 Maurizio Acampa
Maurizio Acampa Pietro E. Lazzerini
Pietro E. Lazzerini Giuseppe Martini
Giuseppe Martini- 1Stroke Unit, Department of Neurological and Neurosensorial Sciences, Azienda Ospedaliera Universitaria Senese, “Santa Maria alle Scotte” General Hospital, Siena, Italy
- 2Department of Medical Sciences, Surgery and Neurosciences, University of Siena, Siena, Italy
Recently, atrial cardiopathy has emerged as possible pathogenic mechanism in cryptogenic stroke and many electrocardiographic (ECG) markers have been proposed in order to detect an altered atrial substrate at an early stage. The autonomic nervous system (ANS) plays a well-known role in determining significant and heterogeneous electrophysiological changes of atrial cardiomyocytes, that promote atrial fibrillation episodes in cardioembolic stroke. Conversely, the role of ANS in atrial cardiopathy and cryptogenic stroke is less known, as well as ANS effects on ECG markers of atrial dysfunction. In this paper, we review the evidence linking ANS dysfunction and atrial cardiopathy as a possible pathogenic factor in cryptogenic stroke.
Introduction
About one third of ischemic strokes occurs without a well-defined etiology and is classified as cryptogenic (1). Different possible pathogenic mechanisms have been proposed (2), including the presence of subclinical atrial fibrillation (AF). Thus, the use of prolonged outpatient cardiac monitoring is currently recommended in order to detect subclinical AF (3) and to provide clues to the mechanism of stroke, leading to appropriate secondary prevention with anticoagulant drugs.
However the relationship between AF and stroke appears more complex than a simple cause-effect mechanism and it seems that AF, atrial substrate, and systemic factors interact in complex ways in the pathway leading to stroke (4). In particular, the lack of direct evidence of a causal association and a temporal relationship between AF and thromboembolic stroke in most patients suggested the hypothesis that atrial cardiopathy may underlie most strokes; thus, AF could represent only a marker of atrial dysfunction (5). Thereby, atrial cardiopathy would represent a continuum, replacing AF as a standalone disease; according to this conceptual model, different races could have different rates of rhythm disorders (AF or atrial flutter) depending on the stage of the atriopathy, with higher risk of stroke at any of these stages (6). In this view, atrial dysfunction, or cardiopathy has emerged as possible pathogenic mechanism in cryptogenic stroke and many ECG markers have been proposed in order to detect atrial substrate at an early stage (5).
We review evidence in favor of a link between atrial cardiopathy, detected with electrocardiographic (ECG) markers, and autonomic nervous system (ANS) dysfunction in order to suggest a possible pathogenic role of ANS in determining atrial substrate that favors cryptogenic stroke occurrence.
Atrial Cardiopathy and Cryptogenic Stroke
Atrial cardiopathy results from different systemic insults (age, obesity, diabetes mellitus, hypertension, and sleep apnea) that promote marked atrial histological abnormalities. These structural atrial alterations include degeneration and apoptosis of myocytes, fibroblast proliferation and differentiation into myofibroblasts with atrial fibrosis, matrix degeneration and formation of non-collagen deposits in the interstitial space (7, 8) (Figure 1).
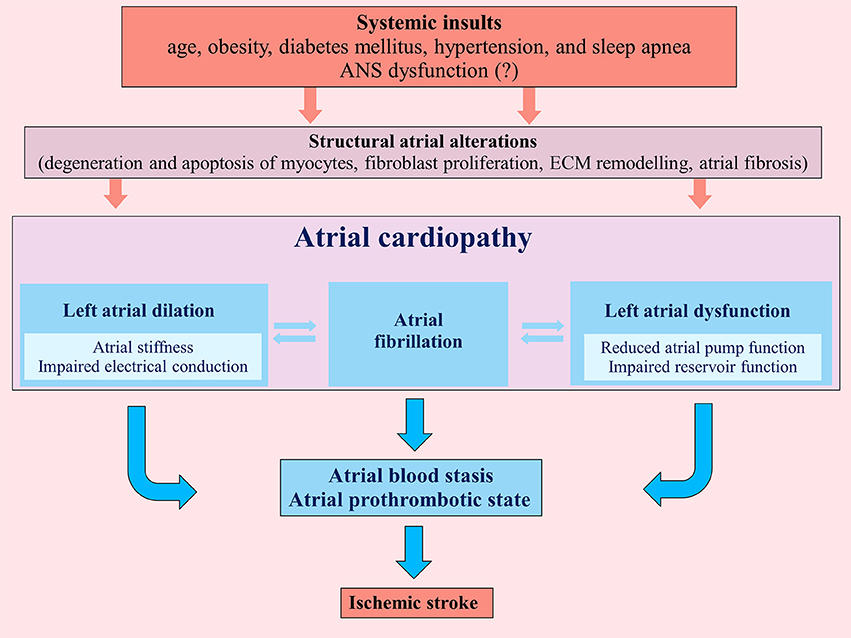
Figure 1. Pathogenic mechanisms determining atrial cardiopathy and favoring an atrial prothrombotic state that can lead to ischemic stroke. ECM, extracellular matrix; ANS, autonomic nervous system.
Myocyte apoptosis promotes reparative fibrosis that replaces myocardial cells (8), whereas fibroblast proliferation induces a reactive fibrosis with an altered ratios of collagen subtypes that separate myocytes, interfering with electrical impulse propagation (8); furthermore, these structural alterations of the atrial myocardium induce the disorganization of connexins (especially Cx43) within junction channels (9).
These patho-histological changes lead to left atrial dilation and dysfunction, determining not only a substrate for AF, but also an atrial prothrombotic milieu that, by itself, represents a possible pathogenic mechanism of cardioembolic ischemic stroke, independently from AF (10) (Figure 1). Indeed atrial and left atrial appendage hypocontractility associated to atrial dilation cause blood stasis in the atrial chambers inducing a thrombotic substrate.
Left atrial dysfunction consists of reduced atrial compliance and relaxation during ventricular systole and impaired pump function during ventricular diastole; in particular atria that exhibit greater fibrotic and apoptotic burdens have impaired conduit, reservoir and contractile function (11). Reduced left atrial compliance is associated with higher clinical recurrence of AF (12) and increased left atrial stiffness has been suggested to be a predictor of cryptogenic stroke in subjects with patent foramen ovale (13). Furthermore, impaired reservoir function (assessed by left atrial reservoir strain with speckle-tracking echocardiography) is associated with cryptogenic stroke, independently of other cardiovascular risk factors (14). Finally, impaired left atrial pump function is significantly depressed in cryptogenic stroke with atrial septal aneurysm (15).
Similarly to left atrial dysfunction, left atrial enlargement is related to the degree of atrial structural pathology and the amount of atrial fibrosis; in particular, moderate-severe left atrial enlargement represents an independent marker of recurrent cardioembolic or cryptogenic stroke (16). Left atrial enlargement is also associated with high risk of AF occurrence (17), but a recent analysis of the Cardiovascular Health Study demonstrated that left atrial enlargement is associated with ischemic stroke, independently from other several confounders such as AF (16).
