 Jose L. Vega1,2*
Jose L. Vega1,2*- 1Department of Neurosciences and Stroke, Novant Health, Forsyth Medical Center, Winston-Salem, NC, United States
- 2TeleNeurologia SAS, Medellin, Colombia
Even though sudden unexpected death in epilepsy (SUDEP) takes the lives of thousands of otherwise healthy epilepsy patients every year, the physiopathology associated with this condition remains unexplained. This article explores important parallels, which exist between the clinical observations and pathological responses associated with SUDEP, and the pathological responses that can develop when a set of autonomic reflexes known as the mammalian dive response (MDR) is deployed. Mostly unknown to physicians, this evolutionarily conserved physiological response to prolonged apnea economizes oxygen for preferential use by the brain. However, the drastic cardiovascular adjustments required for its execution, which include severe bradycardia and the sequestration of a significant portion of the total blood volume inside the cardiopulmonary vasculature, can result in many of the same pathological responses associated with SUDEP. Thus, this article advances the hypothesis that prolonged apneic generalized tonic clonic seizures induce augmented forms of the MDR, which, in the most severe cases, cause SUDEP.
Introduction
Increasingly, neurologists are coming to terms with the reality that epilepsy patients die suddenly 24–28 times more frequently than the general population (1, 2). Victims are often found dead, in bed. Seizures are occasionally witnessed prior to death, but often only a hint of one is discovered (e.g., a tongue bite) (3). Witness accounts indicate that sudden unexpected death in epilepsy (SUDEP), as this enigmatic phenomenon is known, is associated with generalized tonic-clonic seizures (GTCS) (4–7), hypoventilation, peri-ictal apnea (5, 8–10), bradycardia, ictal asystole (10), and postictal generalized electroencephalographic suppression (PGES) (11). From an epidemiological standpoint, SUDEP affects patients who are young and otherwise healthy (12–14), as well as patients who suffer from comorbid organic psychiatric disease (14).
A well-known study of SUDEP cases videotaped inside epilepsy monitoring units (EMU) from around the world demonstrated a consistent pattern that involves GTCS, ictal hyperventilation, cardiorespiratory dysfunction, and terminal apnea followed by bradycardia and asystole (10). Autopsy reports have shown pulmonary edema so consistently, (15–19) that it is considered a pathological hallmark of this condition (5, 10). Pulmonary hemorrhages, cerebral edema and/or cerebral petechial hemorrhages are also found at autopsy, although less frequently (15, 17–19). Heart and lung weights are generally increased, in association with a normal cardiac structure (12, 15, 18), suggesting that SUDEP results from an acute process unrelated to chronic cardiac disease.
As SUDEP is relatively rare and difficult to investigate (20), it is not entirely surprising that these clinical observations and pathological findings remain unexplained. Current theories include a convergence of postictal coma, airway obstruction and hypoventilation (9), central and obstructive apnea (5), susceptibility to sudden death due to the coexistence of genetic cardiac arrhythmogenicity and epilepsy (21), postictal neurovegetative breakdown (10), and imbalances in sympathetic and parasympathetic control over cardiorespiratory function (22). While these theories offer a general view of the suspected physiopathology of SUDEP, a mechanistic hypothesis is lacking.
This article provides a brief description of the mammalian dive response (MDR), a set of autonomic reflexes triggered simultaneously during sustained apnea in order to protect the brain from hypoxia. It also presents remarkable parallels which exist between the physiopathology of this response, and that of SUDEP, and hypothesizes that prolonged apneic seizures induce augmented, ictal, forms of the MDR, which can culminate in sudden death.
What Is the MDR?
Humans share with marine and other terrestrial mammals a multipronged, autonomic response to submersion known as the MDR (23, 24). Its discovery dates back to 1786 when Edmund Goodwyn [1756–1829], an inquisitive medical student at the University of Edinburgh, subjected a toad to conditions of forced water immersion while investigating the physiological correlates of death by drowning (25, 26). He observed that quickly after immersion, the toad's heart rate decelerated gradually, until it ceased. But soon after its removal from the water, the toad took a deep breath, its heart resumed beating, and it walked about “without any expressions of uneasiness” (25). Goodwyn, however, did not realize the importance of this observation, which remained in relative obscurity for several decades until it became part of the theoretical background that led Paul Bert [1833–1886] to find a similar phenomenon in ducks (26, 27). Later, Charles Richet [1850–1935] demonstrated that Bert's observation represented an oxygen-conserving reflex which is triggered when water makes contact with its nostrils and beak (28). In addition, by blocking the reflex with atropine, he was able to attribute its efferent arm to the decelerating action of the vagus nerve on the heart (29). Decades later, Andersen and colleagues performed selective denervation experiments which led them to conclude that the afferent arm of the reflex was mediated by the ophthalmic branch of the trigeminal nerve (30). Today, over 230 years after its first description, we know that this primitive, oxygen-conserving reflex, which has come to be known as “diving bradycardia” (31, 32), is only one aspect of the autonomic nervous system's response to submersion, –the MDR– which also involves apnea, peripheral vasoconstriction, and splenic contractions (24, 31).
The apnea of the MDR was first shown in anesthetized and decorticated ducks subjected to forced water immersion (33), but a similar reflex was subsequently found in several marine and terrestrial species (34–38). This involuntary apnea, which occurs simultaneously with diving bradycardia, is also associated with a profound vasoconstriction that prevents blood from circulating through peripheral tissues, thus conserving most of the available oxygen for preferential use by the brain and heart (34, 35, 39–41). Seals, for instance, experience over 90% reduction in peripheral circulation in association with a nearly 400% increase in cardiopulmonary blood flow during submersion (41). While cardiac output is reduced under these circumstances, cerebral blood flow is preserved (41) or increased (42). A fourth reflex, also triggered simultaneously, was discovered when seals were noted to exhibit hematocrit increases of more than 60% during some dives (43, 44). This effect was later found to stem from a release of resident red blood cells (RBCs) from the spleen, due to contractions promoted by systemic catecholamines (45, 46). The resulting boost in hemoglobin from these contractions helps to maximize brain oxygenation during apnea (47). Thus, the MDR is composed of four oxygen-conserving reflexes –apnea, diving bradycardia, peripheral vasoconstriction, and splenic contractions, –which act simultaneously during submersion to protect the brain from hypoxia (23, 24, 31, 48).
