et al. Sleep disruption and the sequelae associated with traumatic brain injury. Neurosci Biobehav Rev. (2015) 55:68–77. doi: 10.1016/j.neubiorev.2015.04.010
120. Bird SM, Sohrabi HR, Sutton TA, Weinborn M, Rainey-Smith SR, Brown B, et al. Cerebral amyloid-beta accumulation and deposition following traumatic brain injury–a narrative review and meta-analysis of animal studies. Neurosci Biobehav Rev. (2016) 64:215–28. doi: 10.1016/j.neubiorev.2016.01.004
121. Binder S, Baier PC, Molle M, Inostroza M, Born J, Marshall L. Sleep enhances memory consolidation in the hippocampus-dependent object-place recognition task in rats. Neurobiol Learn Mem. (2012) 97:213–9. doi: 10.1016/j.nlm.2011.12.004
122. Acosta-Pena E, Camacho-Abrego I, Melgarejo-Gutierrez M, Flores G, Drucker-Colin R, Garcia-Garcia F. Sleep deprivation induces differential morphological changes in the hippocampus and prefrontal cortex in young and old rats. Synapse (2015) 69:15–25. doi: 10.1002/syn.21779
123. Kyrtsos CR, Baras JS. Modeling the role of the glymphatic pathway and cerebral blood vessel properties in alzheimer's disease pathogenesis. PLoS ONE (2015) 10:e0139574. doi: 10.1371/journal.pone.0139574
124. Branger P, Arenaza-Urquijo EM, Tomadesso C, Mezenge F, Andre C, de Flores R, et al. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging (2016) 41:107–14. doi: 10.1016/j.neurobiolaging.2016.02.009
125. Brown BM, Rainey-Smith SR, Villemagne VL, Weinborn M, Bucks RS, Sohrabi HR, et al. The relationship between sleep quality and brain amyloid burden. Sleep (2016) 39:1063–8. doi: 10.5665/sleep.5756
126. Vanderburg CR, Davis DA, Diamond RE, Kao PF, Delalle I. Capzb2 protein expression in the brains of patients diagnosed with alzheimer's disease and huntington's disease. Transl Neurosci. (2010) 1:55–8. doi: 10.2478/v10134-010-0008-9
127. Cedernaes J, Lampola L, Axelsson EK, Liethof L, Hassanzadeh S, Yeganeh A, et al. A single night of partial sleep loss impairs fasting insulin sensitivity but does not affect cephalic phase insulin release in young men. J Sleep Res. (2016) 25:5–10. doi: 10.1111/jsr.12340
128. Qian J, Scheer F. Circadian system and glucose metabolism: implications for physiology and disease. Trends Endocrinol Metab. (2016) 27:282–93. doi: 10.1016/j.tem.2016.03.005
129. Nedeltcheva AV, Scheer FA. Metabolic effects of sleep disruption, links to obesity and diabetes. Curr Opin Endocrinol Diabetes Obes. (2014) 21:293–8. doi: 10.1097/MED.0000000000000082
130. Hipolide DC, Moreira KM, Barlow KB, Wilson AA, Nobrega JN, Tufik S. Distinct effects of sleep deprivation on binding to norepinephrine and serotonin transporters in rat brain. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29:297–303. doi: 10.1016/j.pnpbp.2004.11.015
131. Longordo F, Kopp C, Luthi A. Consequences of sleep deprivation on neurotransmitter receptor expression and function. Eur J Neurosci. (2009) 29:1810–9. doi: 10.1111/j.1460-9568.2009.06719.x
132. Figlewicz DP, Brot MD, McCall AL, Szot P. Diabetes causes differential changes in CNS noradrenergic and dopaminergic neurons in the rat: a molecular study. Brain Res. (1996) 736:54–60. doi: 10.1016/0006-8993(96)00727-5
133. Umhau JC, Petrulis SG, Diaz R, Rawlings R, George DT. Blood glucose is correlated with cerebrospinal fluid neurotransmitter metabolites. Neuroendocrinology (2003) 78:339–43. doi: 10.1159/000074887
134. Straznicky NE, Grima MT, Sari CI, Eikelis N, Lambert EA, Nestel PJ, et al. Neuroadrenergic dysfunction along the diabetes continuum: a comparative study in obese metabolic syndrome subjects. Diabetes (2012) 61:2506–16. doi: 10.2337/db12-0138
135. Cipolla MJ, Li R, Vitullo L. Perivascular innervation of penetrating brain parenchymal arterioles. J Cardiovasc Pharmacol. (2004) 44:1–8. doi: 10.1097/00005344-200407000-00001
136. Tan DX, Manchester LC, Fuentes-Broto L, Paredes SD, Reiter RJ. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: relation to human obesity. Obes Rev. (2011) 12:167–88. doi: 10.1111/j.1467-789X.2010.00756.x
137. Kumar Jha P, Challet E, Kalsbeek A. Circadian rhythms in glucose and lipid metabolism in nocturnal and diurnal mammals. Mol Cell Endocrinol. (2015) 418(Pt 1):74–88. doi: 10.1016/j.mce.2015.01.024
138. Cajochen C, Kräuchi K, Wirz-Justice A. Role of melatonin in the regulation of human circadian rhythms and sleep. J Neuroendocrinol. (2003) 15:432–7. doi: 10.1046/j.1365-2826.2003.00989.x
139. El-Sherif Y, Tesoriero J, Hogan MV, Wieraszko A. Melatonin regulates neuronal plasticity in the hippocampus. J Neurosci Res. (2003) 72:454–60. doi: 10.1002/jnr.10605
