 Silvia Paciotti
Silvia Paciotti Giovanni Bellomo
Giovanni Bellomo Leonardo Gatticchi
Leonardo Gatticchi Lucilla Parnetti
Lucilla Parnetti- 1Department of Experimental Medicine, University of Perugia, Perugia, Italy
- 2Magnetic Resonance Center (CERM), University of Florence, Sesto Fiorentino, Italy
- 3Laboratory of Clinical Neurochemistry, Department of Medicine, University of Perugia, Perugia, Italy
The accumulation and deposition of α-synuclein aggregates in brain tissue is the main event in the pathogenesis of different neurodegenerative disorders grouped under the term of synucleinopathies. They include Parkinson's disease, dementia with Lewy bodies and multiple system atrophy. To date, the diagnosis of any of these disorders mainly relies on the recognition of clinical symptoms, when the neurodegeneration is already in an advanced phase. In the last years, several efforts have been carried out to develop new diagnostic tools for early diagnosis of synucleinopathies, with special interest to Parkinson's disease. The Protein-Misfolding Cyclic Amplification (PMCA) and the Real-Time Quaking-Induced Conversion (RT-QuIC) are ultrasensitive protein amplification assays for the detection of misfolded protein aggregates. Starting from the successful application in the diagnosis of human prion diseases, these techniques were recently tested for the detection of misfolded α-synuclein in brain homogenates and cerebrospinal fluid samples of patients affected by synucleinopathies. So far, only a few studies on a limited number of samples have been performed to test PMCA and RT-QuIC diagnostic reliability. Neverthless, these assays have shown very high sensitivity and specificity in detecting synucleinopathies even at the pre-clinical stage. Despite the application of PMCA and RT-QuIC for α-synuclein detection in biological fluids is very recent, these techniques seem to have the potential for identifying subjects that will be likely to develop synucleinopathies.
Introduction
Protein-Misfolding Cyclic Amplification (PMCA) and Real-Time Quaking-Induced Conversion (RT-QuIC) represent two ultrasensitive protein amplification methods for detecting pathological protein aggregates in patients affected by protein misfolding disorders (1–3). PMCA and RT-QuIC are assays conceptually similar to a polymerase chain reaction (PCR): a template (protein aggregate) grows at the expense of a substrate (protein monomer) in a cyclic reaction characterized by a growth step followed by an increase in template units. Currently, the need of specific and sensitive early diagnostic tools for synucleinopathies points out the attention on novel approaches. Since α-synuclein (α-syn) follows aggregation mechanisms similar to PrP, PMCA and RT-QuIC assays were tested for the detection of misfolded α-syn in samples of patients affected by synucleinopathies (4–9).
A critical analysis on PMCA and RT-QuIC available data and protocols could help in evaluating whether these techniques could be suitable for the detection of α-syn aggregates in body fluids with high sensitivity and specificity, hopefully at a preclinical stage (4–7). The aim of this review is to provide an overview on existing data on PMCA and RT-QuIC assays, and their possible application for the diagnosis of synucleinopathies.
PMCA and RT-QuIC: A Brief History
The first PMCA protocol was developed by Soto's group in 2001 to detect the misfolded prion protein (PrPSc) (10). The multiplication of the template units was performed by sonication followed by an incubation phase to let the aggregates grow. These steps were repeated several times in a cyclic process to allow the detection of the misfolded proteins in the samples [e.g., brain homogenates (BH), urine, blood, cerebrospinal fluid (CSF) and saliva]; at the end of the process, proteinase K (PK) digestion and western blot (WB) analysis were used to characterize and recognize the presence of pathological aggregates. The PMCA technique was tested in the subsequent years on biological samples coming from animals and patients affected by transmissible spongiform encephalopathy (11, 12). Atarashi et al., taking advantage on PMCA method, developed the QuIC assay by introducing some variants in the protocol (2, 13, 14). In the QuIC, the PrPC substrate coming from hamsters BH was replaced by recombinant PrPC and sonication was replaced with a vigorous intermittent shaking which promoted seeded aggregation of the monomeric substrate (13). Moreover, the WB analysis was substituted by a real-time monitoring (hence the name RT-QuIC) of the fluorescence emitted by the amyloid-sensitive Thioflavin-T dye (ThT) during the aggregation process (2, 14).
Although PMCA and RT-QuIC are both highly sensitive and specific assays, they showed different accuracy in detecting sporadic and variant Creutzfeldt-Jakob disease (CJD), also depending on the nature of the biological samples analyzed (15–17). The success of RT-QuIC in diagnosing prion diseases, led to test this assay for the detection of synucleinopathies (5, 6, 7). For this purpose, an αSyn-PMCA assay, methodologically very similar to a RT-QuIC was also developed by Soto's group (4).
α-Synuclein and Synucleinopathies
α-syn is a small protein (~14 kDa) largely present in the central nervous system at the pre-synaptic neuronal terminals (18, 19). Although α-syn was discovered almost 30 years ago, the physiological role carried out by this protein is not completely understood. It seems to be involved in the regulation of neurotransmitter release, synaptic plasticity and vesicle trafficking, in brain lipid metabolism, remodeling of the membranes, formation of membrane channels, and modification of their activity (20–23).
α-syn is composed of 140 amino acids and it is characterized by 3 distinct regions: N-terminal, central and C-terminal regions. The N-terminus (1–60 residues) contains seven highly conserved hexameric motifs, which form an amphipathic α-helix structure typical of the lipid binding domain of apolipoproteins (24), while the C-terminus (96–140 residues) contains multiple phosphorylation sites and it is enriched in acidic residues. The central domain of α-syn (61–95 residues), known as the non-amyloid-component (NAC), is highly aggregation-prone and plays a key role in cytotoxicity of α-syn (25–27).