Primary or secondary ANS dysfunction could play a pathogenic role in atrial structural alterations leading to atrial cardiopathy. Notably, previous studies support the idea that various systemic insults (age, obesity, diabetes mellitus, hypertension, and sleep apnea) related to atrial cardiopathy could induce a secondary ANS dysfunction (18–22). In this view, ANS could represent a cause or a mediator of the pathogenic mechanisms underlying the atrial cardiopathy.
ECG Markers of Atrial Cardiopathy and Influences of Autonomic Nervous System
Many ECG markers are currently used in cryptogenic stroke in order to detect the presence of a possible atrial cardiopathy (5, 23–25). They detect different aspects of atrial dysfunction and may be, at least in part, associated with ANS alterations (Table 1).
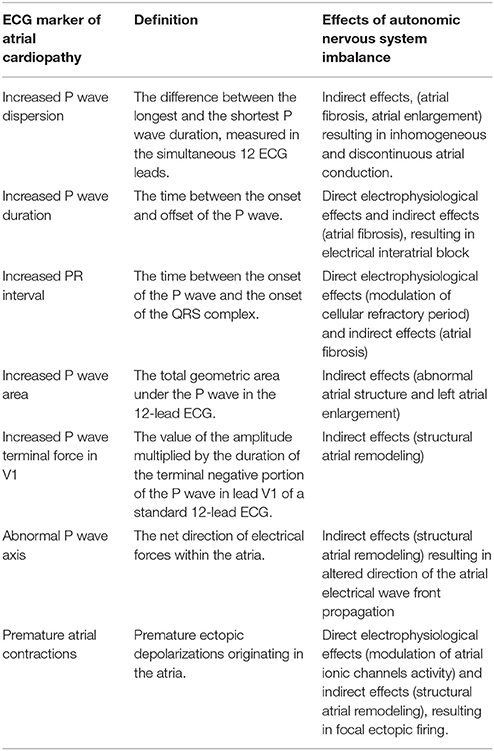
Table 1. Electrocardiographic markers of atrial cardiopathy and influences of autonomic nervous system.
P Wave Dispersion
P wave dispersion results from the difference between the longest and the shortest P wave duration measured in the simultaneous 12 leads on a routine ECG (26) and has been suggested to be a marker of cardioembolism in cryptogenic stroke (27). Different P wave durations in 12-lead ECG reflect regional delays in atrial depolarization; therefore, increased PWD results from inhomogeneous and discontinuous atrial conduction based on inhomogeneous and anisotropic distribution of connections between atrial myocardial fibers (28). In this view, PWD reflects the presence of atrial substrate for AF and previous studies showed a link between high PWD values (>40 ms) and AF in cryptogenic stroke (29–31).
Increased PWD is also associated with impaired left atrial mechanical functions and enlargement in patients with cryptogenic stroke (32).
In fact, the increased collagen deposition and myocardial fibrosis of the left atrial myocardium may result in reduced LA compliance which is reflected by prolongation of the PWD on ECG (33).
The influence of ANS on PWD has been suggested by the effects of chronic traumatic spinal cord injury on ECG markers (34). Autonomic dysfunction after spinal cord injury may contribute to the development of cardiovascular disorders (cardiac arrhythmias and unstable blood pressure), especially in patients with cervical and high thoracic spinal cord injury (above T6) and it is possible that higher PWD values observed in patients with chronic spinal cord injury are linked to atrial structural alterations (34). PWD has also been suggested to be an early sign of cardiac autonomic dysfunction in subjects with neurally-mediated syncope (35).
P Wave Duration
A prolonged P-wave duration (>120 ms) is considered a marker of atrial cardiopathy, reflecting the presence of an interatrial block, determined by a reduced atrial conduction related to a fibro-fatty transformation of atrial walls (36). A previous study demonstrated that in post-mortem atrial tissues from patients who died of cardiovascular causes, the extent of fibrosis, and fatty infiltration is significantly associated with prolonged P wave duration, suggesting that atrial fibrosis of Bachmann's bundle and terminal crest have a major role in prolongation of P wave duration (37). The interatrial block may be partial (when prolonged P wave duration is associated to bimodal P-wave in any lead on the standard 12-lead ECG) or advanced (when prolonged P wave duration is associated with biphasic P-wave in the inferior leads) (38).
Very long P-wave duration in the top fifth percentile is associated with doubling of risk of AF (23). A recent pooled meta-analysis showed that prolonged maximum PWD (evaluated as a categorical variable) increases the risk of ischemic stroke (24). In particular, a previous study demonstrated that in patients with advanced interatrial block there is 1.7-fold increase in the risk of ischemic stroke (39) and this association resulted not attenuated by incident AF events: this evidence suggests that left atrial disease evaluated by means of P wave duration, is possibly an independent risk factor for ischemic stroke.
The acute effects of autonomic stimulation and blockade on P wave duration are well-known electrophysiological effects (40) and are outside the scope of our review article. However, the observation that in non-elite men athletes lifetime training hours are associated with the prolonged signal-averaged P-wave duration and an increased left atrial volume suggested that a prolongation of the P wave duration reflects an altered atrial substrate, determined by the exercise-induced atrial fibrosis (41), possibly induced by increased vagal tone (42).
PR Interval
PR interval is the period of time that extends from the start of the P wave (atrial depolarization) until the start of the QRS complex (ventricular depolarization). The duration of PR interval is normally between 120 and 200 ms. In most cases, a prolonged PR interval (>200 ms) is determined by conduction delay in the atrioventricular (AV) node. Acute effects of ANS on PR interval are well-known, considering that autonomic innervation influences the conduction through the AV junction conduction by modulating the refractory period (43). However, atrial fibrosis may also be considered as the possible cause of PR interval prolongation; indeed, PR interval reproduces the atrial and AV conduction, thus P wave duration represents a relevant part of PR interval and atrial cardiopathy may contribute to the prolongation of the PR interval (44). In this view, prolongation of the PR interval is considered another possible marker of atrial disease (5) and previous studies showed the association between PR interval prolongation and AF in cryptogenic stroke (45, 46). In particular, each prolongation of 10 ms increases 30% the risk of AF in cryptogenic stroke (23); furthermore, PR interval of 200 ms or greater is associated with cryptogenic stroke and may be considered a marker of atrial cardiopathy even in the absence of AF (47).
P Wave Area
P wave area (PWA) is the total geometric area under the P-wave in the 12- lead ECG, representing the product of the duration and amplitude of the P-wave and it is measured in microvolt × milliseconds (48).
PWA is considered a marker of abnormal atrial structure and left atrial enlargement. In the ARIC study, mean P wave area was a predictor of AF (49), in a recent pooled meta-analysis, higher maximal PWA increases of 10% the risk of incident stroke (24).
P Wave Terminal Force in Lead V1
P wave terminal force in lead V1 (PTFV1) is defined as the value of the amplitude multiplied by the duration of the terminal negative deflection of the P wave in lead V1 of a standard 12-lead ECG.
This ECG marker is a predictor of AF; in particular, patients with PTFV1 in the upper 5th percentile have two-fold increased risk of incident AF (47). PTFV1 represents also a strong predictor of paroxysmal AF detection in acute ischemic stroke (50) and even in the absence of detectable AF, abnormal PTFV1 is significantly associated with incident stroke in hypertensive patients (51).
