 Stefania Sgroi
Stefania Sgroi Raffaella Tonini
Raffaella Tonini- Neuromodulation of Cortical and Subcortical Circuits Laboratory, Neuroscience and Brain Technologies Department, Istituto Italiano di Tecnologia, Genoa, Italy
The functional organization of the dorsal striatum is complex, due to the diversity of neural inputs that converge in this structure and its subdivision into direct and indirect output pathways, striosomes and matrix compartments. Among the neurotransmitters that regulate the activity of striatal projection neurons (SPNs), opioid neuropeptides (enkephalin and dynorphin) play a neuromodulatory role in synaptic transmission and plasticity and affect striatal-based behaviors in both normal brain function and pathological states, including Parkinson's disease (PD). We review recent findings on the cell-type-specific effects of opioidergic neurotransmission in the dorsal striatum, focusing on the maladaptive synaptic neuroadaptations that occur in PD and levodopa-induced dyskinesia. Understanding the plethora of molecular and synaptic mechanisms underpinning the opioid-mediated modulation of striatal circuits is critical for the development of pharmacological treatments that can alleviate motor dysfunctions and hyperkinetic responses to dopaminergic stimulant drugs.
Introduction
Opioidergic signaling is involved in several functional aspects of the peripheral and central nervous system and due to the broad distribution of opioid receptors throughout the brain, its activation modulates different neural circuits. Opiate drugs are widely used as analgesic to induce antinociception and to treat pain disorders. However, edonic effects of opiates induce addictive behaviors that entail the involvement of opioidergic system in reward processes (1, 2). Opioid receptors and the endogenous opioid peptides Enkephalin (Enk) and Dynorphin (Dyn) are expressed at striatal circuits, where the opioid system modulates the activity of spiny projection neurons (SPNs) during movement control in both a healthy state and in motor disorders such as Parkinson's disease (PD). In PD, functional changes in striatal pathways are associated with a reorganization of molecular and synaptic mechanisms that counteract the loss of dopaminergic cells. However, aberrant neuroadaptations in the striatal circuit can be responsible for critical aspects of PD, as observed in levodopa-induced dyskinesia (LID). It is still unclear what role opioid transmission plays in striatal circuity and how this system affects neural reorganization, both in PD and in response to dopaminergic treatment. Here, we review recent findings on the cell-type-specific effects of opioid transmission in the dorsal striatum, including the signaling pathways, synaptic and behavioral effects mediated by opioid ligands, as well as their interactions with dopaminergic transmission in both a PD state and in response to dopaminergic treatment with levodopa (L-DOPA).
Anatomy and Physiology of the Basal Ganglia
The basal ganglia (BG) comprise a distributed group of nuclei that include the striatum, which is composed by the caudate and putamen (CPu), the globus pallidus, with the pars externa (GPe) and interna (GPi), the subthalamic nucleus (STN); and the substantia nigra pars compacta (SNpc) and pars reticulata (SNpr). The Striatum and the STN represent the main input nuclei of the BG, while the GP and SNpr are the two output structures projecting to the thalamus and brainstem (3–7). The BG nuclei's connectivity to cortical regions provides a complex network of sensorimotor, limbic and associative information, conferring on the BG a pivotal role in the control of movement as well as in associative learning, emotion and reward-related behavior (8).
Nearly 95% of the striatum is composed of striatal projection neurons (SPNs), which are GABA (γ-aminobutyric acid)-ergic neurons that relay inhibitory efferent transmission and are rich in dopaminergic receptors (DR). These neurons are classified in two subtypes based on their projection targets, neuropeptides expression and DR subtypes (9). SPNs that express the neuropeptide Dyn and bear D1 excitatory receptors (D1Rs) (10) belong to what is termed the direct striatonigral pathway (dSPNs), projecting directly to the GPi/SNpr. On the other hand, SPNs expressing Enk and bearing D2 inhibitory receptors (D2Rs) project to the GPi/SNpr indirectly through the GPe, as part of the indirect striatopallidal pathway (iSPNs) (9, 11). In a healthy state (see Figure 1A), the activation of the direct pathway promotes movement execution by reducing the neural firing of the GPi/SNpr to the thalamus and boosting glutamatergic thalamocortical transmission. In parallel, activation of the indirect pathway reduces movement initiation, exciting GPi/SNpr transmission by inhibiting the GPe and activating the STN, ultimately leading to the inhibition of thalamocortical transmission (4, 12, 13). The concomitant activation of both striatofugal pathways maintains a balance between the direct and indirect pathways, activating specific and voluntary actions through the direct pathway and inhibiting involuntary movements through the indirect pathway (13, 14).
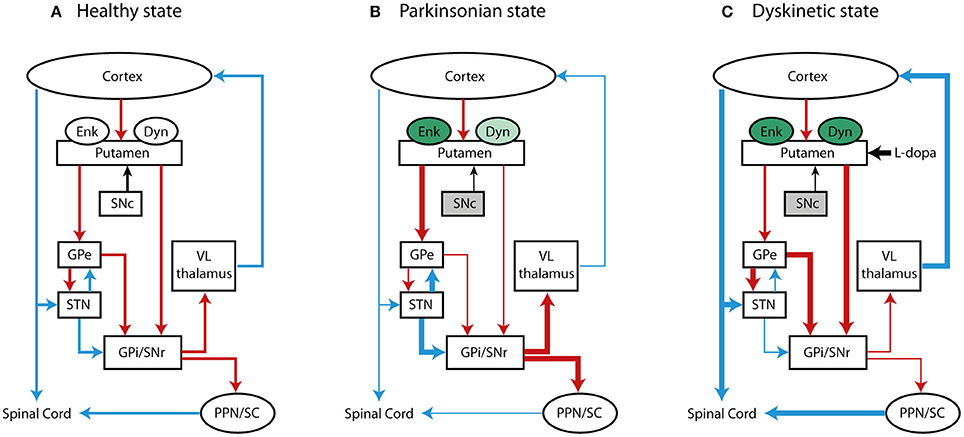
Figure 1. Basal ganglia motor loop in (A) normal, (B) PD, and (C) LID condition. The schematic represents the direct, indirect and hyperdirect pathways projecting to the thalamus and spinal cord and the changes of expression of opiod peptides, Enk and Dyn. Red, blue and black lines indicate GABA-ergic, glutamatergic and dopaminergic projections, respectively. Changes in the rate of neural transmission are indicated with thick (increased activity) and thin (decreased activity) lines. Changes in the expression of Enk and Dyn are depicted in green (increased levels) and light green (decreased levels). The gray color of substantia nigra pars compact (SNc) is representative of PD state due to the loss of dopaminergic cells.