The Human MDR
After decades of animal research on the subject, three legendary physiologists, Irving, Sholander and Grinnell, finally reported “a good human diver” who exhibited significant diving bradycardia while swimming (49). Several years later it was shown that humans can elicit diving bradycardia by combining facial immersion with voluntary apnea (50–52). This effect occurs simultaneously with a marked peripheral vasoconstriction (53, 54), which shunts a significant portion of the total blood volume into the cardiopulmonary vasculature (55–58). This latter component of the human MDR is often referred to as the “blood shift” (55, 56, 59). However, unlike aquatic mammals, whose blood pressure is unchanged by the MDR (60), human blood pressure increases drastically by this response, reaching values as high as 280/200 (61, 62). Such an altered hemodynamic state increases cerebral blood flow (62–65) and impairs cerebral autoregulation (66), despite a concomitant reduction of cardiac output (61, 67). The human MDR also involves splenic contractions that result in transient but modest elevations in circulating RBCs (45, 68, 69). A bystander effect of these contractions, without any apparent physiological benefit to the oxygen-conserving purposes of the MDR, is the release of large numbers of splenic white blood cells (WBC) into the bloodstream (68). More recent work has shown that humans can trigger the MDR through prolonged voluntary apnea, independently of facial immersion (62, 63, 65, 69), and this effect is augmented if apnea is performed with exercise (51, 54, 65, 70). Thus, humans, like all other vertebrate species tested to date, exhibit a complex oxygen-conserving response to prolonged apnea, the MDR, which is characterized by bradycardia, vasoconstriction, hypertension, increased cardiopulmonary and cerebral blood flow, and splenic contractions (48).
Physiopathology of the Human MDR
Even though the MDR represents a beneficial set of reflexes to conserve oxygen and maximize the chances of survival during apneic conditions (23, 24), its deployment poses a major challenge to human physiology. For instance, the overwhelming parasympathetic discharge that causes diving bradycardia can result in heart rates as low as eight beats per minute, and is invariably accompanied by electrocardiographic abnormalities, including pointed T waves, ectopic beats, abnormal P waves, atrioventricular nodal and idioventricular rhythms, AV block, sinus-arrest followed by nodal or ventricular escape, and ectopic ventricular beats (31, 71–73). Combined with a simultaneous, and equally overwhelming sympathetic response, which induces severe peripheral vasoconstriction (51, 54), and hypertension (61, 62, 64) the MDR generates an autonomic conflict that can lead to dangerous conduction abnormalities (74). This has led some to hypothesize that the diving bradycardia of the MDR is to blame for some cases of sudden cardiac death (74, 75), and sudden infant death syndrome (76). Triggered while swimming, diving bradycardia is also thought to induce fatal cardiac arrhythmias in otherwise healthy individuals who harbor subclinical pro-arrhythmogenic conditions (77–80). For instance, an eye-opening study that tested for cardiac channelopathies in drowning victims, found that nearly 30% of them hosted mutations that would have made them susceptible to developing either long QT syndrome (LQTS) or catecholaminergic polymorphic ventricular tachycardia while swimming (79). These data are in sharp contrast with, for instance, the 0.05% estimated incidence of congenital LQTS in the general population (81), and further implicate diving bradycardia in the unmasking of potentially lethal arrhythmias (74, 75, 77–80, 82, 83).
The blood shift is yet another aspect of the MDR thought to induce pathology in humans, due to the abrupt shunting of excess blood into the intrapulmonary vasculature (55–58). Combined with the strong vasoconstriction characteristic of the MDR (53, 54), the blood shift (55, 56, 59) can result in elevated pulmonary capillary transmural pressure, and in the subsequent extravasation of fluid, or blood into the interstitium or into the alveolar space (84) (i.e., pulmonary edema, and pulmonary hemorrhage). This form of pulmonary pathology is often seen in breath-hold divers who induce the MDR by diving to significant depths under apneic conditions (85–87). In this population pulmonary edema becomes clinically manifest as shortness of breath, chest tightness, and the production of a pink, frothy sputum, or frank blood, upon surfacing from a dive (85–87). Rales or crackles, abnormally low arterial oxygen saturation, and reduced pulmonary performance are revealed by physical examination, pulse oximetry and spirometry, respectively (85, 86, 88). A strikingly similar form of pulmonary edema has also been reported in swimmers (89, 90). Thus, two of the main components of the human MDR, diving bradycardia and the blood shift, have been implicated in the development of cardiac arrhythmias, cardiac arrest, pulmonary edema, and pulmonary hemorrhage.
Hypothesis: A Significant Fraction of SUDEP Victims Succumb to an Unfettered Ictal Form of the MDR
As discussed above, a major effect of the MDR is the sequestration of blood inside the cardiopulmonary circulation (23, 24, 35, 55, 56). This is especially important during apneic exercise, when unrestricted blood flow to active muscles could result in a rapid depletion of the limited supply of oxygen available to the brain. It is therefore appropriate that apnea associated with exercise induces an augmented form of the MDR by comparison to that induced by apnea at rest (51, 54, 65, 70). Thus, it is hypothesized that apneic GTCS (91, 92), which by definition exhibit vigorous muscle activity, pose physiological challenges that result in augmented, ictal forms of the MDR. It is further hypothesized that unlike a MDR deployed in a conscious subject, which could be tamed by higher cortical function (75, 93), an ictal MDR deployed during GTCS, or during a period of electrographic suppression (10, 11, 94), has a greater potential to become pernicious, and induce one or more of the common pathological findings of SUDEP, namely pulmonary edema, pulmonary hemorrhage, cerebral edema, cerebral petechial hemorrhage, or cardiac arrest (see Table 1). Specifically, over-distension of pulmonary blood vessels brought on by an augmented blood shift (56, 57, 59), in combination with simultaneous and pronounced vasoconstriction (53, 54, 63, 64), can increase pulmonary capillary transmural pressure to the point of precipitating pulmonary edema and pulmonary hemorrhage (15, 17–19, 84). The magnitude of this effect is likely to correlate with the degree of MDR augmentation produced by specific seizure characteristics. For instance, a non-convulsive seizure that features a brief period of sustained apnea starting at the peak of the lung's inspiratory reserve volume is likely to induce a lesser degree of pulmonary edema or hemorrhage than a convulsive seizure that features a prolonged period of sustained apnea starting at the nadir of the tidal volume. A more severe form of pulmonary edema could occur when a prolonged ictal cry (i.e., a sustained expiratory laryngeal vocalization), precedes the onset of apnea, as the MDR is augmented if triggered at the end of expiration (106, 107). Perhaps the worst pulmonary pathology is to be expected when a prolonged apneic GTCS preceded by an ictal cry is followed by a forceful inhalation against a blocked airway (e.g., from positional causes resulting from the convulsion (9) or from laryngospasm (108)), as this would decrease intra-alveolar pressure further, resulting in the extravasation of fluid and blood into the alveoli (i.e., negative pressure pulmonary edema) (84, 109).
