
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 19 June 2018
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00398
 Moatasem El-Ayadi1,2,3,4
Moatasem El-Ayadi1,2,3,4 Kristof Egervari5,6
Kristof Egervari5,6 Doron Merkler5,6Thomas A. McKee5,6
Doron Merkler5,6Thomas A. McKee5,6 Fabienne Gumy-Pause1,2
Fabienne Gumy-Pause1,2 Damian Stichel7David Capper8,9Torsten Pietsch10Marc Ansari1,2
Damian Stichel7David Capper8,9Torsten Pietsch10Marc Ansari1,2 André O. von Bueren1,2*
André O. von Bueren1,2*Isocitrate Dehydrogenase-1 (IDH1) is a driver gene in several cancers including brain tumors such as low-grade and high-grade gliomas. Mutations of SMARCB1 were described in atypical teratoid rhabdoid tumors and to date have not been associated with the pathogenesis of medulloblastoma. We report concurrent IDH1 and SMARCB1 mutations in a medulloblastoma patient. We searched the catalog of somatic mutations in cancer (COSMIC) database and other mutation databases and -to our knowledge- this is the first reported case of medulloblastoma harboring both mutations together. Our patient is a 13-year-old male presenting with headache and vomiting at diagnosis. MRI revealed left cerebellar expansive lesion with no evidence of metastasis. A histopathological diagnosis of desmoplastic/nodular medulloblastoma was made after complete resection of the tumor. Immunophenotypic characterization and methylation profiling suggested a medulloblastoma with SHH activation. Next generation sequencing of a panel of 400 genes revealed heterozygous somatic IDH1(p.R132C), SMARCB1(p.R201Q), and CDH11(p.L625T) mutations. The patient was treated according to the HIT-SIOP PNET 4 protocol. He is in complete remission more than 2 years after diagnosis. In conclusion, increasing use of high throughput sequencing will certainly increase the frequency with which rare mutations or mutation combinations are identified. The exact frequency of this mutation combination and whether it has any particular therapeutic implications or prognostic relevance requires further investigation.
Medulloblastoma (MB) is a highly malignant embryonal tumor of the cerebellum and represents the most frequent malignant brain tumor of childhood (1). Molecular subgroups of MB had been described in 2012, including the wingless (WNT) group of tumors with WNT signaling pathway activation and the sonic hedgehog (SHH) group with aberrant activation of SHH signaling pathway as well as groups 3 and 4 tumors (2). This was refined by the recent revised WHO classification for tumors of the CNS 2016 in which four genetically defined MB entities have been defined that differ in cell of origin, genetic alterations and pathway activation as well as in histopathological hallmarks and clinical behavior (1). These are MB-WNT, MB-SHH/TP53 wild type, MB-SHH/TP53 mutant and non-WNT/non-SHH MB (with its variants Grp3 and Grp4, not considered as entities but overlapping variants). Several recurrent mutations were recognized in these 4 MB entities including mutations of CTNNB1, PTCH1, SUFU, SMO, TP53, MLL2, SMARCA4, DDX3X, CTDNEP1, KDM6A, TBR1, and other genes (3, 4).
IDH1 is a gene located at 2q33.3 that encodes cytoplasmic isocitrate dehydrogenase, which is involved in the control of cellular oxidative damage. IDH1 is detected as a mutational cancer driver in several cancer types including diffuse low-grade and high-grade gliomas but until recently not in MB (5–8). SMARCB1 (also known as hSNF5/INI1) is a tumor suppressor gene located at 22q11.23. Inactivating mutations of SMARCB1 were previously described as a hallmark event in atypical teratoid rhabdoid tumors (ATRT) (9) and such mutations were ruled out from being involved in the pathogenesis of MB (10).
Here, we report concurrent IDH1 and SMARCB1 mutations -among other mutations- in a case of a pediatric desmoplatic/nodular MB with SHH activation. We searched for reported IDH1 and SMARCB1 mutations in MB within the catalog of somatic mutations in cancer (COSMIC) and other mutation databases. To our knowledge, this is the first reported case of MB that harbor both mutations together; each of these mutations is very rarely reported in MB.
