 Susan M. Tyree
Susan M. Tyree Jeremy C. Borniger
Jeremy C. Borniger Luis de Lecea
Luis de Lecea- Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, United States
The lateral hypothalamus is comprised of a heterogeneous mix of neurons that serve to integrate and regulate sleep, feeding, stress, energy balance, reward, and motivated behavior. Within these populations, the hypocretin/orexin neurons are among the most well studied. Here, we provide an overview on how these neurons act as a central hub integrating sensory and physiological information to tune arousal and motivated behavior accordingly. We give special attention to their role in sleep-wake states and conditions of hyper-arousal, as is the case with stress-induced anxiety. We further discuss their roles in feeding, drug-seeking, and sexual behavior, which are all dependent on the motivational state of the animal. We further emphasize the application of powerful techniques, such as optogenetics, chemogenetics, and fiber photometry, to delineate the role these neurons play in lateral hypothalamic functions.
Introduction
Through decades of neuroanatomical and behavioral work, the lateral hypothalamic area (LHA) has been delineated as a key region for the integration and regulation of sleep, feeding, stress, energy balance, reward, and motivated behavior. Further in-depth analyses of molecular, cellular, and circuit-level functions of the LHA have made progress in gaining a holistic understanding of how the LHA integrates sensory information to influence arousal and behavior. The lateral hypothalamic cell population that has been the most intensely studied thus far is a group of neurons expressing the neurotransmitters hypocretin-1 and -2 (also known as orexin-A and -B). Cells expressing these peptides span the lateral and perifornical/medial hypothalamus (throughout the tuberal hypothalamus). These cells were identified by two groups at essentially the same time. The first approach used directional tag PCR subtraction, a technique adapted to enrich mRNAs that were specifically expressed in the hypothalamus (1). They observed a hypothalamic-specific mRNA (prepro-hypocretin), which encoded two putative protein products (Hcrt-1 and -2). This group named the newly identified neurotransmitters hypocretins after the name was voted on at an annual Society for Neuroscience meeting, the rationale being their restricted hypothalamic localization and their similarities to the gut peptide secretin. They found that these peptides were present in synaptic vesicles, and one of these peptides could stimulate hypothalamic neurons in vitro, suggesting it acted as a neurotransmitter. Another group identified the same peptides via ligand screening of multiple orphan G-protein coupled receptors (GPCRs) using a cell-based reporter system, and, because they promoted feeding, named them “orexins” (2). Over the last 20 years, it has become apparent that the essential purpose of the hypocretins/orexins seems to be for wake maintenance (hypocretin deficiency results in the sleep disorder narcolepsy, discussed below) and not feeding. Therefore, we refer to them as the hypocretins throughout this manuscript.
It is unsurprising that hypocretin neurons are fundamental stabilizers of wakefulness as well as powerful modulators of motivated behavior. The hypocretins were found to be expressed exclusively in an LHA population of glutamatergic cells, and project to most nuclei that regulate sleep/wake behavior (3). Hypocretin (Hcrt) neurons are extremely heterogeneous (Figure 1), this heterogeneity has recently been explored using single cell sequencing to show the large number of genes that are co-expressed within Hcrt cells (4). Hcrt neurons are transcriptionally and electrophysiologically distinct from co-mingled neurons expressing melanin concentrating hormone [MCH; (5, 6)], and they are most strongly active during active wakefulness, decrease activity during quiet wakefulness, and are silent during NREM and REM sleep (7, 8). This is essentially the opposite firing pattern displayed by neurons expressing MCH, which innervate many of the same targets as Hcrt neurons (9, 10). Indeed, evidence suggests inhibitory signaling from Hcrt to MCH neurons prevents simultaneous activation of both neural populations (11). Hcrt and MCH neural populations can be further discriminated based on their expression of key transcription factors [Lhx9; 100% of Hcrt neurons, 3.4% of MCH neurons; and Nkx2.1: 60.1% of MCH neurons and 0% of Hcrt neurons; (4)]. Afferent inputs to Hcrt neurons were mapped using a combination of tract tracing methods, uncovering major projections from the lateral septal nucleus, bed nucleus of the stria terminalis, preoptic area, multiple hypothalamic nuclei, substantia nigra, and ventral tegmental area (VTA), as well as the dorsal raphe (12). Genetic tracing studies revealed cell-type specific inputs arriving from cholinergic neurons in the laterodorsal tegmentum, preoptic GABAergic neurons, as well as 5-HT neurons in the median/paramedian raphe, suggesting a major role for these neurons in functions ranging from neuroendocrine control to arousal and metabolic processes (13).
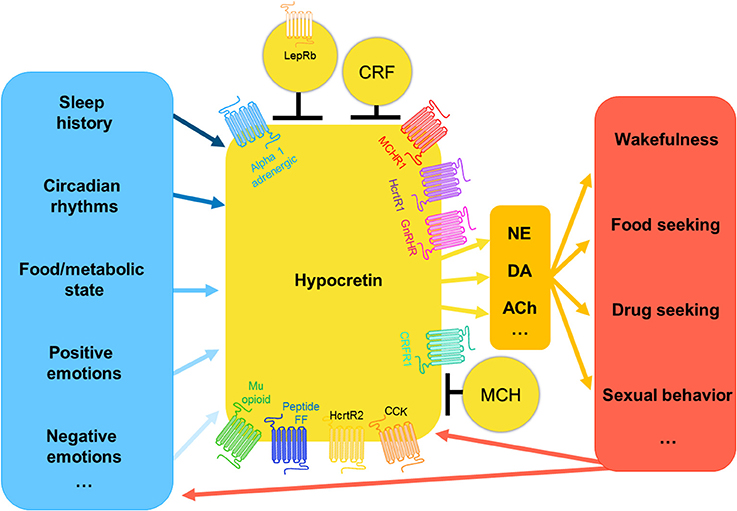
Figure 1. The hypocretin system: inputs and outputs. Hypocretin neurons receive information regarding nutritional state, emotional state, and environmental cues regarding temperature, time of day, and recent sleep history, the weight that these inputs have is very likely variable and interrelated. Hypocretin-expressing neurons are very heterogeneous, they are known to co-express numerous other genes. Additionally, hypocretin-expressing neurons are modulated by the activity of surrounding neuron populations present in the lateral hypothalamus, including melanin-concentrating hormone, leptin receptor-expressing neurons, and corticotropin releasing factor neurons. Hypocretin neurons project to many neuron populations throughout the brain, including norepinephrine, dopaminergic, and cholinergic neurons. It is via these pathways that the hypocretin system modulates the wide range of outputs related to arousal and motivated behavior. In turn, the consequences of these behaviors go on to inform the hypocretin system as to the newly updated status of the animal, and thus determine the next hypocretin-driven behavioral output.
Highlighting its essential role in vigilance state stability is the conservation of the Hcrt peptides throughout the phylogenetic tree. For example, hypothalamic Hcrt expression can be detected in the zebrafish embryo 22 h post-fertilization (14), and Hcrt overexpression causes an insomnia-like phenotype in adult zebrafish (15). Further studies have demonstrated that Hcrt neurons send widespread projections throughout the zebrafish brain that participate in the consolidation of wakefulness, as they do in mammals (16, 17). Indeed, genetic destruction of Hcrt neurons fragments vigilance states in zebrafish (18, 19). In cavefish, Hcrt signaling seems to play a role in the evolution of sleep loss. Hcrt is expressed 3-fold more in cave vs. surface fish, and these fish sleep much less than their surface-dwelling counterparts. Indeed, manipulations that promote sleep inhibit Hcrt expression in cave, but not closely related surface fish (20). Hcrts are also expressed in the avian brain, where they are localized to a single population of neurons spanning the paraventricular and lateral hypothalamus (21, 22).
Arousal Transitions
During World War I, Viennese neurologist Baron Constantin von Economo meticulously analyzed and documented the symptoms of a novel condition he termed “encephalitis lethargica” (23). The causative agent for the condition (presumed viral or autoimmune) remains unknown, but patients showed prolonged states of sleep (>20 h/day) and they could only be awakened with strong and sufficient stimulation (24). He noted that many of these patients had significant damage to the posterior hypothalamus (containing the LHA) and rostral midbrain, while patients showing the opposite problem (prolonged insomnia) had damage to the basal forebrain and preoptic area (25). Based on these observations, he predicted that neurons in the anterior hypothalamus promote sleep while those in the posterior region promote wakefulness. During the following years, his predictions were largely substantiated in a number of species including monkeys (26), rats (27), and cats (28). With the application of the electroencephalogram (EEG; discussed below) to sleep/wake analyses (29), as well as significant advancements in molecular biology, different cell groups within the hypothalamus have been implicated in controlling arousal.
In all mammals (and many non-mammalian vertebrates), sleep stages and structure can be objectively determined via comparative analysis of the electroencephalogram (EEG) and electromyogram (EMG) signals. The EEG reflects large scale changes in electrical activity in the cerebral cortex, as the firing rate of cortical neurons steadily declines during non-rapid eye movement (NREM) sleep in comparison to rapid eye movement (REM) sleep and wakefulness (30–32). Aggregate firing rates of cortical ensembles (reflected in the EEG) can be parsed into conventional bandwidths at approximate frequencies: alpha (9–12 Hz), beta (12–30 Hz), delta (i.e., slow-waves; 0.5–4 Hz), low (30–60 Hz), and high (60–100 Hz) gamma, and theta (5–9 Hz). Synchronization (reflected in high-voltage, low frequency oscillations) of the cortical EEG during NREM sleep depends on a corticothalamocortical loop, which is modulated by local oscillators and is distally controlled by subcortical systems [including those located in the LHA; (33, 34)]. During NREM sleep, delta waves dominate the EEG, and EMG activity (postural tone) is low or absent. During wakefulness, EMG activity is high reflecting muscle activity, and the EEG shows task dependent spectral properties, with low theta frequencies building over time indicating growing sleep propensity (35). REM sleep, also known as paradoxical sleep, is characterized by a wake-like (high theta) EEG spectra composition, but (paradoxically) low or absent EMG activity.
The sleep disorder narcolepsy [characterized by daytime sleepiness, aberrant transitions from wakefulness into REM sleep, and spontaneous loss of muscle tone, referred to as cataplexy; (36)] highlights the essential role of Hcrt in arousal regulation. Loss of Hcrt or Hcrt receptors causes a narcolepsy-like phenotype in mice, including cataplexy and altered sleep/wake cycles (37, 38). Additional canine data identified a genetic defect in Hcrt receptor 2 (HcrtR2) linked to the development of narcolepsy in dogs (36), and several studies have confirmed that degenerative loss of Hcrt neurons is responsible for human narcolepsy (39–41). Degenerative cell loss, rather than reduced Hcrt gene expression is the likely cause because other markers that co-localize within Hcrt neurons are reduced in narcoleptic patients as well (42). Hcrt is a primary stabilizer of wakefulness, as narcoleptics or transgenic mouse models of the disorder display normal amounts of sleep, but with inappropriate transitions between each vigilance state (43, 44). This suggests that Hcrt neurons serve a surprisingly non-redundant role in the regulation of wakefulness, as their loss creates such a marked effect on the stability of arousal.