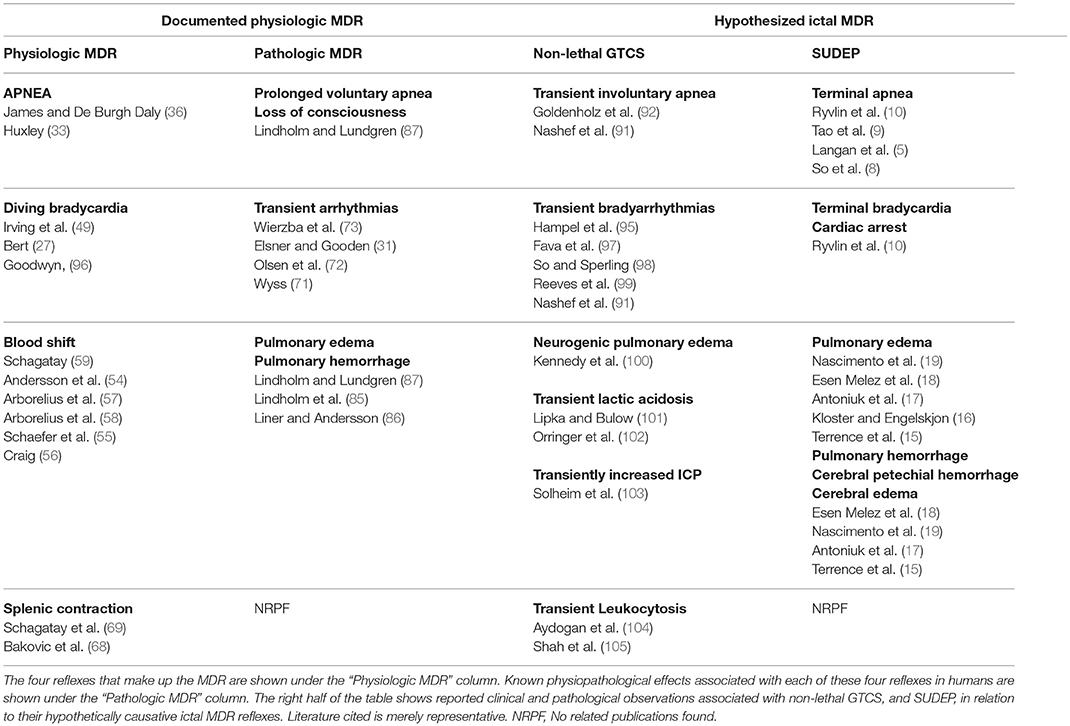
Table 1. Hypothesized pathological effects of ictal MDRs.
Aside from providing an explanation for the pulmonary pathology seen in SUDEP, the expanded intrathoracic blood volume generated by the blood shift of the MDR also accounts for the increased lung weights frequently observed in SUDEP autopsies (15, 17). However, it does not account for the increased heart weight, as blood is typically emptied from the atria and ventricles before the heart is weighed at autopsy. Similarly, this hypothesis does not intuitively explain the presence of focal myocardial fibrosis previously seen in some SUDEP autopsies (19).
If the hypertension that occurs during the human MDR (61) is increased further during an apneic GTCS, it could induce cerebral edema and/or cerebral petechial hemorrhage (17, 18) by increasing cerebral perfusion (62–65) to a critical level that disrupts cerebral vascular autoregulation (66). This notion is consistent with previous demonstrations of increased intracranial pressure (103), and increased sympathetic overactivity during seizures (110, 111), including a recent report of a probable SUDEP victim in whom excess sympathetic activity coexisted with terminal apnea for several minutes preceding death (112). Theoretically, a MDR preceded by intense hyperventilation (i.e., prolonged apnea preceded by hyperventilation), such as that seen in the majority of SUDEP cases reported by Ryvlin and colleagues (10), could further exacerbate this process by lowering arterial CO2 levels, and thus inducing cerebral vasoconstriction before ictal MDR deployment.
While, theoretically, pulmonary edema and/or pulmonary hemorrhage, in combination with cerebral edema or cerebral hemorrhage could lead to SUDEP, the autonomic conflict generated by the simultaneous parasympathetic and sympathetic influences on the heart can also lead to SUDEP by inducing ventricular arrhythmias (74). This pro-arrhythmogenic state (95, 97–99, 113) could provoke sudden death in some epilepsy patients –not always accounted as SUDEP—who consume substances that prolong the QT interval, including recreational ones such as alcohol and amphetamines, and therapeutic ones such as citalopram, and quetiapine (82, 83, 114). Furthermore, the MDR can act as an “effect amplifier” (115), responsible for SUDEP in patients who harbor clinically silent pro-arrhythmogenic gene mutations (116). Finally, the high incidence of SUDEP among the young (12–14) is congruent with reports that the MDR is strongest in younger age groups (73, 117, 118).