Excitatory corticostriatal transmission on SPNs is modulated by dopaminergic input from the SNpc through “diffusion-based volume transmission,” where dopamine (DA) diffuses away from the synapse to reach extrasynaptic receptors and regulate excitability of SPNs (15). However, sparse release sites defined as active zone have been identified in the striatal DA axons to allow for a fast DA release and to generate a localized DA signal (16). Once released, DA exerts a dual effect on striatal neurons (17), exciting the direct pathway by binding to D1Rs and inhibiting the indirect pathway by binding to D2Rs. DA discharge from the dopaminergic neurons of the SNpc is crucial for the initiation and execution of motor sequences (14, 18).
The Opioidergic System: Peptides and Receptors
Enk, Dyn and β-endorphin belong to family of endogenous peptides produced through the proteolytic cleavage of protein precursors such as preproenkephalin-A (PPENK), which forms six copies of methionine-Enk (Met-Enk) and one copy of leucine-Enk; preproenkephalin-B (also known as preprodynorphin), which produces Dyn and endorphin; and finally, proopiomelanocortin, which produces β-endorphin. The endogenous peptides have different degrees of selectivity for the opioid receptors; Enk binds δ opioid receptors (DORs) and μ opioid receptors (MORs), Dyn is selective for κ-opioid receptors (KORs), and β-endorphin binds MORs (1).
Opioid receptors (ORs) are seven-transmembrane receptors and belong to a superfamily of G protein-coupled receptors (GPCRs) with inhibitory activity (Gαi/o) on cellular excitability and synaptic transmission (1). OR activity promotes the activation of G-protein-coupled inwardly rectifying K+ channels, inhibits Ca+ channels and adenylyl cyclase (AC), and reduces neurotransmitter release and neural activity (19, 20). ORs are broadly distributed in the brain, with some structures exhibiting higher expression of a specific type of receptor, while others have three overlapping receptors that can interact locally with one another in synergistic or antagonistic ways (21).
A broad distribution of MORs has been observed in the thalamus, amygdala and locus coeruleus (1), and in the thalamic afferents to the striatum (22). MOR expression has also been observed in CPu striosomal compartments that project to the SNpc (23) (Figure 2). Specifically, MORs are expressed in striosomes both on dSPNs enriched in Dyn precursor and on iSPNs (24), where they colocalize with D2Rs in dendrites (25). MORs are also expressed on striatal cholinergic interneurons (26, 27).
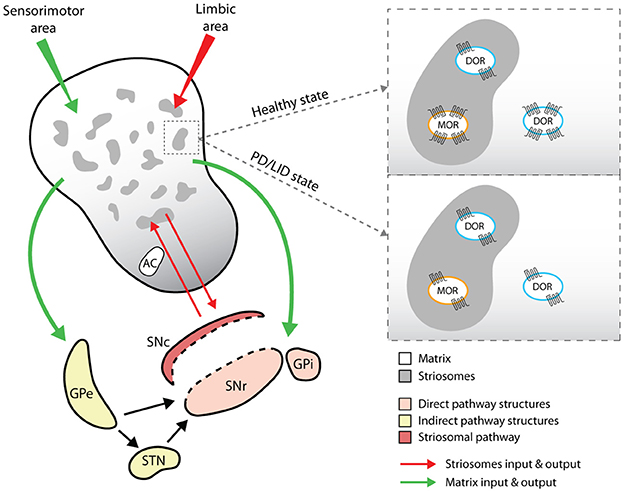
Figure 2. Representative cartoon of opioid receptors distribution and expression in striosomes and matrix compartments of the dorsal striatum. The drawing shows the different input and output pathways related to the striosomes (red lines) and matrix compartments (green lines) and the changes in the expression levels of MOR and DOR. GPe (globus pallidus pars externa) and STN (subthalamic nucleus) indicate the indirect pathway targets; SNr (substantia nigra pars reticulata) and GPi (globus pallidus pars interna) indicate the direct pathway targets; SNc (substantia nigra pars compacta) is the target of the specific pathway arising from the striosomal compartments.
DORs are abundant in layers II, III, IV and V of the cerebral cortex and in the striosomes and matrix compartments of the CPu, with a higher dorsolateral distribution than ventromedial (28). On a cellular level, DORs are expressed within the nucleus and in the somatodendritic area on iSPNs, but not on dSPNs (24, 29).
KORs are predominantly found in the medial sector of the CPu and in the nucleus accumbens and showed a higher coexpression with D1Rs (24). They are also localized presynaptically in the nigrostriatal afferents of the SNpc.
Opioid Receptor Signaling
The activation of opioidergic GPCRs by endogenous opioid peptides or exogenous agonists leads to the dissociation of Gα/βγ subunits that stimulate various intracellular effectors. The inhibitory activity of opioid receptors includes several processes that are selectively initiated by the Gα and Gβγ protein subunits. The Gα subunit inhibits AC by decreasing intracellular cyclic adenosine monophosphate (cAMP) levels and activates the inward-rectifier K+ channel, leading to the hyperpolarization of the cellular membrane and the inhibition of neural activity (30). The inhibition of AC and cAMP by the Gα subunit can also lead to a reduction of Ca2+ conductance (31), although this reduction is predominately induced by the direct binding of Gβγ subunit to the channel, and the consequent decrease in neurotransmitter release. Indeed, activation of KORs on the nigrostriatal afferents of the SNpc reduces the release of DA and influences the kinetics of the DA transporter (32, 33). Intrastriatal injection of a MOR agonist alters extracellular DA levels in the shell and core of the nucleus accumbens and in the rostral and caudal subregions of the dorsal striatum, where the reduction is thought to be due to presynaptic activation of MORs on DA terminals (34, 35). Activation of MORs on striatal cholinergic interneurons reduces Ach release and decreases cholinergic interneurons excitability (26, 27).