Initial microdialysis (45) and cFos immunoreactivity (46) studies demonstrated a circadian modulation of Hcrt neural activity, where Fos expression within Hcrt neurons peaked during the active (dark) phase in rodents. Subsequent juxtacellular recordings in head-fixed or freely moving animals demonstrated that Hcrt activity is largely phasic and firing rates peak prior to sleep-to-wake transitions by approximately 10–20 s (7, 8). Building on these studies, the development of optogenetic technologies for manipulating brain circuits has contributed significantly to the investigation of arousal circuits (47). Indeed, the first primary functional evidence for Hcrt's role in wakefulness was presented in 2007, where the first in vivo use of optogenetics revealed that light-evoked stimulation of Hcrt neurons powerfully promoted wakefulness in mice, an effect dependent on Hcrt (and not other co-released neurotransmitters) signaling (48). The effect was frequency-dependent, as stimulations below 5 Hz failed to alter the probability of sleep-to-wake transitions. Further studies demonstrated that long-term optogenetic silencing of Hcrt neurons promotes slow-wave sleep in mice, although only during the light (inactive) phase (49). These optogenetic findings were recapitulated via chemogenetic experiments where designer receptors exclusively activated by designer drugs (DREADDs) were expressed in Hcrt neurons. Engaging excitatory Gq signaling in Hcrt neurons increased cFos immunoreactivity and promoted wakefulness while inhibitory Gi signaling promoted sleep (50). More recent work has investigated exactly which components of wakefulness for which Hcrt is necessary. For example, Vassali & Franken observed that Hcrt-deficient mice had marked reductions in theta-dominated wakefulness during baseline recordings, an oscillatory pattern associated with goal-driven and exploratory behavior (51). Upon forced wakefulness however, normal amounts of theta-dominated wakefulness persisted. These data suggest that Hcrt is necessary for spontaneous, but not enforced wakefulness, which may rely on other neuromodulators. This avenue of research has led to the development of novel drugs for insomnia. As insomnia is thought to be the result of overactive wake-promoting neural systems (52), Hcrt receptor antagonists have been developed as a targeted therapeutic approach. Suvorexant (Belsomra) is the first dual Hcrt receptor antagonist approved for use by the USDA (in 2014) and is now prescribed for insomnia throughout the USA and Japan.
Efferents Involved in Cortical Arousal
How do these cells in the subcortical hypothalamus promote activation and desynchronization of the cortex? One possibility is that diffuse projections from Hcrt neurons generally promote arousal across the brain. Alternatively, specific projections may be essential for their arousal-promoting properties. Direct actions of the Hcrts on arousal-promoting nuclei has been demonstrated for locus coeruleus (LC) noradrenergic neurons, 5-HT neurons in the dorsal raphe, dopaminergic (DA) neurons within the VTA, histamine neurons in the tuberomammillary nucleus, and cholinergic neurons in the brainstem and basal forebrain (53). Below, we discuss the varying degrees of evidence supporting the role of Hcrt in regulating the arousal-promoting effects of each of these areas.
Locus Coeruleus (LC)
The noradrenergic LC has long been recognized to promote wakefulness and arousal (54, 55), and it receives the densest input from Hcrt neurons (3). Direct administration of Hcrt to the LC depolarizes neurons in this region and increases LC firing rates, which is associated with arousal (56–59). Further studies involving Hcrt stimulation with optogenetic silencing of the LC revealed that it is a critical hub for relaying the signal from the LHA to promote wakefulness (57, 60), as activation of Hcrt neurons with simultaneous inhibition of LC prevented sleep-to-wake transitions. Additionally, increasing the excitability of LC neurons using step function opsins (SFOs) was shown to facilitate Hcrt-mediated wakefulness.
Basal Forebrain (BF)
The basal forebrain is a heterogenous structure composed of cholinergic and non-cholinergic (mostly GABAergic) neurons with differential effects on arousal, spanning the substantia innominata, the vertical and horizontal limbs of the diagonal band, the extended amygdala, ventral pallidum, and the medial septum (61). These areas receive moderate to heavy input from Hcrt neurons (3). The arousal promoting effects of basal forebrain stimulation has been extensively characterized using pharmacological, electrical, and chemical ablation methods (62–65). Using cell-type specific optogenetic tools, stimulation of cholinergic neurons within the basal forebrain was shown to be sufficient for cortical activation and transitions out of NREM sleep (66, 67). Additional targeting of BF GABAergic neurons demonstrated their role in cortical activation and wakefulness (68). BF cholinergic neurons were demonstrated to fire predominantly during cortical activation rather than slow-wave activity and are activated by Hcrt (69). Injection of Hcrt into the basal forebrain stimulates cortical activation and wakefulness (70). Hcrt-1 is released in the BF during wakefulness (71), where it has differential actions on cholinergic and non-cholinergic neurons (72, 73), leading to cortical release of acetylcholine and arousal [for review, see (74)].
Tuberomammillary Nucleus (TMN)
The TMN contains histaminergic neurons that are silent during sleep and begin to fire after wake onset, where histamine release promotes arousal (75–77). Acute silencing of TMN histamine neurons inhibits wakefulness and rapidly induces NREM sleep (78). This area receives moderate to dense input from Hcrt neurons (3). Hcrt was shown to directly activate histaminergic neurons in the TMN (79, 80), and further modulate GABAergic inputs to this region (81). Additional optogenetic studies provided evidence that Hcrt neurons are capable of fast glutamatergic control of histaminergic neurons in the TMN (82). However, Hcrt inputs to TMN histaminergic neurons, in contrast to the LC, do not seem to be important for regulating sleep-to-wake transitions, as stimulation of Hcrt neurons is sufficient to promote awakening in mice lacking histidine decarboxylase [the rate limiting enzyme in histamine synthesis; (83)]. Further optogenetic manipulation of Hcrt fibers in the TMN is necessary to effectively delineate the role this circuit plays in sleep/wake regulation.
Dorsal Raphe Nucleus (DRN)
The DRN has long been implicated in arousal, showing vigilance state-specific changes in firing rates (84). Composed of primarily serotonergic (5-HT) neurons, they are excited by multiple arousal-related peptides, including Hcrt (85–88). Hcrt neurons receive reciprocal input from 5-HT neurons, which directly inhibit them via the 5HT1A receptor, and indirectly via enhancement of GABAergic inputs (89). Direct stimulation of 5-HT-expressing DRN neurons demonstrated that ChR2-mediated excitation of these cells caused an immediate transition from NREM sleep to wakefulness (90), and an additional population of DA neurons in this region was recently implicated in arousal regulation (91). Similar to the TMN, direct (i.e., cell-type and circuit specific) investigation of Hcrt's actions on DRN 5-HT or DA neurons in arousal regulation has yet to be completed.
Laterodorsal Tegmental Nucleus (LDT)
The laterodorsal tegmental nucleus (LDT) is a key pontine site in the regulation of wakefulness (92), primarily composed of cholinergic and non-cholinergic neurons (93). Optogenetic activation of LDT cholinergic neurons increases REM sleep episodes (but not duration) when stimulations were done during NREM sleep (94). Hcrt-1 injections into the LDT promote wakefulness in cats, suggesting that an arousal circuit from Hcrt to the LDT exists (95). Further studies revealed that Hcrt acts on LDT neurons at both pre- and post-synaptic sites. Pre-synaptically, Hcrt increases the amplitude and frequency of spontaneous excitatory post-synaptic currents (EPSCs) via triggering action potentials and enhancing synaptic transmission in glutamatergic nerve terminals (96). Post-synaptically, Hcrt promotes an inward current in tandem with enhanced membrane current noise in both cholinergic and non-cholinergic LDT neurons (96). Similar to other areas studied, application of optogenetic tools to Hcrt projections to LDT neurons has not been described, which would directly integrate the LDT into the Hcrt arousal network.
Ventral Tegmental Area (VTA)
The VTA is composed primarily of DA and GABAergic neurons and has long been recognized as a node regulating arousal and motivation (97, 98). Early electrophysiological studies suggested that VTA and substantia nigra pars compacta neurons do not show behavioral-state dependent alterations in firing rate (97–101). However, subsequent studies challenged this view, where both DA and GABA neurons within the VTA show vigilance-state dependent changes in activity (102, 103). It was only recently that causal evidence was presented demonstrating the role of VTA-DA neurons in vigilance state switching, where DA neurons strongly promote wakefulness and are primarily active during wakefulness and REM sleep (104–106). Hcrt neurons send moderate to dense projections to the VTA, where they activate both DA and non-DA neurons (107, 108). Additionally, Hcrt infusion into the lateral ventricles or directly into the VTA increases DA efflux in the pre-frontal cortex, suggesting that activation of VTA-DA neurons contributes to the vigilance-state modulatory properties of Hcrt (109). Whether a circuit from Hcrt to VTA-DA or GABAergic neurons causally promotes vigilance state switching remains to be determined.
Distributed and Indirect Actions
Hcrt peptides can also have direct effects on cortical neurons. These neurons innervate deep layers of the cerebral cortex (3), and more recent studies suggest that layer VIb cortical neurons directly respond to Hcrt peptides (110, 111). nNOS/NK1R expressing cortical interneurons [which play a putative role in sleep homeostasis (112)] express HcrtR1 mRNA (113). A subpopulation of these interneurons is responsive to Hcrt-1, but lack of Hcrt input to these cells does not influence their ability to detect sleep pressure (114). Indeed, further research is required to gain an understanding of direct Hcrt effects on cortical neural populations.
Recently, brain interstitial ion concentrations have been shown to directly influence sleep and wakefulness, providing an indirect pathway by which Hcrt-ergic signaling may influence arousal (115). Neuromodulators (e.g., dopamine, norepinephrine, hypocretin) all alter membrane potentials to influence spiking properties and intracellular signaling components. In doing so, network-wide changes in ionic concentrations must occur, which in turn may further influence vigilance states to ensure concerted brain activity. Nedergaard and colleagues discovered that independent of synaptic activity, altering the concentrations of extracellular K+, Ca2+, Mg2+, and H+ was able to cause a reversible switch between sleep and wakefulness. This suggests that downstream of neuromodulator actions (e.g., Hcrt), the build-up of certain ionic concentrations in the brain may independently drive sleep pressure and alter the wakefulness threshold.
Regulation of Hypocretin Neural Activity
Hcrt neurons respond to a wide variety of neurotransmitters and hormones (116), including NE, 5-HT, NPY, CCK, ghrelin, acetylcholine, and glutamate (117). These factors contribute to the ability of Hcrt neurons to respond to arousal and metabolic signals to adjust vigilance states accordingly. Hcrt neurons are further activated by corticotropin-releasing factor [CRF; (118)] and thyrotropin-releasing hormone [TRH; (119)], while they are generally inhibited by GABA (120) and adenosine (121). Noradrenergic, serotonergic, and DA inputs negatively regulate Hcrt activity, while histaminergic inputs have little effect (122–125). Other metabolic factors (e.g., ghrelin, leptin, glucose) that regulate their activity are discussed in section Feeding Behaviors and Metabolism.
Do Hcrt receptors (HcrtR1 and HcrtR2) share redundant functions in the regulation of arousal? HcrtR1 binds Hcrt-1 preferentially over Hcrt-2, while HcrtR2 binds both peptides at high affinity (2). These receptors show differential expression patterns across the mammalian brain, with HcrtR1 most abundant in the ventromedial hypothalamus, tenia tecta, hippocampus, dorsal raphe, and LC. HcrtR2 mRNA was localized to the paraventricular hypothalamus, cerebral cortex, nucleus accumbens, paraventricular thalamus, and anterior pretectal nucleus (126). HcrtR2-deficient mice show a phenotype similar to narcolepsy, including fragmented wakefulness, while those deficient in HcrtR1 show only mild sleep alterations (127, 128). Double receptor-knockout mice, however, show an even stronger phenotype than the HcrtR2 knockouts alone, including cataplexy and REM sleep intrusion into wakefulness, suggesting some redundancy in receptor function (37, 129). However, an fMRI study demonstrated that antagonism of HcrtR2 but not HcrtR1 increased REM and NREM sleep time, suggesting distinct roles of the receptors (130). The picture is not so clear though, as HcrtR1 blockade can influence the effects of HcrtR2 antagonism, suggesting complex interactions among Hcrt receptors (131).
Circadian Rhythmicity and Hypocretin
How do Hcrt neurons integrate time-of-day information to appropriately promote arousal and motivated behavior? The circuitry discussed above largely controls vigilance state switching on short to medium (1–30 s) timescales, so there must be some intervening population of cells that conveys longer term (i.e., hours to days) information to Hcrt neurons to regulate their activity. No direct synaptic connections between the master clock located in the suprachiasmatic nuclei (SCN) and Hcrt neurons has been demonstrated (132). However, in dark pulse experiments, where a 6 h pulse is given during the subjective day (a stimulus that promotes arousal and phase advances behavioral rhythms in mice), a pulse of dark is sufficient to release Hcrt neurons from SCN-mediated inhibition (133), providing evidence for light-dependent suppression of Hcrt signaling via the SCN. Because Hcrt neurons receive synaptic input from the dorsomedial and paraventricular hypothalamus (13, 134), which are primary outputs of the SCN (135), polysynaptic communication between the core clock and lateral hypothalamic Hcrt neurons is a likely mediator of this effect.