If, as suggested by the present hypothesis, ictal MDRs are normally triggered by prolonged apneic seizures, then all such seizures should demonstrate a predetermined potential to induce SUDEP, or SUDEP-related pathology, depending on the magnitude of their associated ictal MDR. This notion is supported by several clinical observations familiar to most neurologists (see Table 1). For instance, GTCS induce “neurogenic” pulmonary edema in some patients, through an effect that is directly related to seizure duration (100), suggesting that prolonged, intense, apneic convulsions might be more likely to induce pathogenic forms of the ictal MDR than short, mild ones. If this is true, the possibility arises that “spontaneous” neurogenic pulmonary edema is not actually spontaneous, but instead, is triggered by unwitnessed, prolonged apneic GTCS. After all, this form of pulmonary edema is often reported in patients who are found unconscious or with different degrees of altered mentation (i.e., possibly postictal) before being transported to the hospital, and who harbor epileptogenic pathologies, such as brain tumors (119), hydrocephalus (120), subdural hematomas (121), strokes (122), and subarachnoid hemorrhages (123, 124). Similarly, GTCS often induce a short-lived lactic acidosis (101, 102), which is comparable to that displayed by air-breathing aquatic mammals (34), and humans (61), surfacing from long dives. This lactic acidosis is precipitated by the blood shift, which as stated previously, restricts blood flow to active muscles and forces them to undergo anaerobic metabolism (34, 61). Consequently, it is possible that the magnitude of an ictal blood shift could be responsible for at least some of the characteristics of the transient lactic acidosis seen after GTCS (e.g., peak lactic acid level, duration, etc.). Finally, GTCS occasionally induce a transient leukocytosis (104, 105), which is thought to reflect a release of granulocytes from perivenular locations in response to systemic catecholamines (125). The present hypothesis suggests that the splenic contractions triggered during an ictal MDR (68) could play a role in generating this leukocytosis.
Because frank cessation of breathing is required to trigger the various autonomic components of the MDR, the present hypothesis cannot adequately explain cases in which SUDEP occurs independently of apnea. In addition, this hypothesis fails to explain SUDEP cases characterized by sustained hypotension, as by definition the human blood shift is accompanied by severe hypertension. Unfortunately, the latter cannot be tested against published observations due to a dearth of blood pressure measurements in the SUDEP literature. Moreover, while hypertension appears to predominate over hypotension during non-lethal seizures (111), hypotension can also occur (126), preventing us from drawing any firm conclusions that could support or refute a role for the ictal MDR in SUDEP.
How Can This Hypothesis Be Tested?
As both seizures and their postictal period can exhibit prolonged periods of apnea, (8–10, 91, 92), it should be possible to confirm whether a sequential, and causal, relationship exists between apnea and (1) bradycardia, (2) LQTS, (3) asystole or other arrhythmias, or (4) any combination of these. This can be learned from prospective observations recorded on epilepsy patients monitored for nasal airflow, chest/abdominal excursions, forehead pulse oximetry, and multichannel ECGs at the EMU. Continuous noninvasive blood pressure measurements during and after prolonged apneic GTCS can ascertain whether hypertension occurs in association with bradycardia, and whether it bears a quantifiable relationship with the blood shift, and with lactic acidosis. The latter could be documented via a combination of peripheral arterial Doppler ultrasound studies, echocardiography, chest X-rays, and blood tests performed at different times during the postictal period of prolonged apneic GTCS. If possible, extracranial and intracranial Doppler ultrasound tests can be used to determine the relationship between apnea, hypertension and cerebral blood flow during, and after, prolonged apneic GTCS. Other research questions might aim at revealing whether the magnitude of seizure-induced leukocytosis is related to the magnitude of splenic contractions. This could be accomplished by using ultrasound to correlate the absolute change in splenic size (size before vs. size after GTCS) with the corresponding peak leukocytosis after a GTCS. These studies could result in the identification of post-seizure leukocytosis as a surrogate marker for ictal MDR deployment, which in turn could be used to identify patients at risk of SUDEP. Similar research questions can be asked of post-seizure lactic acidosis. Finally, if this hypothesis were correct, it would be important to understand why some seizures induce ictal MDRs that result in SUDEP, and why others only induce leukocytosis, lactic acidosis, or mild pulmonary edema. This could be related to several factors, including total apneic time, volume of air in the lungs at the onset of apnea, arterial levels of O2 or CO2 at the onset or the end of apnea, intensity of convulsions, and electrographic characteristics, among others. Could such factors be altered during a GTCS to prevent SUDEP?
In unconscious dogs, the MDR can be aborted by artificial lung inflation (127). Could a similar intervention be used in humans to prevent SUDEP? The answer to this and other important questions related to the relationship between SUDEP and MDR physiopathology could bring us closer to understanding SUDEP, and to generating a strategic way to prevent it.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Many thanks to Drs. John Conforti and Ifeanyi Iwuchukwu for their insightful and helpful feedback on this manuscript, and to Barbara Mathis at the Forsyth Medical Center Library for obtaining important background literature.
References
1. Ficker DM, So EL, Shen WK, Annegers JF, O'Brien PC, Cascino GD, et al. Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology (1998) 51:1270–4. doi: 10.1212/WNL.51.5.1270
2. Holst AG, Winkel BG, Risgaard B, Nielsen JB, Rasmussen PV, Haunso S, et al. Epilepsy and risk of death and sudden unexpected death in the young: a nationwide study. Epilepsia (2013) 54:1613–20. doi: 10.1111/epi.12328
3. Nashef L. Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia (1997) 38:S6–8. doi: 10.1111/j.1528-1157.1997.tb06130.x