Besides inhibiting the AC /cAMP, opioid receptors shape several other cellular responses. The interaction with different G proteins, β-arrestins or kinases, can promote the activation of different effectors or signaling pathways (36), or prompt the internalization and desensitization of receptor functional activity (19, 37), with significant changes in behavior (38). The direct activation of opioid receptors and the release of Gβγ subunits can promote the activation of mitogen-activated protein kinases (MAPKs) (19, 39). Notably, activation of MAPK can be also mediated by binding of DA to D1R. In the dorsal striatum of PD animal models, pulsatile replacement of DA, for example by L-DOPA treatment, leads to an overstimulation of the direct striatonigral pathway that promotes the activation of MAPK and its downstream effectors, such as extracellular signal-regulated kinases ERK1/2 or transcription factors (40–42). Increased levels of phosphorylated ERK (p-ERK) or immediate early genes are associated with aberrant cellular responses and dysfunctional behaviors in PD and LID state (43–45). Therefore, opioidergic and dopaminergic receptors could both activate postsynaptic signaling cascades that converge to ultimately promote an increase of proteins and transcriptional factors that affect striatal-based behaviors. However, it is still unclear whether alterations of the striatal motor function arise from a synergic activity of the dopaminergic and opioidergic system or if opioid transmission only modulates the molecular and synaptic mechanisms mediated by dopaminergic transmission.
Compartment-Specificity Localization of Opioid Receptors in the Dorsal Striatum
Beyond the classical division of the striatum into the direct and indirect pathways, this structure is also subdivided into striosomes (defined as striatal bodies) and matrix compartments (Figure 2), which are defined according to neurotransmitter and receptor segregation, afferent and efferent connections (46), signaling cascade activation (47) and neurophysiological features (48). Striosomes represent about 10–15% of the dorsal striatum and are mainly localized in the medial sector of the CPu (29, 49), where they are characterized by acetylcholinesterase (AchE)-poor zones and by immunoreactivity against Enk, substance P and GABA (50). The matrix compartment comprises 85% of the remaining striatum. It is rich in AchE, contains calcium-binding proteins such as parvoalbumin and calbindin, and is directly affiliated with the sensorimotor system (51). Both striosomes and matrix contain dSPNs and iSPNs, although dSPNs are more prevalent in the striosomal compartment and project predominantly to dopaminergic neurons in the SNpc (50, 52).
The matrix and striosomal compartments also receive inputs from different cortical areas; striosomes are related to the limbic area, whereas the matrix is associated with sensorimotor and associative areas (53) (see Figure 2). Overall, this complex striatal subdivision, together with a discrete distribution of neuromodulators between matrix and striosomes compartments, reflects that SPNs functional activity might differ in compartment-specific manner and affect different striatal-based behaviors (54).
While dSPNs and iSPNs are broadly distributed in both striosomes and matrix, opioid-mediated synaptic transmission seems to segregate (46), perhaps due to the different distributions of opioid receptors on dSPNs and iSPNs in these compartments. For example, application of MOR and DOR agonists reduces GABAergic synaptic responses in both dSPNs and iSPNs predominantly in the striosomal compartment, but not in the matrix (29, 48). Specifically, the binding of Enk to DORs located on iSPNs collaterals that synapse on dSPNs, suppresses the inhibition of dSPNs only in the striosomes, but not in matrix, leading to strengthened striosomal dSPNs responses to corticostriatal inputs (29). The behavioral implications of this connectivity might be relevant in PD, where changes in the levels of the endogenous opioid Enk might promote or reduce dSPNs response to cortical inputs, thereby affecting the release of DA through the striatonigral pathway (29).
Opioid-Mediated Neurotransmission and Synaptic Plasticity in the Dorsal Striatum
The first neurophysiological studies on the opioid-mediated neurostransmission at striatal circuits investigated the role of these neuropeptides in the modulation of glutamatergic inputs mainly arising from the cortex. These studies showed that MOR and DOR agonists inhibited glutamatergic inputs to the striatum (55) and more specifically, selective MOR agonists reduced the excitatory inputs at the corticostriatal level in both striosomes and matrix compartments (48, 56). In addition, the application of exogenous MOR and DOR agonists or the release of endogenous opioids induced long-term depression (LTD) on striatal SPNs in both the DLS and the dorsomedial striatum (DMS). Specifically, MOR activation inhibited thalamostriatal excitatory inputs, whereas the activation of DOR inhibited corticostriatal inputs; these results indicate the specificity of opioid-mediated synaptic plasticity in the dorsal striatum (22). Interestingly, applying an exogenous KOR agonist induced LTD more selectively in the DLS than in the DMS, suggesting subregional specificity of KOR-mediated synaptic plasticity (22) (Table 1). This subregional difference between the DLS and DMS might be related to their distinct functional roles in motor control. Behavioral studies demonstrate that the DLS is more connected to the control of body movements rather than to more general control of locomotor activity (58, 59). In PD animals treated with L-DOPA, higher levels of Dyn precursor (PDYN) mRNA, selective for KOR binding, are expressed in the DLS than the DMS (60–62). Moreover, higher PDYN mRNA expression in the lateral striatal portion of the DA-denervated hemisphere correlates only with the severity of dyskinesia, instead of with locomotor variables that define animals' spontaneous motion (60, 62).
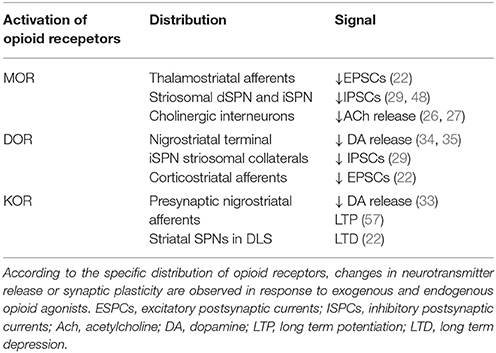
Table 1. Summary of opioid-mediated neurotransmission and synaptic plasticity in the dorsal striatum.
Opioids have been shown to regulate striatal LTD (22). In contrast, their effect on long-term potentiation (LTP) in the dorsal striatum remains unexplored. Most of the studies that have attempted to characterize the role of opioids in LTP have examined different functional areas, such as the ventral tegmental area (63), hippocampus or C-fiber of the spinal dorsal horn (64). A recent study investigated the effect of KOR activation on LTP in the corticostriatal pathway (57), and demonstrated that applying Dyn reduced the release of DA, as expected by binding to KOR on DA nigrostriatal terminals. Moreover, selective activation of the D1R-SPNs that promote the co-release of Dyn also led to impared corticostriatal LTP, likely due to the KOR-mediated reduction in DA release from the nigrostriatal pathway (57) (Table 1).