Hcrt neurons themselves seem to show a circadian rhythm in structural plasticity. In zebrafish, two-photon imaging of Hcrt axons revealed day/night differences in synapse numbers, which was driven by changes in nptx2b, a gene implicated in AMPA receptor clustering (136). Overexpression of this gene in Hcrt neurons further rendered fish resistant to the sleep-promoting effects of melatonin, providing evidence for a behavioral effect of circadian structural plasticity within the Hcrt system. Reciprocally, Hcrt neurons send inputs to the SCN to modulate clock function. SCN neurons express Hcrt receptors (137), suggesting there may be interplay between the two neural populations to coordinate phase coherent output of the SCN coinciding with behavioral arousal. This poses a problem, though, as Hcrt signaling is generally excitable, but SCN neural activity is low during the circadian night when Hcrt activity is high (in nocturnal rodents). A few years ago, it was discovered that unlike other connections, projections from Hcrt to the SCN were suppressive, and the mechanisms mediating this effect varied in a circadian fashion (138). By examining calcium activity in SCN neurons, both spontaneous and in response to Hcrt administration, it was demonstrated that a subset showed reduced (69%) or increased (31% cytosolic Ca2+ levels in response to Hcrt during the day, and 97% reduced Ca2+ activity in response to Hcrt at night. This response depended on HcrtR1 activation, as treatment with the HcrtR1 antagonist (SB334867) prevented this response but did not interfere with NMDA-induced Ca2+ increase. The suppressive effects of Hcrt seems to further depend on GABAergic signaling within the SCN, as the inhibitory effect is blocked when Hcrt is applied in the presence of gabazine and CGP55845 (antagonists of GABAA and GABAB receptors). These observations provide evidence for a reciprocal relationship between the SCN and Hcrt neurons in the lateral hypothalamus, demonstrating suppressive and phase-modulatory actions (through Ca2+) of Hcrt on the circadian clock.
Hyper Arousal in Response to Stressors
The term arousal covers more than mere wakefulness, indeed, it can be considered an umbrella term for multiple waking states, such as restful waking, active waking, and stressed states. Due to the well-studied role for Hcrt initiating arousal from sleep, researchers have also investigated the possibility of a role for Hcrt in the transition from arousal into hyper-arousal associated with stressed states. Supporting evidence for this role has been found in patients suffering from panic symptoms who have elevated levels of Hcrt in their cerebrospinal fluid in comparison to control subjects without substantial panic symptoms (139). Additionally, in animal studies it has been shown that multiple different stressors can trigger changes in Hcrt levels, including restraint stress, cold stress (140), and chronic social stress (141). In line with this finding, studies observing neural activation of Hcrt neurons have shown that stressors such as the forced swim test or indeed, direct infusion of corticotropin-releasing factor result in increased Hcrt activity (118, 142), showing a direct effect of physiological stress responses on Hcrt activity.
In order to further investigate the role of Hcrt in stress researchers have also investigated how manipulation of Hcrt function affects an animal's response to stress. Central administration of Hcrt-1 results in an increase in anxious behaviors as measured in multiple standard laboratory tests of anxiety such as the elevated plus maze, the light-dark box exploration test (143), the open-field test, and novel object exploration (144). Pharmacological administration of Hcrt has also been shown to stimulate hypothalamic-pituitary-adrenal (HPA) axis, resulting in increased concentrations of adrenocorticotropic hormone and corticosterone in plasma (118, 142, 145, 146). In line with these findings, impairing Hcrt function appears to reduce behavioral markers of anxiety and stress responses. Hcrt-deficient mice show a reduction in the exhibition of stress responses, such as changes in blood pressure, heart rate, respiratory changes (147, 148), as well as behavioral responses to resident intruders (147). These results suggest an important role for Hcrt in normal physiological and behavioral stress responses and confirm that these responses are impaired when animals do not have a fully functioning Hcrt system.
Following the development of optogenetic technologies and chemogenetics researchers have been able to genetically target specific neurons populations. Bonnavion et al. (149) used optogenetics to further investigate the role of Hcrt as a potential trigger for HPA axis activation, and showed that optogenetic stimulation of Hcrt neurons is sufficient to induce HPA axis activation which resulted in physiological stress responses including significantly elevated plasma corticosterone secretion and elevated heart rate, suggesting that increased Hcrt activity is a physiologically significant stressor. Bonnavion et al. also observed that this Hcrt-induced stress response could be mediated by nutritional status of the mouse, with food restricted mice showing a significantly higher Hcrt-induced stress response compared to ad libitum-fed controls. Further investigation of interactions between Hcrt, feeding, and stress showed that satiety hormone leptin, which, itself has been shown to be behaviorally anxiolytic (150, 151), modulates HPA axis activation generally, as well as Hcrt-induced HPA axis activation specifically (149). Food interactions with Hcrt activity will be discussed in more depth in section Feeding Behaviors and Metabolism. The development of chemogenetic technologies has also allowed the investigation of the role of Hcrt in resiliency to repeated social stress (152). After discovering that socially resilient rats (as tested in a social defeat assay) showed significantly lower levels of prepro-hypocretin mRNA, (152) went on to chemogenetically inhibit Hcrt neurons during the same social defeat paradigm, and found that reducing Hcrt activity during the social defeat paradigm caused rats to express fewer depressive-like behaviors and increased social interaction, both markers of a socially resilient phenotype. Taken together, these studies have used genetically targeted methods to show that inhibiting Hcrt can reduce stress-related behaviors, and that activation of Hcrt neurons can produce physiological stress responses, suggesting an important role for Hcrt in driving stress responses.
Hypocretin and Immune Stress
The Hcrt system is also influenced by immune stress. Indeed, it has been suggested that narcolepsy may have an autoimmune origin [for review, see (153)]. Specific genotypes in the antigen presentation complex HLA-DQB1 (HLA DQB1*06:02) are strongly associated with narcolepsy (154). Five epitopes from the Hcrt precursors are predicted to bind HLA DQB1*06:02, however, a recent study failed to find evidence of autoreactive CD4+ T-cells targeting Hcrt precursors (155), and a prior study claiming CD4+ T-cell autoimmunity to Hcrt and cross-reactivity to an epitope present in the 2009 H1N1 influenza virus was retracted when the authors failed to replicate their own findings (156). During the H1N1 (aka “Swine flu”) pandemic in 2009, cases of childhood narcolepsy rose dramatically in relation to administration of the vaccine (157). This effect was linked to the adjuvant (AS03, a squalene-based adjuvant from GlaxoSmithKline) co-administered with the vaccine (158, 159), although one study did not find an association (160). Additionally, in a mouse model of H1N1 infection, the virus was shown to enter sleep-wake regulatory regions and promote a narcolepsy-like phenotype (161). Despite this and other associative evidence, neither the essential targets nor a specific molecular mechanism have been identified linking narcolepsy to autoimmunity.
Less well studied is how Hcrt neurons sense the inflammatory milieu to regulate arousal/motivation. Peripheral lipopolysaccharide (LPS) challenge reduces cFos expression in Hcrt neurons as well as Hcrt concentrations in cerebrospinal fluid in rats (162). This is associated with reduced physical activity characteristic of “lethargy.” This, along with the finding that administration (intracerebroventricular) of Hcrt-1 counteracts LPS-induced reductions in locomotor activity, suggests that these neurons sense the inflammatory environment. However, this paper (162) found little evidence for direct targeting of Hcrt neurons by inflammatory cytokines. Instead, LPS seemed to activate upstream neurotensin neurons, which send inhibitory (GABAergic) projections to Hcrt cells to regulate their activity. Another study found that cytotoxic chemotherapy-induced fatigue was associated with reduced Hcrt cFos expression in conjunction with neuroinflammation, and treatment with Hcrt-1 rescued chemotherapy-induced reductions in locomotor activity (163). Inhibition of Hcrt activity by inflammatory mediators seems to be adaptive, as mice lacking Hcrt neurons (ataxin-3 transgenic) show enhanced recovery (NREM sleep) following LPS challenge (164). However, a more recent study demonstrated that peripheral administration of Hcrt-1 improves survival in mice given a lethal dose of LPS (165). An early study suggested that Hcrt-1, but not Hcrt-2, crossed the blood-brain-barrier [BBB; (166)], however, subsequent research has suggested that Hcrt-1 cannot normally cross the BBB except in high-dose administrations (167). Alternatively, systemic inflammation in response to LPS can disrupt BBB function and permit entry of this peptide into the brain. ICV-infusion of Hcrt-1 peptide recapitulates the enhanced survival phenotype shown in mice that received peripheral Hcrt-1. Because Hcrt neurons send projections to sympathetic output nuclei (3), and noradrenergic signaling potently alters peripheral immune function (168), a feedback loop between central and peripheral immune factors may be modulated by Hcrt to gauge arousal/motivation based on inflammatory status.
Motivation
There are a number of VTA-projecting neuron types in the lateral hypothalamus that are considered to play a role in reward processing. Excitotoxic lesions of the LHA produce similar pathologies to that of dopamine depletion, including aphagia (169, 170), suggesting that LHA input to the VTA is critical for modulating motivated behavior. Indeed, both VTA DA and GABAergic neurons receive robust input from diverse LHA cell groups (171, 172). With cell-type specific tools (e.g., Cre-dependent viral vectors carrying optogenetic payloads), the role specific LHA cell types play in VTA-elicited behavior is becoming clearer. For example, by using real time place preference (RTPP) and intracranial self-stimulation (ICSS) experiments, Tye and colleagues were able to demonstrate that LHA-GABAergic and LHA-glutamatergic projections to the VTA promote approach and avoidance behaviors, respectively (173). Furthermore, inhibiting the LHA-GABA projections to the VTA while mice were in a motivated state (i.e., food restricted) reduced feeding time suggesting a reduction in the motivation to eat. These responses were observed to be related to elicited dopamine release in the downstream nucleus accumbens (NAc), as LHA-GABA stimulation enhanced while LHA-glutamatergic stimulation suppressed dopamine release in this brain region [for review, see Tyree et al. (174)].
Feeding Behaviors and Metabolism
Hcrt is known to play a role in feeding behaviors. Indeed, studies have shown that Hcrt administration results in increased food intake [Hcrt-1 and Hcrt-2 i.c.v., (2); Hcrt-1 into NAc shell, (175)] and can increase motivation for food reward in a progressive ratio schedule task (176, 177). Furthermore, temporary antagonism of HcrtR1 (via SB334867) reduces food intake (178), reduces both motivation for (176, 179, 180)and consumption of (177, 180) food reward in a progressive ratio schedule task, and is sufficient to attenuate the effect of Hcrt-1-induced increase in feeding behaviors (181). Taken together, these findings suggest that Hcrt plays a role in driving food-intake and motivating food-seeking behaviors. However, there is some evidence that the nutritional status of the subject, and the type of food reward being presented can both mediate the effectiveness of Hcrt in driving these behaviors, interestingly, a study showed that blocking HcrtR1 significantly reduced the motivation to work for the delivery of cocaine as well as a highly palatable high fat diet, but had no effect on the motivation to work for regular chow food pellets (180).
It is clear that temporary changes in Hcrt activity can modulate feeding and food seeking behaviors, however more chronic manipulation of Hcrt suggests a more complex role for Hcrt in modulating food intake. While it is apparent that Hcrt knockout mice do show a reduction in the intake of food (38, 182) it appears that they still go on to develop obesity later on Hara et al. (38), suggesting that it may not only be food-seeking and food-consumption that is being driven by Hcrt activity, but additional processes underlying food-metabolism as well. This would be in line with previous findings that both gastric acid secretion (183) and metabolic rate (184) are stimulated following Hcrt administration. Additionally, in two obese mouse models (ob/ob and db/db) both strains of mice showed a significant downregulation of the prepro-hypocretin gene (185). These findings could signify that Hcrt may not only be important for driving metabolic processes important for weight maintenance, such as metabolic rate, but that their activity is also affected when metabolic state is altered, for instance in mouse models of obesity.
