 Qian-Qian Li
Qian-Qian Li Lan-Jun Li
Lan-Jun Li Xin-Yu Wang
Xin-Yu Wang Jun Wu
Jun Wu- Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, China
Intracerebral hemorrhage (ICH) is a fatal acute cerebrovascular disease, with a high morbidity and mortality. Following ICH, erythrocytes release heme and several of its metabolites, thereby contributing to brain edema and secondary brain damage. Heme oxygenase is the initial and rate-limiting enzyme of heme catabolism, and the expression of heme oxygenase-1 (HO-1) is rapidly induced following acute brain injury. As HO-1 exerts it effects via various metabolites, its role during ICH remains complex. Therefore, in-depth studies regarding the role of HO-1 in secondary brain damage following ICH may provide a theoretical basis for neuroprotective function after ICH. The present review aims to summarize recent key studies regarding the effects of HO-1 following ICH, as well as its influence on ICH prognosis.
Introduction
Intracerebral hemorrhage (ICH) is in general referred to as non-traumatic spontaneous hemorrhage within the brain parenchyma; it is the second most common cause of stroke, representing approximately 10–15% of all stroke cases (1–5). Relative to ischemic stroke, ICH is associated with increased morbidity and mortality, severely impairing physical and mental health and negatively influencing patient quality of life (6–9). Secondary neurological impairment after ICH is an important cause of the aforementioned phenomena. Degradation of hemin—the oxidized form of heme released by erythrocytes following ICH—can lead to cell and tissue trauma via direct cytotoxic effects, the release of redox-active iron, and the depletion of cellular NADPH and glutathione stores (10). Previous studies have further reported that hemin can enter brain cells (10–13), contributing to brain edema and secondary neurological damage by inducing inflammatory reactions and damaging the blood brain barrier (BBB) (13–15). Because hemin cannot circulate within the central nervous system (CNS), accelerating hemin catabolism may promote hematoma clearance following ICH (16, 17).
Heme oxygenase is the initial and rate-limiting enzyme of heme catabolism. Three isoenzymes of heme oxygenase exist: HO-1, HO-2, and HO-3 (18) (Table 1). HO-1 is an inducible isoform, which has been extensively studied primarily for its ability to respond to oxidative stress, hemorrhage, and trauma (19–21). Previous studies have demonstrated that HO-1 is rapidly induced following ICH, and that HO-1 activators can alleviate BBB dysfunction and neurological impairment after ICH (22, 23). HO-2, the constitutive isoform, is diffusely distributed in the brain and testes (25). Previous reports have indicated that HO-2 can be induced by glucocorticoids as well as oxygen (26–29, 32, 33). The physiological role of HO-3 is not fully understood, but it most likely stems from the effects of HO-2 (30, 31). Researchers have concentrated their attention on the effects of HO-1 due to its potential as a target for intervention in ICH. In this review, we summarize recent studies focused on the relationship between HO-1 and ICH, including the role of HO-1 in secondary brain damage following ICH. We further discuss the vital role of HO-1 in ICH research, emphasizing the importance of identifying novel therapeutic targets for ICH.
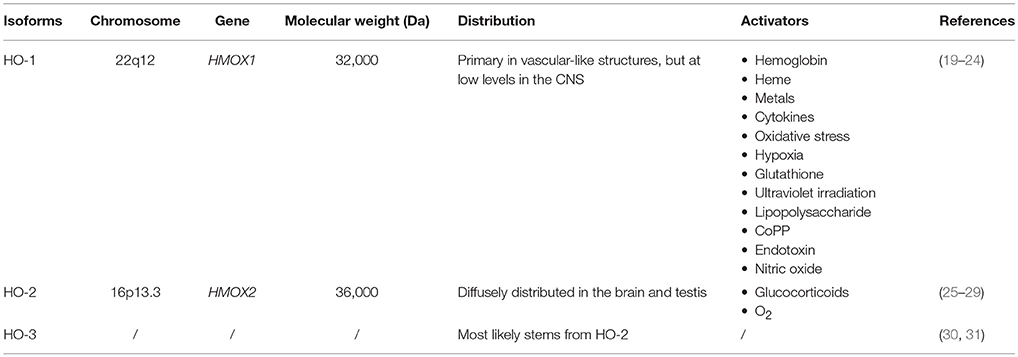
Table 1. The isoforms of heme oxygenase.
Biological Characteristics of HO-1
HO-1 is encoded by the HMOX1 gene, which is located on chromosome 22 and contains four introns and five exons (34). HO-1, also known as heat shock protein 32 (HS32), is an inducible heme oxygenase with 288 amino acids and a molecular weight of 30,000–33,000 Da (24, 35, 36) (Table 1). The HO-1 promoter contains binding sites for various transcription factors such as heat-shock factor, nuclear factor kappa B (NF-κB), activator protein 1 (AP-1), and metal regulatory elements (MREs) (37–40). Originally identified as a liver microsomal protein (41, 42), HO-1 is widely present in systemic tissues, especially in microsomes of the mononuclear macrophage system. The highest HO-1 contents have been observed in the liver, bone marrow, and spleen. Under physiological conditions, HO-1 is expressed primarily in vascular-like structures, but at low levels in the CNS. Expression of HMOX1 is induced in response to a variety of endogenous and exogenous oxidative and inflammatory signals, including hemoglobin, heme, metals, oxidative stress, hypoxia, ultraviolet irradiation, heat, nitric oxide, endotoxin, lipopolysaccharide, cytokines, cobalt protoporphyrin-IX, and glutathione (32, 43) (Table 1). Nuclear transcription factor-erythrocyte 2 related factor (Nrf2) regulates transcriptional activation of HMOX1 (44, 45).
HO-1 is a cytoprotective molecule that is crucial in maintaining cell homeostasis. HO-1 exerts strong anti-oxidant and anti-inflammatory effects by promoting heme catabolism to produce CO and bilirubin. However, it can also produce reductive ferrous iron, and therefore may be cytotoxic (46). Previous studies have revealed that HO-1 exerts strong protective effects in preclinical models of several diseases, including cardiovascular and cerebrovascular disease, diabetes, sepsis, trauma, vascular proliferative diseases, acute lung injury, liver injury, gut ischemia/reperfusion injury, and tumors (47, 48). The following section mainly focuses on the role of HO-1 in ICH.
HO-1 and ICH
Changes in HO-1 Expression After ICH
Preclinical Studies
Numerous studies have verified that HO-1 expression is rapidly induced after acute brain injury in mouse, rat, and rabbit models of traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), and ICH induced by collagenase or autologous blood injection (49–53) (Table 2). Okubo et al. (54) investigated cerebral hematoma development, brain edema formation, BBB disruption, and HO-1 expression after TBI. In their study, elevated HO-1 levels and iron deposition were observed in the ipsilateral hemisphere at 24 h after craniocerebral injury. In addition, the expression of HO-1 in the rat brain began to increase as early as 6 h after an open-skull, weight-drop-induced TBI, increasing continuously during the investigation (55) (Figure 1). These findings suggest that hematoma development upregulates the expression of HO-1 following TBI, which also promotes heme decomposition and iron deposition. Increased expression of HO-1 has also been observed in mouse models of SAH (63). Mouse models of acute ICH developed via the injection of autologous blood into the striatum exhibited increases in HO-1 levels within the first 24 h, exhibiting a 10-fold increase relative to baseline by day 5 and returning to baseline levels on day 8 (57). Similar results were confirmed in collagenase-induced mouse models of ICH (Figure 1): On the first day after ICH, the expression of HO-1 protein significantly increased in the ipsilateral striatum, peaking on day 3 and decreasing by day 7. On days 14 and 28 after ICH, the expression of HO-1 did not significantly differ from that observed in the control group (14).
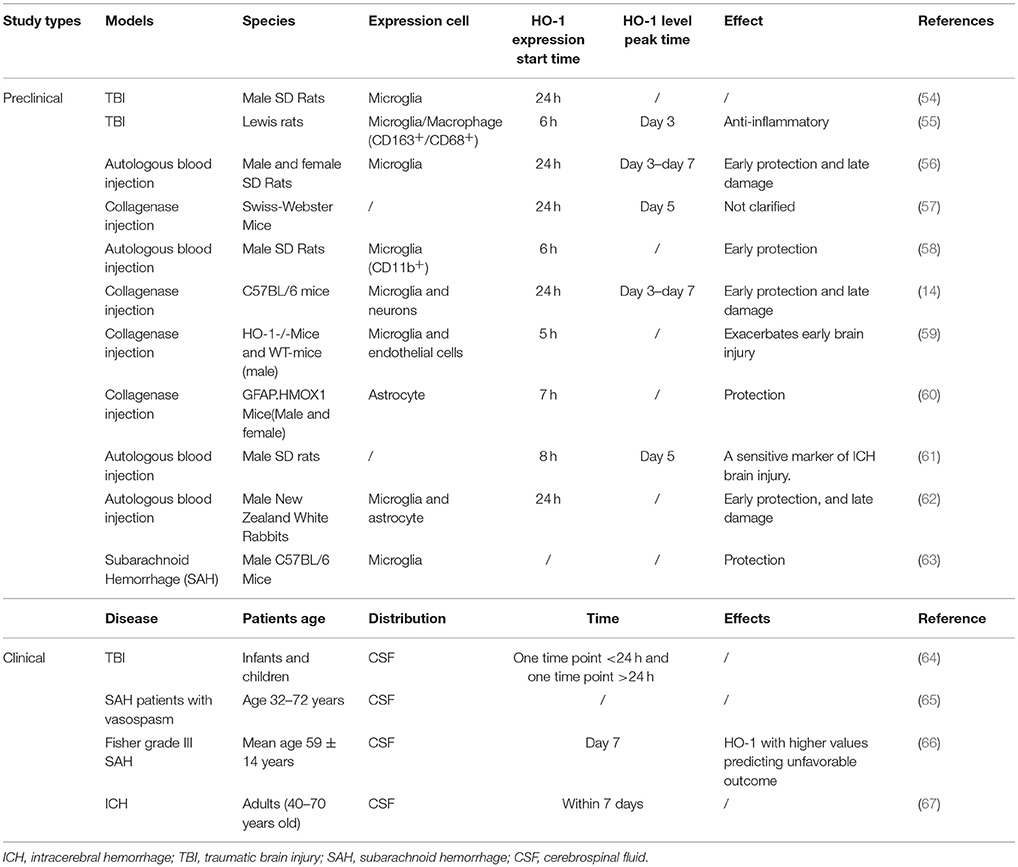
Table 2. Summary of HO-1 in preclinical and clinical studies of intracerebral hemorrhage.