Opioid Neurontransmission in Parkinson's Disease and Levodopa-Induced Dyskinesia
The broad distribution of opioid receptors in the striatum and their interplay with dopaminergic transmission point at critical role for opioidergic neuropeptides in modulating striatal activity and motor control, in particular, both in healthy and pathological states, such as in PD. This is a progressive neurodegenerative disorder characterized by the loss of dopaminergic cells in the SNpc, which results in motor deficits (i.e., bradykinesia, rest tremor, rigidity, and postural and gait impairment) (65, 66). PD patients develop these symptoms only after a significant depletion of striatal DA– by 60 to 80% (67) – likely because of compensatory DA production by surviving neurons or unknown compensatory mechanisms within or outside of the BG (68). Furthermore, the loss of dopaminergic neurons in the SNpc results in a functional imbalance in the two major output pathways of the striatum: hypoactivity in the direct circuit and hyperactivity in the indirect circuit. This imbalance leads to an overstimulation of the GPi/SNpr which decreases thalamic input to motor cortical areas, resulting in reduced movement and classical Parkinson's symptoms (Figure 1B).
Various animal models of PD are used to better understand the disease's pathophysiology, but none of them fully exhibit all PD symptoms, nor do they develop a neurodegenerative state similar to that in PD patients. For this reason, the most suitable animal model depends on the scientific question being investigated (69). Parkinsonian motor deficits due to DA depletion or DA neuronal death are usually recreated in animals through the injection of selective neurotoxins such as 6-hydroxydopamine (6-OHDA) in rat and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mouse or primate, while specific molecular events and protein aggregation are investigated using genetic models of PD-related mutations (70).
PD symptoms can be alleviated with dopaminergic treatments that aim to replace the DA deficiency in the nigrostriatal pathway. No curative treatments exist for PD patients, and currently available therapies are symptomatic. To date, L-DOPA remains the most effective drug for exogenous dopaminergic replacement and for counteracting PD symptoms. However, as the disease progresses and dosages of L-DOPA increase, many patients develop disabling complications, including severe fluctuations in motor function (on-off phenomena) and abnormal involuntary movements called L-DOPA-induced dyskinesia (LID) (71, 72). The pathophysiology of LID has been associated with aberrant activation of the direct striatal pathway and with increased levels of the endogenous opioid neuropeptides Enk and Dyn (Figure 1C). It is still unclear whether opioid transmission can affect the neural reorganization of striatal pathways, and if changes in opioid expression might have a compensatory or synergistic effect on striatal-based behaviors in PD and LID.
Opioid Peptide Expression in PD and LID
Several studies have been conducted in animal models to investigate changes in the expression of endogenous opioids in the dorsal striatum and their association with motor impairment and dyskinetic movements. Indeed, DA and its binding to D1Rs and D2Rs can modulate the striatal levels of mRNA expression of Dyn and Enk neuropeptide precursors (PDYN and PPENK). Changes in PDYN and PPENK mRNA levels have been observed in PD, where DA transmission is lost, and in LID, during the exogenous replacement of DA (Figure 1).
In the striatum of 6-OHDA and MPTP animal models (60, 62, 73, 74), as well as in PD patients (75), the levels of PPENK mRNA expression are increased, irrespective of L-DOPA treatment. The levels of PPENK mRNA remain highly expressed in PD animals also given chronic L-DOPA treatment (76) as well as in PD patients affected by dyskinesia (77), suggesting persistent adaptive changes in the Enk peptide (78).
In contrast, nigrostriatal DA denervation leads to a reduction in the levels of PDYN mRNA (60, 62, 79, 80) that increase under L-DOPA therapy compared to untreated or non-dyskinetic states, consistently across different study models (62, 76, 78, 81). These observations suggest that the expression of opioidergic neuropeptides involved in the modulation of BG output is strictly regulated by striatal DA levels, likely also through the activation of postsynaptic transcription factors that ultimately can promote the expression of multiple genes, including those for opioidergic peptides.
In addition, in dyskinetic PD rat model, high levels of both PPENK and PDYN are overall correlated with L-DOPA-induced locomotor alterations. While there is a more specific association between high levels of PDYN mRNA and dyskinetic movements (60), on the other hand, high expression of PPENK mRNA is also correlated with locomotor hyperactivity, beyond dyskinesia (62). These observations suggest that Enk and Dyn might play different roles in striatal-based behavioral effects and in locomotor alterations in response to dopaminergic treatment.
Opioid Receptor Expression in PD and LID
Along with different levels of opioidergic peptides expression, alterations in the levels of opioidergic receptor immunoreactivity have been observed in both PD patients and animal models. Piccini et al. (82) found reduced opioid receptor binding in the caudate of PD patients, and in the putamen and thalamus of dyskinetic PD patients compared to non-dyskinetic. Similar observations have been described in animal studies, although some differences were found across the various models.
Striatal levels of MOR binding and μ-immunoreactivity were reduced in PD rats (83) and in MPTP-lesioned macaques treated with L-DOPA (80), as well as in PD patients undergoing chronic L-DOPA therapy (84). Lower levels of DOR binding are expressed in the GP and striatal areas of 6-OHDA dyskinetic rats, while an increase of δ-immunoreactivity occurs in the motor and premotor cortex (83) (Figure 2). Consistent with these results, PD patients treated with L-DOPA have reduced levels of DOR binding compared with control patients (84). KOR binding levels are decreased in the striatal areas of dyskinetic PD rats and in the GP of PD rats with and without LID (83); low κ-immunoreactivity is observed only in the GP structure of MPTP-denervated macaques with and without dyskinesia (80).
Even though the exposure to L-DOPA treatment in PD animals and PD patients leads to a reduction in opioid receptor binding levels, Chen and colleagues (85) assessed G protein-coupled receptor signaling as a marker of MOR, DOR and KOR activity in MPTP-lesioned non-human primates. Interestingly, they found a hyperactive transduction signal mediated by all three opioid receptors in the striatum. This suggests that in the parkinsonian state under L-DOPA treatment, although the levels of receptor binding can be decreased, the response to activation of opioid receptors is in fact enhanced.
Pharmacological Implications of Opioids in Motor Function
Elucidating the role of opioidergic transmission in the molecular mechanisms that control motor function is complex, not only due to the striatum's neural heterogeneity, but also because of the broad distribution of opioid receptors throughout the brain. The activation of opioid-mediated postsynaptic signaling cascades likely depends on several factors, including opioid agonists and their response to ORs, the type of ORs activated, and whether receptor stimulation is acute or chronic. Systemic administration of opioidergic drugs might affect different neural circuits and modulate behavioral aspects beyond locomotor activity. Therefore, pharmacological approaches used to distinguish the neural pathways in the control and alteration of movement should be considered critically.