4. Nashef L, Garner S, Sander JW, Fish DR, Shorvon SD. Circumstances of death in sudden death in epilepsy: interviews of bereaved relatives. J Neurol Neurosurg Psychiatry (1998) 64:349–52. doi: 10.1136/jnnp.64.3.349
5. Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry (2000) 68:211–3. doi: 10.1136/jnnp.68.2.211
6. Hesdorffer DC, Tomson T, Benn E, Sander JW, Nilsson L, Langan Y, et al. Combined analysis of risk factors for SUDEP. Epilepsia (2011) 52:1150–9. doi: 10.1111/j.1528-1167.2010.02952.x
7. Ryvlin P, Nashef L, Tomson T. Prevention of sudden unexpected death in epilepsy: a realistic goal? Epilepsia (2013b) 54(Suppl. 2):23–8. doi: 10.1111/epi.12180
8. So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia (2000) 41:1494–7. doi: 10.1111/j.1528-1157.2000.tb00128.x
9. Tao JX, Qian S, Baldwin M, Chen XJ, Rose S, Ebersole SH, et al. SUDEP, suspected positional airway obstruction, and hypoventilation in postictal coma. Epilepsia (2010) 51:2344–7. doi: 10.1111/j.1528-1167.2010.02719.x
10. Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. (2013a) 12:966–77. doi: 10.1016/S1474-4422(13)70214-X
11. Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. (2010) 68:787–96. doi: 10.1002/ana.22101
12. Leestma JE, Walczak T, Hughes JR, Kalelkar MB, Teas SS. A prospective study on sudden unexpected death in epilepsy. Ann Neurol. (1989) 26:195–203. doi: 10.1002/ana.410260203
13. Opeskin K, Berkovic SF. Risk factors for sudden unexpected death in epilepsy: a controlled prospective study based on coroners cases. Seizure (2003) 12:456–64. doi: 10.1016/S1059-1311(02)00352-7
14. Sveinsson O, Andersson T, Carlsson S, Tomson T. The incidence of SUDEP: a nationwide population-based cohort study. Neurology (2017) 89:170–7. doi: 10.1212/WNL.0000000000004094
15. Terrence CF, Rao GR, Perper JA. Neurogenic pulmonary edema in unexpected, unexplained death of epileptic patients. Ann Neurol. (1981) 9:458–64. doi: 10.1002/ana.410090508
16. Kloster R, Engelskjon T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry (1999) 67:439–44. doi: 10.1136/jnnp.67.4.439
17. Antoniuk SA, Oliva LV, Bruck I, Malucelli M, Yabumoto S, Castellano JL. Sudden unexpected, unexplained death in epilepsy autopsied patients. Arq Neuropsiquiatr. (2001) 59:40–5. doi: 10.1590/S0004-282X2001000100009
18. Esen Melez I, Arslan MN, Melez DO, Sanli AN, Koc S. Sudden unexpected death in epilepsy: a retrospective autopsy study of 112 epileptic patients. Noro Psikiyatr Ars. (2017) 54:225–33. doi: 10.5152/npa.2016.14863
19. Nascimento FA, Tseng ZH, Palmiere C, Maleszewski JJ, Shiomi T, McCrillis A, et al. Pulmonary and cardiac pathology in sudden unexpected death in epilepsy (SUDEP). Epilepsy Behav. (2017) 73:119–25. doi: 10.1016/j.yebeh.2017.05.013
20. Devinsky O, Friedman D, Cheng JY, Moffatt E, Kim A, Tseng ZH. Underestimation of sudden deaths among patients with seizures and epilepsy. Neurology (2017) 89:886–92. doi: 10.1212/WNL.0000000000004292
21. Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia (2007) 48:859–71. doi: 10.1111/j.1528-1167.2007.01082.x
22. DeGiorgio CM, Miller P, Meymandi S, Chin A, Epps J, Gordon S, et al. RMSSD, a measure of vagus-mediated heart rate variability, is associated with risk factors for SUDEP: the SUDEP-7 Inventory. Epilepsy Behav. (2010) 19:78–81. doi: 10.1016/j.yebeh.2010.06.011
23. Scholander PF. The master switch of life. Sci Am. (1963) 209:92–106. doi: 10.1038/scientificamerican1263-92
24. Panneton WM. The mammalian diving response: an enigmatic reflex to preserve life? Physiology (Bethesda) (2013) 28:284–97. doi: 10.1152/physiol.00020.2013
26. Vega JL (2017). Edmund Goodwyn and the first description of diving bradycardia. J Appl Physiol. (1985) 123, 275–7. doi: 10.1152/japplphysiol.00221.2017
27. Bert P (1870). Leçons sur la Physiologie Comparée de la Respiration. Paris: J.B. Baillière; Londres: Hippolyte Baillière.
30. Andersen HT. The reflex nature of the physiological adjustments to diving and their afferent pathway. Acta Physiol Scand. (1963) 58:263–73. doi: 10.1111/j.1748-1716.1963.tb02648.x
31. Elsner R, Gooden B. Diving and asphyxia. A comparative study of animals and man. Monogr Physiol Soc. (1983) 40:1–168.
32. Alboni P, Alboni M, Gianfranchi L. Diving bradycardia: a mechanism of defence against hypoxic damage. J Cardiovasc Med (Hagerstown). (2011) 12:422–7. doi: 10.2459/JCM.0b013e328344bcdc
33. Huxley FM. On the reflex nature of apnea in the duck in diving: i. The reflex nature of submersion apnea. Q J Exp Physiol. (1913) 6:147–57. doi: 10.1113/expphysiol.1913.sp000137
34. Scholander PF. Experimental Investigations on the Respiratory Function in Diving Mammals and Birds. Oslo: Jacob Dybwad (1940).
35. Elsner R, Franklin DL, Van Citters RL, Kenney DW. Cardiovascular defense against asphyxia. Science (1966) 153:941–9.
36. James JE, De Burgh Daly M. Reflex respiratory and cardiovascular effects of stimulation of receptors in the nose of the dog. J Physiol. (1972) 220:673–96. doi: 10.1113/jphysiol.1972.sp009729
37. Allison DJ, Powis DA. Early and late hind-limb vascular responses to stimulation of receptors in the nose of the rabbit. J Physiol. (1976) 262:301–17. doi: 10.1113/jphysiol.1976.sp011597
38. Sessle BJ, Greenwood LF, Lund JP, Lucier GE. Effects of upper respiratory tract stimuli on respiration and single respiratory neurons in the adult cat. Exp Neurol. (1978) 61:245–59. doi: 10.1016/0014-4886(78)90244-3
39. Irving L, Solandt O, Solandt D, Fisher K. The respiratory metabolism of the seal and its adjustment to diving. J Cell Comp Physiol. (1935) 7:137–51. doi: 10.1002/jcp.1030070109
40. Grinnell SW, Irving L, Scholander PF. Experiments on the relation between blood flow and heart rate in the diving seal. J Cell Physiol. (1942) 18:341–50. doi: 10.1002/jcp.1030190309
41. Zapol WM, Liggins GC, Schneider RC, Qvist J, Snider MT, Creasy RK, et al. Regional blood flow during simulated diving in the conscious Weddell seal. J Appl Physiol Respir Environ Exerc Physiol. (1979) 47:968–73. doi: 10.1152/jappl.1979.47.5.968
42. Dormer K, Denn M, Stone H. Cerebral blood flow in the sea lion (Zalophus californianus) during voluntary dives. Comp Biochem Physiol Part A Physiol. (1977) 58:11–8. doi: 10.1016/0300-9629(77)90005-6
43. Qvist J, Hill RD, Schneider RC, Falke KJ, Liggins GC, Guppy M, et al. Hemoglobin concentrations and blood gas tensions of free-diving Weddell seals. J Appl Physiol. (1985) 61:1560–9. doi: 10.1152/jappl.1986.61.4.1560
44. Zapol WM, Hill RD, Qvist J, Falke K, Schneider RC, Liggins GC, et al. Arterial gas tensions and hemoglobin concentrations of the freely diving Weddell seal. Undersea Biomed Res. (1989) 16:363–73.