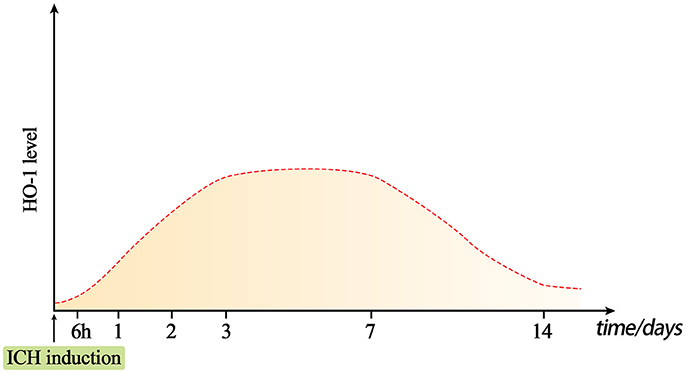
Figure 1. Dynamic changes in HO-1 levels after intracerebral hemorrhage (ICH). Under normal conditions, HO-1 content in the brain is very low. The expression of HO-1 can be rapidly induced approximately 6 h after ICH, and reaches a peak at days 3–7. The level of HO-1 may gradually decrease on day 7, but it will remain present for at least 14 days.
Similar results have been obtained using rat models. Wang and colleagues (56) established a rat model of ICH via the injection of autologous blood. In this model, the number of HO-1-positive cells around the hematoma increased as early as 1 day after ICH. Furthermore, HO-1 mRNA could be detected around the hematoma beginning on day 3, reaching a plateau at days 3 and 7 after ICH and lasting until at least 14 days after ICH. Researchers also observed that HO-1 levels begin to increase significantly at 8 h following autologous blood injection in rat models of ICH, and that such increases in HO-1 are closely related to brain edema and neurological impairment, which may reflect the severity of brain injury to some extent (61). Similar results have also been observed using rabbit models of ICH induced via autologous blood injection (62).
Clinical Studies
In one clinical study involving 48 infants and children with TBI and seven healthy controls, researchers reported that the patient group exhibited increases in cerebrospinal fluid (CSF) levels of HO-1 following TBI, and that such increases were more prominent in infants than in older children (64). Additional studies have reported that CSF levels of HO-1, bilirubin, and peroxidized lipids are significantly higher in patients with SAH who experience vasospasm than in those without vasospasm (65). Although Li et al. (67) were the first to report that serum levels of HO-1 are higher in patients with ICH than in those without, it remains to be clarified whether HO-1 exerts protective or harmful effects in patients with ICH. Additional researchers have investigated the prognostic value of CSF biomarkers related to heme metabolism, including HO-1, oxyhemoglobin, ferritin, and bilirubin in 41 patients with Fisher Grade III aneurysmal SAH. Their study indicated that CSF levels of HO-1 at day 7 post-SAH can be a potent prognostic indicator in this patient population (66).
The results of the aforementioned studies suggest that HO-1 expression is rapidly induced following ICH, likely via one or more of the following mechanisms: (1) Heme released by damaged erythrocytes after ICH directly induces HO-1 expression; (2) HO-1 expression is induced by ischemia and hypoxia around the hematoma after ICH; (3) oxidative stress after ICH leads to upregulation of HO-1 expression via the Nrf2-ARE signaling pathway. As such, HO-1 may represent a useful biomarker of damage due to heme-mediated oxidative stress following ICH (61). Furthermore, the HMOX1 promoter region contains several sites, and various stimuli may act on this region in conjunction with heme to induce HMOX1 gene expression (37, 38, 39, 40). However, the exact molecular mechanisms underlying gene-induced increases in the expression of HO-1 following ICH remain to be elucidated. Further in-depth studies are required to determine how changes in HO-1 expression following ICH can be used to improve the prognosis of patients with ICH.
The Effect of HO-1 Expression at Different Stages After ICH
Although previous studies have demonstrated that HO-1 is rapidly induced after ICH, the role of HO-1 in the early and late stages of ICH remains controversial. Wang et al. (56) investigated collagenase-induced ICH in a mouse model, reporting that HO-1 exerts both anti-oxidant and pro-oxidant properties in ICH. Their results indicated that increases in HO-1 expression on days 1–7 after ICH play a protective role. Moreover, RT-PCR results revealed a positive correlation between copper–zinc superoxide dismutase (Cu/Zn-SOD) and HO-1. Cu/Zn-SOD is a key antioxidant enzyme involved in the detoxification of SOD in the metabolism of normal cells, playing a role in antioxidative stress and exerting cytoprotective effects. Similarly, in other diseases of the CNS (e.g., Parkinson's disease, Alzheimer's disease, etc.), HO-1 acts in conjunction with Cu/Zn-SOD to exert protective effects against oxidative stress (68–70). Conversely, in the late stage of ICH (day 7), overexpression of HO-1 aggravates neurological deficits. RT-PCR have further revealed that HO-1 levels are positively correlated with those of malondialdehyde (MDA), but not with Cu-/Zn-SOD content. MDA is a product of lipid peroxidation, which can represent the production of peroxy radicals. MDA levels can also reflect the degree of oxidation in vivo. Thus, at different times after ICH, positive correlations between HO-1 and Cu-/Zn-SOD dismutase or MDA, respectively, may exert protective and detrimental effects on oxidative stress. Therefore, appropriately up-regulating HO-1 expression may play a neuroprotective role in the early stage of ICH, while down-regulation of ICH expression in later stages may attenuate neurological impairments. Another study demonstrated that HO-1 levels begin to increase 6 h after ICH, exerting neuroprotective effects from 12 h to 7 days. These results suggest that the neuroprotective effects of HO-1 are triggered in the early stages of ICH, and that such effects may be associated with HO-1 induced regulation of Nrf2 and NF-κB entry into the nucleus (58).
However, in one recent study, Zhang et al. (14) investigated the effects of an HO-1 inducer and inhibitor in a collagenase-induced mouse model of ICH. They observed that HO-1 exacerbated brain injury in the early stages of ICH (1–3 days) but promoted hematoma absorption and recovery of neurologic function in the later stages (7–28 days). The authors further reported that HO-1 aggravated brain damage via multiple mechanisms, including microglial activation, BBB damage, inflammatory reactions, neuronal cell death, oxidative damage, white matter injury, and iron accumulation. However, in the late stages of ICH, HO-1 played a neuroprotective role by increasing hematoma absorption and angiogenesis. Additional studies involving HO-1 knockout mice have indicated that HO-1 may exacerbate early neurological impairment after ICH (59).
Results regarding the effects of HO-1 expression at different stages of ICH remain controversial, likely due to factors such as the model selected (collagenase vs. autologous blood injection) and the method of treatment (e.g., anticoagulant heparin, HO-1 inducers and inhibitors, etc.) Future multi-center animal studies should utilize standardized experimental methods to obtain more reliable preclinical results.
At present, there is no effective treatment for ICH. While recombinant factor VIIa (rFVIIa) therapy (71–74), minimally invasive intracranial hematoma removal (75), immunosuppressant therapy (e.g., fingolimod) (74, 76–79), nanomedicine (80, 81), stem cell transplantation (82–86), and treatment with anti-neuroinflammatory, anti-oxidative, and neuroprotective agents (87–89) may produce some therapeutic effects, such treatments have failed to achieve breakthrough results. Some researchers have speculated that there may be a “time window” for effective treatment for ICH, similar to that observed for ischemic stroke. However, such a time window has yet to be defined. Initial studies have revealed that HO-1 expression is rapidly induced following ICH, and that dynamic changes in HO-1 levels can be observed at different stages of ICH, along with differences in the protective and detrimental effects of HO-1. Further studies are required to elucidate the mechanisms underlying the expression of HO-1 after ICH, the time-point at which HO-1-targeted treatments should be administered, and potential biomarkers for predicting patient prognosis. Targeting HO-1 expression after ICH may help to alleviate secondary brain damage and improve prognosis, although further studies regarding the effects of HO-1 during the early and late stages of ICH are required.
Effect of HO-1 Expression on ICH
Previous studies have shown that, after ICH, HO-1 is expressed in both microglia (55, 90–92) and astrocytes (45, 93, 60) (Figure 2), which exert distinctly different effects on ICH (14, 44, 60). Recent research has focused primarily on the relationship between HO-1 expression and microglia, while studies regarding the relationship between HO-1 expression and astrocytes remain relatively rare and controversial.