A 13-year-old boy presented with a history of headache, nausea and vomiting with an acute onset 2 weeks earlier. Magnetic Resonance Imaging (MRI) of the brain and spinal cord revealed left cerebellar expansive lesion with no evidence of metastasis. Cerebrospinal fluid (CSF) examination revealed no evidence of dissemination. He underwent complete surgical resection as confirmed by postoperative imaging. Histopathological analysis including reticulin staining revealed a desmoplastic/nodular MB (confirmed by a central review by T.P.) as shown in Figure 1. Diffuse severe cytological anaplasia was not present. Complementary immunophenotypic characterization as described (11, 12) suggested a MB with SHH activation, TP53 wild-type (Figure 2). Of note, nuclear INI-1 staining was preserved (Figure 1) while P53 immunostaining showed nuclear positivity only in a small proportion of the tumor cells (data not shown). There was no evidence of MYCN or MYCC amplification by fluorescence in-situ hybridization (FISH). Next generation sequencing (NGS) over a panel of 50 genes (Ion AmpliSeq™ Cancer Hotspot Panel v2)1 revealed IDH1 R132C mutation in 46% of cells. NGS was repeated over a panel of 400 genes (Ion AmpliSeq™ Comprehensive Cancer Panel)1 and it revealed IDH1 R132C mutation in 24% of cells as well as SMARCB1-R201G in 30% of cells and CDH11-L625T in 26% of cells (Table A1). The panel was tested on both tumor and normal tissue to confirm the somatic nature of the mutations. Of note, mutations in SMO, PTCH1, SUFU and TP53 were not detected. Infinium Methylation EPIC BeadChip (850k) array revealed highest resemblance to the methylation class MB, subclass SHH A (children and adult). However, the calibrated score was 0.44 so that a clear subgroup assignment could not be done (13). Calculation of a low density copy number profile from the array data indicated a flat genome without relevant chromosomal aberration (Figure 3). Deletions of chromosome 9q (PTCH1) were absent. Assessment of overall CpG methylation and CpG island methylation levels of the tumor showed relative CpG hypermethylation compared to other reference classes of MB (13) (Figure 4). There was no family history indicating a tumor predisposition syndrome.

Figure 1. Histology and immunohistochemistry. Hematoxylin-eosin stained image from a representative area of the tumor shows a small to medium sized primitive cellular population. A desmoplastic micronodular architecture is revealed by reticulin staining. The tumor cells are positive with synaptophysin and weakly with GFAP. INI-1 expression is preserved. The proliferative index (MIB-1) reaches 70%.
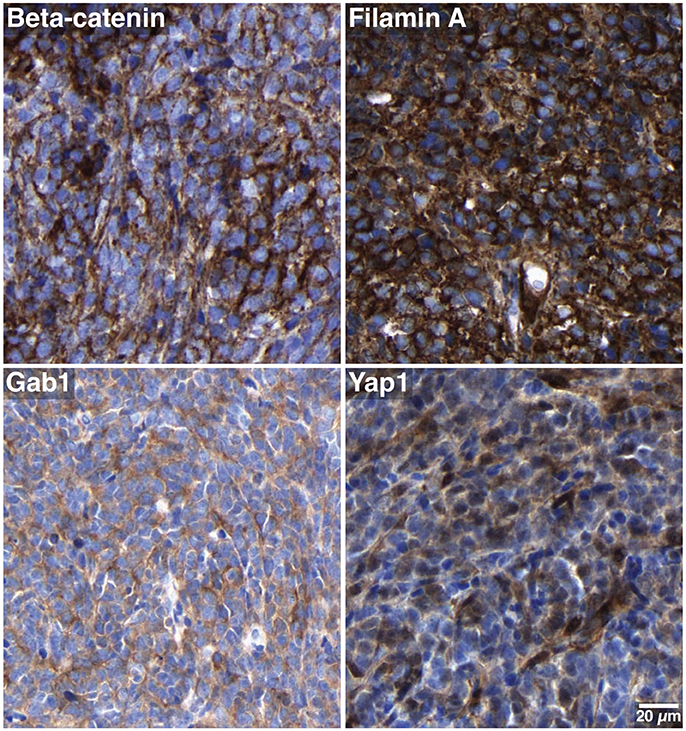
Figure 2. Subgroup-specific immunohistochemical markers. The tumor cells show only cytoplasmic but no nuclear beta-catenin positivity, and are stained with antibodies against filamin A, Gab1 and Yap1. These characteristics are consistent with SHH MB.