Hcrt neurons are specially situated to receive and integrate information on peripheral signals such as dietary content, metabolic status, and immune challenge due to their location in the LHA and there is evidence that altering food-intake also alters Hcrt activity. Hcrt neurons sense energy balance by monitoring glucose concentrations in the extracellular space through the use of a specialized 'tandem-pore' potassium (K+) channel (K2P) (186). Forty-eight hours of fasting can result in the upregulation of hypothalamic prepro-hypocretin mRNA (2, 187), this is consistent with the finding that insulin-induced hypoglycemia also increases the amount of prepro-hypocretin observed in the LHA (188). Conversely, glucodeprivation (induced by i.p. administration of 2-deoxy-D-glucose) decreased prepro-hypocretin (189), or had no effects (187, 190). The evidence of downregulation of prepro-hypocretin in obese mouse models (185) suggests that Hcrt activity is also being driven by dietary signals that are impaired in obesity. Both ob/ob and db/db mouse models are based on a deficiency of, or reduced responsiveness to leptin (191–195) a hormone that is released in response to dietary fat intake (196) and is commonly referred to as a satiety hormone. Hcrt neurons express receptors for leptin (197) and are inhibited by administration of leptin (198). Local GABAergic neurons that express the leptin receptor (LepRb) are additional negative regulators of Hcrt activity (149). Recent experiments using fiber photometry to monitor Hcrt neural activity during feeding behavior revealed that these cells rapidly reduce activity upon eating [even when the 'food' eaten is calorie free; (134)]. These findings suggest that the release of dietary hormones following food intake inhibits Hcrt neuron activation, which has been shown to drive food intake and food seeking. Then, the satiety response resulting from leptin and insulin release following food intake is mediated via the inhibitory effect these hormones have on the Hcrt system.
Hcrt activity is closely linked to not only leptin and insulin, both of which are associated with reducing food intake, but also to the hunger-associated hormone ghrelin which promotes feeding (199–205). Hcrt levels are directly modulated by these food-related hormones—as leptin and glucose levels increase and ghrelin levels decrease (as would occur following food consumption) Hcrt activity increases (117, 186, 206, 207). Additionally, ghrelin-induced feeding can be attenuated via inhibition of Hcrt signaling (208), suggesting that the ghrelin-release driven food-seeking behaviors are mediated via the Hcrt system. Taken together, this evidence suggests that dietary hormone release affects the Hcrt system, and that dietary hormone modulation of feeding behaviors is mediated via the Hcrt system. It is clear that motivation for seeking out food sources should be modulated based on the current nutritional status of the animal, and these inhibitory effects of insulin and leptin, paired with the stimulatory effects of ghrelin on the Hcrt system appear to be informing the Hcrt system as to whether or not the animal requires motivation for food-seeking or food-intake behaviors.
Driving Drug Seeking Behaviors
Given the role Hcrt plays in arousal and motivated states, it is unsurprising that manipulation of Hcrt activity alters many aspects of drug seeking and reward. DiLeone et al. (209) first suggested that Hcrt could be involved in drug abuse, noting that the anatomical projections demonstrated in Peyron et al. (3) implied that these peptides “may also be important in addiction.” Subsequent studies determined that this was indeed the case. For example, chemical activation of LHA Hcrt neurons causes a reinstatement of an extinguished drug-seeking behavior and direct administration of Hcrt to the VTA (a primary reward center in the brain) recapitulates this phenotype (210). It is important to note that Hcrt itself does not drive drug intake intrinsically, but rather drives the motivation to perform behaviors that are associated with drug seeking. Indeed, stress-induced reinstatement of cocaine-seeking behavior is potentiated by Hcrt-1 infusions into the lateral ventricles, an effect dependent on noradrenergic and corticotropin-releasing factor systems (211). Additionally, HcrtR1 antagonism blocks stress-induced reinstatement of previously extinguished drug-seeking behavior, suggesting that the observed effects are due to Hcrt (and not other co-released neurotransmitters) signaling. Supporting this, recent experiments in Hcrt deficient mice demonstrate that they have decreased motivation to perform behaviors required to obtain cocaine rewards (212). Hcrt signaling on VTA DA neurons potentiates neurotransmission via PLC/PKC-dependent insertion of NMDA receptors in VTA DA synapses (213). Additionally, Borgland and colleagues demonstrated that inhibition of HcrtR1 signaling blocks locomotor sensitization to cocaine and attenuates cocaine-induced excitatory currents in VTA DA neurons.
These initial studies have led to a blossoming of research on Hcrt in the context of drug-abuse and addiction, and particularly in relation to the VTA DA system [for review, see Baimel et al. (214)]. Using microdialysis and in vivo voltammetry to directly measure dopamine release, España and colleagues demonstrated that Hcrt-1 increases the effects of cocaine on tonic and phasic dopamine signaling, leading to enhanced motivation to self-administer cocaine (215). Others have shown that HcrtR1 blockade reduces work for cocaine self-administration, and Hcrt-1 strengthens presynaptic glutamatergic inputs to the VTA only in rats that have self-administered cocaine or a high fat diet [but not other highly salient cues like aversive stimuli; (180)]. These data suggest that Hcrt selectively potentiates the motivational drive for stimuli providing positive reinforcement (i.e., cocaine, highly palatable diet).
Using an ICSS paradigm in rats, Hollander and colleagues were able to demonstrate that blockade of HcrtR1 decreased nicotine-self administration and motivation to obtain the drug (but not food). Furthermore, they showed that Hcrt fibers innervate the insular cortex, and local blockade of Hcrt signaling in this brain region, but not the adjacent somatosensory cortex, decreased nicotine self-administration (216). This effect seems to be selective to nicotine (or may be species dependent), as Riday and colleagues were unable to demonstrate that HcrtR1 blockade attenuated cocaine self-administration in Swiss-Webster mice (217). More recently, focus has turned to neurotransmitters co-released by Hcrt neurons, including the kappa-opioid receptor (KOR) agonist dynorphin. Muschamp and colleagues demonstrated that Hcrt and dynorphin have opposite effects on the excitability of VTA DA neurons, are contained within the same pre-synaptic vesicles, and Hcrt co-released in the VTA attenuates the dysphoric effects of dynorphin (218). Alongside this evidence, there has additionally been anecdotal evidence that patients with narcolepsy show very low rates of substance abuse, this has recently been observed in Hcrt deficient mice in an experimental paradigm which showed that Hcrt knock out mice have decreased motivation to obtain cocaine as a reward (212). Taken together, these studies suggest a role for Hcrt in modulating drug-seeking behaviors, as well as drug-rewards, there is also a possibility that these neurons are involved in driving motivation for reward-seeking in general, and not just drug-seeking in particular.
Sexual Behavior
While studies investigating the function of Hcrt have focused in on a variety of types of arousal such as arousal from sleeping, hyper-arousal in stress-states, arousal from illicit drug-use, etc. there is also a role for Hcrt in behaviors associated with sexual arousal. While the lateral hypothalamus in general has also been associated with inducing [via electrical stimulation of the LHA, (219)] or impairing [via lesioning of the LH, (220)] male sexual behavior, there is some evidence that LHA Hcrt neurons may be modulating these sex-behaviors. Studies looking at neural activation of Hcrt neurons via Fos expression showed increased Fos-expressing Hcrt neurons after mating (221, 222), and more generally Hcrt neurons also appeared to respond following exposure to either receptive or non-receptive females (222) as well as contextual cues that they have previously learned to associate with sex via conditioned place preference training (223).
Sexual behavior itself can also be modulated via manipulation of Hcrt neurons specifically, by injecting Hcrt-1 into the medial preoptic area significantly improved male sexual behavior by increasing the speed with which males mount females and ejaculate, as well as increasing the frequency with which they mount and ejaculate (224). Conversely, administration of a HcrtR1 antagonist has been shown to impair male sexual behavior across many standard measures such as increasing the mounting and ejaculation latencies, and reducing the frequency of ejaculation, however mounting frequency did not appear to be affected (221). In contrast to the antagonism of HcrtR1, research looking into the targeted lesioning of Hcrt-2 neurons (via Hcrt-2 saporin) has produced mixed results, rats with Hcrt-2 neuron lesions did not appear to show any consistent impairments to the usual measures of male sexual behavior (222, 223). Additionally, the existence of a line of a Hcrt knockout mice (37) would suggest that Hcrts are not required for successful procreation, but may instead be involved in the motivation or initiation of sexual behaviors.
Integration of Motivation and Arousal by Hcrt
Hcrt has been shown to play important roles in a wide variety of complex behaviors. The LHA Hcrt system integrates sensory information on stress, metabolic, immune, and motivational states to adjust behavioral responses accordingly. Motivation of behaviors is a two-part process, first comes the energizing component which gives the animal the drive necessary to complete a task, followed by a directional component which determines the type of behavior this burst of energy should be focused toward. The earliest electrophysiological studies in which researchers stimulated neural activity in the LHA showed that activity in this brain region could result in numerous different rewarding behaviors (225–227), which turned out to be more dependent on previous experiences in the same setting (228, 229), rather than topographical placement of the electrodes (230). Considering these findings in the scope of the two sub-processes of motivation, it could be that Hcrt neurons in the LHA are important for the energizing or driving of motivation, and the downstream targets activated by the Hcrt neurons play the role of directing the energy toward context-specific behaviors. Considering that arousal is synonymous with energizing, it could be said that the role of Hcrt in motivated behavior is simply another facet of its numerous functions under the umbrella of arousal. However, further research is required to better understand how the Hcrt circuit modulates different facets of arousal states.
Conclusion
Research on the Hcrt peptides has been unusually productive. Within just 15 years of their discovery, they were causally implicated in the etiology of narcolepsy, specific receptor antagonists were developed, and a dual-receptor antagonist formulation was FDA-approved for the treatment of insomnia. Using cutting edge tools like CRISPR/Cas9 in tandem with cre-dependent adeno-associated viral vectors to manipulate specific genes within Hcrt neurons or their projection sites will undoubtedly provide further classification of their actions and potential for modulation. While a lot of previous work has focused on the role of Hcrt in mediating arousal specifically, the actual operations and computations of the Hcrt network are still unknown. These computations could be achieved by studying projection-selective subsets of Hcrt neurons. It is still unknown whether individual cells perform the same computations, or whether projection specific subsets perform different computations. In order to develop a better understanding of the intricacies of the Hcrt network it will be important for researchers to investigate this neural circuit with all of these systems in mind. While this can appear complicated—it is very likely that the motivated behaviors and the environmental inputs that drive them are all interconnected and can modulate other relevant behaviors to the Hcrt system. Up until this point the heterogeneity of Hcrt neurons specifically, and lateral hypothalamic neurons in general, have slowed the progress of research in this region. With recent developments in research technologies such as optogenetic stimulation, calcium recordings, single cell RNA sequencing, chemogenetic neuron manipulation, and advances in computational modeling, a better understanding of the Hcrt system is within reach. These methods hold much promise for the next 20 years of hypocretin research to continue to be as fruitful as the first twenty.
Author Contributions
ST and JB wrote the first draft of this manuscript. LdL contributed editorial help and guidance.