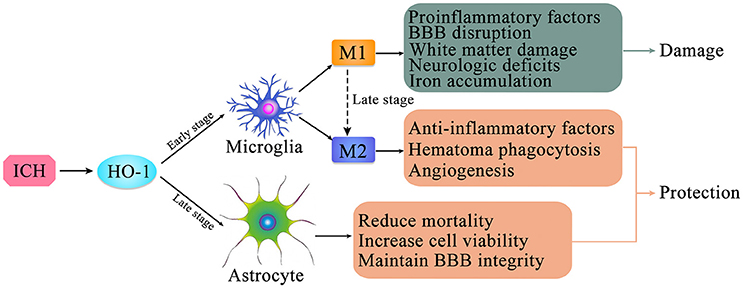
Figure 2. The expression of HO-1 in microglia and astrocytes after intracerebral hemorrhage (ICH). HO-1 is expressed in both microglia and astrocytes after ICH. HO-1 is mainly expressed in microglia in the early stage of ICH; it is primarily expressed in astrocytes in the late stage of ICH. Microglia can be polarized to two phenotypes, M1 and M2, which are in dynamic flux during ICH. After ICH, M1 microglia can produce pro-inflammatory factors, leading to destruction of the blood-brain barrier, brain edema, white matter damage, iron accumulation, and neurological deficits; M2 microglia can produce anti-inflammatory factors and promote hematoma clearance through phagocytosis and promotion of angiogenesis. In the late stage of ICH, high level HO-1 expression in astrocytes may contribute to increased cell viability, maintenance of blood-brain barrier integrity, and reduction of mortality.
Microglia, which are often referred to as phagocytes in the brain, are the primary immune effector cells of the CNS. Microglia can exert both neurotoxic and restorative effects, depending on the type of stimulation, intensity and duration of the stimulus, microenvironment, interactions with other cells, and patient age (87). Zhang et al. (14) revealed that HO-1 is abundantly expressed in microglia in the early stages after ICH, increasing microglial activation and aggravating neurological impairments. The authors performed immunofluorescence experiments to detect changes in HO-1 localization in a rat model of TBI, observing that HO-1 levels began to increase at 6 h after TBI, reaching a peak at 96 h. Immunofluorescence double-staining revealed that most HO-1 cells co-expressed CD68 and CD163, but that HO-1 was not expressed in astrocytes [based on glial fibrillary acidic protein (GFAP) expression]. Therefore, HO-1 expression may define a subtype of activated microglia/macrophage with a specific role in the anti-inflammation processes following TBI (55). In addition, in the early stage of ICH, HO-1 is mainly upregulated in microglia and endothelial cells. Compared with wild-type mice, HO-1 knockout exerts a significant protective effect on early brain function following ICH. These results suggest that this protective effect is related to a reduction in microglia activation, leukocyte infiltration, and the production of reactive oxygen species (ROS) in the early and critical stage after induction of ICH (59). Moreover, the neurological function of wild-type mice improved over time, while the neurological function of HO-1 knockout mice remained constant, suggesting that HO-1 exerted a protective effect in the wild-type group during the ICH recovery phase. Conversely, Schallner et al. (47) reported that microglia regulate blood clearance in SAH via HO-1. Furthermore, the authors reported that microglial HO-1 is necessary for attenuating neuronal cell death, vasospasm, impairments in cognitive function, and for clearance of the cerebral hematoma.
Collectively, these findings indicate that microglial activation following ICH is closely associated with HO-1. Although HO-1 is abundantly expressed in microglia, its role following ICH remains controversial (Figure 2). Previous studies have confirmed that microglial activation occurs following ICH, developing the classic M1-like (pro-inflammatory) or alternative M2-like (anti-inflammatory) phenotype (94, 95), which may fluctuate during the course of ICH. Following ICH, M1 microglia produce a large number of pro-inflammatory factors, leading to destruction of the BBB, brain edema, white matter damage, and neurological deficits. In contrast, M2 microglia produce anti-inflammatory factors and promote hematoma clearance through phagocytosis and angiogenesis (96). However, the detailed mechanisms underlying microglial activation and polarization after ICH remain to be clarified. Further studies regarding the role of microglial in secondary brain injury after ICH may provide insight into the neuroprotective effects of microglial cells. Moreover, inhibiting microglial activation may reduce neurotoxicity and improve the clinical prognosis of patients with ICH.
To date, no studies have investigated the effects of HO-1 expression on M1/M2 polarization and ICH prognosis. Thus, conclusions cannot be drawn based on existing studies. Future studies should focus on the effect of HO-1 expression on microglial polarization following ICH, and whether HO-1 expression differs between M1/M2 microglia.
Several studies have investigated the relationship between HO-1 and astrocytes. Studies involving GFAP·HMOX1 mice [driven by the glial fibrillary acidic protein (GFAP) promoter] have calculated the volume of ICH, mortality, and neurological impairment to evaluate the role of HO-1 after ICH. Such studies have demonstrated that overexpression of HO-1 in astrocytes exerts a strong neuroprotective effect after ICH and improves ICH prognosis (44, 60, 97). Furthermore, Chen-Roetling et al. (44) suggested that selective HO-1 expression in astrocytes reduces mortality, BBB destruction, hematoma-peripheral cell damage, and neurological deficits after ICH (Figure 2).
Kazuhiro et al. used immunohistochemistry to investigate the expression and localization of HO-1 in a rat model of ICH developed via autologous blood injection. Their results revealed that microglia and astrocytes co-induced the expression of HO-1 after ICH. HO-1 was mainly expressed in microglia during the acute phase (1–7 days), and in astrocytes during the subacute and chronic phases (7 days later) (93). Additional studies involving adult rabbits have verified that HO-1 is mainly expressed in microglia following acute ICH and in astrocytes during the late stages of ICH, while expression in neurons and oligodendrocytes is less common (62). Interestingly, astrocytes are also involved in microglia M1 to M2 phenotype conversion after ICH (98, 99), with a complicated internal mechanism (Figure 2).
Taken together, these findings demonstrate that the effects of HO-1 induction on nerve function differ substantially according to cell type. Further in-depth studies are required, regarding the effects of astrocytes on the function of microglia polarization, phagocytosis, angiogenesis, and other functions, to determine whether mechanisms other than those involving microglia and astrocytes play a role in HO-1 expression during ICH. These will greatly improve our understanding of ICH pathology and may provide novel strategies for follow-up treatment of ICH.
The Role of Catalytic Decomposition Products of HO-1 After ICH
The development of brain edema following ICH is a complex pathophysiological process involving several mechanisms, including toxicity associated with heme degradation-related products catalyzed by HO-l. HO-1 can catalyze the production of biliverdin, CO, and free iron (Figure 3).
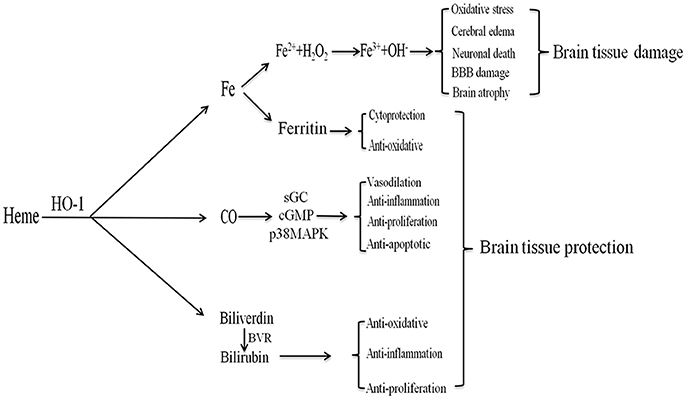
Figure 3. The products of heme metabolism catalyzed by HO-1 (heme oxygenase (HO-1) metabolizes heme). After intracerebral hemorrhage (ICH), HO-1 can catalyze heme to produce biliverdin, carbon monoxide (CO), and free iron. Iron can produce ferritin to exert cytoprotection and anti-oxidative effects; it can also be oxidized to Fe3+, which contributes to brain edema, oxidative stress, neuronal death, blood-brain barrier damage, and brain atrophy. CO can play anti-apoptotic, anti-inflammatory, anti-proliferative, and vasodilating effects through the sGC/cGMP and p38MAPK pathways. Biliverdin is rapidly converted to bilirubin under the BVR, which has anti-inflammatory, anti-oxidative and anti-proliferative effects.
Biliverdin
HO-1 catalyzes free heme and produces equimolar biliverdin, which is rapidly converted to bilirubin via a catalytic reaction involving biliverdin reductase (31). Bilirubin, the most abundant endogenous antioxidant in human serum, is significantly elevated in patients with ICH (100). Furthermore, bilirubin can scavenge free radicals, inhibit the modification of low-density lipoprotein oxidation by free radicals, resist lipid peroxidation, and exert anti-inflammatory, anti-proliferation, and cytoprotective effects (32, 28, 101). The serum bilirubin level reflects the intensity of oxidative stress and HO-1 expression in response to oxidative stress in various diseases (100). However, tissue damage occurs when the heme content is high and when hyperheme is formed, exerting a detrimental effect on prognosis in patients with ICH. As the role of bilirubin after ICH is 2-fold, it is therefore important to moderately regulate bilirubin expression while avoiding potential hazards when investigating its impact on brain function following ICH.