A recent metanalysis (24) showed that higher PTFV1 values increase of 59% the risk of ischemic stroke and the association between increased PTFV1 and embolic stroke of undetermined cause (52) suggested the hypothesis that left atrial cardiopathy could be involved in the pathogenesis of cryptogenic stroke even in the absence of recognized AF (53).
PTFV1 represents a sign of left atrial enlargement, but it is also associated with left atrial fibrosis, dilation and elevating filling pressure, reflecting also a delayed interatrial conduction (54). In particular, PRIMERI study demonstrated that both components of PTFV1 (amplitude and duration of the terminal portion of P wave) are differently associated with atrial alterations: the duration is strongly associated with atrial fibrosis, while the amplitude is significantly associated with indices of atrial mechanical function such as left atrial volume and left atrial strain.
Currently no studies showed an influence of ANS on PTFV1, but indirect evidence suggests a possible link between autonomic activity and PTFV1. Indeed, repeated exposure to mental stress may promote an adverse atrial remodeling and acute mental stress alters left atrial electrophysiology, increasing abnormal values of PTFV1 (55); the pathophysiology that links mental stress with PTFV1 is unknown, but a recent study suggested the possible role of ANS alterations (56).
Furthermore, sleep disordered breathing is associated with subclinical left atrial disease, as indicated by PTFV1 (57); even in this case, it is possible to hypothesize a role of ANS, because an impaired cardiac autonomic modulation in the sense of sympathetic overflow and weaker parasympathetic modulation are considered clinical hallmarks of sleep disorder breathing (58).
P Wave Axis
The P-wave axis represents the net direction of electrical forces within the atria. Axis values between 0 and 75° are considered normal (48). P wave axis may be influenced by the anatomy, size and positioning of the atria within the thoracic cavity (48). Furthermore, mechanical and metabolic insults to the atria may induce structural remodeling and an abnormal electrical conduction, resulting in an altered direction of the atrial electrical wave front propagation. Therefore, an abnormal P wave axis represents a significant predictor of incident AF (59–62). Recently, the results of the ARIC study (63) showed also that an abnormal P wave axis is associated with ischemic stroke, predisposing to cardiac thromboembolism, regardless of AF occurrence.
Premature Atrial Contractions
Atrial premature contractions (APCs) are premature supraventricular ectopic depolarizations originating in the atria and represent a risk marker for AF. Previous studies showed that frequent APCs and non-sustained runs of atrial tachyarrhythmia are associated with AF occurrence in cryptogenic stroke (64, 65).
In particular, APCs detected on a routine 12-lead ECG are associated with an increased risk of AF development, independently from race or sex (66) and with an increased risk for non-lacunar ischemic strokes, especially in women (67). Autonomic nervous system imbalance represents a well-known factor that is able to induce significant and heterogeneous changes of atrial electrophysiology; in particular adrenergic activation can lead to focal ectopic firing, modulating cardiac ionic channels activity and promoting atrial structural remodeling (68).
Pathogenic Role of ANS in Developing Atrial Cardiopathy
It is well known the role of ANS in inducing atrial tachyarrhythmias; in fact, the ANS may determine significant and heterogeneous electrophysiological changes of atrial cardiomyocytes, causing atrial fibrillation episodes (68), but these effects are out of scope for our review article. Furthermore, changes of ANS activity may trigger different signaling pathways that are able to determine an atrial derangement, promoting structural alterations. In this section, we hypothesize the possible pathogenic role of ANS in developing these structural alterations (Figure 2).
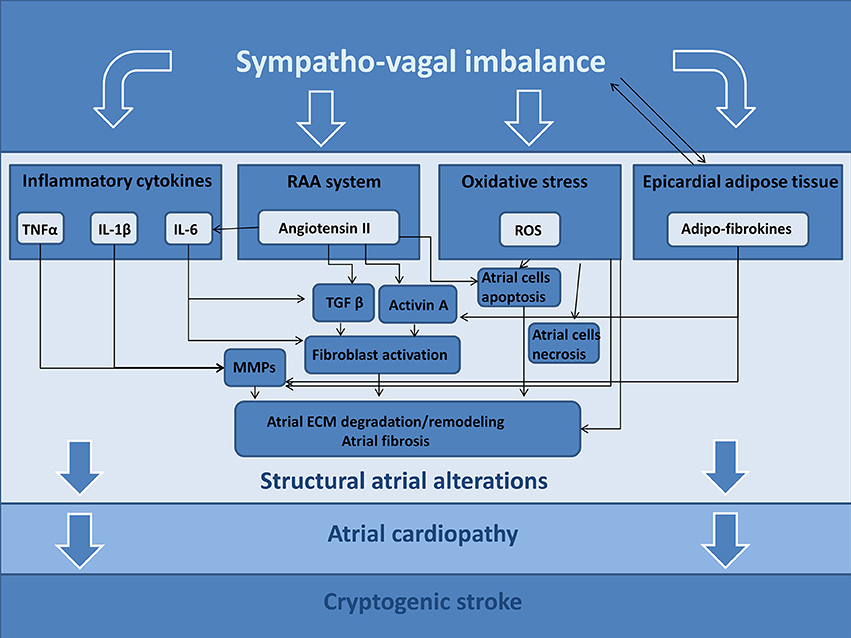
Figure 2. Possible ANS-related mechanisms promoting atrial cardiopathy and cryptogenic stroke. TNFα, tumor necrosis factor alpha; IL-1β, Interleukin 1 Beta; IL-6, Interleukin 6; RAA, renin-angiotensin-aldosterone; ROS, reactive oxygen species; MMPs, matrix metalloproteinases; ECM, extracellular matrix.
ANS and Proinflammatory Cytokines
Autonomic nervous system has been suggested to affect inflammatory responses, balancing inflammatory and anti-inflammatory mediators (69), according to the so-called “inflammatory reflex” (70). In reality, the influence and the role of the ANS in inflammation are complicated by the fact that sympathetic nervous system may elicit pro-inflammatory, as well as anti-inflammatory responses according to different environments (60). A further complicating matter is the effect of local inflammation that, by itself, promotes restructuring of neuronal innervation and shifting in adrenergic receptor subtype expression (71).
Previous studies showed that ANS imbalance may induce the expression of pro-inflammatory cytokine expression in the atrial myocardial cells: in rats, a 7-day stimulation with isoproterenol leads to myocardial gene expression and protein production of tumor necrosis factor-α (TNF α), interleukin-1β (IL-1β), interleukin-6 (IL-6) (72), while beta-adrenergic blockade reduces myocardial expression of TNF-α and IL-1β, cytokines that promote impaired contractile function and chamber enlargement (73). Conversely, vagal activity exerts antiinflammatory effects on the heart, decreasing TNF-α and IL-1β (74, 75).
These cytokines are especially important in extracellular matrix (ECM) degradation; indeed, IL-1β increases the release of matrix metalloproteinases (MMP 2, 3, and 9) while TNF-α augments IL-1β-stimulated release of MMP-9 (76), that promote the net degradation of damaged ECM.