Considering the enhanced expression of endogenous opioid peptides in the striatum of PD animal models and in PD patients, selective agonists and antagonists to ORs have been used to counteract akinesia in PD and to reduce the development of dyskinesia in response to L-DOPA treatment (Table 2). MOR antagonists (cyprodine and ADL5510) alleviated LID in MPTP-lesioned non-human primates without interfering with the antiparkinsonian effects of L-DOPA (86, 87). A selective DOR antagonist (naltrindole) has a similar effect, reducing dyskinetic movements in MPTP-lesioned marmoset and 6-OHDA rats treated with L-DOPA (86, 88), although there is an akinetic effect on motor activity in a PD model without DA treatment (89). A selective DOR agonist (SNC-80) increased locomotor activity in naive and PD animals, but its potential therapeutic applications are limited by its convulsive effects (90–92). In contrast, a κ-receptor antagonist (norBNI) did not induce any anti-dyskinetic effect in MPTP-lesioned macaques (86); yet a selective κ-receptor agonist (U50, 488) reduced LID in PD rats and monkeys, but impaired the anti-parkinsonian effects of L-DOPA treatment (93). In line with these effects, the synthetic opioid analgesic nalbuphine, acting as both a KOR agonist and a MOR antagonist, reduced LID in a non-human primate model of PD and decreased the levels of specific molecular markers associated with the development of dyskinesia (94). Also noteworthy is the effect of the non-selective antagonist naloxone, which reduced LID in 6-OHDA rats (95, 96), although results in MPTP-lesioned macaques and PD patients were inconclusive (97, 98).
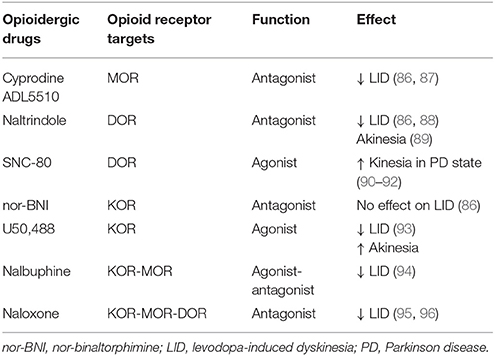
Table 2. Summary of opioidergic drugs used as pharmacological intervention to counteract parkinsonian symptoms and dyskinetic movements in PD animal model.
The literature makes it clear that different pharmacological responses are expected across animal models and in human patients, likely due to the greater neural organization and connectivity in primates and humans. The lost of DA in PD and its exogenous replacement by L-DOPA lead to changes in the expression of opioid peptides and receptor immunoreactivity that reflect a strong interaction between dopaminergic and opioidergic systems in the BG motor circuit. However, it is still debated whether changes in the opioid transmission occur to compensate for DA denervation and L-DOPA treatment, or whether these changes interact with the molecular and synaptic mechanisms associated with altered neural responses in motor diseases.
Concluding Remarks
The recent advances in understanding the striatal functionality highlight the strong impact of opioidergic transmission to modulate synaptic plasticity and cellular responses of the SPNs. The studies here reviewed, demonstrate that opioid receptors have a regional (ventral vs. dorsal striatum), compartmental (striosomes vs. matrix) and cellular (dSPNs vs. iSPNs) specificity that affects the striatal activity in response to different inputs. Such specificity reflects the complexity of striatal organization and the efforts to find selective opioidergic treatments that can modulate specific neural pathways. Although the literature points out the inhibitory effect of opioid agonists on synaptic transmission and neurotransmitters release, it is still debated how opioid receptors interact with dopaminergic receptors and whether they share common mechanisms to activate postsynaptic signaling cascades and downstream effectors. The interaction between opioidergic and dopaminergic pathways becomes crucial in PD and LID where the high levels of endogenous opioids occurs in parallel with aberrant dopaminergic transmission, and are associated with alterated striatal-based behaviors. Since the broad distribution of opioid receptors throughout the brain, pharmacological approaches should aim to selectively target defined receptor subtypes, in a cell-type- and input-specific manner. The use of chemogenetic or optogenetic approaches are therefore crucial to dissect opioidergic neurotransmission within the striatum and its interaction with dopaminergic system. This would be instrumental to develop specific pharmacological treatments able to restore maladaptive changes without interfering with other neuronal pathways.
Author Contributions
SS wrote the manuscript. RT conceived the review contribution, supervised the writing and critically edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer GF declared a past collaboration with one of the authors RT to the handling Editor.
Acknowledgments
This work was supported by Fondazione Istituto Italiano di Tecnologia. This review includes some sessions of PhD dissertation thesis - SS. (2015): Role of striatal opioidergic neuropeptides on locomotor hypersensitivity and dyskinesia in Parkinsonian rats (unpublished)-University of Bern and Laboratory for Biomedical Neurosciences (LBN), Switzerland.
References
1. Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev (2009) 89:1379–412. doi: 10.1152/physrev.00005.2009
2. Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. (2013) 36:195–206. doi: 10.1016/j.tins.2012.11.002
3. Crossman AR. Neural mechanisms in disorders of movement. Comp Biochem Physiol Comp Physiol. (1989) 93:141–9.
4. DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci (1990) 13:281–5.
5. Redgrave P, Rodriguez M, Smith Y, Rodriguez-Oroz MC, Lehericy S, Bergman H, et al. Goal-directed and habitual control in the basal ganglia: implications for Parkinson's disease. Nat Rev Neurosci. (2010) 11:760–72. doi: 10.1038/nrn2915
6. Lei W, Jiao Y, Del Mar N, Reiner A. Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J Neurosci. (2004) 24:8289–99. doi: 10.1523/JNEUROSCI.1990-04.2004
7. Barroso-Chinea P, Bezard E. Basal Ganglia circuits underlying the pathophysiology of levodopa-induced dyskinesia. Front Neuroanat. (2010) 4:131. doi: 10.3389/fnana.2010.00131
8. Obeso JA, Rodriguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Mov Disord. (2008) 23 Suppl. 3:S548–59. doi: 10.1002/mds.22062
9. Gerfen CR, Young WS. Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res (1988) 460:161–7.
10. Fallon JH, Leslie FM, Cone RI. Dynorphin-containing pathways in the substantia nigra and ventral tegmentum: a double labeling study using combined immunofluorescence and retrograde tracing. Neuropeptides (1985) 5:457–60.