Funding
ST is currently funded by the Philip Wrightson Postdoctoral Fellowship, awarded by the New Zealand Neurological Foundation. JB is currently funded by a BRAIN Initiative F32MH11543. LdL is supported by grants from the NIH (5R01MH087592; R01MH1026380, 1R01AG047671) and Boehringer Ingelheim.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. De Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. (1998) 95:322–7. doi: 10.1073/pnas.95.1.322
2. Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell (1998) 92:573–85. doi: 10.1016/S0092-8674(00)80949-6
3. Peyron C, Tighe DK, Van Den Pol AN, De Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. (1998) 18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998
4. Mickelsen LE, Kolling FW, Chimileski BR, Fujita A, Norris C, Chen K, et al. Neurochemical heterogeneity among lateral hypothalamic hypocretin/orexin and melanin-concentrating hormone neurons identified through single-cell gene expression analysis. eNeuro (2017) 4:1–24. doi: 10.1523/ENEURO.0013-17.2017
5. Broberger C, De Lecea L, Sutcliffe JG, Hökfelt T. Hypocretin/orexin-and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and Agouti gene-related protein systems. J Comp Neurol. (1998) 402:460–74. doi: 10.1002/(SICI)1096-9861(19981228)402:4<460::AID-CNE3>.0.CO;2-S
6. Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, et al. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. (1998) 402:442–59. doi: 10.1002/(SICI)1096-9861(19981228)402:4<442::AID-CNE2>3.0.CO;2-R
7. Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. (2005) 25:6716–20. doi: 10.1523/JNEUROSCI.1887-05.2005
8. Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron (2005) 46:787–98. doi: 10.1016/j.neuron.2005.04.035
9. Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Leger L, et al. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci. (2003) 4:19. doi: 10.1186/1471-2202-4-19
10. Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci USA. (2009) 106:2418–22. doi: 10.1073/pnas.0811400106
11. Apergis-Schoute J, Iordanidou P, Faure C, Jego S, Schone C, Aitta-Aho T, et al. Optogenetic evidence for inhibitory signaling from orexin to MCH neurons via local microcircuits. J Neurosci. (2015) 35:5435–41. doi: 10.1523/JNEUROSCI.5269-14.2015
12. Yoshida K, Mccormack S, Espana RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. (2006) 494:845–61. doi: 10.1002/cne.20859
13. Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, et al. Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron (2005) 46:297–308. doi: 10.1016/j.neuron.2005.03.010
14. Faraco JH, Appelbaum L, Marin W, Gaus SE, Mourrain P, Mignot E. Regulation of hypocretin (orexin) expression in embryonic zebrafish. J Biol Chem. (2006) 281:29753–61. doi: 10.1074/jbc.M605811200
15. Prober DA, Rihel J, Onah AA, Sung RJ, Schier AF. Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J Neurosci. (2006) 26:13400–10. doi: 10.1523/JNEUROSCI.4332-06.2006
16. Yelin-Bekerman L, Elbaz I, Diber A, Dahary D, Gibbs-Bar L, Alon S, et al. Hypocretin neuron-specific transcriptome profiling identifies the sleep modulator Kcnh4a. Elife (2015) 4:e08638. doi: 10.7554/eLife.08638
17. Elbaz I, Zada D, Tovin A, Braun T, Lerer-Goldshtein T, Wang G, et al. Sleep-dependent structural synaptic plasticity of inhibitory synapses in the dendrites of hypocretin/orexin neurons. Mol Neurobiol. (2017) 54:6581–97. doi: 10.1007/s12035-016-0175-x
18. Elbaz I, Yelin-Bekerman L, Nicenboim J, Vatine G, Appelbaum L. Genetic ablation of hypocretin neurons alters behavioral state transitions in zebrafish. J Neurosci. (2012) 32:12961–72. doi: 10.1523/JNEUROSCI.1284-12.2012
19. Chen A, Chiu CN, Mosser EA, Kahn S, Spence R, Prober DA. QRFP and its receptors regulate locomotor activity and sleep in zebrafish. J Neurosci. (2016) 36:1823–40. doi: 10.1523/JNEUROSCI.2579-15.2016
20. Jaggard JB, Stahl BA, Lloyd E, Prober DA, Duboue ER, Keene AC. Hypocretin underlies the evolution of sleep loss in the Mexican cavefish. Elife (2018) 7:e32637. doi: 10.7554/eLife.32637
21. Ohkubo T, Boswell T, Lumineau S. Molecular cloning of chicken prepro-orexin cDNA and preferential expression in the chicken hypothalamus. Biochim Biophys Acta (2002) 1577:476–80. doi: 10.1016/S0167-4781(02)00483-9
22. Singletary KG, Deviche P, Strand C, Delville Y. Distribution of orexin/hypocretin immunoreactivity in the brain of a male songbird, the house finch, Carpodacus mexicanus (vol (2007) 32, pg 81. J Chem Neuroanat. (2006) 33:100–9. doi: 10.1016/j.jchemneu.2006.12.002
24. Dickman MS. von Economo encephalitis. Arch Neurol. (2001) 58:1696–8. doi: 10.1001/archneur.58.10.1696
25. Economo CV. Encephalitis lethargica: its sequelae and treatment. Oxford: Oxford University Press (1931).
26. Ranson SW, Ranson M. Pallidofugal fibers in the monkey. Arch Neurol Psychiatry (1939) 42:1059–67. doi: 10.1001/archneurpsyc.1939.02270240097006
27. Nauta WJ. Hypothalamic regulation of sleep in rats; an experimental study. J Neurophysiol. (1946) 9:285–316. doi: 10.1152/jn.1946.9.4.285
28. Swett CP, Hobson JA. The effects of posterior hypothalamic lesions on behavioral and electrographic manifestations of sleep and waking in cats. Arch Ital Biol. (1968) 106:283–93.
29. Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. (1949) 1:455–73. doi: 10.1016/0013-4694(49)90219-9
30. Loomis AL, Harvey EN, Hobart G. Potential rhythms of the cerebral cortex during sleep. Science (1935) 81:597–8. doi: 10.1126/science.81.2111.597
31. Szymusiak R, Gvilia I, Mcginty D. Hypothalamic control of sleep. Sleep Med. (2007) 8:291–301. doi: 10.1016/j.sleep.2007.03.013
32. Siegel JM. SLEEP - OPINION Sleep viewed as a state of adaptive inactivity. Nat Rev Neurosci. (2009) 10:747–53. doi: 10.1038/nrn2697
33. Crunelli V, Soltesz I, Tóth TI, Turner J, Leresche N. 31 - intrinsic low-frequency oscillations of thalamocortical cells and their modulation by synaptic potentials. Minciacchi D, Molinari M, Macchi G, Jones EG, editors. In: Thalamic Networks for Relay and Modulation. Pergamon: Elsevier Ltd. (1993). p. 375–384.
34. Crunelli V, Hughes SW. The slow (< 1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci. (2010) 13:9–17. doi: 10.1038/nn.2445
35. Vyazovskiy VV, Tobler I. Theta activity in the waking EEG is a marker of sleep propensity in the rat. Brain Res. (2005) 1050:64–71. doi: 10.1016/j.brainres.2005.05.022
36. Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin XY, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell (1999) 98:365–76. doi: 10.1016/S0092-8674(00)81965-0
37. Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell (1999) 98:437–51. doi: 10.1016/S0092-8674(00)81973-X
38. Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron (2001) 30:345–54. doi: 10.1016/S0896-6273(01)00293-8
39. Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet (2000) 355:39–40. doi: 10.1016/S0140-6736(99)05582-8
40. Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. (2000) 6:991–7. doi: 10.1038/79690
41. Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology (2001) 57:2253–8. doi: 10.1212/WNL.57.12.2253
42. Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology (2005) 65:1184–8. doi: 10.1212/01.WNL.0000168173.71940.ab
43. De Lecea L, Sutcliffe JG. The hypocretins and sleep. FEBS J. (2005) 272:5675–88. doi: 10.1111/j.1742-4658.2005.04981.x
44. Branch AF, Navidi W, Tabuchi S, Terao A, Yamanaka A, Scammell TE, et al. Progressive loss of the orexin neurons reveals dual effects on wakefulness. Sleep (2016) 39:369–77. doi: 10.5665/sleep.5446
45. Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light–dark cycle and sleep–wake activities. Eur J Neurosci. (2001) 14:1075–81. doi: 10.1046/j.0953-816x.2001.01725.x
46. Estabrooke IV, Mccarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, et al. Fos expression in orexin neurons varies with behavioral state. J Neurosci. (2001) 21:1656–62. doi: 10.1523/JNEUROSCI.21-05-01656.2001
47. Tyree SM, De Lecea L. Optogenetic investigation of arousal circuits. Int J Mol Sci. (2017) 18:E1773. doi: 10.3390/ijms18081773
48. Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, De Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature (2007) 450:420–4. doi: 10.1038/nature06310
49. Tsunematsu T, Tabuchi S, Tanaka KF, Boyden ES, Tominaga M, Yamanaka A. Long-lasting silencing of orexin/hypocretin neurons using archaerhodopsin induces slow-wave sleep in mice. Behav Brain Res. (2013) 255:64–74. doi: 10.1016/j.bbr.2013.05.021
50. Sasaki K, Suzuki M, Mieda M, Tsujino N, Roth B, Sakurai T. Pharmacogenetic modulation of orexin neurons alters sleep/wakefulness states in mice. PLoS ONE (2011) 6:e20360. doi: 10.1371/journal.pone.0020360
51. Vassalli A, Franken P. Hypocretin (orexin) is critical in sustaining theta/gamma-rich waking behaviors that drive sleep need. Proc Natl Acad Sci USA. (2017) 114:E5464–73. doi: 10.1073/pnas.1700983114
52. Riemann D, Nissen C, Palagini L, Otte A, Perlis ML, Spiegelhalder K. The neurobiology, investigation, and treatment of chronic insomnia. Lancet Neurol. (2015) 14:547–558. doi: 10.1016/S1474-4422(15)00021-6
53. van den Pol AN. Physiological characteristics of hypocretin/orexin neurons. In: Hypocretins Boston, MA: Springer (2005). p. 123–36.