At cellular level, α-syn is predominantly present as unfolded soluble monomer with not well-defined secondary or tertiary structures (28–30). Nevertheless, several factors like post-translational modifications (31–33), oxidative stress (28), fatty acids concentration (34–36), proteolysis (37, 38), phospholipids and metal ions (28, 29) can promote the misfolding of α-syn with the consequent formation of oligomers and amyloid-like fibrils (39, 40). α-syn amyloid-like fibrils are composed of several protofilaments containing cross β-sheet secondary structure in which individual β-strands run perpendicular to the fiber axis (41, 42). The α-syn aggregation kinetics is similar to that of the Aβ peptide (43, 44). It is characterized by an initial lag-phase which reflects the seed formation (nucleation phase) and a subsequent growth phase that culminates in a steady state (45).
Aggregated α-syn is involved in the pathogenesis of different neurodegenerative disorders known as synucleinopathies (46–48), which include Parkinson's disease (PD) (49), dementia with Lewy bodies (DLB) (50) and multiple system atrophy (MSA) (51). Fibrillary α-syn is the major constituent of Lewy bodies (LBs) and Lewy neurites (LNs), which represent the main histopathological hallmarks of PD and DLB (46, 47). Differently, in MSA, aggregated α-syn is found in oligodendrocytes as glial cytoplasmic inclusions (48).
The diagnostic value of α-syn as biomarker of synucleinopathies has been extensively investigated (52–56). Several studies have been performed to measure the levels of α-syn species (total, oligomeric and phosphorylated) in body fluids using different techniques: ELISA (57–60), multiplex immunoassays (61, 62), and Förster's resonance energy transfer (63). The heterogeneity of the applied methods partly justifies some ambiguous outcome obtained so far from the available studies. Furthermore, the lower concentration of the oligomeric/fibrillary α-syn species with respect to the monomeric α-syn form and the complexity to develop selective antibodies having high affinity and avidity to the misfolded α-syn species, make it difficult the detection of these species by using the most common antibodies-based assays (52, 64, 65).
The detection of pathogenic aggregates could help in diagnosis, both in terms of specificity and timeliness of diagnosis, since α-syn aggregation is an early phenomenon preceding the onset of clinical symptoms (66).
RT-QuIC and PMCA Assays: Basic Concepts
The RT-QuIC and PMCA techniques are based on the amplification of a preformed quantity of misfolded proteins present in biological fluids or tissue samples. Samples are incubated, at a defined temperature, in a buffer solution containing the monomeric substrate. Preformed aggregates (seeds) work as templates polymerizing at their extremities at the expense of the monomer (Figure 1A). By introducing a shaking/sonication step, the grown aggregates are then fragmented to generate more polymerization points (67). Incubation and fragmentation cycles are repeated multiple times to achieve an exponential amplification of the aggregates. Apart from the basic polymerization and fragmentation processes, also surface catalyzed nucleation should be considered in the aggregation kinetics (68). This mechanism consists in the formation of new nuclei of misfolded proteins on the surface of preformed fibrils and it has been recently proposed for PrPSc (69), Aβ peptides (68, 70), and α-syn (71).
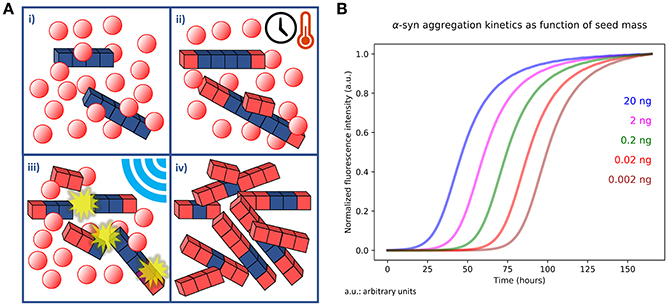
Figure 1. (A) Outline of PMCA and RT-QuIC kinetic assays. (i) An aliquot of tissue homogenate or a biological fluid containing a quantity of aggregates (blue cubes) is dissolved in a buffer containing abundant monomer in solution (red spheres). (ii) The sample is incubated for a defined time at specific temperature. In this phase the preformed aggregates undergo polymerization at their extremities and catalyze the formation of new nuclei on their surfaces. Monomers that undergo misfolding are depicted as red cubes. (iii) The number of available points for polymerization is increased by performing sonication or shaking of the sample, thus fragmenting the fibrils grown in the previous step. The steps (ii) and (iii) are repeated several times. (iv) At the end of the procedure the initial quantity of misfolded and aggregated protein is exponentially amplified at the expense of the monomer present in solution. (B) Simulation of a PMCA or RT-QuIC experiment. The simulation was performed by integrating differential equations describing polymerization, secondary catalyzed nucleation and fragmentation kinetics in presence of different quantities (20, 2, 0.2, 0.02, and 0.002 ng) of preformed aggregates (seeds). The simulation consisted in cycles of 30 min in which fragmentation kinetics was kept active only for 1 min (shaking) and turned off for 29 min (incubation). The cycles were repeated for a total time of 150 h. Normalized fluorescence intensity was calculated by considering it proportional to the total mass of fibrillary aggregates formed at a certain time.