11. Cuello AC, Paxinos G. Evidence for a long Leu-enkephalin striopallidal pathway in rat brain. Nature (1978) 271:178–80.
12. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. (1989) 12:366–75.
13. Obeso JA, Marin C, Rodriguez-Oroz C, Blesa J, Benitez-Temino B, Mena-Segovia J, et al. The basal ganglia in Parkinson's disease: current concepts and unexplained observations. Ann Neurol. (2008) 64 Suppl. 2:S30–46. doi: 10.1002/ana.21481
14. Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature (2013) 494:238–42. doi: 10.1038/nature11846
15. Rice ME, Cragg SJ. Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev. (2008) 58:303–13. doi: 10.1016/j.brainresrev.2008.02.004
16. Liu C, Kershberg L, Wang J, Schneeberger S, Kaeser PS. Dopamine secretion is mediated by sparse active zone-like release sites. Cell (2018) 172:706–18e15. doi: 10.1016/j.cell.2018.01.008
17. West AR, Grace AA. Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J. Neurosci. (2002) 22:294–304. doi: 10.1523/JNEUROSCI.22-01-00294.2002
18. da Silva JA, Tecuapetla F, Paixao V, Costa RM. Dopamine neuron activity before action initiation gates and invigorates future movements. Nature (2018) 554:244–8. doi: 10.1038/nature25457
19. Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. (2001) 81:299–343. doi: 10.1152/physrev.2001.81.1.299
20. Dacher M, Nugent FS. Opiates and plasticity. Neuropharmacology (2011) 61:1088–96. doi: 10.1016/j.neuropharm.2011.01.028
21. Smith AP, Lee NM. Opioid receptor interactions: local and nonlocal, symmetric and asymmetric, physical and functional. Life Sci. (2003) 73:1873–93. doi: 10.1016/S0024-3205(03)00549-6
22. Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. (2014) 17:540–8. doi: 10.1038/nn.3652
23. Tajima K, Fukuda T. Region-specific diversity of striosomes in the mouse striatum revealed by the differential immunoreactivities for mu-opioid receptor, substance P, and enkephalin. Neuroscience (2013) 241:215–28. doi: 10.1016/j.neuroscience.2013.03.012
24. Oude Ophuis RJ, Boender AJ, van Rozen AJ, Adan RA. Cannabinoid, melanocortin and opioid receptor expression on DRD1 and DRD2 subpopulations in rat striatum. Front Neuroanat (2014) 8:14. doi: 10.3389/fnana.2014.00014
25. Ambrose LM, Unterwald EM, Van Bockstaele EJ. Ultrastructural evidence for co-localization of dopamine D2 and micro-opioid receptors in the rat dorsolateral striatum. Anat Record Part A Discov Mol Cell Evol Biol (2004) 279:583–91. doi: 10.1002/ar.a.20054
26. Ponterio G, Tassone A, Sciamanna G, Riahi E, Vanni V, Bonsi P, et al. Powerful inhibitory action of mu opioid receptors (MOR) on cholinergic interneuron excitability in the dorsal striatum. Neuropharmacology (2013) 75:78–85. doi: 10.1016/j.neuropharm.2013.07.006
27. Jabourian M, Venance L, Bourgoin S, Ozon S, Perez S, Godeheu G, et al. Functional mu opioid receptors are expressed in cholinergic interneurons of the rat dorsal striatum: territorial specificity and diurnal variation. Eur J Neurosci. (2005) 21:3301–9. doi: 10.1111/j.1460-9568.2005.04154.x
28. Cahill CM, McClellan KA, Morinville A, Hoffert C, Hubatsch D, O'Donnell D, et al. Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J Comp Neurol (2001) 440:65–84. doi: 10.1002/cne.1370
29. Banghart MR, Neufeld SQ, Wong NC, Sabatini BL. Enkephalin disinhibits mu opioid receptor-rich striatal patches via delta opioid receptors. Neuron (2015) 88:1227–39. doi: 10.1016/j.neuron.2015.11.010
30. North RA, Williams JT, Surprenant A, Christie MJ. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA. (1987) 84:5487–91.
31. Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology (2011) 115:1363–81. doi: 10.1097/ALN.0b013e318238bba6
32. Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. (1995) 18:22–9.
33. Kivell B, Uzelac Z, Sundaramurthy S, Rajamanickam J, Ewald A, Chefer V, et al. Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism. Neuropharmacology (2014) 86:228–40. doi: 10.1016/j.neuropharm.2014.07.016
34. Hipolito L, Sanchez-Catalan MJ, Zanolini I, Polache A, Granero L. Shell/core differences in mu- and delta-opioid receptor modulation of dopamine efflux in nucleus accumbens. Neuropharmacology (2008) 55:183–9. doi: 10.1016/j.neuropharm.2008.05.012
35. Campos-Jurado Y, Marti-Prats L, Zornoza T, Polache A, Granero L, Cano-Cebrian MJ. Regional differences in mu-opioid receptor-dependent modulation of basal dopamine transmission in rat striatum. Neurosci Lett. (2017) 638:102–8. doi: 10.1016/j.neulet.2016.12.024
36. Costa-Neto CM, Parreiras ESLT, Bouvier M. A Pluridimensional View of Biased Agonism. Mol Pharmacol. (2016) 90:587–95. doi: 10.1124/mol.116.105940
37. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. (2012) 52:179–97. doi: 10.1146/annurev.pharmtox.010909.105800
38. Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, et al. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS ONE (2009) 4:e5425. doi: 10.1371/journal.pone.0005425
39. Kramer HK, Onoprishvili I, Andria ML, Hanna K, Sheinkman K, Haddad LB, et al. Delta opioid activation of the mitogen-activated protein kinase cascade does not require transphosphorylation of receptor tyrosine kinases. BMC Pharmacol. (2002) 2:5. doi: 10.1186/1471-2210-2-5
40. Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA. (2005) 102:491–6. doi: 10.1073/pnas.0408305102
41. Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, et al. Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. (2005) 280:27662–9. doi: 10.1074/jbc.M502593200
42. Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem. (2006) 99:381–92. doi: 10.1111/j.1471-4159.2006.04124.x
43. Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry (2007) 62:800–10. doi: 10.1016/j.biopsych.2006.11.032
44. Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. (2007) 27:6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007
45. Fasano S, Bezard E, D'Antoni A, Francardo V, Indrigo M, Qin L, et al. Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc Natl Acad Sci USA. (2010) 107:21824–9. doi: 10.1073/pnas.1012071107
46. Lopez-Huerta VG, Nakano Y, Bausenwein J, Jaidar O, Lazarus M, Cherassse Y, et al. Erratum to: The neostriatum: two entities, one structure? Brain Struct Funct. (2016) 221:1789. doi: 10.1007/s00429-015-1017-8
47. Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. (1990) 13:244–54.