54. Berridge CW, Waterhouse BD. The locus coeruleus–noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. (2003) 42:33–84. doi: 10.1016/S0165-0173(03)00143-7
55. Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. (2005) 28:403–50. doi: 10.1146/annurev.neuro.28.061604.135709
56. Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA. (1999) 96:10911–6. doi: 10.1073/pnas.96.19.10911
57. Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, Kilduff TS, et al. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol. (1999) 415:145–59. doi: 10.1002/(SICI)1096-9861(19991213)415:2<145::AID-CNE1>3.0.CO;2-2
58. Ivanov A, Aston-Jones G. Hypocretin/orexin depolarizes and decreases potassium conductance in locus coeruleus neurons. Neuroreport (2000) 11:1755–8. doi: 10.1097/00001756-200006050-00031
59. Gompf HS, Aston-Jones G. Role of orexin input in the diurnal rhythm of locus coeruleus impulse activity. Brain Res. (2008) 1224:43–52. doi: 10.1016/j.brainres.2008.05.060
60. Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, De Lecea L. Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc Natl Acad Sci USA. (2012) 109:E2635–44. doi: 10.1073/pnas.1202526109
61. Jones BE. Activity, modulation and role of basal forebrain cholinergic neurons innervating the cerebral cortex. Prog Brain Res. (2004) 145:157–69. doi: 10.1016/S0079-6123(03)45011-5
62. Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. (1988) 8:4007–26. doi: 10.1523/JNEUROSCI.08-11-04007.1988
63. Jiménez-Capdeville ME, Dykes RW, Myasnikov AA. Differential control of cortical activity by the basal forebrain in rats: a role for both cholinergic and inhibitory influences. J Comp Neurol (1997) 381:53–67. doi: 10.1002/(SICI)1096-9861(19970428)381:1<53::AID-CNE5>3.0.CO;2-J
64. Cape EG, Jones BE. Effects of glutamate agonist versus procaine microinjections into the basal forebrain cholinergic cell area upon gamma and theta EEG activity and sleep–wake state. Eur J Neurosci. (2000) 12:2166–84. doi: 10.1046/j.1460-9568.2000.00099.x
65. Cape EG, Manns ID, Alonso A, Beaudet A, Jones BE. Neurotensin-induced bursting of cholinergic basal forebrain neurons promotes gamma and theta cortical activity together with waking and paradoxical sleep. J Neurosci. (2000) 20:8452–61. doi: 10.1523/JNEUROSCI.20-22-08452.2000
66. Han Y, Shi YF, Xi W, Zhou R, Tan ZB, Wang H, et al. Selective activation of cholinergic basal forebrain neurons induces immediate sleep-wake transitions. Curr Biol. (2014) 24:693–8. doi: 10.1016/j.cub.2014.02.011
67. Irmak SO, De Lecea L. Basal forebrain cholinergic modulation of sleep transitions. Sleep (2014) 37:1941–51. doi: 10.5665/sleep.4246
68. Anaclet C, Pedersen NP, Ferrari LL, Venner A, Bass CE, Arrigoni E, et al. Basal forebrain control of wakefulness and cortical rhythms. Nat Commun. (2015) 6:8744. doi: 10.1038/ncomms9744
69. Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, et al. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience (2001) 108:177–81. doi: 10.1016/S0306-4522(01)00512-7
70. Espana RA, Baldo BA, Kelley AE, Berridge CW. Wake-promoting and sleep-suppressing actions of hypocretin (orexin): basal forebrain sites of action. Neuroscience (2001) 106:699–715. doi: 10.1016/S0306-4522(01)00319-0
71. Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, John J, et al. Release of hypocretin (orexin) during waking and sleep states. J Neurosci. (2002) 22:5282–6. doi: 10.1523/JNEUROSCI.22-13-05282.2002
72. Wu X, Gao J, Yan J, Owyang C, Li Y. Hypothalamus–brain stem circuitry responsible for vagal efferent signaling to the pancreas evoked by hypoglycemia in rat. J Neurophysiol. (2004) 91:1734–47. doi: 10.1152/jn.00791.2003
73. Blanco-Centurion C, Xu M, Murillo-Rodriguez E, Gerashchenko D, Shiromani AM, Salin-Pascual RJ, et al. Adenosine and sleep homeostasis in the Basal forebrain. J Neurosci. (2006) 26:8092–100. doi: 10.1523/JNEUROSCI.2181-06.2006
74. Arrigoni E, Mochizuki T, Scammell TE. Activation of the basal forebrain by the orexin/hypocretin neurones. Acta Physiol. (2010) 198:223–35. doi: 10.1111/j.1748-1716.2009.02036.x
75. Lin JS, Sakai K, Jouvet M. Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology (1988) 27:111–22. doi: 10.1016/0028-3908(88)90159-1
76. Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. (2003) 4:121–30. doi: 10.1038/nrn1034
77. Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. (2006) 26:10292–8. doi: 10.1523/JNEUROSCI.2341-06.2006
78. Fujita A, Bonnavion P, Wilson MH, Mickelsen LE, Bloit J, De Lecea L, et al. Hypothalamic tuberomammillary nucleus neurons: electrophysiological diversity and essential role in arousal stability. J Neurosci. (2017) 37:9574–92. doi: 10.1523/JNEUROSCI.0580-17.2017
79. Bayer L, Eggermann E, Serafin M, Saint-Mleux B, Machard D, Jones B, et al. Orexins (hypocretins) directly excite tuberomammillary neurons. Eur J Neurosci. (2001) 14:1571–5. doi: 10.1046/j.0953-816x.2001.01777.x
80. Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. (2001) 21:9273–9. doi: 10.1523/JNEUROSCI.21-23-09273.2001
81. Eriksson KS, Sergeeva OA, Selbach O, Haas HL. Orexin (hypocretin)/dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur J Neurosci. (2004) 19:1278–84. doi: 10.1111/j.1460-9568.2004.03243.x
82. Schone C, Cao ZF, Apergis-Schoute J, Adamantidis A, Sakurai T, Burdakov D. Optogenetic probing of fast glutamatergic transmission from hypocretin/orexin to histamine neurons in situ. J Neurosci. (2012) 32:12437–43. doi: 10.1523/JNEUROSCI.0706-12.2012
83. Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, De Lecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci. (2009) 29:10939–49. doi: 10.1523/JNEUROSCI.1205-09.2009
84. Mcginty DJ, Harper RM. Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res. (1976) 101:569–75. doi: 10.1016/0006-8993(76)90480-7
85. Brown RE, Sergeeva OA, Eriksson KS, Haas HL. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci. (2002) 22:8850–9. doi: 10.1523/JNEUROSCI.22-20-08850.2002
86. Liu RJ, Van Den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci. (2002) 22:9453–64. doi: 10.1523/JNEUROSCI.22-21-09453.2002
87. Kohlmeier KA, Inoue T, Leonard CS. Hypocretin/orexin peptide signaling in the ascending arousal system: elevation of intracellular calcium in the mouse dorsal raphe and laterodorsal tegmentum. J Neurophysiol. (2004) 92:221–35. doi: 10.1152/jn.00076.2004
88. Kohlmeier KA, Watanabe S, Tyler CJ, Burlet S, Leonard CS. Dual orexin actions on dorsal raphe and laterodorsal tegmentum neurons: noisy cation current activation and selective enhancement of Ca2+ transients mediated by L-type calcium channels. J Neurophysiol. (2008) 100:2265–81. doi: 10.1152/jn.01388.2007
89. Chowdhury S, Yamanaka A. Optogenetic activation of serotonergic terminals facilitates GABAergic inhibitory input to orexin/hypocretin neurons. Sci Rep. (2016) 6:36039. doi: 10.1038/srep36039
90. Moriya R, Kanamaru M, Ookuma N, Tanaka KF, Izumizaki M, Onimaru H, et al. (2017). The effect of activating serotonergic neurons in the dorsal raphe nucleus on control of vigilance state. Eur Resp. 50:OA1753 doi: 10.1183/1393003.congress-2017.OA1753
91. Cho JR, Treweek JB, Robinson JE, Xiao C, Bremner LR, Greenbaum A, et al. Dorsal raphe dopamine neurons modulate arousal and promote wakefulness by salient stimuli. Neuron (2017) 94:1205.e8–19. doi: 10.1016/j.neuron.2017.05.020
92. Webster HH, Jones BE. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II. Effects upon sleep-waking states. Brain Res. (1988) 458:285-302.
93. Satoh K, Fibiger HC. Cholinergic neurons of the laterodorsal tegmental nucleus: efferent and afferent connections. J Comp Neurol. (1986) 253:277–302. doi: 10.1002/cne.902530302
94. Van Dort CJ, Zachs DP, Kenny JD, Zheng S, Goldblum RR, Gelwan NA, et al. Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc. Natl. Acad. Sci. (2015) 112:584-89.
95. Xi MC, Morales FR, Chase MH. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res. (2001) 901:59–264. doi: 10.1016/S0006-8993(01)02317-4
96. Burlet S, Tyler CJ, Leonard CS. Direct and indirect excitation of laterodorsal tegmental neurons by Hypocretin/Orexin peptides: implications for wakefulness and narcolepsy. J Neurosci. (2002) 22:2862–72. doi: 10.1523/JNEUROSCI.22-07-02862.2002
97. Miller JD, Farber J, Gatz P, Roffwarg H, German DC. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat. Brain Res. (1983) 273:133–41. doi: 10.1016/0006-8993(83)91101-0
98. Boutrel B, Koob GF. What keeps us awake: the neuropharmacology of stimulants and wakefulness promoting medications. Sleep (2004) 27:1181–94. doi: 10.1093/sleep/27.6.1181
99. Trulson ME, Preussler DW, Howell GA. Activity of substantia nigra units across the sleep-waking cycle in freely moving cats. Neurosci Lett. (1981) 26:183–8. doi: 10.1016/0304-3940(81)90346-3
100. Steinfels GF, Heym J, Strecker RE, Jacobs BL. Behavioral correlates of dopaminergic unit activity in freely moving cats. Brain Res. (1983) 258:217–28. doi: 10.1016/0006-8993(83)91145-9
101. Trulson ME, Preussler DW. Dopamine-containing ventral tegmental area neurons in freely moving cats: activity during the sleep-waking cycle and effects of stress. Exp Neurol. (1984) 83:367–77. doi: 10.1016/S0014-4886(84)90105-5
102. Lee RS, Steffensen SC, Henriksen SJ. Discharge profiles of ventral tegmental area GABA neurons during movement, anesthesia, and the sleep-wake cycle. J Neurosci. (2001) 21:1757–66. doi: 10.1523/JNEUROSCI.21-05-01757.2001
103. Dahan L, Astier B, Vautrelle N, Urbain N, Kocsis B, Chouvet G. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology (2007) 32:1232–41. doi: 10.1038/sj.npp.1301251
104. Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, De Lecea L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci. (2016) 19:1356–66. doi: 10.1038/nn.4377
105. Taylor AMW, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L. et al. Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology (2016) 41:949–59. doi: 10.1038/npp.2015.221
106. Oishi Y, Suzuki Y, Takahashi K, Yonezawa T, Kanda T, Takata Y, et al. Activation of ventral tegmental area dopamine neurons produces wakefulness through dopamine D2-like receptors in mice. Brain Struct Funct. (2017) 222:2907–15. doi: 10.1007/s00429-017-1365-7
107. Fadel J, Deutch AY. Anatomical substrates of orexin–dopamine interactions: lateral hypothalamic projections to the ventral tegmental area. Neuroscience (2002) 111:379–87. doi: 10.1016/S0306-4522(02)00017-9
108. Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE. Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci. (2003) 23:7–11. doi: 10.1523/JNEUROSCI.23-01-00007.2003
109. Vittoz NM, Berridge CW. Hypocretin/orexin selectively increases dopamine efflux within the prefrontal cortex: involvement of the ventral tegmental area. Neuropsychopharmacology (2006) 31:384–95. doi: 10.1038/sj.npp.1300807
110. Hay YA, Andjelic S, Badr S, Lambolez B. Orexin-dependent activation of layer VIb enhances cortical network activity and integration of non-specific thalamocortical inputs. Brain Struct Funct. (2015) 220:3497–12. doi: 10.1007/s00429-014-0869-7
111. Wenger Combremont AL, Bayer L, Dupre A, Muhlethaler M, Serafin M. Slow bursting neurons of mouse cortical layer 6b are depolarized by hypocretin/orexin and major transmitters of arousal. Front Neurol. (2016) 7:88. doi: 10.3389/fneur.2016.00088
112. Morairty SR, Dittrich L, Pasumarthi RK, Valladao D, Heiss JE, Gerashchenko D, et al. A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc Natl Acad Sci USA. (2013) 110:20272–7. doi: 10.1073/pnas.1314762110
113. Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. (2016) 19:335–46. doi: 10.1038/nn.4216
114. Williams RH, Black SW, Thomas AM, Piquet J, Cauli B, Kilduff TS. (2018). Excitation of cortical nNOS/NK1R neurons by hypocretin 1 is independent of sleep homeostasis. Cereb Cortex. doi: 10.1093/cercor/bhy015. [Epub ahead of print].