Furthermore, TNF-α induces apoptosis in cardiomyocytes (77) and apoptosis, in turn, is thought to cause atrial fibrosis by inducing a reparative fibrosis process that replaces the degenerating myocardial cells (78).
Both, TNF-α and IL-1β enhance also fibroblast migration (76), while IL-6 facilitates the conversion of cardiac fibroblasts to myofibroblasts (79), inducing the production of collagen and promoting myocardial fibrosis.
The pro-fibrotic effects of IL-6 are mediated in part by the transforming growth factor β1 (TGF-β1)/Smad3 pathway. TGF-β1 is a member of the transforming growth factor superfamily of cytokines that is able to modulate inflammatory process in atrial cells, mediating cell proliferation and differentiation; it is a potent stimulator of collagen-producing cardiac fibroblasts, inducing the production of growth factors (in particular, connective tissue growth factor), angiogenic factors (such as Platelet-Derived Growth Factor), extracellular matrix proteins, and proteases (80) and finally increasing atrial interstitial fibrosis.
Targeted gene-based reduction of TGF-β1 signaling in the posterior left atrium decreased the extent of replacement fibrosis, improving atrial dysfunction (81).
In addition to structural atrial alterations, proinflammatory cytokines (especially IL-6) may drive a prothrombotic state, leading to the increased risk of atrial thrombogenesis and, subsequently, potentially fatal thromboembolism (82, 83): proposed mechanisms linking inflammation to thrombosis include endothelial activation and/or damage, production of tissue factor from monocytes, increased platelet activation, and increased expression of fibrinogen (84).
ANS and Epicardial Adipose Tissue
Epicardial adipose tissue (EAT) has an important endocrine and inflammatory function, and its close relationship to the atrial myocardium suggests a role in the pathogenesis of metabolic-related cardiac diseases (85). EAT is a local source of various hormones, cytokines, and vasoactive substances affecting the myocardium (86); in particular it represents the source of several proinflammatory mediators, including IL-1β, IL-6, TNF α, monocyte chemoattractant protein-1 and adipo-fibrokines (87).
The topographic distribution of the EAT surrounding the left atrium and the lack of fascia between EAT and the myocardium enable molecules secreted by the EAT to diffuse into the myocardium, in a paracrin-like manner (88, 89). The ANS inputs to the heart converge at several locations and are typically embedded in the epicardial fat pads, forming ganglionated plexi (GP) that contain autonomic ganglia and nerves (90); thus, EAT contains both adrenergic and cholinergic nerves which interact with the extrinsic sympathetic and parasympathetic nervous system. Significant interplay occurs between the epicardial fat and the ANS; in particular, a sympathovagal imbalance is related to EAT activity and thickness (91).
The excess of EAT has a significant impact on atrial remodeling and cardiac function (92). Secretome from human EAT contains high levels of MMPs that contribute to extracellular matrix remodeling of the myocardium (76). In addition, the expression of adiponectin, a protective adipokine, is significantly lower in the EAT of patients with coronary artery disease, with evidence of inflammatory cells in the atrial tissue and local inflammatory response that may contribute to developing atrial cardiopathy. Furthermore, a recent study found that EAT secretes high levels of Activin A, an adipo-fibrokine that is able to induce atrial fibrosis: activin A may represent a possible mediator of atrial profibrotic effect and it can be neutralized by anti-Activin A antibodies (93).
ANS and Oxidative Stress
Oxidative stress occurs when the formation of reactive oxygen species (ROS) and reactive nitrogen species exceeds the body's ability to metabolize them. These reactive molecules are able to determine cardiac damage by means of different molecular pathways (94): direct oxidative damage of myofibrillar proteins (95), increased inflammation by NF-κB and c-jun activation (96), TGF-β, MAPK and ERK1/ERK2 activation (96), increased activity of MMPs in cardiac fibroblasts (97), regulation of cardiomyocyte apoptosis (98). Therefore, oxidative stress represents a pathogenic mechanism promoting atrial fibrosis and structural cardiac remodeling (99).
The role of ANS in regulating oxidative stress is supported by previous evidence (100–102), showing that the increase in adrenergic drive may result in catecholamine excitotoxicity, increased oxidative stress and free radical myocardium injury. Furthermore, elevated sympathetic activity related to mental stress can lead to increased oxidative and nitrosative damage of myocardiocytes in rats (101) and sympathetic imbalance may also cause a decrease in functional respirasomes, leading to mitochondrial dysfunction and contributing to maintain and amplify the oxidative stress (102).
ANS and Renin-Angiotensin-Aldosterone System
The renin-angiotensin-aldosterone (RAA) system exerts a well-known role in atrial fibrosis (103). In particular Angiotensin II is a well characterized profibrotic molecule and may cause atrial fibrosis and atrial dilation by means of different mechanisms (104, 105). Angiotensin II plays an important role in the pathogenesis of atrial fibrosis via the following different mechanisms: gene expressions of profibrotic cytokines (106), including TGF1-β (107), activation of cardiac fibroblast function, upregulation of ECM protein synthesis (108), induction of atrial myocardial apoptosis (109) and activation of activin A/ALK4 activin receptor-like kinase 4 /smad2/3 pathway that plays an important role in the pathogenesis of atrial fibrosis (110).
Conversely, angiotensin converting enzyme inhibitors, angiotensin receptor blockers and aldosterone antagonists attenuate atrial fibrosis determining beneficial improvement in atrial structural alterations (105).
The ANS may modulate this system: previous studies concerning renal sympathetic denervation (RSDN) showed that interruption of afferent and efferent sympathetic signaling between the kidney and central sympathetic nervous system may reduce atrial fibrosis and atrial sympathetic nerve sprouting and these effects seem, at least in part, mediated by the suppression of renin-angiotensin-aldosteron system (111, 112).
Furthermore RSDN exerts antioxidant effects and a decrease in the local activity of the sympathetic nervous system and Angiotensin II activity (113), attenuating myocardial fibrosis and left atrial enlargement (114) and improving inflammation, apoptosis, and gap junction expression (115).
Conclusions
In last years, atrial cardiopathy has emerged as possible pathogenic mechanism in cryptogenic stroke and many ECG markers are currently used in these patients in order to detect at an early stage the presence of a possible atrial involvement. Dissecting the role of ANS in developing atrial structural alterations leading to atrial cardiopathy may lead in the future to the development of novel therapeutic strategies to prevent atrial dysfunction and ischemic stroke.