Co
CO is an important gas conductor and vasodilator with anti-apoptotic, anti-inflammatory, and anti-proliferative properties (43, 102, 103). Mice pretreated with CO exhibit reduced production of the serum inflammatory cytokines tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), and IL-6, along with increased production of the anti-inflammatory cytokine IL-10. In addition, these mice exhibited reduced organ damage and prolonged survival time (104). CO can play a protective role by regulating multiple signaling pathways, including the soluble guanylate cyclase (sGC) and mitogen-activated protein kinase (MAPK) pathways (105–107). The p38MAPK pathway in particular is closely associated with anti-inflammatory, anti-apoptotic, and anti-proliferative effects (105, 104, 108–110). In addition, CO lowers blood pressure by regulating the release of neurotransmitters. HO-1/CO also plays an important role in the central regulation of pulse pressure in the moving axis (32). Schallner et al. (47) demonstrated that the HO-1/CO axis increases the phagocytic activity of microglia and promotes erythrocyte clearance following ICH. When HO-1 is limited in microglia, phagocytosis is impaired, resulting in increased neuronal damage and cognitive impairment. These effects can be attenuated via the administration of exogenous CO. The HO-1/CO axis may also play an immunomodulatory role in regulating the function of antigen-presenting cells, dendritic cells, and regulatory T cells (43).
Taken together, these studies indicate that the HO-1/CO axis plays an important role in the subsequent pathophysiological changes associated with ICH. Regulation of the HO-1/CO axis to alleviate damage to nerve functions after ICH may represent a novel target for achieving neuroprotection following ICH.
Iron
Iron, which is catalyzed by HO-1, is another product of heme degradation. Iron is an essential cofactor for various cytochromes and redox-dependent proteins. However, numerous studies have indicated that iron overload can cause brain damage and irreversible neurological deficits through various pathways after ICH, including lipid peroxidation and the formation of free radicals (111–114). Iron deposition can occur within a short period of time after ICH, lasting at least 14 days in many cases (115, 116). Additional studies have demonstrated that intracerebral iron overload can cause cerebral edema, oxidative stress, BBB damage, neuronal death, brain atrophy, and combined ischemia after ICH (117). In addition, iron-induced oxidation can lead to DNA damage in the brain after ICH, and may be a direct cause of post-ICH edema. HO-derived iron can exert both anti-oxidative and cytoprotective effects. Indeed, HO-1-induced increases in ROS production and lipid oxidation are due in part to iron accumulation during the early stages of ICH. One MRI study revealed that iron content in the brain and hematoma volume influence the extent of the surrounding edema after ICH (118). Therefore, inhibiting iron accumulation may attenuate delayed injury following ICH, representing a promising target for ICH therapy (54, 119).
Deferoxamine (DFO) is an iron chelator and an inhibitor of microglial activation. Previous studies have confirmed that DFO can reduce secondary impairments in neurological function due to ICH (e.g., brain edema, neuronal death, brain atrophy, and neurological deficits) in young rats (101, 120, 121), aged rats (122, 123), pigs (124), and patients (74, 125, 126). Studies have also revealed that both iron overload and aquaporin 4 (AQP4) play a key role in the development of brain edema following ICH. Moreover, such studies have indicated that AQP4 expression is influenced by iron concentration. Notably, DFO treatment significantly inhibits AQP4 upregulation and reduces cerebral edema in rat models of ICH (119, 120). Minocycline, another iron-chelating agent, can exert neuroprotective effects by attenuating microglial activation and matrix metalloproteinases in the CNS in models of stroke, TBI, and neurodegenerative disease (127, 128). Previous researchers have also reported that minocycline can reduce iron overload and iron-induced neurological deficits after ICH in male rats (128, 129). Recent studies have further demonstrated that minocycline attenuates secondary brain injury and iron overload after ICH in aged female rats (6). Importantly, early clinical trials reported that a 400 mg dose of minocycline was safe and achieved neuroprotective serum concentrations in patients with ICH (130). Additional studies have indicated that valproate (VPA) may also play a neuroprotective role by down-regulating the expression of HO-1, thereby reducing iron release (12).
Taken together, these findings indicate that iron plays a vital yet complex role in functional neurological impairments following ICH. Iron-chelating agents can attenuate cerebral edema, neurological deficits, and brain atrophy following ICH. Indeed, the prevention of iron-mediated toxicity represents a promising therapeutic strategy for ICH (15, 131).
Anti-Inflammatory and Antioxidant Effects of HO-1 After ich
The inflammatory response after ICH plays an important role in brain edema and is positively correlated with disease severity (132). NF-κB is an important transcription factor that mediates inflammatory responses. Previous studies have revealed that HO-1 inhibits NF-κB and attenuates inflammation (133), directly interferes with the nuclear localization signal of NF-κB (134), and inhibits activation of NF-κB by regulating the GSK-3β signaling pathway (135). HO-1 also inhibits the activation of NF-κB by inhibiting the expression of inflammatory factors, including TNF-α and IL-1β (136). Previous studies have further demonstrated that fenofibrate plays an important role in neuronal protection by increasing the expression of HO-1 and decreasing the expression of NF-κB after ICH (137).
Heme released by hematoma decomposition after ICH induces oxidative damage by producing ROS and reducing glutathione (GSH) antioxidant reserves (112). Nrf2 is a key modulator of the cellular defense mechanism against oxidative stress, which positively regulates the expression of HO-1 at the transcriptional level (138, 139). Under physiological conditions, Nrf2 is sequestered in the cytoplasm by binding to Kelch-like ECH-related protein 1 (Keap1) and Cullin 3 (Figure 4). Under oxidative stress, conformational changes in Keap1 lead to the release of Nrf2, which allows Nrf2 to be transferred to the nucleus. Nuclear-translocated Nrf2 binds to the antioxidant response element located in the promoter region of the cell protection gene, and activates the transcription of antioxidant genes, including HMOX1 (138–140). This activation then leads to the production of phase II enzymes and antioxidant proteins, including HO-1, superoxide dismutase 2 (SOD2), chloramphenical acetyl transferase (CAT), glutathione S-transferase (GST), and NAD(P)H quinone dehydrogenase 1 (NQO-1) (141–143). During this process, Bach1 competes with Nrf2 for binding with Maf proteins (Bach1/Maf/ARE), resulting in decreased expression of antioxidant enzymes. Bach1 negatively regulates Nrf2-mediated transcription of antioxidant genes, and the removal of Bach1 is a prerequisite for transcriptional activation of antioxidant genes such as HMOX1 (144, 145). Wang et al. (146) demonstrated that knockout of the Nrf2 gene leads to decreased neurological function in mouse models if ICH, relative to levels observed in control mice. These findings indicate that Nrf2 may alleviate ICH-induced early brain damage by protecting against leukocyte-mediated free radical oxidative damage.
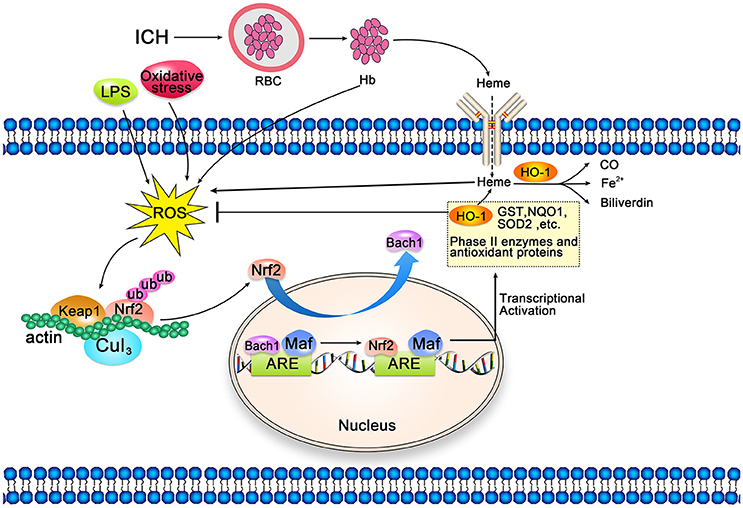
Figure 4. Schematic representation of cellular protection mechanism conferred by Nrf2-ARE pathway after intracerebral hemorrhage (ICH). Under normal conditions, Nrf2 forms a complex with actin-bound Keap1 and Cullin 3, which are isolated in the cytoplasm. Heme released after ICH, along with LPS and oxidative stress, can induce reactive oxygen species (ROS), which can induce and mediate dissociation of the Nrf2-Keap1 complex. Then, Nrf2 translocates to the nucleus and heterodimerizes with Maf. Subsequently, Nrf2 binds to antioxidant response elements (ARE) in the promoter regions of HO-1 and other target genes, activating transcription. Bach1 is an inhibitor of the Nrf2-ARE signaling pathway; thus, the removal of Bach1 is a prerequisite for transcription of this pathway.
Yang et al. (147) demonstrated that hemoglobin induces upregulation and nuclear translocation of Nrf2 in astrocytes, which may contribute to upregulation of HO-1 and thereby increase the ability to defend against heme-induced toxicity. Since heme is a product of hemoglobin degradation, this phenomenon can be regarded as an adaptive self-defense mechanism in the pathological process of ICH. Therefore, the Nrf2-HO-1 pathway can be used as a target for neuroprotection after ICH. Chen-Roetling et al. first proposed the use of Nrf2 activators for the treatment of ICH (45). The main Nrf2 activators include sulforaphane (148, 149), curcumin (150, 151), hemin (23), dimethyl fumarate (DMF) (152, 153), and tert-butylhydroquinone (TBHQ) (154). The beneficial effects of these Nrf2 activators have been validated in rodent models of ICH induced via collagenase or autologous blood injection (45). DMF and hemin have long been approved for the treatment of psoriasis (45, 155) and acute porphyria (156, 157), respectively, which may provide insight into the clinical application of Nf2 activators for the treatment of ICH.
The results of the aforementioned studies indicate that HO-1 not only exerts anti-inflammatory and anti-oxidant effects via its catalytic products bilirubin and CO, but also plays an anti-inflammatory role by inhibiting the expression of inflammatory factors and blocking the NF-κB signaling pathway. Such studies have also demonstrated that Nrf2 regulates HO-1 expression by enhancing its anti-inflammatory and anti-oxidant effects. Thus, regulating the NF-κB and Nrf2-HO-1 signaling pathways may represent a novel strategy for ensuring neuroprotection following ICH.