In PMCA, WB analysis is used to detect the amplified PrPSc (10), while in the RT-QuIC and αSyn-PMCA the detection of the misfolded aggregates is performed by recording the fluorescence of the ThT dye. ThT fluorescence (excitation at 450 nm and emission at 480 nm) is enhanced upon binding to fibrils. Compared to WB, ThT fluorescence assay has the limitation to be sensitive only to fibrillary aggregates rich in cross-beta sheet motifs (72). However, ThT assay in multi-well plates has the advantage to be less time-consuming; moreover, the intermittent shaking can be directly performed inside fluorometers and thus easily automated. The recorded fluorescence of ThT in RT-QuIC and αSyn-PMCA is proportional to the mass of fibrillary aggregates present in the sample and its trend gives information about the aggregation kinetics of the monomer. Fluorescence acquisition allows mapping an aggregation curve describing a lag-phase (time with stationary fluorescence), an exponential phase (increase in fluorescence) and a plateau. A simulated example of an ideal output of a RT-QuIC experiment is shown in Figure 1B. The process produces sigmoid-like profiles (73, 74) whose lag-times, slopes and stationary points depend on the experimental conditions (temperature, shaking cycles and strength, pH, buffer, etc.). Particularly, the length of the lag-phase correlates to the amount of seeds in the samples (75). However, since the lag-phase is a threshold value established by the investigator, the t50, named the time necessary to reach the 50% of the maximum fluorescence, is often used as a quantitative and objective measurement of the amplification process. The approximate linear relation between the t50 (or the lag-time) and the logarithm of the seed quantities has been shown for different pathogenic proteins like PrPsc (75) α-syn (4, 7, 76), Aβ1-40 (77), and Aβ1-42 (78). Sometimes, deviations from the ideal lineshape, like multiple inflection points or a decrease of the signal at the end of the reaction are present (6, 79). These abnormalities might be caused by sample heterogeneity (amyloids tend to form a suspension in aqueous solution) or by the entrapment of ThT in large aggregates, respectively (80). Thus, most of the authors prefers to define a lag-phase threshold, in which controls do not exhibit aggregation, while positive samples display an increase in fluorescence intensity that exceed the established threshold (e.g., 5–10 times higher than average baseline fluorescence) (4–7, 75, 81). Apart from the length of the lag-phase, Kang et al. (82) suggested that also differences in amyloid formation rate, ThT fluorescence maxima and integrated area under the curve show discrimination between seeded and unseeded samples, thus these features could be also suitable for αSyn-PMCA and RT-QuIC data analysis.
Protocols
Several physical (temperature and sonication/shaking), chemical (ionic strength, pH, monomer concentration, detergents), and exogenous factors were described to affect α-syn aggregation kinetics (83, 84). The most recent implementations in PMCA and RT-QuIC protocols, specifically applied to the detection of α-syn aggregates for the diagnosis of synucleinopathies, are reported in Table 1 and discussed below.
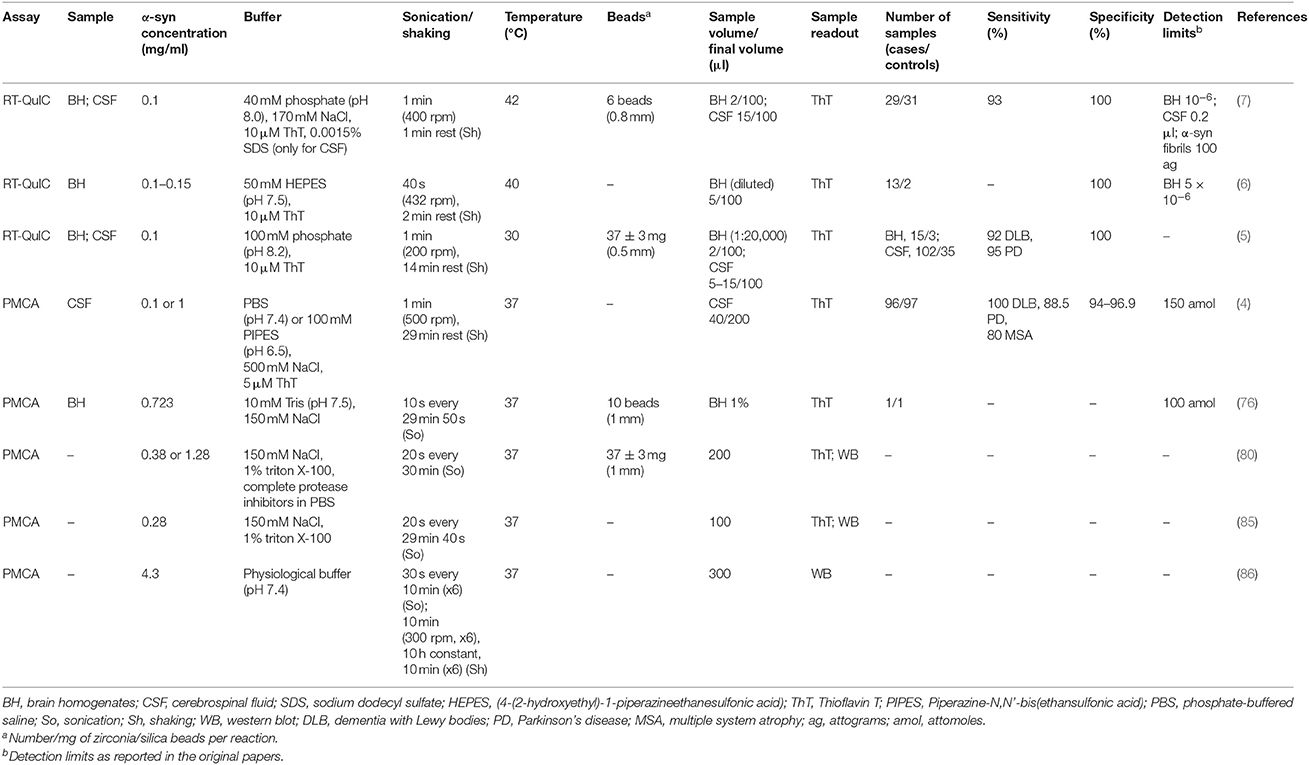
Table 1. PMCA and RT-QuIC assays protocols.
αSyn-PMCA and RT-QuIC Substrate
In vitro aggregation assay usually requires large amounts of highly purified monomeric α-syn as reaction substrate for fibrils polymerization. Large quantities of recombinant α-syn are obtained by using Escherichia coli cultures. The expressed protein can be purified by different chromatographic procedures (7, 87–89). The purity of α-syn preparations can be evaluated by SDS-PAGE followed by silver staining and then confirmed by mass spectrometry. The quality of the initial α-syn monomer solution is a critical factor in determining the successful application of αSyn-PMCA and RT-QuIC techniques. α-syn monomer solution can be filtered with a 100 kDa cutoff filter device (4) in order to remove any preformed aggregates generated during the purification process. To use the optimal amount of substrate in αSyn-PMCA or RT-QuIC, the concentration of the purified α-syn is assessed by spectrophotometric measurement of absorbance at 280 nm (83, 86).