Author Contributions
All the authors have directly contributed to drafting of the manuscript or revising it critically for important intellectual content and final approval of the manuscript submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Saver JL. Clinical practice. Cryptogenic stroke. N Engl J Med. (2016) 374:2065–74. doi: 10.1056/NEJMcp1503946
2. Yaghi S, Bernstein RA, Passman R, Okin PM, Furie KL. Cryptogenic stroke: research and practice. Circ Res. (2017) 120:527–40. doi: 10.1161/CIRCRESAHA.116.308447
3. Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, and Council on Peripheral Vascular Disease. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2014) 45:2160–236. doi: 10.1161/STR.0000000000000024
4. Kamel H, Okin PM, Elkind MSV, Iadecola C. Atrial fibrillation and mechanisms of stroke. Time for a new model. Stroke (2016) 47:895–900. doi: 10.1161/STROKEAHA.115.012004
5. Yaghi S, Kamel H, Elkind MSV. Atrial cardiopathy: a mechanism of cryptogenic stroke. Expert Rev Cardiovasc Ther. (2017) 15:591–9. doi: 10.1080/14779072.2017.1355238
6. Soliman EZ. Race and atrial flutter: a needed update to understand the atrial fibrillation race paradox. Future Cardiol. (2017) 13:423–7. doi: 10.2217/fca-2017-0049
7. Stirbys P. Neuro-atriomyodegenerative origin of atrial fibrillation and superimposed conventional risk factors: continued search to configure the genuine etiology of “eternal arrhythmia”. J Atr Fibrillat. (2016) 9:6–11. doi: 10.4022/jafib.1503
8. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. (2008) 51:802–9. doi: 10.1016/j.jacc.2007.09.064
9. Rucker-Martin C, Milliez P, Tan S, Decrouy X, Recouvreur M, Vranckx R, et al. Chronic hemodynamic overload of the atria is an important factor for gap junction remodeling in human and rat hearts. Cardiovasc Res. (2006) 72:69–79. doi: 10.1016/j.cardiores.2006.06.016
10. Delgado V, Di Biase L, Leung M, Romero J, Tops LF, Casadei B, et al. Structure and function of the left atrium and left atrial appendage: af and stroke implications. J Am Coll Cardiol. (2017) 70:3157–72. doi: 10.1016/j.jacc.2017.10.063
11. Gasparovic H, Cikes M, Kopjar T, Hlupic L, Velagic V, Milicic D, et al. Atrial apoptosis and fibrosis adversely affect atrial conduit, reservoir and contractile functions. Interact Cardiovasc Thorac Surg. (2014) 19:223–30. doi: 10.1093/icvts/ivu095
12. Park J, Yang P-S, Kim T-H, Uhm J-S, Kim JY, Joung B, et al. Low left atrial compliance contributes to the clinical recurrence of atrial fibrillation after catheter ablation in patients with structurally and functionally normal heart. PLoS ONE (2015) 10:e0143853. doi: 10.1371/journal.pone.0143853
13. Mahfouz RA, Alawady WS, Salem A, Abdelghafar AS. Atrial dyssynchrony and left atrial stiffness are risk markers for cryptogenic stroke in patients with patent foramen ovale. Echocardiography (2017) 34:1888–94. doi: 10.1111/echo.13721
14. Leong DP, Joyce E, Debonnaire P, Katsanos S, Holman ER, Schalij MJ, et al. Left atrial dysfunction in the pathogenesis of cryptogenic stroke: novel insights from speckle-tracking echocardiography. J Am Soc Echocardiogr. (2017) 30:71–9.e1. doi: 10.1016/j.echo.2016.09.013
15. Na JO, Shin SY, Lim HE, Choi CU, Kim SH, Kim JW, et al. Impaired transport function of the left atrium and left atrial appendage in cryptogenic stroke patients with atrial septal aneurysm and without patent foramen ovale. Eur J Echocardiogr. (2011) 12:140–7. doi: 10.1093/ejechocard/jeq164
16. Yaghi S, Moon YP, Mora-McLaughlin C, Willey JZ, Cheung K, Di Tullio MR, et al. Left atrial enlargement and stroke recurrence: the Northern Manhattan Stroke Study. Stroke (2015) 46:1488–93. doi: 10.1161/STROKEAHA.115.008711
17. Wozakowska-Kaplon B. Changes in left atrial size in patients with persistent atrial fibrillation: A prospective echocardiographic study with a 5-year follow-up period. Int J Cardiol. (2005) 101:47–52. doi: 10.1016/j.ijcard.2004.03.010
18. Santulli G, Iaccarino G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas (2016) 93:65–72. doi: 10.1016/j.maturitas.2016.03.022
19. Han C, Rice MW, Cai D. Neuroinflammatory and autonomic mechanisms in diabetes and hypertension. Am J Physiol Endocrinol Metab. (2016) 311:E32–41. doi: 10.1152/ajpendo.00012.2016
20. Baker SE, Limberg JK, Ranadive SM, Joyner MJ. Neurovascular control of blood pressure is influenced by aging, sex, and sex hormones. Am J Physiol Regul Integr Comp Physiol. (2016) 311:R1271–5. doi: 10.1152/ajpregu.00288.2016
21. Guarino D, Nannipieri M, Iervasi G, Taddei S, Bruno RM. The role of the autonomic nervous system in the pathophysiology of obesity. Front Physiol. (2017) 8:665. doi: 10.3389/fphys.2017.00665
22. Tobaldini E, Costantino G, Solbiati M, Cogliati C, Kara T, Nobili L, et al. Sleep, sleep deprivation, autonomic nervous system and cardiovascular diseases. Neurosci Biobehav Rev. (2017) 74(Pt B):321–9. doi: 10.1016/j.neubiorev.2016.07.004
23. Thijs V. Atrial fibrillation detection: fishing for an irregular heartbeat before and after stroke. Stroke (2017) 48:2671–7. doi: 10.1161/STROKEAHA.117.017083
24. He J, Tse G, Korantzopoulos P, Letsas KP, Ali-Hasan-Al-Saegh S, Kamel H, et al. P-Wave indices and risk of ischemic stroke: a systematic review and meta-analysis. Stroke (2017) 48:2066–72. doi: 10.1161/STROKEAHA.117.017293
25. Acampa M, Lazzerini PE, Martini G. Letter by Acampa et al Regarding Article, “Underutilization of Ambulatory ECG Monitoring After Stroke and transient ischemic attack: missed opportunities for atrial fibrillation detection”. Stroke (2016) 47:e277. doi: 10.1161/STROKEAHA.116.014947
26. Acampa M, Lazzerini PE, Martini G. How to identify patients at risk of silent atrial fibrillation after cryptogenic stroke: potential role of p wave dispersion. J Stroke (2017) 19:239–41. doi: 10.5853/jos.2016.01620
27. Acampa M, Guideri F, Tassi R, Dello Buono D, Celli L, di Toro Mammarella L, et al. P wave dispersion in cryptogenic stroke: a risk factor for cardioembolism? Int J Cardiol. (2015) 190:202–4. doi: 10.1016/j.ijcard.2015.04.185
28. Okutucu S, Aytemir K, Oto A. P-wave dispersion: What we know till now? JRSM Cardiovasc Dis. (2016) 5:2048004016639443. doi: 10.1177/2048004016639443
29. Dogan U, Dogan EA, Tekinalp M, Tokgoz OS, Aribas A, Akilli H, et al. P-wave dispersion for predicting paroxysmal atrial fibrillation in acute ischemic stroke. Int J Med Sci. (2012) 9:108–14. doi: 10.7150/ijms.9.108
30. Elansary M, Hamdi M, Zaghla H, Ragab. P-wave dispersion and left atrial indices as predictors of paroxysmal atrial fibrillation in patients with non-hemorrhagic cerebrovascular strokes and transient ischemic attacks. Egypt Heart J. (2014) 66: 369–74. doi: 10.1016/j.ehj.2013.10.004
31. Acampa M, Lazzerini PE, Guideri F, Tassi R, Lo Monaco A, Martini G. Inflammation and Atrial electrical remodeling in patients with embolic strokes of undetermined source. Heart Lung Circ. (2018). doi: 10.1016/j.hlc.2018.04.294. [Epub ahead of print].