45. Hurford WE, Hong SK, Park YS, Ahn DW, Shiraki K, Mohri M, et al. Splenic contraction during breath-hold diving in the Korean ama. J Appl Physiol. (1990) 69:932–6. doi: 10.1152/jappl.1990.69.3.932
46. Hochachka PW, Liggins GC, Guyton GP, Schneider RC, Stanek KS, Hurford WE, et al. Hormonal regulatory adjustments during voluntary diving in Weddell seals. Comp Biochem Physiol B Biochem Mol Biol. (1995) 112:361–75. doi: 10.1016/0305-0491(96)85239-4
47. Castellini MA, Baskurt O, Castellini JM, Meiselman HJ. Blood rheology in marine mammals. Front Physiol. (2010) 1:146. doi: 10.3389/fphys.2010.00146
48. Foster GE, Sheel AW. The human diving response, its function, and its control. Scand J Med Sci Sports (2005) 15:3–12. doi: 10.1111/j.1600-0838.2005.00440.x
49. Irving L, Scholander PF, Grinnell SW. The respiration of the porpoise, tursiops truncatus. J Cell Physiol. (1941) 17:145–68. doi: 10.1002/jcp.1030170203
50. Campbell LB, Gooden BA, Horowitz JD. Cardiovascular responses to partial and total immersion in man. J Physiol. (1969) 202:239–50. doi: 10.1113/jphysiol.1969.sp008807
51. Stromme SB, Kerem D, Elsner R. Diving bradycardia during rest and exercise and its relation to physical fitness. J Appl Physiol. (1970) 28:614–21. doi: 10.1152/jappl.1970.28.5.614
52. Schuitema K, Holm B. The role of different facial areas in eliciting human diving bradycardia. Acta Physiol Scand. (1988) 132:119–20. doi: 10.1111/j.1748-1716.1988.tb08306.x
53. Elsner R, Gooden BA, Robinson SM. Arterial blood gas changes and the diving response in man. Aust J Exp Biol Med Sci. (1971) 49:435–44. doi: 10.1038/icb.1971.47
54. Andersson JP, Liner MH, Runow E, Schagatay EK. Diving response and arterial oxygen saturation during apnea and exercise in breath-hold divers. J Appl Physiol. (2002) 93:882–6. doi: 10.1152/japplphysiol.00863.2001
55. Schaefer KE, Allison RD, Dougherty JH, Carey CR, Walker R, Yost F, et al. Pulmonary and circulatory adjustments determining the limits of depths in breathhold diving. Science (1968) 162:1020–3.
56. Craig ABJr. Depth limits of breath hold diving (an example of Fennology). Respir Physiol. (1968) 5:14–22. doi: 10.1016/0034-5687(68)90073-X
57. Arborelius M Jr, Balldin UI, Lila B, Lundgren CE. Regional lung function in man during immersion with the head above water. Aerosp Med. (1972a) 43:701–7.
58. Arborelius MJr, Ballidin UI, Lilja B, Lundgren CE. Hemodynamic changes in man during immersion with the head above water. Aerosp Med. (1972b) 43:592–8.
59. Schagatay E. Predicting performance in competitive apnea diving. Part III deep diving. Diving Hyperb Med. (2011) 41:216–28.
60. Hochachka PW. Brain, lung, and heart functions during diving and recovery. Science (1981) 212:509–14.
61. Ferrigno M, Ferretti G, Ellis A, Warkander D, Costa M, Cerretelli P, et al. Cardiovascular changes during deep breath-hold dives in a pressure chamber. J Appl Physiol. (1997) 83:1282–90. doi: 10.1152/jappl.1997.83.4.1282
62. Willie CK, Ainslie PN, Drvis I, MacLeod DB, Bain AR, Madden D, et al. Regulation of brain blood flow and oxygen delivery in elite breath-hold divers. J Cereb Blood Flow Metab. (2015) 35:66–73. doi: 10.1038/jcbfm.2014.170
63. Palada I, Obad A, Bakovic D, Valic Z, Ivancev V, Dujic Z. Cerebral and peripheral hemodynamics and oxygenation during maximal dry breath-holds. Respir Physiol Neurobiol. (2007) 157:374–81. doi: 10.1016/j.resp.2007.02.002
64. Joulia F, Lemaitre F, Fontanari P, Mille ML, Barthelemy P. Circulatory effects of apnoea in elite breath-hold divers. Acta Physiol (Oxf). (2009) 197:75–82. doi: 10.1111/j.1748-1716.2009.01982.x
65. Kjeld T, Pott FC, Secher NH. Facial immersion in cold water enhances cerebral blood velocity during breath-hold exercise in humans. J Appl Physiol. (2009) 106:1243–8. doi: 10.1152/japplphysiol.90370.2008
66. Cross TJ, Kavanagh JJ, Breskovic T, Johnson BD, Dujic Z. Dynamic cerebral autoregulation is acutely impaired during maximal apnoea in trained divers. PLoS ONE (2014) 9:e87598. doi: 10.1371/journal.pone.0087598
67. Marabotti C, Belardinelli A, L'Abbate A, Scalzini A, Chiesa F, Cialoni D, et al. Cardiac function during breath-hold diving in humans: an echocardiographic study. Undersea Hyperb Med. (2008) 35:83–90.
68. Bakovic D, Eterovic D, Saratlija-Novakovic Z, Palada I, Valic Z, Bilopavlovic N, et al. Effect of human splenic contraction on variation in circulating blood cell counts. Clin Exp Pharmacol Physiol. (2005) 32:944–51. doi: 10.1111/j.1440-1681.2005.04289.x
69. Schagatay E, Andersson JP, Nielsen B. Hematological response and diving response during apnea and apnea with face immersion. Eur J Appl Physiol. (2007) 101:125–32. doi: 10.1007/s00421-007-0483-y
70. Lindholm P, Sundblad P, Linnarsson D. Oxygen-conserving effects of apnea in exercising men. J Appl Physiol. (1999) 87:2122–7. doi: 10.1152/jappl.1999.87.6.2122
71. Wyss V. Swimming under water in apnea and the nature of the ECG. Boll Soc Ital Biol Sper. (1956) 32:506–9.
72. Olsen CR, Fanestil DD, Scholander PF. Some effects of breath holding and apneic underwater diving on cardiac rhythm in man. J Appl Physiol. (1962) 17:461–6. doi: 10.1152/jappl.1962.17.3.461
73. Wierzba TH, Musial P, Cwikalowska H. Variable profile of individual heart rate responses to cold water immersion apnea in healthy late adolescent men. Georgian Med News (2011) (196–197):28–38.