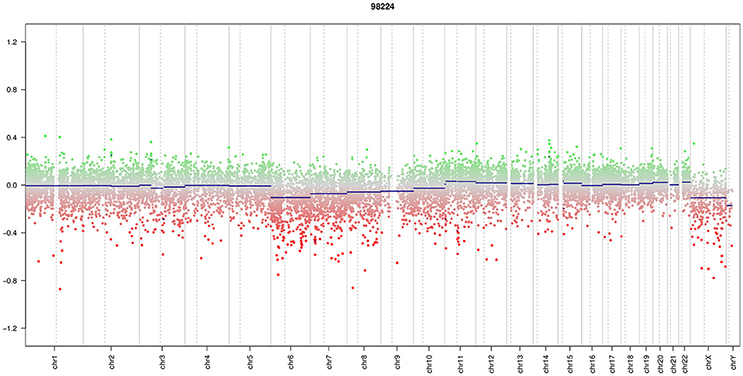
Figure 3. Copy number profile. Array data showed a low density copy number profile which indicates an almost flat genome without relevant chromosomal aberration.
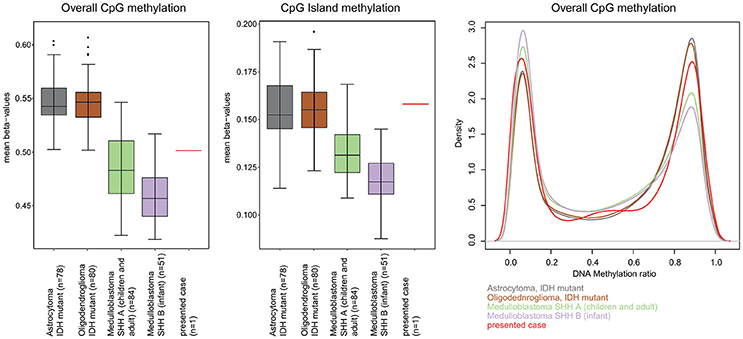
Figure 4. Assessment of overall CpG methylation and CpG island methylation levels. (Left) The distribution of mean beta-values for all CpG-probes of the EPIC methylation array is displayed as box-plots for IDH mutant astrocytoma, IDH mutant oligodendroglioma, medulloblastoma SHH A (children and adult), medulloblastoma SHH B (infant) and the presented case. IDH mutant astrocytoma and oligodendroglioma are given for comparison because of the known CpG Island Methylator Phenotype (CIMP) associated with IDH mutations in these tumors. The overall CpG methylation of our presented cases was not higher than that in other medulloblastoma groups and lower than that in the CIMP tumors. (Middle) When just the CpG island associated CpG sites were analyzed, the mean beta value of our case was clearly higher than that of other medulloblastoma of the same age group and was well in the range of the other CIMP tumors indicating some degree of CpG island hypermethylation. (Right) Density plot analysis of the distribution of beta-values (“methylation ratio”) of all CpG probes also indicated a moderate relative hypermethylation.
The patient started treatment according to the HIT-SIOP PNET 4 protocol on the standard radiotherapy (RT) arm (14) where he received cranio-spinal RT (23.4 Gy) with boost to the posterior fossa up to 55.8 Gy in 30 fractions over 42 days. Adjuvant chemotherapy started 6 weeks after the end of RT and it includes eight cycles of maintenance chemotherapy (14). Treatment was well tolerated with no major life-threatening adverse events or dose limiting toxicities. To date, the patient is in complete remission more than 2 years since diagnosis.