32. Vural MG, Cetin S, Yilmaz M, Akdemir R, Gunduz H. Relation between left atrial remodeling in young patients with cryptogenic stroke and normal inter-atrial anatomy. J Stroke (2015) 17:312–9. doi: 10.5853/jos.2015.17.3.312
33. Abou R, Leung M, Tonsbeek AM, Podlesnikar T, Maan AC, Schalij MJ, et al. Effect of aging on left atrial compliance and electromechanical properties in subjects without structural heart disease. Am J Cardiol. (2017) 120:140–7. doi: 10.1016/j.amjcard.2017.03.243
34. Yasar E, Yilmaz B, Yasar AS, Goktepe AS, Alaca R, Mohur H. Effect of autonomic dysfunction on p-wave dispersion in patients with chronic spinal cord injury. Am J Phys Med Rehabil. (2010) 89:824–30. doi: 10.1097/PHM.0b013e3181f1ba2c
35. Köse MD, Bag Ö, Güven B, Meşe T, Öztürk A, Tavli V. P-wave dispersion: an indicator of cardiac autonomic dysfunction in children with neurocardiogenic syncope. Pediatr Cardiol. (2014) 35:596–600. doi: 10.1007/s00246-013-0825-y
36. Scott CC, Leier CV, Kilman JW, Vasko JS, Unverferth DV. The effect of left atrial histology and dimension on P wave morphology. J Electrocardiol. (1983) 16:363–6. doi: 10.1016/S0022-0736(83)80086-7
37. Huo Y, Mitrofanova L, Orshanskaya V, Holmberg P, Holmqvist F, Platonov PG. P-wave characteristics and histological atrial abnormality. J Electrocardiol. (2014) 47:275–80. doi: 10.1016/j.jelectrocard.2014.01.011
38. Bayes de Luna A, Cladellas M, Oter R, Torner P, Guindo J, Marti V, et al. Interatrial conduction block and retrograde activation of the left atrium and paroxysmal supraventricular tachyarrhythmia. Eur Heart J. (1988) 9:1112–8.
39. O'Neal WT, Kamel H, Zhang ZM, Chen LY, Alonso A, Soliman EZ. Advanced interatrial block and ischemic stroke: the Atherosclerosis Risk in Communities Study. Neurology (2016) 87:352–6. doi: 10.1212/WNL.0000000000003445
40. Cheema AN, Ahmed MW, Kadish AH, Goldberger JJ. Effects of autonomic stimulation and blockade on signal-averaged P wave duration. J Am Coll Cardiol. (1995) 26:497–502. doi: 10.1016/0735-1097(95)80028-F
41. Wilhelm M, Roten L, Tanner H, Wilhelm I, Schmid JP, Saner H. Atrial remodeling, autonomic tone, and lifetime training hours in nonelite athletes. Am J Cardiol. (2011) 108:580–5. doi: 10.1016/j.amjcard.2011.03.086
42. Wilhelm M, Brem MH, Rost C, Klinghammer L, Hennig FF, Daniel WG, et al. Early repolarization, left ventricular diastolic function, and left atrial size in professional soccer players. Am J Cardiol. (2010) 106:569–74. doi: 10.1016/j.amjcard.2010.03.072
43. Bagliani G, Della Rocca DG, Di Biase L, Padeletti L. PR Interval and Junctional Zone. Card Electrophysiol Clin. (2017) 9:411–33. doi: 10.1016/j.ccep.2017.05.003
44. Schumacher K, Dagres N, Hindricks G, Husser D, Bollmann A, Kornej J. Characteristics of PR interval as predictor for atrial fibrillation: association with biomarkers and outcomes. Clin Res Cardiol. (2017) 106:767–75. doi: 10.1007/s00392-017-1109-y
45. Thijs VN, Brachmann J, Morillo CA, Passman RS, Sanna T, Bernstein RA, et al. Predictors for atrial fibrillation detection after cryptogenic stroke: Results from CRYSTAL AF. Neurology (2016) 86:261–9. doi: 10.1212/WNL.0000000000002282
46. Carrazco C, Golyan D, Kahen M, Black K, Libman RB, Katz JM. Prevalence and risk factors for paroxysmal atrial fibrillation and flutter detection after cryptogenic ischemic stroke. J Stroke Cerebrovasc Dis. (2018) 27:203–9. doi: 10.1016/j.jstrokecerebrovasdis.2017.08.022
47. Montalvo M, Tadi P, Merkler A, Gialdini G, Martin-Schild S, Navalkele D, et al. PR interval prolongation and cryptogenic stroke: a multicenter retrospective study. J Stroke Cerebrovasc Dis. (2017) 26:2416–20. doi: 10.1016/j.jstrokecerebrovasdis.2017.05.036
48. German DM, Kabir MM, Dewland TA, Henrikson CA, Tereshchenko LG. Atrial fibrillation predictors: importance of the electrocardiogram. Ann Noninvasive Electrocardiol. (2016) 21:20–9. doi: 10.1111/anec.12321
49. Soliman EZ, Prineas RJ, Case LD, Zhang ZM, Goff DC. Ethnic distribution of ECG predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the Atherosclerosis Risk in Communities (ARIC) study. Stroke (2009) 40:1204–11. doi: 10.1161/STROKEAHA.108.534735
50. Goda T, Sugiyama Y, Ohara N, Ikegami T, Watanabe K, Kobayashi J, et al. P-Wave terminal force in lead V1 predicts paroxysmal atrial fibrillation in acute ischemic stroke. J Stroke Cerebrovasc Dis. (2017) 26:1912–5. doi: 10.1016/j.jstrokecerebrovasdis.2017.06.031
51. Okin PM, Kamel H, Kjeldsen SE, Devereux RB. Electrocardiographic left atrial abnormalities and risk of incident stroke in hypertensive patients with electrocardiographic left ventricular hypertrophy. J Hypertens. (2016) 34:1831–7. doi: 10.1097/HJH.0000000000000989
52. Lattanzi S, Cagnetti C, Pulcini A, Morelli M, Maffei S, Provinciali L, et al. The P-wave terminal force in embolic strokes of undetermined source. J Neurol Sci. (2017) 375:175–178. doi: 10.1016/j.jns.2017.01.063
53. Kamel H, O'Neal WT, Okin PM, Loehr LR, Alonso A, Soliman EZ. Electrocardiographic left atrial abnormality and stroke subtype in the atherosclerosis risk in communities study. Ann Neurol. (2015) 78:670-8. doi: 10.1002/ana.24482
54. Tiffany Win T, Ambale Venkatesh B, Volpe GJ, Mewton N, Rizzi P, Sharma RK, et al. Associations of electrocardiographic P-wave characteristics with left atrial function, and diffuse left ventricular fibrosis defined by cardiac magnetic resonance: the PRIMERI study. Heart Rhythm (2015) 12:155–62. doi: 10.1016/j.hrthm.2014.09.044
55. O'Neal WT, Hammadah M, Sandesara PB, Almuwaqqat Z, Samman-Tahhan A, Gafeer MM, et al. The association between acute mental stress and abnormal left atrial electrophysiology. J Cardiovasc Electrophysiol. (2017) 28:1151–7. doi: 10.1111/jce.13295
56. Guan L, Collet JP, Mazowita G, Claydon V. Autonomic nervous system and stress to predict secondary ischemic events after transient ischemic attack or minor stroke: possible implications of heart rate variability. Front Neurol. (2018) 9:90. doi: 10.3389/fneur.2018.00090.