The Relationship Between HO-1 and ICH-Related Risk Factors
Most cases of ICH are caused by hypertension, arteriosclerosis, vascular rupture, and obesity, the latter of which is the main risk factor for diabetes, cardio-cerebrovascular diseases, and hypertension. Several studies have indicated that the induction of HO-1 and its catalytic products reduces blood pressure, delays the development of hypertension (43, 158), and reduces damage to the target organ in models of cardiovascular and cerebrovascular disease. In addition, chronic induction of HO-1 reduces body weight and corrects hyperglycemia and hyperinsulinemia (158). Zhao et al. (159) demonstrated a unique role of HO-1 in anti-oxidative stress, anti-inflammation, anti-apoptosis, and enhancement of autophagy in mouse models of diabetes with high expression of HO-1, Tg-HO-1, and mutant HO-1 (Tg-mutHO-1).
Cerebral vasospasm is one of the main causes of death after ICH. Several previous studies have revealed that HO-1 exerts anti-vasculopathy effects (18, 160–162). Suzuki et al. (162) used mouse models to confirm that inducing gene expression of HO-1 exerts anti-vasospasm effects, which are important for the prevention and treatment of delayed cerebral vasospasm. Overexpression of HO-1 after experimental SAH can inhibit heme-induced arterial contraction and reduce vasospasm (18). Moreover, previous studies have indicated that introduction of the 11R-HO-1 protein into cerebral arteries prevents vasospasm, which may aid in the development of novel treatment strategies for ICH (160).
Outlook
One major intriguing question regarding ICH treatment remains: Is there a time window during which ICH treatment must be applied to achieve significant results, similar to findings observed for ischemic stroke? Further, how should this time window be judged and defined? Because HO-1 expression is rapidly induced following ICH, exhibiting dynamic changes in structure and function throughout the course of the disease, it represents a highly promising entry point for determining whether such a time window exists. Indeed, HO-1 expression may be an appropriate biomarker for determining not only the time window for ICH treatment, but also patient prognosis. Elucidating the intrinsic mechanisms underlying the effects of HO-1 during ICH may enable the development of therapeutic strategies for ICH. Future studies should explore the following aspects: (a) the effect of HO-1 on microglial polarization after ICH; (b) HO-1 expression in different types of microglia and its influence on ICH prognosis (potentially via transcriptomics and proteomics); (c) gene-based interventions aimed at regulating HO-1 protein expression; (d) targeting transcriptional activators of HO-1, such as the Nrf2/Keap1 pathway, to affect HO-1 protein expression; (e) targeting transcriptional repressors of HO-1, such as Bach1 and micro-RNAs (miR217 and miR377), to affect HO-1 protein expression; (f) the multiple pathways that jointly regulate the level of HO-1 protein expression. At present, the key mechanism underlying secondary impairments in neurological function following ICH remain unknown, and there are no adequately effective treatments for ICH. Therefore, future studies should focus on multiple possible intervention targets. These in-depth studies of HO-1 will provide insight into novel strategies for the preservation of neurological function following ICH.
Author Contributions
Q-QL drafted the manuscript. JW repeatly modified the text structure and details. L-JL, X-YW, and Y-YS participate in the literature review and discussion about article writing and revision. All the authors revised and approved final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China to JW (grant number U1604181) and the Department of Science and Technology of Henan Province to JW (grant number 142300410390).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kim JY, Bae HJ. Spontaneous intracerebral hemorrhage: management. J Stroke (2017) 19:28–39. doi: 10.5853/jos.2016.01935
2. Steiner T, Al-Shahi Salman R, Beer R, Christensen H, Cordonnier C, Csiba L, et al. European Stroke Organisation (ESO) guidelines for the management of spontaneous intracerebral hemorrhage. Int J Stroke (2014) 9:840–55. doi: 10.1111/ijs.12309
3. Donnan GA, Hankey GJ, Davis SM. Intracerebral haemorrhage: a need for more data and new research directions. Lancet Neurol. (2010) 9:167–76. doi: 10.1016/S1474-4422(10)70001-6
4. Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. (2009) 8:355–69. doi: 10.1016/S1474-4422(09)70025-0
5. Li Y, Yang R, Li Z, Tian B, Zhang X, Wang J, et al. Urokinase vs. tissue-type plasminogen activator for thrombolytic evacuation of spontaneous intracerebral hemorrhage in basal ganglia. Front Neurol. (2017) 8:371. doi: 10.3389/fneur.2017.00371
6. Hemphill JC, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, et al. Guidelines for the management of spontaneous intracerebral hemorrhage. A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke (2015) 46:2032–60. doi: 10.1161/str.0000000000000069
7. Ziai W, Nyquist P, Hanley DF. Surgical strategies for spontaneous intracerebral hemorrhage. Semin Neurol. (2016) 36:261–8. doi: 10.1055/s-0036-1582131
8. Thabet AM, Kottapally M, Hemphill JC III. Management of intracerebral hemorrhage. Handb Clin Neurol. (2017) 140:177–94. doi: 10.1016/B978-0-444-63600-3.00011-8
9. Nakagawa K, King SL, Seto TB. Optimal hematoma volume cut points to predict functional outcome after basal ganglia and thalamic hemorrhages. Front Neurol. (2018) 9:291. doi: 10.3389/fneur.2018.00291
10. Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. (2009) 14:228–35. doi: 10.1179/135100009X12525712409931
11. Chen L, Zhang X, Chen-Roetling J, Regan RF. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J Neurosurg. (2011) 114:1159–67. doi: 10.3171/2010.10.JNS10861
12. Kwon KJ, Kim JN, Kim MK, Kim SY, Cho KS, Jeon SJ, et al. Neuroprotective effects of valproic acid against hemin toxicity: possible involvement of the down-regulation of heme oxygenase-1 by regulating ubiquitin-proteasomal pathway. Neurochem Int. (2013) 62:240–50. doi: 10.1016/j.neuint.2012.12.019
13. Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation (2012) 9:46. doi: 10.1186/1742-2094-9-46
14. Zhang Z, Song Y, Zhang Z, Li D, Zhu H, Liang R, et al. Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J Cereb Blood Flow Metab. (2017) 37:25–38. doi: 10.1177/0271678X16655814
15. Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. (2006) 5:53–63. doi: 10.1016/S1474-4422(05)70283-0
16. Chen-Roetling J, Lu X, Regan RF. Targeting heme oxygenase after intracerebral hemorrhage. Ther Targets Neurol Dis. (2015) 2:474. doi: 10.14800/ttnd.474
17. Smith A. Homeostasis of heme in health and disease: current aspects of the structural biology of heme-protein interactions and of gene regulation. DNA Cell Biol. (2002) 21:245–9. doi: 10.1089/104454902753759663
18. Ono S, Komuro T, Macdonald RL. Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. J Neurosurg. (2002) 96:1094–102. doi: 10.3171/jns.2002.96.6.1094
19. Dwyer BE, Nishimura RN, Lu SY. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res Mol Brain Res. (1995) 30:37–47.
20. Matz PG, Weinstein PR, Sharp FR. Heme oxygenase-1 and heat shock protein 70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery (1997) 40:152–60; discussion 160–2.
21. Fukuda K, Panter SS, Sharp FR, Noble LJ. Induction of heme oxygenase-1 (HO-1) after traumatic brain injury in the rat. Neurosci Lett. (1995) 199:127–30.
22. Skowronska M, Zielinska M, Wojcik-Stanaszek L, Ruszkiewicz J, Milatovic D, Aschner M, et al. Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases. J Neurochem. (2012) 121:125–34. doi: 10.1111/j.1471-4159.2012.07669.x
23. Lu X, Jing CR, Regan RF. Systemic hemin therapy attenuates blood-brain barrier disruption after intracerebral hemorrhage. Neurobiol Dis. (2014) 70:245–51. doi: 10.1016/j.nbd.2014.06.005
24. Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA. (1989) 86:99–103.
25. Wang J, Dore S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience (2008) 155:1133–41. doi: 10.1016/j.neuroscience.2008.07.004
26. Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci USA. (1996) 93:795–8.
27. Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, et al. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science (2004) 306:2093–7. doi: 10.1126/science.1105010
28. Poulos TL. Heme enzyme structure and function. Chem Rev. (2014) 114:3919–62. doi: 10.1021/cr400415k
29. Raju VS, McCoubrey WK Jr., Maines MD. Regulation of heme oxygenase-2 by glucocorticoids in neonatal rat brain: characterization of a functional glucocorticoid response element. Biochim Biophys Acta (1997) 1351:89–104.
30. Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, et al. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene (2004) 336:241–50. doi: 10.1016/j.gene.2004.04.002
31. Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. (1997) 37:517–54. doi: 10.1146/annurev.pharmtox.37.1.517
32. Ayer A, Zarjou A, Agarwal A, Stocker R. Heme oxygenases in cardiovascular health and disease. Physiol Rev. (2016) 96:1449–508. doi: 10.1152/physrev.00003.2016
33. Maines MD, Eke BC, Zhao X. Corticosterone promotes increased heme oxygenase-2 protein and transcript expression in the newborn rat brain. Brain Res. (1996) 722:83–94.
34. Kuwano A, Ikeda H, Takeda K, Nakai H, Kondo I, Shibahara S. Mapping of the human gene for inducible heme oxygenase to chromosome 22q12. Tohoku J Exp Med. (1994) 172:389–92.