140. Bob P, Fedor-Freybergh P. Melatonin, consciousness, and traumatic stress. J Pineal Res. (2008) 44:341–7. doi: 10.1111/j.1600-079X.2007.00540.x
141. Wang LM, Suthana NA, Chaudhury D, Weaver DR, Colwell CS. Melatonin inhibits hippocampal long-term potentiation. Eur J Neurosci. (2005) 22:2231–7. doi: 10.1111/j.1460-9568.2005.04408.x
142. Ozcan M, Yilmaz B, Carpenter DO. Effects of melatonin on synaptic transmission and long-term potentiation in two areas of mouse hippocampus. Brain Res. (2006) 1111:90–4. doi: 10.1016/j.brainres.2006.06.117
143. Vanecek J, Klein DC. Melatonin inhibition of GnRH-induced LH release from neonatal rat gonadotroph: involvement of Ca2+ not cAMP. Am J Physiol. (1995) 269(1 Pt 1):E85–90. doi: 10.1152/ajpendo.1995.269.1.E85
144. Escames G, Macias M, Leon J, Garcia J, Khaldy H, Martin M, et al. Calcium-dependent effects of melatonin inhibition of glutamatergic response in rat striatum. J Neuroendocrinol. (2001) 13:459–66. doi: 10.1046/j.1365-2826.2001.00656.x
145. Cheng XP, Sun H, Ye ZY, Zhou JN. Melatonin modulates the GABAergic response in cultured rat hippocampal neurons. J Pharmacol Sci. (2012) 119:177–85. doi: 10.1254/jphs.11183FP
146. Chang YS, Lin MH, Lee JH, Lee PL, Dai YS, Chu KH, et al. Melatonin supplementation for children with atopic dermatitis and sleep disturbance: a randomized clinical trial. JAMA Pediatr. (2016) 170:35–42. doi: 10.1001/jamapediatrics.2015.3092
147. Lahteenmaki R, Puustinen J, Vahlberg T, Lyles A, Neuvonen PJ, Partinen M, et al. Melatonin for sedative withdrawal in older patients with primary insomnia: a randomized double-blind placebo-controlled trial. Br J Clin Pharmacol. (2014) 77:975–85. doi: 10.1111/bcp.12294
148. Amstrup AK, Sikjaer T, Mosekilde L, Rejnmark L. The effect of melatonin treatment on postural stability, muscle strength, and quality of life and sleep in postmenopausal women: a randomized controlled trial. Nutr J. (2015) 14:102. doi: 10.1186/s12937-015-0093-1
149. Touitou Y, Bogdan A. Promoting adjustment of the sleep-wake cycle by chronobiotics. Physiol Behav. (2007) 90:294–300. doi: 10.1016/j.physbeh.2006.09.001
150. Shan Z, Ma H, Xie M, Yan P, Guo Y, Bao W, et al. Sleep duration and risk of type 2 diabetes: a meta-analysis of prospective studies. Diabetes Care (2015) 38:529–37. doi: 10.2337/dc14-2073
151. Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Magi R, Reschen ME, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. (2015) 47:1415–25. doi: 10.1038/ng.3437
152. Peschke E, Frese T, Chankiewitz E, Peschke D, Preiss U, Schneyer U, et al. Diabetic Goto Kakizaki rats as well as type 2 diabetic patients show a decreased diurnal serum melatonin level and an increased pancreatic melatonin-receptor status. J Pineal Res. (2006) 40:135–43. doi: 10.1111/j.1600-079X.2005.00287.x
153. Rios-Lugo MJ, Cano P, Jimenez-Ortega V, Fernandez-Mateos MP, Scacchi PA, Cardinali DP, et al. Melatonin effect on plasma adiponectin, leptin, insulin, glucose, triglycerides and cholesterol in normal and high fat-fed rats. J Pineal Res. (2010) 49:342–8. doi: 10.1111/j.1600-079X.2010.00798.x
154. Agil A, Rosado I, Ruiz R, Figueroa A, Zen N, Fernandez-Vazquez G. Melatonin improves glucose homeostasis in young Zucker diabetic fatty rats. J Pineal Res. (2012) 52:203–10. doi: 10.1111/j.1600-079X.2011.00928.x
155. Peschke E, Bahr I, Muhlbauer E. Melatonin and pancreatic islets: interrelationships between melatonin, insulin and glucagon. Int J Mol Sci. (2013) 14:6981–7015. doi: 10.3390/ijms14046981
156. Wu YH, Swaab DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer's disease. Sleep Med. (2007) 8:623–36. doi: 10.1016/j.sleep.2006.11.010
157. Poeggeler B, Miravalle L, Zagorski MG, Wisniewski T, Chyan YJ, Zhang Y, et al. Melatonin reverses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Abeta peptide. Biochemistry (2001) 40:14995–5001. doi: 10.1021/bi0114269
158. Francioso A, Punzi P, Boffi A, Lori C, Martire S, Giordano C, et al. beta-sheet interfering molecules acting against beta-amyloid aggregation and fibrillogenesis. Bioorg Med Chem. (2015) 23:1671–83. doi: 10.1016/j.bmc.2015.02.041
159. Blask DE, Brainard GC, Dauchy RT, Hanifin JP, Davidson LK, Krause JA, et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. (2005) 65:11174–84. doi: 10.1158/0008-5472.CAN-05-1945
160. Stevens RG. Light-at-night, circadian disruption and breast cancer: assessment of existing evidence. Int J Epidemiol. (2009) 38:963–70. doi: 10.1093/ije/dyp178
161. Babaei-Balderlou F, Zare S. Melatonin improves spatial navigation memory in male diabetic rats. Vet Res Forum. (2012) 3:187–92.