57. Kwon Y, Misialek JR, Duprez D, Alonso A, Jacobs DR Jr, Heckbert SR, et al. Association between sleep disordered breathing and electrocardiographic markers of atrial abnormalities: the MESA study. Europace (2017) 19:1759–66. doi: 10.1093/europace/euw328
58. Floras JS. Sympathetic nervous system in patients with sleep related breathing disorders. Curr Hypertens Rev. (2016) 12:18–26. doi: 10.2174/1573402112666160114093359
59. Rangel MO, O'Neal WT, Soliman EZ. Usefulness of the electrocardiographic P-Wave Axis as a Predictor of Atrial Fibrillation. Am J Cardiol. (2016) 117:100–4. doi: 10.1016/j.amjcard.2015.10.013
60. Perez MV, Dewey FE, Marcus R, Ashley EA, Al-Ahmad AA, Wang PJ, et al. Electrocardiographic predictors of atrial fibrillation. Am Heart J. (2009) 158:622–8. doi: 10.1016/j.ahj.2009.08.002
61. Maheshwari A, Norby FL, Soliman EZ, Koene R, Rooney M, O'Neal WT, et al. Refining prediction of atrial fibrillation risk in the general population with analysis of P-Wave Axis (from the Atherosclerosis Risk in Communities Study). Am J Cardiol. (2017) 120:1980–4. doi: 10.1016/j.amjcard.2017.08.015
62. Spach MS. Mounting evidence that fibrosis generates a major mechanism for atrial fibrillation. Circ Res. (2007) 101:743–5. doi: 10.1161/CIRCRESAHA.107.163956
63. Maheshwari A, Norby FL, Soliman EZ, Koene RJ, Rooney MR, O'Neal WT, et al. Abnormal P-Wave Axis and Ischemic Stroke: The ARIC Study (Atherosclerosis Risk In Communities). Stroke (2017) 48:2060–5. doi: 10.1161/STROKEAHA.117.017226
64. Gladstone DJ, Dorian P, Spring M, Panzov V, Mamdani M, Healey JS, et al. EMBRACE Steering Committee and Investigators. Atrial premature beats predict atrial fibrillation in cryptogenic stroke: results from the EMBRACE trial. Stroke (2015) 46:936–41. doi: 10.1161/STROKEAHA.115.008714
65. Kochhäuser S, Dechering DG, Dittrich R, Reinke F, Ritter MA, Ramtin S, et al. Supraventricular premature beats and short atrial runs predict atrial fibrillation in continuously monitored patients with cryptogenic stroke. Stroke (2014) 45:884–6. doi: 10.1161/STROKEAHA.113.003788
66. O'Neal WT, Kamel H, Judd SE, Safford MM, Vaccarino V, Howard VJ, et al. Usefulness of Atrial premature complexes on routine electrocardiogram to determine the risk of atrial fibrillation (from the REGARDS Study). Am J Cardiol. (2017) 120:782–5. doi: 10.1016/j.amjcard.2017.06.007
67. O'Neal WT, Kamel H, Kleindorfer D, Judd SE, Howard G, Howard VJ, et al. Premature atrial contractions on the screening electrocardiogram and risk of ischemic stroke: the reasons for geographic and racial differences in stroke study. Neuroepidemiology (2016) 47:53–8. doi: 10.1159/000448619
68. Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. (2014) 114:1500–15. doi: 10.1161/CIRCRESAHA.114.303772
69. Chobanyan-Jürgens K, Jordan J. Autonomic nervous system activity and inflammation: good ideas, good treatments, or both? Am J Physiol Heart Circ Physiol. (2015) 309:H1999-2001. doi: 10.1152/ajpheart.00826.2015
71. Pongratz G, Straub RH. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat Rev Rheumatol. (2013) 9:117–126. doi: 10.1038/nrrheum.2012.181
72. Murray DR, Prabhu SD, Chandrasekar B. Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation (2000) 101:2338–41. doi: 10.1161/01.CIR.101.20.2338
73. Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. Beta-adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation (2000) 101:2103–9. doi: 10.1161/01.CIR.101.17.2103
74. Zhao M, He X, Bi XY, Yu XJ, Gil Wier W, Zang WJ. Vagal stimulation triggers peripheral vascular protection through the cholinergic anti-inflammatory pathway in a rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. (2013) 108:345. doi: 10.1007/s00395-013-0345-1
75. Olshansky B. Vagus nerve modulation of inflammation: cardiovascular implications. Trends Cardiovasc Med. (2016) 26:1–11. doi: 10.1016/j.tcm.2015.03.016
76. Brown RD, Jones GM, Laird RE, Hudson P, Long CS. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem Biophys Res Commun. (2007) 362:200–5. doi: 10.1016/j.bbrc.2007.08.003
77. Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. (1996) 98:2854–65.
78. Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. (1999) 79:215–62. doi: 10.1152/physrev.1999.79.1.215
79. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension (2010) 56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635
80. Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE (2012) 7:e35144. doi: 10.1371/journal.pone.0035144
81. Svystonyuk DA, Ngu JM, Mewhort HE, Lipon BD, Teng G, Guzzardi DG, et al. Fibroblast growth factor-2 regulates human cardiac myofibroblast-mediated extracellular matrix remodeling. J Transl Med. (2015) 13:147. doi: 10.1186/s12967-015-0510-4.
82. Rolda'n V, Marin F, Blann AD, Garcia A, Marco P, Sogorb F, et al. Interleukin-6, endothelial activation and thrombogenesis in chronic atrial fibrillation. Eur Heart J. (2003) 24:1373–80. doi: 10.1016/S0195-668X(03)00239-2
83. Conway DS, Buggins P, Hughes E, Lip GY. Relationship of interleukin-6 and C-reactive protein to the prothrombotic state in chronic atrial fibrillation. J Am Coll Cardiol. (2004) 43:2075–82. doi: 10.1016/j.jacc.2003.11.062
84. Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. (2012) 60:2263–70. doi: 10.1016/j.jacc.2012.04.063
85. Mahabadi AA, Massaro JM, Rosito GA, Levy D, Murabito JM, Wolf PA, et al. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: the Framingham Heart Study. Eur Heart J. (2009) 30:850–6. doi: 10.1093/eurheartj/ehn573
86. Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation (2003) 108:2460–66. doi: 10.1161/01.CIR.0000099542.57313.C5
87. Chatterjee TL, Stol LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, et al. Proinflammatory phenotype of perivascular adipocytes influence of high-fat feeding. Circ Res. (2009) 104:416–8. doi: 10.1161/CIRCRESAHA.108.182998
88. Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc Res. (2015) 108:62–73. doi: 10.1093/cvr/cvv208
89. Sacks HS. Fain JN. Human epicardial adipose tissue: a review. Am Heart J. (2007) 153:907–17. doi: 10.1016/j.ahj.2007.03.019
90. Armour JA, Murphy DA, Yuan BX, Macdonald S, Hopkins DA. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat Rec. (1997) 247:289–98.