115. Ding F, O'donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science (2016) 352:550–5. doi: 10.1126/science.aad4821
116. Inutsuka A, Yamanaka A. The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front Endocrinol. (2013) 4:18. doi: 10.3389/fendo.2013.00018
117. Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. (2007) 8:171–81. doi: 10.1038/nrn2092
118. Winsky-Sommerer R, Yamanaka A, Diano S, Borok E, Roberts AJ, Sakurai T, et al. Interaction between the corticotropin-releasing factor system and hypocretins (Orexins): a novel circuit mediating stress response. J Neurosci. (2004) 24:11439–48. doi: 10.1523/JNEUROSCI.3459-04.2004
119. Hara J, Gerashchenko D, Wisor JP, Sakurai T, Xie XM, Kilduff TS. Thyrotropin-releasing hormone increases behavioral arousal through modulation of hypocretin/orexin neurons. J Neurosci. (2009) 29:3705–14. doi: 10.1523/JNEUROSCI.0431-09.2009
120. Xie X, Crowder TL, Yamanaka A, Morairty S, Lewinter RD, Sakurai T, et al. GABAB receptor-mediated modulation of hypocretin/orexin neurones in mouse hypothalamus. J Physiol. (2006) 574:399–414. doi: 10.1113/jphysiol.2006.108266
121. Liu ZW, Gao XB. Adenosine inhibits activity of hypocretin/orexin neurons by the A1 receptor in the lateral hypothalamus: a possible sleep-promoting effect. J Neurophysiol. (2007) 97:837–48. doi: 10.1152/jn.00873.2006
122. Li Y, Gao XB, Sakurai T, Van Den Pol AN. Hypocretin/Orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron (2002) 36:1169–81. doi: 10.1016/S0896-6273(02)01132-7
123. Yamanaka A, Muraki Y, Tsujino N, Goto K, Sakurai T. Regulation of orexin neurons by the monoaminergic and cholinergic systems. Biochem Biophys Res Commun. (2003) 303:120–9. doi: 10.1016/S0006-291X(03)00299-7
124. Yamanaka A, Muraki Y, Ichiki K, Tsujino N, Kilduff TS, Goto K, et al. Orexin neurons are directly and indirectly regulated by catecholamines in a complex manner. J Neurophysiol. (2006) 96:284–98. doi: 10.1152/jn.01361.2005
125. Kumar S, Szymusiak R, Bashir T, Rai S, Mcginty D, Alam MN. Effects of serotonin on perifornical-lateral hypothalamic area neurons in rat. Eur J Neurosci. (2007) 25:201–12. doi: 10.1111/j.1460-9568.2006.05268.x
126. Trivedi P, Yu H, Macneil DJ, Van Der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. (1998) 438:71–5. doi: 10.1016/S0014-5793(98)01266-6
127. Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. (2001) 24:429–58. doi: 10.1146/annurev.neuro.24.1.429
128. Mieda M, Hasegawa E, Kisanuki YY, Sinton CM, Yanagisawa M, Sakurai T. Differential roles of orexin receptor-1 and−2 in the regulation of non-REM and REM sleep. J Neurosci. (2011) 31:6518–26. doi: 10.1523/JNEUROSCI.6506-10.2011
129. Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, et al. Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of Non-REM and REM sleep regulatory processes. Neuron (2003) 38:715–30. doi: 10.1016/S0896-6273(03)00330-1
130. Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M, et al. Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS ONE (2011) 6:e16406. doi: 10.1371/journal.pone.0016406
131. Dugovic C, Shelton JE, Aluisio LE, Fraser IC, Jiang X, Sutton SW, et al. Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp Ther. (2009) 330:142–51. doi: 10.1124/jpet.109.152009
132. Sakurai T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev. (2005) 9:231–41. doi: 10.1016/j.smrv.2004.07.007
133. Marston OJ, Williams RH, Canal MM, Samuels RE, Upton N, Piggins HD. Circadian and dark-pulse activation of orexin/hypocretin neurons. Mol Brain (2008) 1:19. doi: 10.1186/1756-6606-1-19
134. Gonzalez JA, Iordanidou P, Strom M, Adamantidis A, Burdakov D. Awake dynamics and brain-wide direct inputs of hypothalamic MCH and orexin networks. Nat Commun. (2016) 7:11395. doi: 10.1038/ncomms11395
135. Kalsbeek A, Palm IF, La Fleur SE, Scheer FA, Perreau-Lenz S, Ruiter M, et al. SCN outputs and the hypothalamic balance of life. J Biol Rhythms (2006) 21:458–69. doi: 10.1177/0748730406293854
136. Appelbaum L, Wang G, Yokogawa T, Skariah GM, Smith SJ, Mourrain P, et al. Circadian and homeostatic regulation of structural synaptic plasticity in hypocretin neurons. Neuron (2010) 68:87–98. doi: 10.1016/j.neuron.2010.09.006
137. Backberg M, Hervieu G, Wilson S, Meister B. Orexin receptor-1 (OX-R1) immunoreactivity in chemically identified neurons of the hypothalamus: focus on orexin targets involved in control of food and water intake. Eur J Neurosci. (2002) 15:315–28. doi: 10.1046/j.0953-816x.2001.01859.x
138. Belle MD, Hughes AT, Bechtold DA, Cunningham P, Pierucci M, Burdakov D, et al. Acute suppressive and long-term phase modulation actions of orexin on the mammalian circadian clock. J Neurosci. (2014) 34:3607–21. doi: 10.1523/JNEUROSCI.3388-13.2014
139. Johnson PL, Truitt W, Fitz SD, Minick PE, Dietrich A, Sanghani S, et al. A key role for orexin in panic anxiety. Nat Med. (2010) 16:111–5. doi: 10.1038/nm.2075
140. Ida T, Nakahara K, Murakami T, Hanada R, Nakazato M, Murakami N. Possible involvement of orexin in the stress reaction in rats. Biochem Biophys Res Commun. (2000) 270:318–23. doi: 10.1006/bbrc.2000.2412
141. Arendt DH, Hassell J, Li H, Achua JK, Guarnieri DJ, Dileone RJ, et al. Anxiolytic function of the orexin 2/hypocretin A receptor in the basolateral amygdala. Psychoneuroendocrinology (2014) 40:17–26. doi: 10.1016/j.psyneuen.2013.10.010
142. Chang H, Saito T, Ohiwa N, Tateoka M, Deocaris CC, Fujikawa T, et al. Inhibitory effects of an orexin-2 receptor antagonist on orexin A- and stress-induced ACTH responses in conscious rats. Neurosci Res. (2007) 57:462–6. doi: 10.1016/j.neures.2006.11.009
143. Suzuki M, Beuckmann CT, Shikata K, Ogura H, Sawai T. Orexin-A (hypocretin-1) is possibly involved in generation of anxiety-like behavior. Brain Res. (2005) 1044:116–21. doi: 10.1016/j.brainres.2005.03.002
144. Li Y, Li S, Wei C, Wang H, Sui N, Kirouac GJ. Orexins in the paraventricular nucleus of the thalamus mediate anxiety-like responses in rats. Psychopharmacology (2010) 212:251–65. doi: 10.1007/s00213-010-1948-y
145. Jaszberenyi M, Bujdoso E, Pataki I, Telegdy G. Effects of orexins on the hypothalamic-pituitary-adrenal system. J Neuroendocrinol. (2000) 12:1174–8. doi: 10.1046/j.1365-2826.2000.00572.x
146. Kuru M, Ueta Y, Serino R, Nakazato M, Yamamoto Y, Shibuya I, et al. Centrally administered orexin/hypocretin activates HPA axis in rats. Neuroreport (2000) 11:1977–80. doi: 10.1097/00001756-200006260-00034
147. Kayaba Y, Nakamura A, Kasuya Y, Ohuchi T, Yanagisawa M, Komuro I, et al. Attenuated defense response and low basal blood pressure in orexin knockout mice. Am J Physiol Regul Integr Comp Physiol. (2003) 285, R581–93. doi: 10.1152/ajpregu.00671.2002
148. Zhang W, Sakurai T, Fukuda Y, Kuwaki T. Orexin neuron-mediated skeletal muscle vasodilation and shift of baroreflex during defense response in mice. Am J Physiol Regul Integr Comp Physiol. (2006) 290:R1654–63. doi: 10.1152/ajpregu.00704.2005
149. Bonnavion P, Jackson AC, Carter ME, De Lecea L. Antagonistic interplay between hypocretin and leptin in the lateral hypothalamus regulates stress responses. Nat Commun. (2015) 6:6266. doi: 10.1038/ncomms7266
150. Liu J, Garza JC, Bronner J, Kim CS, Zhang W, Lu XY. Acute administration of leptin produces anxiolytic-like effects: a comparison with fluoxetine. Psychopharmacology (2010) 207:535–45. doi: 10.1007/s00213-009-1684-3
151. Tyree SM, Munn RG, Mcnaughton N. Anxiolytic-like effects of leptin on fixed interval responding. Pharmacol Biochem Behav (2016) 148:15–20. doi: 10.1016/j.pbb.2016.05.005
152. Grafe LA, Eacret D, Dobkin J, Bhatnagar S. Reduced orexin system function contributes to resilience to repeated social stress. eNeuro (2018) 5:1–13. doi: 10.1523/ENEURO.0273-17.2018
153. Mahlios J, De La Herran-Arita AK, Mignot E. The autoimmune basis of narcolepsy. Curr Opin Neurobiol. (2013) 23:767–73. doi: 10.1016/j.conb.2013.04.013
154. Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep (1997) 20:1012–20.
155. Kornum BR, Burgdorf KS, Holm A, Ullum H, Jennum P, Knudsen S. Absence of autoreactive CD4(+) T-cells targeting HLA-DQA1(star)01:02/DQB1(star)06:02 restricted hypocretin/orexin epitopes in narcolepsy type 1 when detected by EliSpot. J Neuroimmunol. (2017) 309:7–11. doi: 10.1016/j.jneuroim.2017.05.001
156. De la Herrán-Arita AK, Kornum BR, Mahlios J, Jiang W, Lin L, Hou T, et al. Retraction of the Research Article: “CD4+ T cell autoimmunity to hypocretin/orexin and cross-reactivity to a 2009 H1N1 influenza A epitope in narcolepsy”. Sci Transl Med. (2014) 6:247rt1. doi: 10.1126/scitranslmed.3009995
157. Partinen M, Saarenpaa-Heikkila O, Ilveskoski I, Hublin C, Linna M, Olsen P, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS ONE (2012) 7:e33723. doi: 10.1371/journal.pone.0033723
158. Nohynek H, Jokinen J, Partinen M, Vaarala O, Kirjavainen T, Sundman J, et al. AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS ONE (2012) 7:e33536. doi: 10.1371/journal.pone.0033536
159. Wijnans L, Lecomte C, De Vries C, Weibel D, Sammon C, Hviid A, et al. The incidence of narcolepsy in Europe: before, during, and after the influenza A(H1N1)pdm09 pandemic and vaccination campaigns. Vaccine (2013) 31:1246–54. doi: 10.1016/j.vaccine.2012.12.015
160. Duffy J, Weintraub E, Vellozzi C, Destefano F, Vaccine Safety D. Narcolepsy and influenza A(H1N1) pandemic 2009 vaccination in the United States. Neurology (2014) 83:1823–30. doi: 10.1212/WNL.0000000000000987
161. Tesoriero C, Codita A, Zhang M.-D., Cherninsky A, Karlsson H, Grassi-Zucconi G, et al. H1N1 influenza virus induces narcolepsy-like sleep disruption and targets sleep–wake regulatory neurons in mice. Proc Natl Acad Sci USA. (2016) 113:E368-77. doi: 10.1073/pnas.1521463112
162. Grossberg AJ, Zhu XX, Leinninger GM, Levasseur PR, Braun TP, Myers MG, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci (2011) 31:11376–86. doi: 10.1523/JNEUROSCI.2311-11.2011
163. Weymann KB, Wood LJ, Zhu X, Marks DL. A role for orexin in cytotoxic chemotherapy-induced fatigue. Brain Behav Immun. (2014) 37:84–94. doi: 10.1016/j.bbi.2013.11.003
164. Tanaka S, Toyoda H, Honda Y, Seki Y, Sakurai T, Honda K, et al. Hypocretin/orexin prevents recovery from sickness. Biomed Rep. (2015) 3:648–50. doi: 10.3892/br.2015.491
165. Ogawa Y, Irukayama-Tomobe Y, Murakoshi N, Kiyama M, Ishikawa Y, Hosokawa N, et al. Peripherally administered orexin improves survival of mice with endotoxin shock. Elife (2016) 5:e21055. doi: 10.7554/eLife.21055
166. Kastin AJ, Akerstrom V. Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J Pharmacol Exp Ther. (1999) 289:219–23.