Temperature, pH, and Buffer Composition
Reaction temperature is one of the most well established factors driving α-syn aggregation (39, 90). Generally, in PMCA or RT-QuIC assay, the temperature is set at 37°C. Thirty-seven degree celsius is compatible with a balance between obtaining a short lag-phase, a stable elongation rate, and a minor evaporation of the sample. Similarly, the decrease of pH values toward the isoelectric point of α-syn (pI = 4.67) contributes to the neutralization of protein net charge, that enhances hydrophobicity and boosts the fibrillization process (91). Moreover, the rate of aggregates formation is enhanced by the increase in ionic strength of the reaction buffer (84).
Interestingly, Shahnawaz et al. reported an inhibitory effect of CSF for α-syn aggregation (4); the causes of this behavior are not yet well understood, although Padayachee et al. observed a similar effect also for Aβ (92). Shahnawaz et al. introduced the buffer with the best results in terms of α-syn aggregation timescales and sensitivity in the presence of CSF. By using this buffer, they were able to reduce significantly the lag-phase for positive samples and to decrease the detection sensitivity threshold to femtograms of preformed α-syn seeds. In addition, detergents can be added to reaction buffers to ensure the complete recovery of insoluble amorphous aggregates, together with soluble forms of α-syn fibrils when sonication rather than shaking is used (80, 85). Notably, generation of different species of α-syn aggregates is likely to be linked to different synucleinopathies (93, 94). The application of PMCA protocol allows to amplify brain-derived fibrils with conserved conformation of the original seed (85).
Incubation and Agitation Cycles
The introduction of incubation and agitation cycles played a key-role from the first implementation of PMCA to the last RT-QuIC. In the first version of PMCA (10) the sample was sonicated every hour (five pulses of 1 s each), while in the last RT-QuIC implementations, the sonication step has been replaced by automatic shaking in well plates. Particularly, in the works regarding α-syn, Jung et al. (85), Herva et al. (80), and Roostaee et al. (86) performed sonication on their samples for non-diagnostic applications (Table 1). Conversely, Fairfoul et al. (5), Shahnawaz et al. (4), Sano et al. (6), and Groveman et al. (7) applied the following cycles: 1 min shaking (200 rpm) with 14 min of incubation, 60 s shaking (432 rpm) with 2 min of incubation, 40 s shaking (500 rpm) with 29 min of incubation, and 1 min shaking (400 rpm) with 1 min of incubation, respectively. Shaking is one of the most important promoting factors of α-syn aggregation (83, 84). Nevertheless, it is also important to let the sample rest for some time to promote elongation phase: Herva et al. (80) noticed that alternating cycles of incubation and agitation produced a shorter lag-phase compared to continuous agitation. Furthermore, the addition of zirconia/silica beads to the samples increases the fragmentation and diffusion rates and improves the reproducibility of the assay (5, 80, 83).
αSyn-PMCA and RT-QuIC Studies in Diagnostic Cohorts
Currently, only a few studies have been performed to test the accuracy of PMCA and RT-QuIC as diagnostic tools for synucleinopathies. Groveman et al. performed RT-QuIC on CSF samples from 29 patients affected by synucleinopathies (12 PD and 17 DLB) and 31 non-synucleinopathy controls [including 16 patients affected by Alzheimer's disease (AD)] (7). Almost all synucleinopathy CSF samples (27 out of 29) gave positive RT-QuIC, whereas none of the non-synucleinopathy controls met the criteria to be considered positive (93% sensitivity and 100% specificity). In this work, an end-point dilution assay was also performed to quantify the RT-QuIC seeding activity in PD (n = 1) and DLB (n = 3) BH and DLB (n = 5) CSF samples by calculating the concentration of seeding activity units (SD50). The estimated SD50 was 105-106 per mg of brain tissue and 4–54 per 15 μl of CSF. These results indicate that CSF samples have seeding activities higher than the minimum detectable level of 1 SD50.
Fairfoul et al. tested the RT-QuIC technology on BH from patients affected by DLB, AD, CJD, and control subjects (5). None of the reactions seeded with BH from patients affected by CJD or AD as well as from control subjects gave positive results after 120 h from the beginning of the reaction. The same group analyzed CSF samples from the OPTIMA (Oxford Project to Investigate Memory and Ageing) cohort with the aim to investigate RT-QuIC sensitivity. The study included patients with clinically and neuropathologically confirmed diagnosis of DLB (n = 12), PD (n = 2), progressive supranuclear palsy (PSP) (n = 2), corticobasal degeneration (CBD) (n = 3), DLB with AD pathology (n = 17), AD with incidental LBs (n = 13), pure AD (n = 30), and controls (n = 20). DLB and PD patients were diagnosed with a 92 and 95% sensitivity, respectively, and with a specificity of 100%. A sensitivity of 65% was observed for patients affected by mixed AD/DLB pathology. None of the patients affected by PSP, CBD, or pure AD, resulted positive to RT-QuIC. A validation study was also carried out in CSF samples from 20 patients diagnosed as PD, 15 control subjects, and 3 subjects affected by rapid eye movement sleep behavior disorder (RBD), a condition at high risk of developing synucleinopathies. Out of 20, 19 PD patients resulted positive (sensitivity = 95%, specificity = 100%), whereas all controls were found negative. The three RBD also showed a positive RT-QuIC response, suggesting the suitability of this approach for early diagnosis.