91. Balcioglu AS, Çiçek D, Akinci S, Eldem HO, Bal UA, Okyay K, et al. Arrhythmogenic evidence for epicardial adipose tissue: heart rate variability and turbulence are influenced by epicardial fat thickness. Pacing Clin Electrophysiol. (2015) 38:99–106. doi: 10.1111/pace.12512
92. Fernandes-Cardoso A, Santos-Furtado M, Grindler J, Ferreira LA, Andrade JL, Santo MA. Epicardial fat thickness correlates with P-wave duration, left atrial size and decreased left ventricular systolic function in morbid obesity. Nutr Metab Cardiovasc Dis. (2017) 27:731–8. doi: 10.1016/j.numecd.2017.05.009
93. Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. (2015) 36:795–805a. doi: 10.1093/eurheartj/eht099
94. Rubattu S, Mennuni S, Testa M, Mennuni M, Pierelli G, Pagliaro B, et al. Pathogenesis of chronic cardiorenal syndrome: is there a role for oxidative stress? Int J Mol Sci. (2013) 14:23011–32. doi: 10.3390/ijms141123011
95. Rosca M.G., Hoppel C.L. Mitochondria in heart failure. Cardiovasc Res. (2010) 88:40–50. doi: 10.1093/cvr/cvq240
96. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. (2003) 35:615–21. doi: 10.1016/S0022-2828(03)00084-1
97. Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. (2001) 280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53
98. Lin YK, Chen YA, Lee TI, Chen YC, Chen SA, Chen YJ. Aging modulates the substrate and triggers remodeling in atrial fibrillation. Circ J. (2018) 82:1237–44. doi: 10.1253/circj.CJ-17-0242
99. Antonopoulos AS, Goliopoulou A, Oikonomou E, Tsalamandris S, Papamikroulis GA, Lazaros G, et al. Redox state in atrial fibrillation pathogenesis and relevant therapeutic approaches. Curr Med Chem. (2017). doi: 10.2174/0929867324666170718130408. [Epub ahead of print].
100. Basantsova NY, Tibekina LM, Shishkin AN. [A role of the autonomic nervous system in cerebro-cardiac disorders]. Zh Nevrol Psikhiatr Im S S Korsakova (2017) 117:153–60. doi: 10.17116/jnevro2017117111153-160
101. Mercanoglu G, Safran N, Uzun H, Eroglu L. Chronic emotional stress exposure increases infarct size in rats: the role of oxidative and nitrosative damage in response to sympathetic hyperactivity. Methods Find Exp Clin Pharmacol. (2008) 30:745–52. doi: 10.1358/mf.2008.30.10.1316822
102. Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. (2008) 80:30–9. doi: 10.1093/cvr/cvn184
103. Weber KT, Brilla CG, Campbell SE, Guarda E, Zhou G, Sriram K. Myocardial fibrosis: role of angiotensin II and aldosterone. Basic Res Cardiol. (1993) 88(Suppl. 1):107–24. doi: 10.1007/978-3-642-72497-8_8
104. Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr, Kasi VS, et al. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol. (2004) 165:1019–32. doi: 10.1016/S0002-9440(10)63363-9
105. Thanigaimani S, Lau DH, Agbaedeng T, Elliott AD, Mahajan R, Sanders P. Molecular mechanisms of atrial fibrosis: implications for the clinic. Expert Rev Cardiovasc Ther. (2017) 15:247–56. doi: 10.1080/14779072.2017.1299005
106. Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. (2006) 20:953–70. doi: 10.1210/me.2004-0536
107. Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ. Angiotensin II stimulates the autocrine production of transforming growth factor beta 1 in adult rat cardiac fibroblasts. J Mol Cell Cardiol. (1995) 27:2347–57. doi: 10.1016/S0022-2828(95)91983-X
108. Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol. (1994) 26:809–20. doi: 10.1006/jmcc.1994.1098
109. Zheng L, Jia X, Zhang C, Wang D, Cao Z, Wang J, et al. Angiotensin II in atrial structural remodeling: the role of Ang II/JAK/STAT3 signaling pathway. Am J Transl Res. (2015) 7:1021–31.
110. Wang Q, Yu Y, Zhang P, Chen Y, Li C, Chen J, et al. The crucial role of activin A/ALK4 pathway in the pathogenesis of Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation. Basic Res Cardiol. (2017) 112:47. doi: 10.1007/s00395-017-0634-1
111. Linz D, Hohl M, Nickel A, Mahfoud F, Wagner M, Ewen S, et al. Effect of renal denervation on neurohumoral activation triggering atrial fibrillation in obstructive sleep apnea. Hypertension (2013) 62:767–74. doi: 10.1161/HYPERTENSIONAHA.113.01728
112. Liu Q, Lu D, Wang S, Wang K, Zhang Q, Wang Y, et al. Renal denervation significantly attenuates cardiorenal fibrosis in rats with sustained pressure overload. J Am Soc Hypertens. (2016) 10:587–96.e4. doi: 10.1016/j.jash.2016.05.006
113. Feng Q, Lu C, Wang L, Song L, Li C, Uppada RC. Effects of renal denervation on cardiac oxidative stress and local activity of the sympathetic nervous system and renin-angiotensin system in acute myocardial infracted dogs. BMC Cardiovasc Disord. (2017) 17:65. doi: 10.1186/s12872-017-0498-1
114. Li ZZ, Jiang H, Chen D, Liu Q, Geng J, Guo JQ, et al. Renal sympathetic denervation improves cardiac dysfunction in rats with chronic pressure overload. Physiol Res. (2015) 64:653–62.
Keywords: ischemic stroke, ECG, P wave, P wave dispersion, autonomic nervous system, atrial fibrosis, atrial dilation, atrial cardiopathy
Citation: Acampa M, Lazzerini PE and Martini G (2018) Atrial Cardiopathy and Sympatho-Vagal Imbalance in Cryptogenic Stroke: Pathogenic Mechanisms and Effects on Electrocardiographic Markers. Front. Neurol. 9:469. doi: 10.3389/fneur.2018.00469
Received: 27 January 2018; Accepted: 31 May 2018;
Published: 19 June 2018.
Edited by:
Tijana Bojić, Vinča Nuclear Institute, University of Belgrade, SerbiaReviewed by:
Elsayed Z. Soliman, Wake Forest School of Medicine, United StatesChiu-Ming Ho, Taipei Veterans General Hospital, Taiwan
Copyright © 2018 Acampa, Lazzerini and Martini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Acampa, TS5BY2FtcGFAYW8tc2llbmEudG9zY2FuYS5pdA==