Several mutation databases were searched, namely; the catalog of somatic mutations in cancer (COSMIC) database (15), the cBioPortal for cancer genomics database (16), the integrative onco genomics (IntOGen) database for mutational cancer drivers (17), the cancer genetics web database (18) as well as the medulloblastoma advanced genomics international consortium (MAGIC) project (19). We checked for reported IDH1 and SMARCB1 mutations in MB samples.
The COSMIC database reports on IDH1 mutation in only 5 tumor samples from 4 patients with MB; three samples from two adult cases (20, 21) and two samples from two pediatric patients [(8), ICGC PedBrain Tumor Project; unpublished data]. Searching other databases didn't reveal any additional reported cases.
As for the SMARCB1 mutation, the COSMIC database reports only on 9 mutated samples of MB from four different studies (10, 22–24). Similarly, we couldn't find any additional reported cases in other mutation databases. The specific mutation (SMARCB1-R201G) was only reported in a single case of ovarian cancer, and wasn't previously reported in any central nervous system (CNS) tumor.
There was no reported case of a MB harboring both mutations of IDH1 and SMARCB1 in any of the above mentioned databases.
Our 13-year-old patient had a desmoplastic/nodular MB with SHH activation and without TP53 alteration. Around 25% of MB show SHH activation, most of which show desmoplastic/nodular histology, though classic or large-cell/anaplastic (LCA) histologies are seen too (25). SHH-MB frequently arise from external granule cell progenitors in a cerebellar hemisphere (26, 27). Childhood SHH-MB frequently harbor TP53 mutations (48%), PTCH1 mutations (36%) or SMO mutations. TP53 mutations are commonly germline (80%) as part of Li-Fraumeni syndrome, and they can carry MYCN (42%) and/or GLI2 amplifications (28). None of these mutations were found in our case, and MYCN amplification was ruled out by FISH. In contrast to SHH -TP53 mutant MB which represent the most aggressive MB entity, SHH-TP53 wild-type MB usually show a more favorable clinical course. This might -in part- explain the apparently good outcome of our patient who is still alive and in complete remission for more than 2 years since diagnosis. It remains less evident which genetic event cause the SHH activation in this case.
Several studies investigated IDH1 mutations among different types of cancers (6, 29–32). Hayden et al. analyzed the combined results of five different studies with more than 2,650 tumor samples of different cancer types including 1,603 samples of brain tumors (33). They found 37% of CNS tumors with IDH1 mutations—all involving codon 132—with highest frequency of mutations found in diffuse astrocytic or oligodendroglial tumors (81%) (33). Notably, no IDH1 mutation was found among 113 cases of MB (33). Similarly, the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project examined samples of 125 pediatric MB patients using whole genome sequencing (WGS) and whole-exome sequencing (WES) (24). None of the examined cases showed IDH1 mutations. However, another large sequencing study on 92 MB samples reported a single case with IDH1 R132C mutation in a 13 years old boy with WNT-MB (8). An adult study about CIC and FUBP1 mutations in primary brain tumors reported another case of MB with an IDH1 R132H mutation (21). In 2015, Snuderl et al. reported an adult SHH-MB case with IDH1 R132S mutation using deep sequencing. They also identified another case with an IDH1 R132H mutation using immunohistochemistry. Interestingly, a recent analysis of 491 sequenced MB samples identified six IDH1-R132C mutations with five of them in SHH activated MB (4).
Somatic mutation of the IDH1 gene was found to establish CpG island methylator phenotype (CIMP) in IDH-mutant astrocytomas and oligodendrogliomas, as well as secondary glioblastomas arising from these tumors (34). CIMP represents a distinct molecular phenotype in glioblastoma (35), that is associated with the proneural subgroup, where it shows extensive, coordinated hypermethylation at specific loci (35, 36). Notably, in their analysis, Northcott et al. found that IDH1-mutant SHH-MBs were determined to be CIMP+, highlighting the fact that these mutations play an epigenetic role comparable to those reported in other IDH1/2-mutant cancers (4). In line with this, we also observed a CpG island hypermethylation, further indicating a functional role of IDH mutation in reforming the DNA methylation pattern of MB. As a possible consequence of this reformed methylome, when analyzed with the DNA methylation classifier (13), the case only receives a relatively low classifier score (0.44), far below what is typically observed for SHH medulloblastoma (Figure 4).