74. Shattock MJ, Tipton MJ. 'Autonomic conflict': a different way to die during cold water immersion? J Physiol. (2012) 590:3219–30. doi: 10.1113/jphysiol.2012.229864
75. Wolf S. The bradycardia of the dive reflex–a possible mechanism of sudden death. Trans Am Clin Climatol Assoc. (1964) 76:192–200.
76. Lobban CD. The oxygen-conserving dive reflex re-examined as the principal contributory factor in sudden infant death. Med Hypotheses (1995) 44:273–7. doi: 10.1016/0306-9877(95)90179-5
77. Ackerman M, Tester D, Porter C. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. (1999) 74:1088–94. doi: 10.4065/74.11.1088
78. Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. (1999) 84:876–9. doi: 10.1016/S0002-9149(99)00458-0
79. Tester DJ, Medeiros-Domingo A, Will ML, Ackerman MJ. Unexplained drownings and the cardiac channelopathies: a molecular autopsy series. Mayo Clin Proc. (2011) 86:941–7. doi: 10.4065/mcp.2011.0373
80. Baskar S, Aziz PF. Genotype-phenotype correlation in long QT syndrome. Glob Cardiol Sci Pract. (2015) 2015:26. doi: 10.5339/gcsp.2015.26
81. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation (2009) 120:1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209
82. Vincenzi FF, Lunetta P. Citalopram-induced long qt syndrome and the mammalian dive reflex. Drug Saf Case Rep. (2015) 2:12. doi: 10.1007/s40800-015-0013-5
83. Vincenzi FF. Drug-induced long QT syndrome increases the risk of drowning. Med Hypotheses (2016) 87:11–3. doi: 10.1016/j.mehy.2015.12.015
84. West JB. Invited review: pulmonary capillary stress failure. J Appl Physiol. (2000) 89:2483–9. doi: 10.1152/jappl.2000.89.6.2483
85. Lindholm P, Ekborn A, Oberg D, Gennser M. Pulmonary edema and hemoptysis after breath-hold diving at residual volume. J Appl Physiol. (2008) 104:912–7. doi: 10.1152/japplphysiol.01127.2007
86. Liner MH, Andersson JP. Pulmonary edema after competitive breath-hold diving. J Appl Physiol. (2008) 104:986–90. doi: 10.1152/japplphysiol.00641.2007
87. Lindholm P, Lundgren CE. The physiology and pathophysiology of human breath-hold diving. J Appl Physiol. (2009) 106:284–92. doi: 10.1152/japplphysiol.90991.2008
88. Fitz-Clarke JR. Adverse events in competitive breath-hold diving. Undersea Hyperb Med. (2006) 33:55–62.
89. Adir Y, Shupak A, Gil A, Peled N, Keynan Y, Domachevsky L, et al. Swimming-induced pulmonary edema: clinical presentation and serial lung function. Chest (2004) 126:394–9. doi: 10.1378/chest.126.2.394
90. Grunig H, Nikolaidis PT, Moon RE, Knechtle B. Diagnosis of swimming induced pulmonary edema-a review. Front Physiol. (2017) 8:652. doi: 10.3389/fphys.2017.00652
91. Nashef L, Walker F, Allen P, Sander JW, Shorvon SD, Fish DR. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry (1996) 60:297–300. doi: 10.1136/jnnp.60.3.297
92. Goldenholz DM, Kuhn A, Austermuehle A, Bachler M, Mayer C, Wassertheurer S, et al. Long-term monitoring of cardiorespiratory patterns in drug-resistant epilepsy. Epilepsia (2017) 58:77–84. doi: 10.1111/epi.13606
93. Ross A, Steptoe A. Attenuation of the diving reflex in man by mental stimulation. J Physiol. (1980) 302:387–93. doi: 10.1113/jphysiol.1980.sp013250
94. Lhatoo SD, Nei M, Raghavan M, Sperling M, Zonjy B, Lacuey N, et al. Nonseizure SUDEP: sudden unexpected death in epilepsy without preceding epileptic seizures. Epilepsia (2016) 57:1161–8. doi: 10.1111/epi.13419
95. Hampel KG, Thijs RD, Elger CE, Surges R. Recurrence risk of ictal asystole in epilepsy. Neurology (2017) 89:785–91. doi: 10.1212/WNL.0000000000004266
96. Goodwyn E. Dissertatio Medica de Morteque Submersorum Investigandis. Department of Medicine, University of Edinburgh, Edinburgh (1786).