Shahnawaz et al. used the αSyn-PMCA for detecting α-syn aggregates in CSF samples from different synucleinopathies (PD n = 76, DLB n = 10, MSA n = 10) and other miscellaneous neurological disorders (n = 97) including other neurodegenerative diseases not belonging to synucleinopathies (AD, frontotemporal dementia, PSP, ataxia) (4). Out of 76 PD patients, 67 (88%) resulted positive to αSyn-PMCA, whereas 61 out of 65 (94%) patients affected by other neurological disorders resulted negative. Notably, two samples, which were clinically diagnosed as PD after some years from sample collection, resulted positive, indicating the ability of αSyn-PMCA to identify patients even at the prodromal stage. All DLB patients and 8 out of 10 MSA cases were positive at αSyn-PMCA. Out of 14 AD patients, 5 showed positive results. This result might not be considered as false-positive, since α-syn inclusions are not rare in AD brain (95, 96). For this reason, sensitivity and specificity were calculated by excluding AD patients from the analysis. Sensitivity was 88.5% for PD, 100% for DLB and 80% for MSA. Specificity was 94%, reaching 97% when considering patients affected by neurological, but not neurodegenerative, disorders. In this study, the possible correlation between the disease severity and αSyn-PMCA kinetic parameters was also investigated in PD group. A significant negative correlation between the t50 in αSyn-PMCA and the Hoehn and Yahr scale was found. The reduction of the lag-phase suggests the presence of higher concentration of α-syn aggregates in CSF samples of advanced PD cases, thus allowing the monitoring of disease progression. However, these data need to be confirmed in a larger cohort.
Finally, Nishida's group investigated the presence of prion-like seeding of misfolded α-syn in brain samples from patients affected by DLB (n = 7), CJD (n = 3), Gerstmann-Sträussler-Scheinker disease (n = 1), pure AD (n = 2), and controls. They found positive results only in BH from DLB patients (6).
Conclusion and Future Directions
The first trials of PMCA on PrPSc date back to 2001 but only recently the αSyn-PMCA and RT-QuIC techniques have been applied for the amplification and detection of aggregates of misfolded α-syn. The positive results obtained from different studies confirm that αSyn-PMCA and RT-QuIC are suitable assays for detecting α-syn aggregates in CSF samples. Furthermore, the high sensitivity and specificity of these techniques in detecting synucleinopathies, even at the pre-clinical stage, suggest their possible use as diagnostic tools. Although the combined analysis of α-syn aggregates with other CSF biomarkers (e.g., Aβ42, t-Tau and p-Tau) can be used in the cases of uncertain diagnosis (e.g., patients affected by mixed AD/DLB pathology), in-depth investigations are still necessary to perform a differential diagnosis among different synucleinopathies. The study of α-syn aggregation kinetics, the characterization of the fibrillary aggregate structure (e.g., by PK digestion, WB analysis, X-ray scattering and solid-state NMR) (42, 97–100), as well as the detection of other soluble or insoluble α-syn non-fibrillary aggregates might be suitable to this purpose (42, 94, 97, 98, 101).
Furthermore, the possibility to assess the SD50 in CSF samples, might be relevant for determining prognosis in patients even at the early stage of disease (4, 7). So far, αSyn-PMCA and RT-QuIC has been performed mainly in CSF samples; however, based on the encouraging results obtained in the diagnosis of prion disease in both human and animals (102–107), other more “easily accessible” biological fluids like blood, plasma, serum, urine and saliva, as well as peripheral tissues obtained from biopsies (e.g., nasal mucosa, gastrointestinal tract and skin) have the potential to be used as samples for the detection of misfolded α-syn.
Further developments are still needed to standardize operating procedures, decrease the duration of the assays, and increase their sensitivity. To this purpose, testing different shaking cycles and incubation temperatures will be crucial. The reproducibility of the method has also to be improved in order to uniform lag-times, maximum of fluorescence intensity and lineshapes among replicates.
In conclusion, αSyn-PMCA and RT-QuIC have the potential to be effective tools for the diagnosis of synucleinopathies. It will be exciting to follow the growth of scientific reports about this goal in the next future.
Author Contributions
SP, GB, LG, and LP wrote the paper. GB prepared illustrations. LP revised the text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. (2003) 4:49–60. doi: 10.1038/nrn1007
2. Atarashi R, Sano K, Satoh K, Nishida N. Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion (2011) 5:150–3. doi: 10.4161/pri.5.3.16893
3. Properzi F, Pocchiari M. Identification of misfolded proteins in body fluids for the diagnosis of prion diseases. Int J Cell Biol. (2013) 2013:839329. doi: 10.1155/2013/839329
4. Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a biochemical diagnosis of parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. (2017) 74:163–72. doi: 10.1001/jamaneurol.2016.4547
5. Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. (2016) 3:812–8. doi: 10.1002/acn3.338
6. Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, et al. Prion-like seeding of misfolded α-synuclein in the brains of dementia with Lewy body patients in RT-QUIC. Mol Neurobiol. (2017) 55:3916–30. doi: 10.1007/s12035-017-0624-1
7. Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun. (2018) 6:7. doi: 10.1186/s40478-018-0508-2
8. Bernis ME, Babila JT, Breid S, Wüsten KA, Wüllner U, Tamgüney G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun. (2015) 3:75. doi: 10.1186/s40478-015-0254-7
9. Tamgüney G, Korczyn AD. A critical review of the prion hypothesis of human synucleinopathies. Cell Tissue Res. (2017). doi: 10.1007/s00441-017-2712-y. [Epub ahead of print].
10. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature (2001) 411:810–3. doi: 10.1038/35081095
11. Soto C, Anderes L, Suardi S, Cardone F, Castilla J, Frossard M-J, et al. Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett. (2005) 579:638–42. doi: 10.1016/j.febslet.2004.12.035
12. Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE (2009) 4:e4848. doi: 10.1371/journal.pone.0004848
13. Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods (2008) 5:211–2. doi: 10.1038/nmeth0308-211
14. Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. (2011) 17:175–8. doi: 10.1038/nm.2294
15. Orrù CD, Groveman BR, Hughson AG, Manca M, Raymond LD, Raymond GJ, et al. RT-QuIC assays for prion disease detection and diagnostics. Methods Mol Biol. (2017) 1658:185–203. doi: 10.1007/978-1-4939-7244-9_14
16. Rubenstein R, Chang B. Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS ONE (2013) 8:e66352. doi: 10.1371/journal.pone.0066352
17. Henderson DM, Manca M, Haley NJ, Denkers ND, Nalls AV, Mathiason CK, et al. Rapid antemortem detection of CWD prions in deer saliva. PLoS ONE (2013) 8:e74377. doi: 10.1371/journal.pone.0074377
18. Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. (2001) 345:27–32. doi: 10.1016/0014-5793(94)00395-5
19. Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, et al. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. J Neurosci. (2014) 34:2037–50. doi: 10.1523/JNEUROSCI.2581-13.2014
20. Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Mol Brain Res. (1991) 11:335–43. doi: 10.1016/0169-328X(91)90043-W
21. Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. (2012) 3:e350. doi: 10.1038/cddis.2012.94
22. Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. (2010) 285:32486–93. doi: 10.1074/jbc.M110.139576
23. Ottolini D, Calí T, Szabò I, Brini M. Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications. Biol Chem. (2017) 398:77–100. doi: 10.1515/hsz-2016-0201
24. Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. (1998) 21:249–54. doi: 10.1016/S0166-2236(97)01213-7
25. Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA. (2009) 106:20051–56. doi: 10.1073/pnas.0908005106
26. Han H, Weinreb PH, Lansbury PT. The core Alzheimer's peptide NAC forms amyloid fibrils which seed and are seeded by β-amyloid: is NAC a common trigger or target in neurodegenerative disease? Cell Chem Biol. (1995) 2:163–9. doi: 10.1016/1074-5521(95)90071-3
27. Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:11282–6. doi: 10.1073/pnas.90.23.11282
28. Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, et al. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport (1999) 10:717–21. doi: 10.1097/00001756-199903170-00011
29. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT. NACP, a protein implicated in alzheimer's disease and learning, is natively unfolded. Biochemistry (1996) 35:13709–15. doi: 10.1021/bi961799n
30. Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature (2016) 530:45–50. doi: 10.1038/nature16531
31. Ruzafa D, Hernandez-Gomez YS, Bisello G, Broersen K, Morel B, Conejero-Lara F. The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of α-Synuclein. PLoS ONE (2017) 12:e0178576. doi: 10.1371/journal.pone.0178576
32. Andringa G, Lam KY, Chegary M, Wang X, Chase TN, Bennett MC. Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson's disease. FASEB J. (2004) 18:932–4. doi: 10.1096/fj.03-0829fje
33. Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim H-Y, Lamberto GR, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. (2010) 30:3184–98. doi: 10.1523/JNEUROSCI.5922-09.2010
34. Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Sisk A, et al. Abnormal distribution of the non-Abeta component of Alzheimer's disease amyloid precursor/alpha-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. (1998) 78:1169–77.
35. Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. (2001) 276:41958–62. doi: 10.1074/jbc.M105022200
36. Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron (2003) 37:583–95. doi: 10.1016/S0896-6273(03)00024-2
37. Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci USA. (2005) 102:2162–7. doi: 10.1073/pnas.0406976102
38. Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. (2007) 170:1725–38. doi: 10.2353/ajpath.2007.061232
39. Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. (2001) 276:10737–44. doi: 10.1074/jbc.M010907200
40. Volles MJ, Lansbury PT. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry (2003) 42:7871–8. doi: 10.1021/bi030086j
41. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci USA. (2000) 97:4897–4902. doi: 10.1073/pnas.97.9.4897
42. Vilar M, Chou HT, Lührs T, Maji SK, Riek-Loher D, Verel R, et al. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci USA. (2008) 105:8637–42. doi: 10.1073/pnas.0712179105
43. Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci USA. (1997) 94:7942–7.
44. Naiki H, Gejyo F. Kinetic analysis of amyloid fibril formation. Methods Enzymol (1999) 309:305–18.
45. Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. (1999) 274:19509–12.
46. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388:839–40. doi: 10.1038/42166
47. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. (1998) 95:6469–73.
48. Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. (1998) 44:415–22. doi: 10.1002/ana.410440324
49. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primer (2017) 3:17013. doi: 10.1038/nrdp.2017.13
50. McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology (2017) 89:88–100. doi: 10.1212/WNL.0000000000004058
51. Whittaker HT, Qui Y, Bettencourt C, Houlden H. Multiple system atrophy: genetic risks and alpha-synuclein mutations. F1000Research (2017) 6:2072. doi: 10.12688/f1000research.12193.1
52. Parnetti L, Cicognola C, Eusebi P, Chiasserini D. Value of cerebrospinal fluid α-synuclein species as biomarker in Parkinson's diagnosis and prognosis. Biomark Med. (2016) 10:35–49. doi: 10.2217/bmm.15.107
53. Farotti L, Paciotti S, Tasegian A, Eusebi P, Parnetti L. Discovery, validation and optimization of cerebrospinal fluid biomarkers for use in Parkinson's disease. Expert Rev Mol Diagn. (2017) 17:771–80. doi: 10.1080/14737159.2017.1341312
54. Simonsen AH, Kuiperij B, El-Agnaf OMA, Engelborghs S, Herukka SK, Parnetti L, et al. The utility of α-synuclein as biofluid marker in neurodegenerative diseases: a systematic review of the literature. Biomark Med. (2016) 10:19–34. doi: 10.2217/BMM.14.105
55. Visanji NP, Mollenhauer B, Beach TG, Adler CH, Coffey CS, Kopil CM, et al. The Systemic Synuclein Sampling Study: toward a biomarker for Parkinson's disease. Biomark Med. (2017) 11:359–68. doi: 10.2217/bmm-2016-0366
56. Shah A, Hiew KW, Han P, Parsons RB, Chang R, Legido-Quigley C. Alpha-synuclein in bio fluids and tissues as a potential biomarker for Parkinson's disease. Alzheimers Park Dement. (2017) 2.