SMARCB1/INI1 encodes a chromatin remodeling protein (SNF5) that has been demonstrated to interact with various key proteins in several signaling pathways (37). SNF5 is identified as one of the key regulators of SHH signaling pathway through interacting with the glioma-associated oncogene family zinc finger-1 (GLI1), a crucial effector of SHH signaling pathway (38). Biallelic inactivation of SMARCB1/INI1 results in aberrant expression or loss of SNF5 which is primarily seen in malignant rhabdoid tumors and ATRT (9) and recently reported in other tumor types such as epithelioid sarcoma, renal medullary carcinoma and schwannomatosis (37). Such SMARCB1/INI1-deficient tumors were found to show gene expression patterns associated with both activated SHH signaling pathway and GLI1 overexpression signatures, similar to SHH-activated MB (38).
Of notice, our case harbored a heterozygous missense somatic mutation (SMARCB1-R201G) that wasn't previously reported in any CNS tumor. This mutation is not predicted as inactivating mutation. Germline SMARCB1 mutations are commonly associated with ATRT and renal rhabdoid tumors, in contrast to extra-renal rhabdoid tumors which typically harbor somatic biallelic deletions of SMARCB1 (39). Interestingly, missense mutations of SMARCB1—whether germline or somatic—are virtually absent in all malignant rhabdoid tumors (39).
Sévenet et al. examined 299 different tumors for the prevalence of SMARCB1 mutations (22). Among other brain tumors, they reported five out of 36 cases of MB with mutated SMARCB1 gene (22). A contradictory study by Biegel et al. suggested that tumors harboring this mutation (especially in young age) are mostly ATRT misdiagnosed as MB (23). Indeed, two out of four MB/PNET samples with SMARCB1 mutations were reclassified as ATRT after pathology revision, while other two samples (one MB and one PNET) had insufficient material for revision (23). Notably, three of the five cases reported by Sévenet et al. were less than 3 years old, and no pathology revision was performed in the study (22). In 2002, a dedicated study to settle this contradiction confirmed the absence of SMARCB1 mutation in 90 MB samples (10).
Our case is diagnosed as a desmoplastic/nodular MB with characteristic SHH activation by virtue of detection of SHH targets (GAB1, p75NGFR) by immunohistochemistry. NGS identified a somatic missense SMARCB1 mutation, yet there was no loss of SNF5 nuclear expression, so diagnosis of ATRT was ruled out.
Our case shows that IDH1 and missense SMARCB1 mutations can be found concurrently in pediatric MB. While IDH1 mutation represents the molecular basis of CIMP in gliomas, such role can't be confirmed in our MB case. Similarly, SMARCB1 deletions / mutations result in loss of SNF5 expression and malignant rhabdoid tumor phenotype, yet in our case; nuclear expression of SNF5/INI1 was preserved. Whole exome and whole genome sequencing are becoming frequently used as clinical diagnostic tests at treatment institutions, and presumably they augment the accuracy and certainty of many diagnoses. However, the identification of rare mutations as an incidental finding will also increase, which might in-turn add to the diagnostic challenges in certain cases. The diagnostic significance as well as the clinical relevance of these mutations in the setup of histopathologically and genetically defined diagnosis need further investigation. Furthermore, such mutations need to be interpreted in the context of the different genetically defined entities of MB and larger studies are needed to explore what role these rare mutations might play in MB.
A written informed consent was obtained from the patient and his parents for the publication of this case report.