167. Fujiki N, Yoshida Y, Ripley B, Mignot E, Nishino S. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin-ligand-deficient narcoleptic dog. Sleep (2003) 26:953–9. doi: 10.1093/sleep/26.8.953
168. Pavlov VA, Tracey KJ. Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci. (2017) 20:156–66. doi: 10.1038/nn.4477
169. Grossman SP, Dacey D, Halaris AE, Collier T, Routtenberg A. Aphagia and adipsia after preferential destruction of nerve cell bodies in hypothalamus. Science (1978) 202:537–9. doi: 10.1126/science.705344
170. Stricker EM, Swerdloff AF, Zigmond MJ. Intrahypothalamic injections of kainic acid produce feeding and drinking deficits in rats. Brain Res. (1978) 158:470–3. doi: 10.1016/0006-8993(78)90692-3
171. Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron (2012) 74:858–73. doi: 10.1016/j.neuron.2012.03.017
172. Beier KT, Steinberg EE, Deloach KE, Xie S, Miyamichi K, Schwarz L, et al. Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell (2015) 162:622–34. doi: 10.1016/j.cell.2015.07.015
173. Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, et al. Inhibitory input from the lateral hypothalamus to the ventral tegmental area disinhibits dopamine neurons and promotes behavioral activation. Neuron (2016) 90:1286–98. doi: 10.1016/j.neuron.2016.04.035
174. Tyree SM, De Lecea L. Lateral Hypothalamic control of the ventral tegmental area: reward evaluation and the driving of motivated behavior. Front Syst Neurosci. (2017) 11:50. doi: 10.3389/fnsys.2017.00050
175. Thorpe AJ, Kotz CM. Orexin A in the nucleus accumbens stimulates feeding and locomotor activity. Brain Res. (2005) 1050:156–62. doi: 10.1016/j.brainres.2005.05.045
176. Thorpe AJ, Cleary JP, Levine AS, Kotz CM. Centrally administered orexin A increases motivation for sweet pellets in rats. Psychopharmacology (2005) 182:75–83. doi: 10.1007/s00213-005-0040-5
177. Choi DL, Davis JF, Fitzgerald ME, Benoit SC. The role of orexin-A in food motivation, reward-based feeding behavior and food-induced neuronal activation in rats. Neuroscience (2010) 167:11–20. doi: 10.1016/j.neuroscience.2010.02.002
178. Haynes AC, Jackson B, Chapman H, Tadayyon M, Johns A, Porter RA, et al. A selective orexin-1 receptor antagonist reduces food consumption in male and female rats. Regul Pept. (2000) 96:45–51. doi: 10.1016/S0167-0115(00)00199-3
179. Nair SG, Golden SA, Shaham Y. Differential effects of the hypocretin 1 receptor antagonist SB 334867 on high-fat food self-administration and reinstatement of food seeking in rats. Br J Pharmacol. (2008) 154:406–16. doi: 10.1038/bjp.2008.3
180. Borgland SL, Chang SJ, Bowers MS, Thompson JL, Vittoz N, Floresco SB, et al. Orexin A/hypocretin-1 selectively promotes motivation for positive reinforcers. J Neurosci. (2009) 29:11215–25. doi: 10.1523/JNEUROSCI.6096-08.2009
181. Rodgers RJ, Halford JC, Nunes De Souza RL, Canto De Souza AL, Piper DC, Arch JR, et al. SB-334867, a selective orexin-1 receptor antagonist, enhances behavioural satiety and blocks the hyperphagic effect of orexin-A in rats. Eur J Neurosci. (2001) 13:1444–52. doi: 10.1046/j.0953-816x.2001.01518.x
182. Matsuo E, Mochizuki A, Nakayama K, Nakamura S, Yamamoto T, Shioda S, et al. Decreased intake of sucrose solutions in orexin knockout mice. J Mol Neurosci. (2011) 43:217–24. doi: 10.1007/s12031-010-9475-1
183. Takahashi N, Okumura T, Yamada H, Kohgo Y. Stimulation of gastric acid secretion by centrally administered orexin-A in conscious rats. Biochem Biophys Res Commun. (1999) 254:623–7. doi: 10.1006/bbrc.1998.9994
184. Lubkin M, Stricker-Krongrad A. Independent feeding and metabolic actions of orexins in mice. Biochem Biophys Res Commun. (1998) 253:241–5. doi: 10.1006/bbrc.1998.9750
185. Yamamoto Y, Ueta Y, Date Y, Nakazato M, Hara Y, Serino R, et al. Down regulation of the prepro-orexin gene expression in genetically obese mice. Brain Res Mol Brain Res. (1999) 65:14–22. doi: 10.1016/S0169-328X(98)00320-9
186. Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'kelly I, et al. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron (2006) 50:711–22. doi: 10.1016/j.neuron.2006.04.032
187. Cai XJ, Widdowson PS, Harrold J, Wilson S, Buckingham RE, Arch JR, et al. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes (1999) 48:2132–7. doi: 10.2337/diabetes.48.11.2132
188. Griffond B, Risold PY, Jacquemard C, Colard C, Fellmann D. Insulin-induced hypoglycemia increases preprohypocretin (orexin) mRNA in the rat lateral hypothalamic area. Neurosci Lett. (1999) 262:77–80. doi: 10.1016/S0304-3940(98)00976-8
189. Bayer L, Colard C, Nguyen NU, Risold PY, Fellmann D, Griffond B. Alteration of the expression of the hypocretin (orexin) gene by 2-deoxyglucose in the rat lateral hypothalamic area. Neuroreport (2000) 11:531–3. doi: 10.1097/00001756-200002280-00021
190. Sergeyev V, Broberger C, Gorbatyuk O, Hokfelt T. Effect of 2-mercaptoacetate and 2-deoxy-D-glucose administration on the expression of NPY, AGRP, POMC, MCH and hypocretin/orexin in the rat hypothalamus. Neuroreport (2000) 11:117–21. doi: 10.1097/00001756-200001170-00023
191. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
192. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell (1996) 84:491–5. doi: 10.1016/S0092-8674(00)81294-5
193. Chua SC, Chung WK, Wupeng XS, Zhang YY, Liu SM, Tartaglia L, et al. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science (1996) 271:994–6. doi: 10.1126/science.271.5251.994
194. Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science (1996) 274:1704–7. doi: 10.1126/science.274.5293.1704
195. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature (1996) 379:632–5. doi: 10.1038/379632a0
196. Havel PJ. Role of adipose tissue in body-weight regulation: mechanisms regulating leptin production and energy balance. Proc Nutr Soc. (2000) 59:359–71. doi: 10.1017/S0029665100000410
197. Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, et al. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. (2009) 10:89–98. doi: 10.1016/j.cmet.2009.06.011
198. Hakansson M, De Lecea L, Sutcliffe JG, Yanagisawa M, Meister B. Leptin receptor- and STAT3-immunoreactivities in hypocretin/orexin neurones of the lateral hypothalamus. J Neuroendocrinol. (1999) 11:653–63. doi: 10.1046/j.1365-2826.1999.00378.x
199. Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology (2000) 141:4325–8. doi: 10.1210/endo.141.11.7873
200. Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology (2001) 120:337–45. doi: 10.1053/gast.2001.22158
201. Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, et al. A role for ghrelin in the central regulation of feeding. Nature (2001) 409:194–8. doi: 10.1038/35051587
202. Shintani M, Ogawa Y, Ebihara K, Aizawa-Abe M, Miyanaga F, Takaya K, et al. Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes (2001) 50:227–32. doi: 10.2337/diabetes.50.2.227
203. Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature (2000) 407:908. doi: 10.1038/35038090
204. Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes (2001) 50:2540–7. doi: 10.2337/diabetes.50.11.2540
205. Lawrence CB, Snape AC, Baudoin FM, Luckman SM. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology (2002) 143:155–62. doi: 10.1210/endo.143.1.8561
206. Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. (2005) 25:2429–33. doi: 10.1523/JNEUROSCI.4925-04.2005
207. Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D. Adaptive sugar sensors in hypothalamic feeding circuits. Proc Natl Acad Sci USA. (2008) 105:11975–80. doi: 10.1073/pnas.0802687105
208. Toshinai K, Date Y, Murakami N, Shimada M, Mondal MS, Shimbara T, et al. Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology (2003) 144:1506–12. doi: 10.1210/en.2002-220788
209. Dileone RJ, Georgescu D, Nestler EJ. Lateral hypothalamic neuropeptides in reward and drug addiction. Life Sci. (2003) 73:759–68. doi: 10.1016/S0024-3205(03)00408-9
210. Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature (2005) 437:556–9. doi: 10.1038/nature04071
211. Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, et al. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci USA. (2005) 102:19168–73. doi: 10.1073/pnas.0507480102
212. Steiner N, Rossetti C, Sakurai T, Yanagisawa M, De Lecea L, Magistretti PJ, et al. Hypocretin/orexin deficiency decreases cocaine abuse liability. Neuropharmacology (2018) 133:395–403. doi: 10.1016/j.neuropharm.2018.02.010
213. Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron (2006) 49:589–601. doi: 10.1016/j.neuron.2006.01.016
214. Baimel C, Bartlett SE, Chiou LC, Lawrence AJ, Muschamp JW, Patkar O, et al. Orexin/hypocretin role in reward: implications for opioid and other addictions. Br J Pharmacol. (2015) 172:334–48. doi: 10.1111/bph.12639
215. Espana RA, Melchior JR, Roberts DC, Jones SR. Hypocretin 1/orexin A in the ventral tegmental area enhances dopamine responses to cocaine and promotes cocaine self-administration. Psychopharmacology (2011) 214:415–26. doi: 10.1007/s00213-010-2048-8
216. Hollander JA, Lu Q, Cameron MD, Kamenecka TM, Kenny PJ. Insular hypocretin transmission regulates nicotine reward. Proc Natl Acad Sci USA. (2008) 105:19480–5. doi: 10.1073/pnas.0808023105
217. Riday TT, Fish EW, Robinson JE, Jarrett TM, Mcguigan MM, Malanga C. Orexin-1 receptor antagonism does not reduce the rewarding potency of cocaine in Swiss–Webster mice. Brain Res. (2012) 1431:53–61. doi: 10.1016/j.brainres.2011.11.003
218. Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, et al. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proc Natl Acad Sci USA. (2014) 111:E1648-55. doi: 10.1073/pnas.1315542111
219. Vaughan E, Fisher AE. Male sexual behavior induced by intracranial electrical stimulation. Science (1962) 137:758–60. doi: 10.1126/science.137.3532.758-a
220. Hansen S, Kohler C, Goldstein M, Steinbusch HV. Effects of ibotenic acid-induced neuronal degeneration in the medial preoptic area and the lateral hypothalamic area on sexual behavior in the male rat. Brain Res. (1982) 239:213–32. doi: 10.1016/0006-8993(82)90843-5
221. Muschamp JW, Dominguez JM, Sato SM, Shen RY, Hull EM. A role for hypocretin (orexin) in male sexual behavior. J Neurosci (2007) 27:2837–45. doi: 10.1523/JNEUROSCI.4121-06.2007
222. Di Sebastiano AR, Yong-Yow S, Wagner L, Lehman MN, Coolen LM. Orexin mediates initiation of sexual behavior in sexually naive male rats, but is not critical for sexual performance. Horm Behav. (2010) 58:397–404. doi: 10.1016/j.yhbeh.2010.06.004
223. Di Sebastiano AR, Wilson-Perez HE, Lehman MN, Coolen LM. Lesions of orexin neurons block conditioned place preference for sexual behavior in male rats. Horm Behav. (2011) 59:1–8. doi: 10.1016/j.yhbeh.2010.09.006
224. Gulia KK, Mallick HN, Kumar VM. Orexin a (hypocretin-1) application at the medial preoptic area potentiates male sexual behavior in rats. Neuroscience (2003) 116:921–3. doi: 10.1016/S0306-4522(02)00877-1
225. Roberts WW, Carey RJ. Rewarding effect of performance of gnawing aroused by hypothalamic stimulation in the rat. J Comp Physiol Psychol. (1965) 59:317–24. doi: 10.1037/h0022030
226. Caggiula AR, Hoebel BG. “Copulation-Reward Site” in the posterior hypothalamus. Science (1966) 153:1284–5. doi: 10.1126/science.153.3741.1284
227. Mogenson GJ, Stevenson JA. Drinking induced by electrical stimulation of the lateral hypothalamus. Exp Neurol. (1967) 17:119–27. doi: 10.1016/0014-4886(67)90139-2
228. Valenstein ES, Cox VC, Kakolewski JW. Modification of motivated behavior elicited by electrical stimulation of the hypothalamus. Science (1968) 159:1119–21. doi: 10.1126/science.159.3819.1119
229. Wise RA. Hypothalamic motivational systems: fixed or plastic neural circuits? Science (1968) 162:377–9.
Keywords: hypocretin, lateral hypothalamus, arousal, motivation, reward
Citation: Tyree SM, Borniger JC and de Lecea L (2018) Hypocretin as a Hub for Arousal and Motivation. Front. Neurol. 9:413. doi: 10.3389/fneur.2018.00413
Received: 16 March 2018; Accepted: 18 May 2018;
Published: 06 June 2018.
Edited by:
Benjamin Boutrel, Laboratory on the Neurobiology of Addictive and Eating Disorders-CHUV, SwitzerlandReviewed by:
Rodrigo España, College of Medicine, Drexel University, United StatesRhiannan Hope Williams, Helmholtz Zentrum München-Deutsches Forschungszentrum für Gesundheit und Umwelt, Germany
Copyright © 2018 Tyree, Borniger and de Lecea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis de Lecea, bGxlY2VhQHN0YW5mb3JkLmVkdQ==
†These authors have contributed equally to this work and are co-first authors.