162. DePietro RH, Knutson KL, Spampinato L, Anderson SL, Meltzer DO, Van Cauter E, et al. Association between inpatient sleep loss and hyperglycemia of hospitalization. Diabetes Care (2017) 40:188–93. doi: 10.2337/dc16-1683
163. Knutson KL, Ryden AM, Mander BA, Van Cauter E. Role of sleep duration and quality in the risk and severity of type 2 diabetes mellitus. Arch Intern Med. (2006) 166:1768–74. doi: 10.1001/archinte.166.16.1768
164. Anothaisintawee T, Reutrakul S, Van Cauter E, Thakkinstian A. Sleep disturbances compared to traditional risk factors for diabetes development: systematic review and meta-analysis. Sleep Med Rev. (2016) 30:11–24. doi: 10.1016/j.smrv.2015.10.002
165. Vgontzas AN, Liao D, Pejovic S, Calhoun S, Karataraki M, Bixler EO. Insomnia with objective short sleep duration is associated with type 2 diabetes: a population-based study. Diabetes Care (2009) 32:1980–5. doi: 10.2337/dc09-0284
166. Maneshi MM, Maki B, Gnanasambandam R, Belin S, Popescu GK, Sachs F, et al. Mechanical stress activates NMDA receptors in the absence of agonists. Sci Rep. (2017) 7:39610. doi: 10.1038/srep39610
167. Yin M, Pu T, Wang L, Marshall C, Wu T, Xiao M. Astroglial water channel aquaporin 4-mediated glymphatic clearance function: a determined factor for time-sensitive treatment of aerobic exercise in patients with Alzheimer's disease. Med Hypotheses (2018) 119:18–21. doi: 10.1016/j.mehy.2018.07.016
168. Burfeind KG, Murchison CF, Westaway SK, Simon MJ, Erten-Lyons D, Kaye JA, et al. The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer's disease. Alzheimers Dement (N Y) (2017) 3:348–59. doi: 10.1016/j.trci.2017.05.001
169. Rainey-Smith SR, Mazzucchelli GN, Villemagne VL, Brown BM, Porter T, Weinborn M, et al. Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl Psychiatry (2018) 8:47. doi: 10.1038/s41398-018-0094-x
170. Kiviniemi V, Wang X, Korhonen V, Keinanen T, Tuovinen T, Autio J, et al. Ultra-fast magnetic resonance encephalography of physiological brain activity - glymphatic pulsation mechanisms? J Cereb Blood Flow Metab. (2016) 36:1033–45. doi: 10.1177/0271678X15622047
171. van den Berg R, Mook-Kanamori DO, Donga E, van Dijk M, van Dijk JG, Lammers GJ, et al. A single night of sleep curtailment increases plasma acylcarnitines: Novel insights in the relationship between sleep and insulin resistance. Arch Biochem Biophys. (2016) 589:145–51. doi: 10.1016/j.abb.2015.09.017
Keywords: glymphatic system, diabetes-induced dementia, sleep, cognitive decline, norepinephrine
Citation: Kim Y-K, Nam KI and Song J (2018) The Glymphatic System in Diabetes-Induced Dementia. Front. Neurol. 9:867. doi: 10.3389/fneur.2018.00867
Received: 17 July 2018; Accepted: 26 September 2018;
Published: 31 October 2018.
Edited by:
Victor B. Fenik, VA Greater Los Angeles Healthcare System, United StatesReviewed by:
Eling D. de Bruin, Karolinska Institutet (KI), SwedenCopyright © 2018 Kim, Nam and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwang Il Nam, YXRsYXNAY2hvbm5hbS5hYy5rcg==
Juhyun Song, anVoeXVuc29uZ0BjaG9ubmFtLmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.