AvB and ME-A design of the study. AvB, FG-P, and MA patient management with regards of diagnosis, treatment, and follow-up. AvB and ME-A collection and interpretation of patient data. ME-A and AvB wrote the first draft of the manuscript. TP, KE, DC, DS, DM, TM, ME-A, and AvB contributed to the histopathological diagnosis, methylation profiling, gene sequencing, and their interpretation of the presented case. Manuscript editing all authors. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to this child and his family for sharing tumor material and information, and thus contributing to the research efforts to understand and find cures for pediatric medulloblastoma. We would like to thank the whole healthcare team who provided excellent care of our patient. We would like to thank the CANSEARCH Foundation for continuous support. ME received a postdoctoral fellowship granted by the Egyptian ministry of Higher Education and Scientific Research.
1. ^https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-workflow/ion-torrent-next-generation-sequencing-select-targets/ampliseq-target-selection/ready-to-use-panels.html
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, et al. (eds). WHO Classification of Tumours of the Central Nervous System. 4th Edn. Lyon (2016).
2. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho Y-J, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. (2012) 123:465–72. doi: 10.1007/s00401-011-0922-z
3. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science (2011) 331:435–9. doi: 10.1126/science.1198056
4. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature (2017) 547:311–7. doi: 10.1038/nature22973
5. Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DTW, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. (2009) 11:341–7. doi: 10.1215/15228517-2009-025
6. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. (2008) 116:597–602. doi: 10.1007/s00401-008-0455-2
7. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. (2009) 118:469–74. doi: 10.1007/s00401-009-0561-9
8. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature (2012) 488:106–10. doi: 10.1038/nature11329
9. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. (1999) 59:74–9.
10. Kraus JA, Oster C, Sörensen N, Berthold F, Schlegel U, Tonn JC, et al. Human medulloblastomas lack point mutations and homozygous deletions of the hSNF5/INI1 tumour suppressor gene. Neuropathol Appl Neurobiol. (2002) 28:136–41. doi: 10.1046/j.1365-2990.2002.00388.x
11. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. (2011) 121:381–96. doi: 10.1007/s00401-011-0800-8
12. Pietsch T, Haberler C. Update on the integrated histopathological and genetic classification of medulloblastoma – a practical diagnostic guideline. Clin Neuropathol. (2016) 35:344–52. doi: 10.5414/NP300999
13. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature (2018) 555:469–74. doi: 10.1038/nature26000
14. Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. (2012) 30:3187–93. doi: 10.1200/JCO.2011.39.8719
15. COSMIC. Catalogue of Somatic Mutations in Cancer. Available online at: http://cancer.sanger.ac.uk/cosmic. (cited January 1, 2018).
16. cBioPortalfor Cancer Genomics. Available online at: http://www.cbioportal.org/. (cited January 1, 2018).
17. IntOGen - mutational cancer drivers database. Available online at: https://www.intogen.org/search. (cited January 1, 2018).
18. Cancer Genetics Web. Available online at: http://www.cancerindex.org/geneweb/Medulloblastoma.htm. (cited January 1, 2018).
19. Canada's Michael Smith Genome Sciences Centre. Available online at: http://www.bcgsc.ca/project. (cited January 1, 2018).
20. Snuderl M, Triscott J, Northcott PA, Shih HA, Kong E, Robinson H, et al. Deep sequencing identifies IDH1 R132S mutation in adult medulloblastoma. J Clin Oncol. (2015) 33:e27–31. doi: 10.1200/JCO.2013.49.4864