48. Miura M, Saino-Saito S, Masuda M, Kobayashi K, Aosaki T. Compartment-specific modulation of GABAergic synaptic transmission by mu-opioid receptor in the mouse striatum with green fluorescent protein-expressing dopamine islands. J Neurosci. (2007) 27:9721–8. doi: 10.1523/JNEUROSCI.2993-07.2007
49. Graybiel AM, Ragsdale CW. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci USA. (1978) 75:5723–6.
50. Gerfen CR. The neostriatal mosaic: compartmentalization of corticostriatal input and striatonigral output systems. Nature (1984) 311:461–4.
51. Gerfen CR, Baimbridge KG, Miller JJ. The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc Natl Acad Sci USA. (1985) 82:8780–4.
52. Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, et al. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur J Neurosci. (2011) 33:668–77. doi: 10.1111/j.1460-9568.2010.07564.x
53. Crittenden JR, Graybiel AM. Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat. (2011) 5:59. doi: 10.3389/fnana.2011.00059
54. Brimblecombe KR, Cragg SJ. The striosome and matrix compartments of the striatum: a path through the labyrinth from neurochemistry toward function. ACS Chem Neurosci. (2017) 8:235–42. doi: 10.1021/acschemneuro.6b00333
55. Jiang ZG, North RA. Pre- and postsynaptic inhibition by opioids in rat striatum. J Neurosci. (1992) 12:356–61.
56. Blomeley CP, Bracci E. Opioidergic interactions between striatal projection neurons. J Neurosci. (2011) 31:13346–56. doi: 10.1523/JNEUROSCI.1775-11.2011
57. Hawes SL, Salinas AG, Lovinger DM, Blackwell KT. Long-term plasticity of corticostriatal synapses is modulated by pathway-specific co-release of opioids through kappa-opioid receptors. J Physiol. (2017) 595:5637–52. doi: 10.1113/JP274190
58. Pisa M. Motor functions of the striatum in the rat: critical role of the lateral region in tongue and forelimb reaching. Neuroscience (1988) 24:453–63.
59. Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. (1999) 6:461–74. doi: 10.1006/nbdi.1999.0259
60. Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. (1998) 10:2694–706.
61. Capper-Loup C, Frey CM, Rebell D, Kaelin-Lang A. Adaptive gene expression changes on the healthy side of parkinsonian rats. Neuroscience (2013) 233:157–65. doi: 10.1016/j.neuroscience.2012.12.027
62. Sgroi S, Capper-Loup C, Paganetti P, Kaelin-Lang A. Enkephalin and dynorphin neuropeptides are differently correlated with locomotor hypersensitivity and levodopa-induced dyskinesia in parkinsonian rats. Exp Neurol. (2016) 280:80–8. doi: 10.1016/j.expneurol.2016.03.024
63. Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature (2007) 446:1086–90. doi: 10.1038/nature05726
64. Drdla R, Gassner M, Gingl E, Sandkuhler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science (2009) 325:207–10. doi: 10.1126/science.1171759
65. Massano J, Bhatia KP. Clinical approach to Parkinson's disease: features, diagnosis, and principles of management. Cold Spring Harbor Perspect Med (2012) 2:a008870. doi: 10.1101/cshperspect.a008870
66. Sgroi S, Kaelin-Lang A, Capper-Loup C. Spontaneous locomotor activity and L-DOPA-induced dyskinesia are not linked in 6-OHDA parkinsonian rats. Front Behav Neurosci. (2014) 8:331. doi: 10.3389/fnbeh.2014.00331
67. Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. (1973) 20:415–55.
68. Bezard E, Gross CE, Brotchie JM. Presymptomatic compensation in Parkinson's disease is not dopamine-mediated. Trends Neurosci (2003) 26:215–21. doi: 10.1016/S0166-2236(03)00038-9
69. Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: how relevant is the rat? Nat Rev Neurosci. (2002) 3:574–9. doi: 10.1038/nrn877
70. Blesa J, Phani S, Jackson-Lewis V, Przedborski S. Classic and new animal models of Parkinson's disease. J Biomed Biotechnol. (2012) 2012:845618. doi: 10.1155/2012/845618
71. Granerus AK. Factors influencing the occurrence of “on-off” symptoms during long-term treatment with L-dopa. Acta Med Scand. (1978) 203:75–85.
72. Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord. (2007) 22:1379–89. doi: 10.1002/mds.21475
73. Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science (1990) 250:1429–32.
74. Herrero MT, Augood SJ, Hirsch EC, Javoy-Agid F, Luquin MR, Agid Y, et al. Effects of L-DOPA on preproenkephalin and preprotachykinin gene expression in the MPTP-treated monkey striatum. Neuroscience (1995) 68:1189–98.
75. Nisbet AP, Foster OJ, Kingsbury A, Eve DJ, Daniel SE, Marsden CD, et al. Preproenkephalin and preprotachykinin messenger RNA expression in normal human basal ganglia and in Parkinson's disease. Neuroscience (1995) 66:361–76.