97. Fava I, Del Gaudio S, Volpe E, Paolisso G, Rizzo MR. The ictal bradycardia syndrome: a case report. Epilepsy Behav Case Rep. (2015) 4:9–12. doi: 10.1016/j.ebcr.2015.04.002
98. So NK, Sperling MR. Ictal asystole and SUDEP. Neurology (2007) 69:423–4. doi: 10.1212/01.wnl.0000268698.04032.bc
99. Reeves AL, Nollet KE, Klass DW, Sharbrough FW, So EL. The ictal bradycardia syndrome. Epilepsia (1996) 37:983–7. doi: 10.1111/j.1528-1157.1996.tb00536.x
100. Kennedy JD, Hardin KA, Parikh P, Li CS, Seyal M. Pulmonary edema following generalized tonic clonic seizures is directly associated with seizure duration. Seizure (2015) 27:19–24. doi: 10.1016/j.seizure.2015.02.023
101. Lipka K, Bulow HH. Lactic acidosis following convulsions. Acta Anaesthesiol Scand. (2003) 47:616–8. doi: 10.1034/j.1399-6576.2003.00115.x
102. Orringer CE, Eustace JC, Wunsch CD, Gardner LB. Natural history of lactic acidosis after grand-mal seizures. A model for the study of an anion-gap acidosis not associated with hyperkalemia. N Engl J Med. (1977) 297:796–9. doi: 10.1056/NEJM197710132971502
103. Solheim O, Vik A, Gulati S, Eide PK. Rapid and severe rise in static and pulsatile intracranial pressures during a generalized epileptic seizure. Seizure (2008) 17:740–3. doi: 10.1016/j.seizure.2008.05.006
104. Aydogan M, Aydogan A, Kara B, Basim B, Erdogan S. Transient peripheral leukocytosis in children with afebrile seizures. J Child Neurol. (2007) 22:77–9. doi: 10.1177/0883073807299961
105. Shah AK, Shein N, Fuerst D, Yangala R, Shah J, Watson C. Peripheral WBC count and serum prolactin level in various seizure types and nonepileptic events. Epilepsia (2001) 42:1472–5. doi: 10.1046/j.1528-1157.2001.11901.x
106. Elsner RW, Garey WF, Scholander PF. Selective ischemia in diving man. Am Heart J. (1963) 65:571–3. doi: 10.1016/0002-8703(63)90110-8
107. Asmussen E, Kristiansson NG. The “diving bradycardia” in exercising man. Acta Physiol Scand. (1968) 73:527–35. doi: 10.1111/j.1365-201X.1968.tb10892.x
108. Tavee J, Morris HIII. Severe postictal laryngospasm as a potential mechanism for sudden unexpected death in epilepsy: a near-miss in an EMU. Epilepsia (2008) 49:2113–7. doi: 10.1111/j.1528-1167.2008.01781.x
109. Lorch DG, Sahn SA. Post-extubation pulmonary edema following anesthesia induced by upper airway obstruction. Are certain patients at increased risk? Chest (1986) 90:802–5. doi: 10.1378/chest.90.6.802
110. Drake MEJr, Andrews JM, Castleberry CM. Electrophysiologic assessment of autonomic function in epilepsy. Seizure (1998) 7:91–6. doi: 10.1016/S1059-1311(98)80062-9
111. Devinsky O. Effects of seizures on autonomic and cardiovascular function. Epilepsy Curr. (2004) 4:43–6. doi: 10.1111/j.1535-7597.2004.42001.x
112. Picard RW, Migliorini M, Caborni C, Onorati F, Regalia G, Friedman D, et al. Wrist sensor reveals sympathetic hyperactivity and hypoventilation before probable SUDEP. Neurology (2017) 89:633–5. doi: 10.1212/WNL.0000000000004208
113. Morita ME, Cendes F. Recurrent ictal asystole: are we doing enough to prevent and treat it? Neurology (2017) 89:756–7. doi: 10.1212/WNL.0000000000004276
114. Kim A, Lim KS, Lee H, Chung H, Yoon SH, Yu KS, et al. A thorough QT study to evaluate the QTc prolongation potential of two neuropsychiatric drugs, quetiapine and escitalopram, in healthy volunteers. Int Clin Psychopharmacol. (2016) 31:210–7. doi: 10.1097/YIC.0000000000000124
115. Vlachos K, Georgopoulos S, Efremidis M, Sideris A, Letsas KP. An update on risk factors for drug-induced arrhythmias. Expert Rev Clin Pharmacol. (2016) 9:117–27. doi: 10.1586/17512433.2016.1100073
116. Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. (2016) 79:522–34. doi: 10.1002/ana.24596
117. Kaijser L, Sachs C. Autonomic cardiovascular responses in old age. Clin Physiol. (1985) 5:347–57. doi: 10.1111/j.1475-097X.1985.tb00755.x
118. Goksor E, Rosengren L, Wennergren G. Bradycardic response during submersion in infant swimming. Acta Paediatr. (2002) 91:307–12. doi: 10.1111/j.1651-2227.2002.tb01720.x
119. Keplinger JE, Webster JRJr. Acute pulmonary edema occurring with brain tumor: report of a case. Q Bull Northwest Univ Med Sch. (1959) 33:340–2.
120. Findler G, Cotev S. Neurogenic pulmonary edema associated with a colloid cyst in the third ventricle. Case report J Neurosurg. (1980) 52:395–8. doi: 10.3171/jns.1980.52.3.0395
121. Raha A, Wadehra A, Sandhu K, Dasgupta A. Acute subdural hematoma causing neurogenic pulmonary edema following lumbar spine surgery. J Neurosurg Anesthesiol. (2017) 29:63–4. doi: 10.1097/ANA.0000000000000254
122. Tsui P, Po H. Neurogenic pulmonary edema following acute ischemic stroke: a case report and literature review. Neurology Asia (2014) 19:195–8.
123. Mayer SA, Fink ME, Homma S, Sherman D, LiMandri G, Lennihan L, et al. Cardiac injury associated with neurogenic pulmonary edema following subarachnoid hemorrhage. Neurology (1994) 44:815–20. doi: 10.1212/WNL.44.5.815
124. Muroi C, Keller M, Pangalu A, Fortunati M, Yonekawa Y, Keller E. Neurogenic pulmonary edema in patients with subarachnoid hemorrhage. J Neurosurg Anesthesiol. (2008) 20:188–92. doi: 10.1097/ANA.0b013e3181778156
125. Athens JW, Raab SO, Haab OP, Mauer AM, Ashenbrucker H, Cartwright GE, et al. Leukokinetic studies. III the distribution of granulocytes in the blood of normal subjects J Clin Invest. (1961) 40:159–64. doi: 10.1172/JCI104230
126. Van Buren JM. Some autonomic concomitants of ictal automatism; a study of temporal lobe attacks. Brain (1958) 81:505–28. doi: 10.1093/brain/81.4.505
Keywords: sudden unexpected death in epilepsy, SUDEP, mammalian dive response, MDR, pulmonary edema, diving bradycardia, apnea, demargination
Citation: Vega JL (2018) Ictal Mammalian Dive Response: A Likely Cause of Sudden Unexpected Death in Epilepsy. Front. Neurol. 9:677. doi: 10.3389/fneur.2018.00677
Received: 21 March 2018; Accepted: 27 July 2018;
Published: 17 August 2018.
Edited by:
David M. Labiner, College of Medicine, University of Arizona, United StatesReviewed by:
Luiz Eduardo Betting, Universidade Estadual Paulista Júlio de Mesquita Filho (UNESP), BrazilFrank F. Vincenzi, University of Washington, United States
Copyright © 2018 Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose L. Vega, ZXVyZWthLnZAZ21haWwuY29t