57. Compta Y, Valente T, Saura J, Segura B, Iranzo Á, Serradell M, et al. Correlates of cerebrospinal fluid levels of oligomeric- and total-α-synuclein in premotor, motor and dementia stages of Parkinson's disease. J Neurol. (2015) 262:294–306. doi: 10.1007/s00415-014-7560-z
58. Hall S, Surova Y, Öhrfelt A, Zetterberg H, Lindqvist D, Hansson O. CSF biomarkers and clinical progression of Parkinson disease. Neurology (2015) 84:57–63. doi: 10.1212/WNL.0000000000001098
59. Førland MG, Öhrfelt A, Oftedal LS, Tysnes OB, Larsen JP, Blennow K, et al. Validation of a new assay for α-synuclein detection in cerebrospinal fluid. Clin Chem Lab Med. (2017) 55:254–60. doi: 10.1515/cclm-2016-0409
60. Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry (2015) 86:1240–7. doi: 10.1136/jnnp-2014-309562
61. Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, et al. Phosphorylated α-synuclein in Parkinson's disease. Sci Transl Med. (2012) 4:121ra20. doi: 10.1126/scitranslmed.3002566
62. Hall S, Öhrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. (2012) 69:1445–52. doi: 10.1001/archneurol.2012.1654
63. Bidinosti M, Shimshek DR, Mollenhauer B, Marcellin D, Schweizer T, Lotz GP, et al. Novel one-step immunoassays to quantify α-synuclein: applications for biomarker development and high-throughput screening. J Biol Chem. (2012) 287:33691–705. doi: 10.1074/jbc.M112.379792
64. Eusebi P, Giannandrea D, Biscetti L, Abraha I, Chiasserini D, Orso M, et al. Diagnostic utility of CSF α-synuclein species in Parkinson's disease: protocol for a systematic review and meta-analysis. BMJ Open (2016) 6:e011113. doi: 10.1136/bmjopen-2016-011113
65. Mollenhauer B, El-Agnaf OMA, Marcus K, Trenkwalder C, Schlossmacher MG. Quantification of α-synuclein in cerebrospinal fluid as a biomarker candidate: review of the literature and considerations for future studies. Biomark Med. (2010) 4:683–9. doi: 10.2217/bmm.10.90
66. Braak H, Bohl JR, Müller CM, Rüb U, de Vos RAI, Del Tredici K. Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord (2006) 21:2042–51. doi: 10.1002/mds.21065
67. Pöschel T, Brilliantov NV, Frömmel C. Kinetics of prion growth. Biophys J. (2003) 85:3460–74. doi: 10.1016/S0006-3495(03)74767-5
68. Linse S. Monomer-dependent secondary nucleation in amyloid formation. Biophys Rev. (2017) 9:329–38. doi: 10.1007/s12551-017-0289-z
69. Orgel LE. Prion replication and secondary nucleation. Chem Biol. (1996) 3:413–4. doi: 10.1016/S1074-5521(96)90087-3
70. Meisl G, Kirkegaard JB, Arosio P, Michaels TCT, Vendruscolo M, Dobson CM, et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat Protoc. (2016) 11:252. doi: 10.1038/nprot.2016.010
71. Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, et al. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. (2017) 50:e6. doi: 10.1017/S0033583516000172
72. Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta (2010) 1804:1405–12. doi: 10.1016/j.bbapap.2010.04.001
74. Cohen SIA, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM, Knowles TPJ. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys. (2011) 135:065105. doi: 10.1063/1.3608916
75. Henderson DM, Davenport KA, Haley NJ, Denkers ND, Mathiason CK, Hoover EA. Quantitative assessment of prion infectivity in tissues and body fluids by real-time quaking-induced conversion. J Gen Virol. (2015) 96:210–9. doi: 10.1099/vir.0.069906-0
76. Becker K, Wang X, Vander Stel K, Chu Y, Kordower J, Ma J. Detecting alpha synuclein seeding activity in formaldehyde-fixed MSA patient tissue by PMCA. Mol Neurobiol. (2018). doi: 10.1007/s12035-018-1007-y. [Epub ahead of print].
77. Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, et al. A Kinetic aggregation assay enabling selective and sensitive Aβ amyloid quantification in cells and tissues. Biochemistry (2011) 50:1607–17. doi: 10.1021/bi1013744
78. Arosio P, Cukalevski R, Frohm B, Knowles TPJ, Linse S. Quantification of the concentration of Aβ42 propagons during the lag phase by an amyloid chain reaction assay. J Am Chem Soc. (2014) 136:219–25. doi: 10.1021/ja408765u
79. Peden AH, McGuire LI, Appleford NEJ, Mallinson G, Wilham JM, Orrú CD, et al. Sensitive and specific detection of sporadic Creutzfeldt–Jakob disease brain prion protein using real-time quaking-induced conversion. J Gen Virol. (2012) 93:438–49. doi: 10.1099/vir.0.033365-0
80. Herva ME, Zibaee S, Fraser G, Barker RA, Goedert M, Spillantini MG. Anti-amyloid compounds inhibit α-synuclein aggregation induced by protein misfolding cyclic amplification (PMCA). J Biol Chem. (2014) 289:11897–905. doi: 10.1074/jbc.M113.542340
81. Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer's disease. Cell Rep. (2014) 7:261–8. doi: 10.1016/j.celrep.2014.02.031
82. Kang HE, Mo Y, Abd Rahim R, Lee HM, Ryou C. Prion diagnosis: application of real-time quaking-induced conversion. BioMed Res Int. (2017) 2017:5413936. doi: 10.1155/2017/5413936
83. Giehm L, Otzen DE. Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal Biochem. (2010) 400:270–81. doi: 10.1016/j.ab.2010.02.001
84. Narkiewicz J, Giachin G, Legname G. In vitro aggregation assays for the characterization of α-synuclein prion-like properties. Prion (2014) 8:19–32. doi: 10.4161/pri.28125
85. Jung BC, Lim Y-J, Bae E-J, Lee JS, Choi MS, Lee MK, et al. Amplification of distinct α-synuclein fibril conformers through protein misfolding cyclic amplification. Exp Mol Med. (2017) 49:e314. doi: 10.1038/emm.2017.1
86. Roostaee A, Côté S, Roucou X. Aggregation and amyloid fibril formation induced by chemical dimerization of recombinant prion protein in physiological-like conditions. J Biol Chem. (2009) 284:30907–16. doi: 10.1074/jbc.M109.057950
87. Volpicelli-Daley LA, Luk KC, Lee VMY. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. (2014) 9:2135–46. doi: 10.1038/nprot.2014.143
88. Paslawski W, Lorenzen N, Otzen DE. Formation and characterization of α-synuclein oligomers. Methods Mol Biol. (2016) 1345:133–50. doi: 10.1007/978-1-4939-2978-8_9
89. Huang C, Ren G, Zhou H, Wang C. A new method for purification of recombinant human alpha-synuclein in Escherichia coli. Protein Expr Purif. (2005) 42:173–7. doi: 10.1016/j.pep.2005.02.014
90. Ariesandi W, Chang CF, Chen TE, Chen YR. Temperature-dependent structural changes of Parkinson's alpha-synuclein reveal the role of pre-existing oligomers in alpha-synuclein fibrillization. PLoS ONE (2013) 8:e53487. doi: 10.1371/journal.pone.0053487
91. Gould N, Mor DE, Lightfoot R, Malkus K, Giasson B, Ischiropoulos H. Evidence of native α-synuclein conformers in the human brain. J Biol Chem. (2014) 289:7929–34. doi: 10.1074/jbc.C113.538249
92. Padayachee ER, Zetterberg H, Portelius E, Borén J, Molinuevo JL, Andreasen N, et al. Cerebrospinal fluid-induced retardation of amyloid β aggregation correlates with Alzheimer's disease and the APOE ε4 allele. Brain Res. (2016) 1651:11–6. doi: 10.1016/j.brainres.2016.09.022
93. Melki R. Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J Park Dis. (2015) 5:217–27. doi: 10.3233/JPD-150543
94. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature (2015) 522:340–4. doi: 10.1038/nature14547
95. Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, et al. Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. (2015) 129:729–48. doi: 10.1007/s00401-015-1406-3
96. Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. (2000) 10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x
97. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell (2013) 154:103–17. doi: 10.1016/j.cell.2013.05.057
98. Peng C, Gathagan RJ, Lee VMY. Distinct α-Synuclein strains and implications for heterogeneity among α-Synucleinopathies. Neurobiol Dis. (2018) 109:209–18. doi: 10.1016/j.nbd.2017.07.018
99. Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. (2007) 2:18. doi: 10.1186/1750-1326-2-18
100. Saverioni D, Notari S, Capellari S, Poggiolini I, Giese A, Kretzschmar HA, et al. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem. (2013) 288:27972–85. doi: 10.1074/jbc.M113.477547
101. Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. (2013) 4:2575. doi: 10.1038/ncomms3575
102. Escada PA, Lima C, da Silva JM. The human olfactory mucosa. Eur Arch Otorhinolaryngol. (2009) 266:1675–80. doi: 10.1007/s00405-009-1073-x
103. Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, Groveman BR, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. (2014) 371:519–29. doi: 10.1056/NEJMoa1315200
104. Haley NJ, Motter AV de, Carver S, Henderson D, Davenport K, Seelig DM, et al. Prion-seeding activity in cerebrospinal fluid of deer with chronic wasting disease. PLoS ONE (2013) 8:e81488. doi: 10.1371/journal.pone.0081488
105. Haley NJ, Carver S, Hoon-Hanks LL, Henderson DM, Davenport KA, Bunting E, et al. Detection of chronic wasting disease in the lymph nodes of free-ranging cervids by real-time quaking-induced conversion. J Clin Microbiol. (2014) 52:3237–43. doi: 10.1128/JCM.01258-14
106. Haley NJ, Siepker C, Walter WD, Thomsen BV, Greenlee JJ, Lehmkuhl AD, et al. Antemortem detection of chronic wasting disease prions in nasal brush collections and rectal biopsy specimens from white-tailed deer by real-time quaking-induced conversion. J Clin Microbiol. (2016) 54:1108–16. doi: 10.1128/JCM.02699-15
107. Haley NJ, Siepker C, Hoon-Hanks LL, Mitchell G, Walter WD, Manca M, et al. Seeded amplification of chronic wasting disease prions in nasal brushings and recto-anal mucosa-associated lymphoid tissues from elk by real-time quaking-induced conversion. J Clin Microbiol. (2016) 54:1117–26. doi: 10.1128/JCM.02700-15
Keywords: PMCA, RT-QuIC, α-synuclein, synucleinopathies, early diagnosis
Citation: Paciotti S, Bellomo G, Gatticchi L and Parnetti L (2018) Are We Ready for Detecting α-Synuclein Prone to Aggregation in Patients? The Case of “Protein-Misfolding Cyclic Amplification” and “Real-Time Quaking-Induced Conversion” as Diagnostic Tools. Front. Neurol. 9:415. doi: 10.3389/fneur.2018.00415
Received: 20 March 2018; Accepted: 22 May 2018;
Published: 06 June 2018.
Edited by:
Claudio Soto, University of Texas Health Science Center at Houston, United StatesReviewed by:
Luis Concha-Marambio, Amprion Inc., United StatesAlison Jane Ellen Green, University of Edinburgh, United Kingdom
Copyright © 2018 Paciotti, Bellomo, Gatticchi and Parnetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucilla Parnetti, bHVjaWxsYS5wYXJuZXR0aUB1bmlwZy5pdA==
†These authors have contributed equally to this work.