35. Dwyer BE, Nishimura RN, De Vellis J, Yoshida T. Heme oxygenase is a heat shock protein and PEST protein in rat astroglial cells. Glia (1992) 5:300–5. doi: 10.1002/glia.440050407
36. Ewing JF, Maines MD. Rapid induction of heme oxygenase 1 mRNA and protein by hyperthermia in rat brain: heme oxygenase 2 is not a heat shock protein. Proc Natl Acad Sci USA. (1991) 88:5364–8.
37. Waza AA, Hamid Z, Ali S, Bhat SA, Bhat MA. A review on heme oxygenase-1 induction: is it a necessary evil. Inflamm Res. (2018) 67:579–88. doi: 10.1007/s00011-018-1151-x
38. Yan L, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-κB and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA. (1994) 91:5987–91.
39. Müller RM, Taguchi H, Shibahara S. Nucleotide sequence and organization of the rat heme oxygenase gene. J Biol Chem. (1987) 262:6795–802.
40. Weber CM, Eke BC, Maines MD. Corticosterone regulates heme oxygenase-2 and NO synthase transcription and protein expression in rat brain. J Neurochem. (1994) 63:953–62.
41. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA. (1968) 61:748–55.
42. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. (1988) 2:2557–68.
43. Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. (2016) 167:7–34. doi: 10.1016/j.trsl.2015.06.011
44. Chen-Roetling J, Kamalapathy P, Cao Y, Song W, Schipper HM, Regan RF. Astrocyte heme oxygenase-1 reduces mortality and improves outcome after collagenase-induced intracerebral hemorrhage. Neurobiol Dis. (2017) 102:140–6. doi: 10.1016/j.nbd.2017.03.008
45. Chen-Roetling J, Regan RF. Targeting the Nrf2-heme oxygenase-1 axis after intracerebral hemorrhage. Curr Pharm Des. (2017) 22:2226–2237. doi: 10.2174/1381612822666161027150616
46. Zou X, Gao J, Zheng Y, Wang X, Chen C, Cao K, et al. Zeaxanthin induces Nrf2-mediated phase II enzymes in protection of cell death. Cell Death Dis. (2014) 5:e1218. doi: 10.1038/cddis.2014.190
47. Schallner N, Pandit R, LeBlanc R III, Thomas AJ, Ogilvy CS, Zuckerbraun BS, et al. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest. (2015) 125:2609–25. doi: 10.1172/JCI78443
48. Wegiel B, Nemeth Z, Correa-Costa M, Bulmer AC, Otterbein LE. Heme oxygenase-1: a metabolic nike. Antioxid Redox Signal (2014) 20:1709–22. doi: 10.1089/ars.2013.5667
49. Fukuda K, Richmon JD, Sato M, Sharp FR, Panter SS, Noble LJ. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. (1996) 736:68–75.
50. Geddes JW, Pettigrew LC, Holtz ML, Craddock SD, Maines MD. Permanent focal and transient global cerebral ischemia increase glial and neuronal expression of heme oxygenase-1, but not heme oxygenase-2, protein in rat brain. Neurosci Lett. (1996) 210:205–8.
51. Shu L, Wang C, Wang J, Zhang Y, Zhang X, Yang Y, et al. The neuroprotection of hypoxic preconditioning on rat brain against traumatic brain injury by up-regulated transcription factor Nrf2 and HO-1 expression. Neurosci Lett. (2016) 611:74–80. doi: 10.1016/j.neulet.2015.11.012
52. Lou J, Cao G, Li R, Liu J, Dong Z, Xu L. Beta-caryophyllene attenuates focal cerebral ischemia-reperfusion injury by Nrf2/HO-1 pathway in rats. Neurochem Res. (2016) 41:1291–304. doi: 10.1007/s11064-016-1826-z
53. Li Z, Wang Y, Zeng G, Zheng X, Wang W, Ling Y, et al. Increased miR-155 and heme oxygenase-1 expression is involved in the protective effects of formononetin in traumatic brain injury in rats. Am J Transl Res. (2017) 9:5653–61.
54. Okubo S, Xi G, Keep RF, Muraszko KM, Hua Y. Cerebral hemorrhage, brain edema, and heme oxygenase-1 expression after experimental traumatic brain injury. Acta Neurochir Suppl. (2013) 118:83–7. doi: 10.1007/978-3-7091-1434-6_14
55. Liu Y, Zhang Z, Luo B, Schluesener HJ, Zhang Z. Lesional accumulation of heme oxygenase-1+ microglia/macrophages in rat traumatic brain injury. Neuroreport (2013) 24:281–6. doi: 10.1097/WNR.0b013e32835f2810
56. Wang G, Yang Q, Li G, Wang L, Hu W, Tang Q, et al. Time course of heme oxygenase-1 and oxidative stress after experimental intracerebral hemorrhage. Acta Neurochir (Wien). (2011) 153:319–25. doi: 10.1007/s00701-010-0750-2
57. Chen M, Regan RF. Time course of increased heme oxygenase activity and expression after experimental intracerebral hemorrhage: correlation with oxidative injury. J Neurochem. (2007) 103:2015–21. doi: 10.1111/j.1471-4159.2007.04885.x
58. Yin XP, Wu D, Zhou J, Chen ZY, Bao B, Xie L. Heme oxygenase 1 plays role of neuron-protection by regulating Nrf2-ARE signaling post intracerebral hemorrhage. Int J Clin Exp Pathol. (2015) 8:10156–63. doi: 10.3390/ijms19082260
59. Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain (2007) 130:1643–52. doi: 10.1093/brain/awm095
60. Chen-Roetling J, Song W, Schipper HM, Regan CS, Regan RF. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke (2015) 46:1093–8. doi: 10.1161/STROKEAHA.115.008686
61. Shang H, Yang D, Zhang W, Li T, Ren X, Wang X, et al. Time course of Keap1-Nrf2 pathway expression after experimental intracerebral haemorrhage: correlation with brain oedema and neurological deficit. Free Radic Res. (2013) 47:368–75. doi: 10.3109/10715762.2013.778403
62. Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. (2004) 63:587–97. doi: 10.1093/jnen/63.6.587
63. Schallner N, Lieberum JL, Gallo D, LeBlanc RH III, Fuller PM, Hanafy KA, et al. Carbon monoxide preserves circadian rhythm to reduce the severity of subarachnoid hemorrhage in mice. Stroke (2017) 48:2565–73. doi: 10.1161/STROKEAHA.116.016165
64. Cousar JL, Lai Y, Marco CD, Bayir H, Adelson PD, Janesko-Feldman KL, et al. Heme oxygenase 1 in cerebrospinal fluid from infants and children after severe traumatic brain injury. Dev Neurosci. (2006) 28:342–7. doi: 10.1159/000094160
65. Pyne-Geithman GJ, Morgan CJ, Wagner K, Dulaney EM, Carrozzella J, Kanter DS, et al. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. (2005) 25:1070–7. doi: 10.1038/sj.jcbfm.9600101
66. Wang KC, Tang SC, Lee JE, Lai DM, Huang SJ, Hsieh ST, et al. Prognostic value of intrathecal heme oxygenase-1 concentration in patients with Fisher Grade III aneurysmal subarachnoid hemorrhage. J Neurosurg. (2014) 121:1388–93. doi: 10.3171/2014.7.JNS131704
67. Li X, Li C, Hou L, He M, Song G, Ren S, et al. Higher level of serum heme oxygenase-1 in patients with intracerebral hemorrhage. Int Surg. (2015) 100:1220–4. doi: 10.9738/INTSURG-D-14-00086.1
68. Du X, Xu H, Jiang H, Xie J. Akt/Nrf2 activated upregulation of heme oxygenase-1 involves in the role of Rg1 against ferrous iron-induced neurotoxicity in SK-N-SH cells. Neurotox Res. (2013) 24:71–9. doi: 10.1007/s12640-012-9362-3
69. Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, et al. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer's disease. Neurobiol Aging (2008) 29:51–70. doi: 10.1016/j.neurobiolaging.2006.09.012
70. Taniguchi N, Takahashi M, Kizuka Y, Kitazume S, Shuvaev VV, Ookawara T, et al. Glycation vs. glycosylation: a tale of two different chemistries and biology in Alzheimer's disease. Glycoconj J. (2016) 33:487–97. doi: 10.1007/s10719-016-9690-2
71. Simpson E, Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Recombinant factor VIIa for the prevention and treatment of bleeding in patients without haemophilia. Cochrane Database Syst Rev. (2012). CD005011. doi: 10.1002/14651858.CD005011.pub4
72. Mayer SA. Recombinant activated factor VII for acute intracerebral hemorrhage. Stroke (2007) 38:763–7. doi: 10.1161/01.STR.0000254499.46122.22
73. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. (2008) 358:2127–37. doi: 10.1056/NEJMoa0707534
74. Mittal MK, Lackamp A. Intracerebral hemorrhage: perihemorrhagic edema and secondary hematoma expansion: from bench work to ongoing controversies. Front Neurol. (2016) 7:210. doi: 10.3389/fneur.2016.00210
75. Wu G, Wang L, Hong Z, Li C, Long X, Sheng F. Effects of minimally invasive procedures for removal of intracranial hematoma on matrix metalloproteinase expression and blood-brain barrier permeability in perihematomal brain tissues. Neurol Res. (2011) 33:300–6. doi: 10.1179/016164110X12759951866993
76. Zhang J, Shi K, Li Z, Li M, Han Y, Wang L, et al. Organ- and cell-specific immune responses are associated with the outcomes of intracerebral hemorrhage. FASEB J. (2018) 32:220–9. doi: 10.1096/fj.201700324R
77. Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. (2014) 71:1092–101. doi: 10.1001/jamaneurol.2014.1065
78. Lu L, Barfejani AH, Qin T, Dong Q, Ayata C, Waeber C. Fingolimod exerts neuroprotective effects in a mouse model of intracerebral hemorrhage. Brain Res. (2014) 1555:89–96. doi: 10.1016/j.brainres.2014.01.048
79. Rolland WB, Lekic T, Krafft PR, Hasegawa Y, Altay O, Hartman R, et al. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol. (2013) 241:45–55. doi: 10.1016/j.expneurol.2012.12.009
80. Elizarova OS, Litvinova SA, Balaban'ian V, Barskov IV, Novikova SV, Stel'mashuk EV, et al. Neuroprotective effect of recombinant human erythropoietin-loaded poly(lactic-co-glycolic) acid nanoparticles in rats with intracerebral posttraumatic hematoma. Eksp Klin Farmakol. (2012) 75:7–10.