21. Sahm F, Koelsche C, Meyer J, Pusch S, Lindenberg K, Mueller W, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. (2012) 123:853–60. doi: 10.1007/s00401-012-0993-5
22. Sevenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D, et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet. (1999) 8:2359–68. doi: 10.1093/hmg/8.13.2359
23. Biegel JA, Fogelgren B, Zhou JY, James CD, Janss AJ, Allen JC, et al. Mutations of the INI1 rhabdoid tumor suppressor gene in medulloblastomas and primitive neuroectodermal tumors of the central nervous system. Clin Cancer Res. (2000) 6:2759–63. Available online at: http://clincancerres.aacrjournals.org/content/6/7/2759.long
24. Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Cho Y, et al. ICGC PedBrain: dissecting the genomic complexity underlying medulloblastoma. Nature (2012) 488:100–5. doi: 10.1038/nature11284
25. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. (2012) 123:473–84. doi: 10.1007/s00401-012-0958-8
26. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature (2010) 468:1095–9. doi: 10.1038/nature09587
27. Bühren J, Christoph AH, Buslei R, Albrecht S, Wiestler OD, Pietsch T. Expression of the neurotrophin receptor p75NTR in medulloblastomas is correlated with distinct histological and clinical features: evidence for a medulloblastoma subtype derived from the external granule cell layer. J Neuropathol Exp Neurol. (2000) 59:229–40. doi: 10.1093/jnen/59.3.229
28. Kool M, Jones DTW, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell (2014) 25:393–405. doi: 10.1016/j.ccr.2014.02.004
29. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. (2009) 174:1149–53. doi: 10.2353/ajpath.2009.080958
30. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi: 10.1126/science.1164382
31. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. IDH1 mutations at residue p.R132 (IDH1 R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. (2009) 30:7–11. doi: 10.1002/humu.20937
32. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 Mutations in Gliomas. N Engl J Med. (2009) 360:765–73. doi: 10.1056/NEJMoa0808710
33. Hayden JT, Frühwald MC, Hasselblatt M, Ellison DW, Bailey S, Clifford SC. Frequent IDH1 mutations in supratentorial primitive neuroectodermal tumors (sPNET) of adults but not children. Cell Cycle (2009) 8:1806–7. doi: 10.4161/cc.8.11.8594
34. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi: 10.1038/nature10866
35. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell (2010) 17:510–22. doi: 10.1016/j.ccr.2010.03.017
36. Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. (2007) 39:237–42. doi: 10.1038/ng1972
37. Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. (2017) 108:547–52. doi: 10.1111/cas.13173
38. Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. (2010) 16:1429–33. doi: 10.1038/nm.2251
39. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. (2014) 166C:350–66. doi: 10.1002/ajmg.c.31410
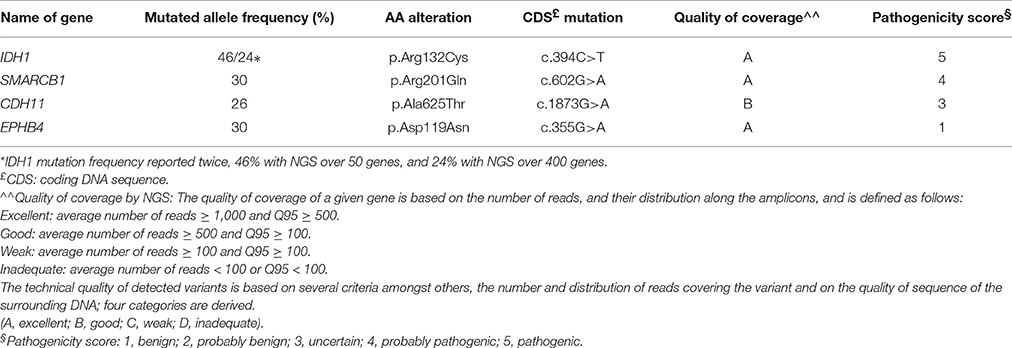
Table A1. Sequencing results.
Keywords: medulloblastoma, IDH-1, SMARCB1, SHH activation, pediatric, mutation
Citation: El-Ayadi M, Egervari K, Merkler D, McKee TA, Gumy-Pause F, Stichel D, Capper D, Pietsch T, Ansari M and von Bueren AO (2018) Concurrent IDH1 and SMARCB1 Mutations in Pediatric Medulloblastoma: A Case Report. Front. Neurol. 9:398. doi: 10.3389/fneur.2018.00398
Received: 05 April 2018; Accepted: 15 May 2018;
Published: 19 June 2018.
Edited by:
Shawn Hervey-Jumper, University of California, San Francisco, United StatesReviewed by:
Todd Charles Hollon, University of Michigan Health System, United StatesCopyright © 2018 El-Ayadi, Egervari, Merkler, McKee, Gumy-Pause, Stichel, Capper, Pietsch, Ansari and von Bueren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André O. von Bueren, YW5kcmUudm9uYnVyZW5AaGN1Z2UuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.