76. Westin JE, Andersson M, Lundblad M, Cenci MA. Persistent changes in striatal gene expression induced by long-term L-DOPA treatment in a rat model of Parkinson's disease. Eur J Neurosci (2001) 14:1171–6. doi: 10.1046/j.0953-816x.2001.01743.x
77. Calon F, Birdi S, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T. Increase of preproenkephalin mRNA levels in the putamen of Parkinson disease patients with levodopa-induced dyskinesias. J Neuropathol Exp Neurol. (2002) 61:186–96. doi: 10.1093/jnen/61.2.186
78. Tamim MK, Samadi P, Morissette M, Gregoire L, Ouattara B, Levesque D, et al. Effect of non-dopaminergic drug treatment on Levodopa induced dyskinesias in MPTP monkeys: common implication of striatal neuropeptides. Neuropharmacology (2010) 58:286–96. doi: 10.1016/j.neuropharm.2009.06.030
79. Tel BC, Zeng BY, Cannizzaro C, Pearce RK, Rose S, Jenner P. Alterations in striatal neuropeptide mRNA produced by repeated administration of L-DOPA, ropinirole or bromocriptine correlate with dyskinesia induction in MPTP-treated common marmosets. Neuroscience (2002) 115:1047–58. doi: 10.1016/S0306-4522(02)00535-3
80. Aubert I, Guigoni C, Li Q, Dovero S, Bioulac BH, Gross CE, et al. Enhanced preproenkephalin-B-derived opioid transmission in striatum and subthalamic nucleus converges upon globus pallidus internalis in L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry (2007) 61:836–44. doi: 10.1016/j.biopsych.2006.06.038
81. Bishop C, Krolewski DM, Eskow KL, Barnum CJ, Dupre KB, Deak T, et al. Contribution of the striatum to the effects of 5-HT1A receptor stimulation in L-DOPA-treated hemiparkinsonian rats. J Neurosci Res. (2009) 87:1645–58. doi: 10.1002/jnr.21978
82. Piccini P, Weeks RA, Brooks DJ. Alterations in opioid receptor binding in Parkinson's disease patients with levodopa-induced dyskinesias. Ann Neurol. (1997) 42:720–6. doi: 10.1002/ana.410420508
83. Johansson PA, Andersson M, Andersson KE, Cenci MA. Alterations in cortical and basal ganglia levels of opioid receptor binding in a rat model of l-DOPA-induced dyskinesia. Neurobiol Dis. (2001) 8:220–39. doi: 10.1006/nbdi.2000.0372
84. Fernandez A, de Ceballos ML, Jenner P, Marsden CD. Neurotensin, substance P, delta and mu opioid receptors are decreased in basal ganglia of Parkinson's disease patients. Neuroscience (1994) 61:73–9.
85. Chen L, Togasaki DM, Langston JW, Di Monte DA, Quik M. Enhanced striatal opioid receptor-mediated G-protein activation in L-DOPA-treated dyskinetic monkeys. Neuroscience (2005) 132:409–20. doi: 10.1016/j.neuroscience.2004.10.026
86. Henry B, Fox SH, Crossman AR, Brotchie JM. Mu- and delta-opioid receptor antagonists reduce levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson's disease. Exp Neurol. (2001) 171:139–46. doi: 10.1006/exnr.2001.7727
87. Koprich JB, Fox SH, Johnston TH, Goodman A, Le Bourdonnec B, Dolle RE, et al. The selective mu-opioid receptor antagonist ADL5510 reduces levodopa-induced dyskinesia without affecting antiparkinsonian action in MPTP-lesioned macaque model of Parkinson's disease. Mov Disord. (2011) 26:1225–33. doi: 10.1002/mds.23631
88. Billet F, Costentin J, Dourmap N. Influence of corticostriatal delta-opioid receptors on abnormal involuntary movements induced by L-DOPA in hemiparkinsonian rats. Exp Neurol. (2012) 236:339–50. doi: 10.1016/j.expneurol.2012.04.017
89. Mabrouk OS, Volta M, Marti M, Morari M. Stimulation of delta opioid receptors located in substantia nigra reticulata but not globus pallidus or striatum restores motor activity in 6-hydroxydopamine lesioned rats: new insights into the role of delta receptors in parkinsonism. J Neurochem. (2008) 107:1647–59. doi: 10.1111/j.1471-4159.2008.05727.x
90. Spina L, Longoni R, Mulas A, Chang KJ, Di Chiara G. Dopamine-dependent behavioural stimulation by non-peptide delta opioids BW373U86 and SNC 80: 1. Locomotion, rearing and stereotypies in intact rats. Behav Pharmacol. (1998) 9:1–8.
91. Hudzik TJ, Howell A, Payza K, Cross AJ. Antiparkinson potential of delta-opioid receptor agonists. Eur J Pharmacol. (2000) 396:101–7. doi: 10.1016/S0014-2999(00)00209-0
92. Nozaki C, Le Bourdonnec B, Reiss D, Windh RT, Little PJ, Dolle RE, et al. delta-Opioid mechanisms for ADL5747 and ADL5859 effects in mice: analgesia, locomotion, and receptor internalization. J Pharmacol Exp Ther. (2012) 342:799–807. doi: 10.1124/jpet.111.188987
93. Cox H, Togasaki DM, Chen L, Langston JW, Di Monte DA, Quik M. The selective kappa-opioid receptor agonist U50,488 reduces L-dopa-induced dyskinesias but worsens parkinsonism in MPTP-treated primates. Exp Neurol. (2007) 205:101–7. doi: 10.1016/j.expneurol.2007.01.024
94. Potts LF, Park ES, Woo JM, Dyavar Shetty BL, Singh A, Braithwaite SP, et al. Dual kappa-agonist/mu-antagonist opioid receptor modulation reduces levodopa-induced dyskinesia and corrects dysregulated striatal changes in the nonhuman primate model of Parkinson disease. Ann Neurol. (2015) 77:930–41. doi: 10.1002/ana.24375
95. Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. (2002) 15:120–32. doi: 10.1046/j.0953-816x.2001.01843.x
96. Klintenberg R, Svenningsson P, Gunne L, Andren PE. Naloxone reduces levodopa-induced dyskinesias and apomorphine-induced rotations in primate models of parkinsonism. J Neural Trans. (2002) 109:1295–307. doi: 10.1007/s00702-002-0715-6
97. Gomez-Mancilla B, Bedard PJ. Effect of nondopaminergic drugs on L-dopa-induced dyskinesias in MPTP-treated monkeys. Clin Neuropharmacol. (1993) 16:418–27.
Keywords: opioids, dopamine, striatum, Parkinson's disease, signaling pathway, synaptic plasticity
Citation: Sgroi S and Tonini R (2018) Opioidergic Modulation of Striatal Circuits, Implications in Parkinson's Disease and Levodopa Induced Dyskinesia. Front. Neurol. 9:524. doi: 10.3389/fneur.2018.00524
Received: 30 March 2018; Accepted: 13 June 2018;
Published: 05 July 2018.
Edited by:
Graziella Madeo, National Institutes of Health (NIH), United StatesReviewed by:
Gilberto Fisone, Karolinska Institutet (KI), SwedenGiuseppina Martella, Università degli Studi di Roma Tor Vergata, Italy
Copyright © 2018 Sgroi and Tonini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaella Tonini, UmFmZmFlbGxhLnRvbmluaUBpaXQuaXQ=