81. Balaban'ian V, Solev IN, Elizarova OS, Garibova TL, Litvinova SA, Voronina TA. Neuroprotector effect of human recombinant erythropoietin sorbed on polymer nanoparticles studied on model of intracerebral post-traumatic hematoma (hemorrhagic stroke). Eksp Klin Farmakol. (2011) 74:17–22.
82. Shimamura N, Kakuta K, Wang L, Naraoka M, Uchida H, Wakao S, et al. Neuro-regeneration therapy using human Muse cells is highly effective in a mouse intracerebral hemorrhage model. Exp Brain Res. (2017) 235:565–72. doi: 10.1007/s00221-016-4818-y
83. Chen M, Li X, Zhang X, He X, Lai L, Liu Y, et al. The inhibitory effect of mesenchymal stem cell on blood-brain barrier disruption following intracerebral hemorrhage in rats: contribution of TSG-6. J Neuroinflammation (2015) 12:61. doi: 10.1186/s12974-015-0284-x
84. Gao L, Lu Q, Huang LJ, Ruan LH, Yang JJ, Huang WL, et al. Transplanted neural stem cells modulate regulatory T, gammadelta T cells and corresponding cytokines after intracerebral hemorrhage in rats. Int J Mol Sci. (2014) 15:4431–41. doi: 10.3390/ijms15034431
85. Wakai T, Sakata H, Narasimhan P, Yoshioka H, Kinouchi H, Chan PH. Transplantation of neural stem cells that overexpress SOD1 enhances amelioration of intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. (2014) 34:441–9. doi: 10.1038/jcbfm.2013.215
86. Stroemer P, Hope A, Patel S, Pollock K, Sinden J. Development of a human neural stem cell line for use in recovery from disability after stroke. Front Biosci. (2008) 13:2290–2. doi: 10.2741/2842
87. Siawdebrah F, Nyanzu M, Ni H, Lin X, Xu Z, Ruan L, et al. Preclinical studies and translational applications of intracerebral hemorrhage. Biomed Res Int. (2017) 2017:1–18. doi: 10.1155/2017/5135429
88. Askenase MH, Sansing LH. Stages of the inflammatory response in pathology and tissue repair after intracerebral hemorrhage. Semin Neurol. (2016) 36:288–97. doi: 10.1055/s-0036-1582132
89. Zhou Y, Wang Y, Wang J, Anne SR, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
90. Matz P, Turner C, Weinstein PR, Massa SM, Panter SS, Sharp FR. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. (1996) 713:211–22.
91. Turner CP, Panter SS, Sharp FR. Anti-oxidants prevent focal rat brain injury as assessed by induction of heat shock proteins (HSP70, HO-1/HSP32, HSP47) following subarachnoid injections of lysed blood. Brain Res Mol Brain Res. (1999) 65:87–102.
92. Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, et al. The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. (2013) 19:1135. doi: 10.1089/ars.2012.4671
93. Nakaso K, Kitayama M, Mizuta E, Fukuda H, Ishii T, Nakashima K, et al. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci Lett. (2000) 293:49–52. doi: 10.1016/S0304-3940(00)01491-9
94. Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, et al. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav Immun. (2017) 61:326–39. doi: 10.1016/j.bbi.2016.12.012
95. Taylor RA, Chang CF, Goods BA, Hammond MD, Grory BM, Ai Y, et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest. (2017) 127:280–92. doi: 10.1172/JCI88647
96. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. (2017) 54:1874–86. doi: 10.1007/s12035-016-9785-6
97. Benvenistizarom L, Regan RF. Astrocyte-specific heme oxygenase-1 hyperexpression attenuates heme-mediated oxidative injury. Neurobiol Dis. (2007) 26:688–95. doi: 10.1016/j.nbd.2007.03.006
98. Hernandez-Rabaza V, Cabrera-Pastor A, Taoro-Gonzalez L, Gonzalez-Usano A, Agusti A, Balzano T, et al. Neuroinflammation increases GABAergic tone and impairs cognitive and motor function in hyperammonemia by increasing GAT-3 membrane expression. Reversal by sulforaphane by promoting M2 polarization of microglia. J Neuroinflammation (2016) 13:83. doi: 10.1186/s12974-016-0549-z
99. Sun L-X, Lin Z-B, Lu J, Li W-D, Niu Y-D, Sun Y, et al. The improvement of M1 polarization in macrophages by glycopeptide derived from Ganoderma lucidum. Immunol Res. (2017) 65:658–65. doi: 10.1007/s12026-017-8893-3
100. Dohi K, Mochizuki Y, Satoh K, Jimbo H, Hayashi M, Toyoda I, et al. Transient elevation of serum bilirubin (a heme oxygenase-1 metabolite) level in hemorrhagic stroke: bilirubin is a marker of oxidant stress. Acta Neurochir Suppl. (2003) 86:247–9.
101. Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. (2004) 100:672. doi: 10.3171/jns.2004.100.4.0672
102. Miró O, Casademont J, Barrientos A, Urbanomárquez A, Cardellach F. Mitochondrial cytochrome c oxidase inhibition during acute carbon monoxide poisoning. Pharmacol Toxicol. (1998) 82:199–202.
103. Takagi T, Naito Y, Uchiyama K, Suzuki T, Hirata I, Mizushima K, et al. Carbon monoxide liberated from carbon monoxide-releasing molecule exerts an anti-inflammatory effect on dextran sulfate sodium-induced colitis in mice. Dig Dis Sci. (2011) 56:1663–71. doi: 10.1007/s10620-010-1484-y
104. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6:422–8. doi: 10.1038/74680
105. Kim HP, Ryter SW, Choi AM. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol. (2006) 46:411–49. doi: 10.1146/annurev.pharmtox.46.120604.141053
106. Ryter SW, Morse D, Choi AM. Carbon monoxide: to boldly go where NO has gone before. Sci STKE (2004) 2004:RE6. doi: 10.1126/stke.2302004re6
107. Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci USA. (1995) 92:1475–9.
108. Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. (2003) 9:183–90. doi: 10.1038/nm817
109. Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, et al. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol. (2003) 163:2555–63. doi: 10.1016/S0002-9440(10)63610-3
110. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AMK, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. (2000) 192:1015–26. doi: 10.1084/jem.192.7.1015
111. Haque ME, Gabr RE, Zhao X, Hasan KM, Valenzuela A, Narayana PA, et al. Serial quantitative neuroimaging of iron in the intracerebral hemorrhage pig model. J Cereb Blood Flow Metab. (2018) 38:375–81. doi: 10.1177/0271678X17751548
112. Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. (2012) 11:720–31. doi: 10.1016/S1474-4422(12)70104-7
113. Wilkinson DA, Keep RF, Hua Y, Xi G. Hematoma clearance as a therapeutic target in intracerebral hemorrhage: from macro to micro. J Cereb Blood Flow Metab. (2018) 38:741–5. doi: 10.1177/0271678X17753590
114. Hurttila H, Koponen JK, Kansanen E, Jyrkkänen HK, Kivelä A, Kylätie R, et al. Oxidative stress-inducible lentiviral vectors for gene therapy. Gene Ther. (2008) 15:1271. doi: 10.1038/gt.2008.75
115. Wang G, Shao A, Hu W, Xue F, Zhao H, Jin X, et al. Changes of ferrous iron and its transporters after intracerebral hemorrhage in rats. Int J Clin Exp Pathol. (2015) 8:10671–9.
116. Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke (2003) 34:2964–9. doi: 10.1161/01.STR.0000103140.52838.45
117. Wu H, Wu T, Li M, Wang J. Efficacy of the lipid-soluble iron chelator 2,2′-dipyridyl against hemorrhagic brain injury. Neurobiol Dis. (2012) 45:388–94. doi: 10.1016/j.nbd.2011.08.028
118. Lou M, Lieb K, Selim M. The relationship between hematoma iron content and perihematoma edema: an MRI study. Cerebrovasc Dis. (2009) 27:266–71. doi: 10.1159/000199464
119. Qing WG, Dong YQ, Ping TQ, Lai LG, Fang LD, Min HW, et al. Brain edema after intracerebral hemorrhage in rats: the role of iron overload and aquaporin 4. J Neurosurg. (2009) 110:462–8. doi: 10.3171/2008.4.JNS17512
120. Hua Y, Nakamura T, Keep RF, Wu J, Schallert T, Hoff JT, et al. Long-term effects of experimental intracerebral hemorrhage: the role of iron. J Neurosurg. (2006) 104:305–12. doi: 10.3171/jns.2006.104.2.305
121. Song S, Hua Y, Keep RF, Hoff JT, Xi G. A new hippocampal model for examining intracerebral hemorrhage-related neuronal death: effects of deferoxamine on hemoglobin-induced neuronal death. Stroke (2007) 38:2861–3. doi: 10.1161/STROKEAHA.107.488015
122. Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G. Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke (2010) 41:375–82. doi: 10.1161/STROKEAHA.109.569830
123. Okauchi M, Hua Y, Keep RF, Morgenstern L, Xi G. Effects of deferoxamine on intracerebral hemorrhage-induced brain injury in aged rats. Stroke (2009) 40:1858–63. doi: 10.1161/STROKEAHA.108.535765
124. Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke (2009) 40:2241–3. doi: 10.1161/STROKEAHA.108.539536
125. Selim M, Yeatts S, Goldstein JN, Gomes J, Greenberg S, Morgenstern LB, et al. Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke (2011) 42:3067–74. doi: 10.1161/STROKEAHA.111.617589
126. Yu Y, Zhao W, Zhu C, Kong Z, Xu Y, Liu G, et al. The clinical effect of deferoxamine mesylate on edema after intracerebral hemorrhage. PLoS ONE (2015) 10:e0122371. doi: 10.1371/journal.pone.0122371
127. Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke (2008) 39:3372–7. doi: 10.1161/STROKEAHA.108.514026
128. Zhao F, Hua Y, He Y, Keep RF, Xi G. Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke (2011) 42:3587–93. doi: 10.1161/STROKEAHA.111.623926
129. Cao S, Hua Y, Keep RF, Chaudhary N, Xi G. Minocycline effects on intracerebral hemorrhage-induced iron overload in aged rats: brain iron quantification with magnetic resonance imaging. Stroke (2018) 49:995–1002. doi: 10.1161/STROKEAHA.117.019860
130. Fouda AY, Newsome AS, Spellicy S, Waller JL, Zhi W, Hess DC, et al. Minocycline in acute cerebral hemorrhage: an early phase randomized trial. Stroke (2018) 48:2885–7. doi: 10.1161/STROKEAHA.117.018658
131. Leclerc JL, Lampert AS, Loyola AC, Schlakman B, Vasilopoulos T, Svendsen P, et al. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J Cereb Blood Flow Metab. (2017) 38:262–73. doi: 10.1177/0271678X17701459
132. Di Napoli M, Parry-Jones AR, Smith CJ, Hopkins SJ, Slevin M, Masotti L, et al. C-reactive protein predicts hematoma growth in intracerebral hemorrhage. Stroke (2014) 45:59–65. doi: 10.1161/STROKEAHA.113.001721
133. Lee JE, Cho SM, Park E, Lee SM, Kim Y, Auh JH, et al. Anti-inflammatory effects of Rubus coreanus Miquel through inhibition of NF-kappaB and MAP kinase. Nutr Res Pract. (2014) 8:501–8. doi: 10.4162/nrp.2014.8.5.501
134. Cho YJ, Kim SJ. Effect of quercetin on the production of nitric oxide in murine macrophages stimulated with lipopolysaccharide from Prevotella intermedia. J Periodontal Implant Sci. (2013) 43:191–7. doi: 10.5051/jpis.2013.43.4.191
135. Uddin MJ, Jeong SO, Zheng M, Chen Y, Cho GJ, Chung HT, et al. Carbon monoxide attenuates dextran sulfate sodium-induced colitis via inhibition of GSK-3beta signaling. Oxid Med Cell Longev. (2013) 2013:210563. doi: 10.1155/2013/210563
136. Lee JW, Choi YJ, Park JH, Sim JY, Kwon YS, Lee HJ, et al. 3,4,5-Trihydroxycinnamic acid inhibits lipopolysaccharide-induced inflammatory response through the activation of Nrf2 pathway in BV2 microglial cells. Biomol Ther (Seoul) (2013) 21:60–5. doi: 10.4062/biomolther.2012.091
137. Wang Y, Yu M, Ma Y, Wang R, Liu W, Xia W, et al. Fenofibrate increases heme oxygenase 1 expression and astrocyte proliferation while limits neuronal injury during intracerebral hemorrhage. Curr Neurovasc Res. (2017) 14:11–8. doi: 10.2174/1567202613666161014161943
138. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. (2016) 73:3221–47. doi: 10.1007/s00018-016-2223-0
139. Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. (2012) 2:200. doi: 10.3389/fonc.2012.00200
140. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
141. Bajpai VK, Alam MB, Quan KT, Kwon KR, Ju MK, Choi HJ, et al. Antioxidant efficacy and the upregulation of Nrf2-mediated HO-1 expression by (+)-lariciresinol, a lignan isolated from Rubia philippinensis, through the activation of p38. Sci Rep. (2017) 7:46035. doi: 10.1038/srep46035
142. Zhang CX, Wang T, Ma JF, Liu Y, Zhou ZG, Wang DC. Protective effect of CDDO-ethyl amide against high-glucose-induced oxidative injury via the Nrf2/HO-1 pathway. Spine J. (2017) 17:1017–25. doi: 10.1016/j.spinee.2017.03.015
143. Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells (2011) 16:123–40. doi: 10.1111/j.1365-2443.2010.01473.x
144. Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. (2013) 100:30–47. doi: 10.1016/j.pneurobio.2012.09.003
145. Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. (2007) 35:7074. doi: 10.1093/nar/gkm638
146. Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. (2007) 43:408–14. doi: 10.1016/j.freeradbiomed.2007.04.020
147. Yang Y, Xi Z, Xue Y, Ren J, Sun Y, Wang B, et al. Hemoglobin pretreatment endows rat cortical astrocytes resistance to hemin-induced toxicity via Nrf2/HO-1 pathway. Exp Cell Res. (2017) 361:217–24. doi: 10.1016/j.yexcr.2017.10.020
148. Yin X, Chen Z, Zhou J, Wu D, Bao B. Mechanisms underlying the perifocal neuroprotective effect of the Nrf2-ARE signaling pathway after intracranial hemorrhage. J Apoplexy Nerv Dis. (2015) 9:5973–86. doi: 10.2147/DDDT.S79399
149. Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke (2007) 38:3280–6. doi: 10.1161/STROKEAHA.107.486506
150. King MD, Mccracken DJ, Wade FM, Meiler SE, Jr AC, Dhandapani KM. Attenuation of hematoma size and neurological injury with curcumin following intracerebral hemorrhage in mice. J Neurosurg. (2011) 115:116–23. doi: 10.3171/2011.2.JNS10784
151. Sun Y, Dai M, Wang Y, Wang W, Sun Q, Yang GY, et al. Neuroprotection and sensorimotor functional improvement by curcumin after intracerebral hemorrhage in mice. J Neurotrauma (2011) 28:2513–21. doi: 10.1089/neu.2011.1958
152. Iniaghe LO, Krafft PR, Klebe DW, Omogbai EKI, Zhang JH, Jiping T. Dimethyl fumarate confers neuroprotection by casein kinase 2 phosphorylation of Nrf2 in murine intracerebral hemorrhage. Neurobiol Dis. (2015) 82:349–58. doi: 10.1016/j.nbd.2015.07.001
153. Zhao X, Sun G, Zhang J, Ting SM, Gonzales N, Aronowski J. Dimethyl fumarate protects brain from damage produced by intracerebral hemorrhage by mechanism involving Nrf2. Stroke (2015) 46:1923–8. doi: 10.1161/STROKEAHA.115.009398
154. Sukumari-Ramesh S, Alleyne CH Jr. Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J Mol Neurosci. (2016) 58:525–31. doi: 10.1007/s12031-016-0722-y
155. Hoefnagel JJ, Thio HB, Willemze R, Bouwes Bavinck JN. Long-term safety aspects of systemic therapy with fumaric acid esters in severe psoriasis. Br J Dermatol. (2015) 149:363–9. doi: 10.1046/j.1365-2133.2003.05433.x
156. Dhar GJ, Bossenmaier I, Petryka Z, Cardinal R, Watson C. Effects of hematin in hepatic porphyria. Ann Intern Med. (1975) 83:20–30.
157. Lamon JM, Frykholm BC, Hess RA, Tschudy DP. Hematin therapy for acute porphyria. Medicine (1979) 58:252.
158. Hosick PA, Stec DE. Heme oxygenase, a novel target for the treatment of hypertension and obesity? Am J Physiol Regul Integr Comp Physiol. (2012) 302:R207–14. doi: 10.1152/ajpregu.00517.2011
159. Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE (2013) 8:e75927. doi: 10.1371/journal.pone.0075927
160. Ogawa T, Hanggi D, Wu Y, Michiue H, Tomizawa K, Ono S, et al. Protein therapy using heme-oxygenase-1 fused to a polyarginine transduction domain attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. (2011) 31:2231–42. doi: 10.1038/jcbfm.2011.87
161. Shimada Y, Tsunoda H, Zang L, Hirano M, Oka T, Tanaka T. Synergistic induction of heme oxygenase-1 by nicaraven after subarachnoid hemorrhage to prevent delayed cerebral vasospasm. Eur J Pharmacol. (2009) 620:16–20. doi: 10.1016/j.ejphar.2009.08.009
Keywords: heme oxygenase-1, intracerebral hemorrhage, heat shock protein 32, heme, neurological impairment, microglia
Citation: Li Q-Q, Li L-J, Wang X-Y, Sun Y-Y and Wu J (2018) Research Progress in Understanding the Relationship Between Heme Oxygenase-1 and Intracerebral Hemorrhage. Front. Neurol. 9:682. doi: 10.3389/fneur.2018.00682
Received: 07 May 2018; Accepted: 30 July 2018;
Published: 20 August 2018.
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Jaime Ramos-Cejudo, Langone Medical Center, New York University, United StatesJong Eun Lee, College of Medicine, Yonsei University, South Korea
Copyright © 2018 Li, Li, Wang, Sun and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Wu, d2p1bjM2NUAxNjMuY29t